

Abstract
The N-terminal transcription activation domain of p53 is intrinsically unstructured. We show in vitro and in vivo that this domain initiates p53 degradation by the 20 S proteasome in a ubiquitin-independent fashion. The decay of metabolically labeled p53 follows biphasic kinetics with an immediate fast phase that is ubiquitin-independent and a second slower phase that is ubiquitin-dependent. The 20 S proteasome executes the first phase by default, whereas the second phase requires the 26 S proteasome. p53 N-terminal binding proteins, such as Hdmx, can selectively block the first phase of degradation. Remarkably, γ-irradiation inhibits both p53 decay phases, whereas UV selectively negates the second phase, giving rise to discrete levels of p53 accumulation. Our data of a single protein experiencing double mode degradation mechanisms each with unique kinetics provide the mechanistic basis for programmable protein homeostasis (proteostasis).
The process of ubiquitin-dependent (UD)2 degradation is a key mechanism in regulating the level of mature and functional proteins that are often found in large multiprotein complexes. It has become apparent that certain proteins are also subjected to proteasomal degradation in a ubiquitin-independent (UI) manner, which at least in vitro is executed by the 20 S proteasome (1). The 20 S proteasome is the catalytic chamber that lacks the two regulatory caps (19 S) that are present in the 26 S proteasome. Without the regulatory domains, proteins cannot be unfolded in an ATP-dependent manner as is the case with the 26 S proteasome. In vitro the 20 S proteasomes degrade proteins that are intrinsically unstructured, whereas structured proteins are not very susceptible to this degradation (2, 3), suggesting that 20 S proteasomal substrates are naturally intrinsically unstructured proteins (IUPs). There are many unstructured proteins (disordered) in the genome and even more with unstructured domains. Studies based on various in silico predictors for protein disorder estimate that at least 25% of the sequences in SwissProt contain long disordered regions and that in eukaryotes, this value may be as high as 63% (4). Different prediction methods also suggested that unstructured proteins are more susceptible to degradation (5). An important question is whether the unstructured proteins are subjected to 20 S proteasomal degradation in the cells as well. Supporting this possibility is that at least a fraction of certain proteins is subjected to UI degradation, a process that might be executed by the 20 S proteasome. In a recent study, it was shown that as much as 20% of cellular proteins can undergo 20 S proteasomal degradation (6). There are also a growing number of proteins that have been shown to undergo 20 S proteasomal degradation in vitro and in vivo and are subjected to UI degradation (7–10). Additionally, it appears that proteins that have been reported to undergo UI degradation are unstructured or have one or more long unstructured segments. Thus, it is very likely that IUPs are susceptible to degradation by both UD and UI mechanisms. In the emerging field of proteostasis (11, 12), the UI degradation pathway might be a key regulator IUP homeostasis.
IUPs lacking defined tertiary structure fulfill multiple roles by associating with many partners, as reviewed by Dyson and Wright (13). Many key regulators in the cells either are completely unstructured or have large unstructured domains. IUPs may escape 20 S proteasomal degradation when associated with a second protein (14–16). IκBα, for example, has unstructured domains at the N and C termini (17). Interestingly, the basal turnover of IκBα is regulated by UI degradation (18). IκBα can be degraded by the 20 S proteasome in vitro, and the binding of p65 can prevent IκBα degradation by the 20 S proteasome (19, 20). Upon binding to NF-kB, IκBα escapes degradation by default and is stabilized. Under this conditions, IκBα can be destabilized by IκB kinase-mediated phosphorylation followed by ubiquitination and degradation (21, 22).
p53 is a tumor suppressor whose cellular level is maintained mostly by the rate of its degradation (23). There are two distinct proteasomal degradation pathways of p53, the UD and UI pathways. In the UI pathway, p53 undergoes degradation by the 20 S proteasome (7, 24, 25). Interestingly, UI degradation of p53 can be blocked by NQO1, an NADH-regulated enzyme (24), which is in association with the 20 S proteasomes (7). In the UD pathway, p53 is ubiquitinated by an E3 ligase such as Mdm2 (26, 27), Pirh2 (28), or COP1 (29), and following this ubiquitination, p53 is recognized and degraded by the 26 S proteasome. Mdm2, the most studied p53 E3 ligase, binds p53 at the N terminus, resulting in the ubiquitination of the C terminus of p53. Hdmx is a paralog of Mdm2, and although it shares high sequence and structural homology with Mdm2, it does not have E3 ligase activity and thus cannot target p53 for degradation. Hdmx has been shown to stabilize p53 on the protein level (30–33). However, despite the ability of Hdmx to stabilize p53, Hdmx still acts as a repressor of p53 because, like Mdm2, the binding of Hdmx to the N terminus of p53 prevents its transcription activation (34–37). Thus, Hdmx in some cases has a conflicting activity: increasing p53 level on one hand and inhibiting p53 activity on the other hand.
p53 is highly unstructured at the N and C termini (38). Our goal was to determine whether one of these unstructured domains is responsible for p53 susceptibility to UI 20 S proteasomal degradation. The N terminus of p53 contains the transcription activation domains and the binding site for Mdm2/Hdmx (residues 1–42), the second transcription activation domain, and the proline-rich domain (residues 64–92), whereas the C terminus contains the oligomerization domain (residues 307–355) and three nuclear localization sequence motifs (residues 356–393). The unstructured N terminus transcription activation domain of p53 generates a large number of protein-protein interactions and is subjected to multiple modifications (39–41). We report here that this domain is responsible for the susceptibility of p53 to 20 S proteasomal degradation in vitro and in vivo. Thus, cellular p53 level is regulated by a double mode degradation mechanism, namely 26 S proteasomal, E3 ligase-mediated UD degradation and 20 S proteasomal degradation by default. We demonstrate that p53 undergoes biphasic decay kinetics and further correlate the biphasic kinetics curve with the UI (20 S proteasome) and UD degradation processes. Remarkably, utilizing Hdmx as a model protein, we show that N terminus-binding proteins rescue p53 from the first UI degradation phase, exemplifying the capacity of the cell to differentially regulate the two pathways of degradation and control p53 homeostasis (proteostasis). Furthermore, the fact that p53 is subjected to two distinct degradation pathways, each with specific kinetics, permits programmable p53 discrete steady state levels tailored to cope with different types of stress.
EXPERIMENTAL PROCEDURES
Plasmids and Transfections
The plasmids used are as follows: pRc/CMV p53 encoding wild-type human p53, pcDNA3.1 WT p53, 1–363, 1–312, 40–393(Δ40), 97-393(Δ97), all with an HA tag at the C terminus pcDNA3.1 Hdmx-HA and Mdm2. Lentiviral vectors with shRNAmir against 26 S proteasome subunit Rpn2 and control shRNAmir were purchased from Open Biosystems. Transducing lentivirus particles were produced according to the manufacturer's protocol.
Nondenaturing PAGE
Cells were collected and homogenized in buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm EDTA, 1 mm dithiothreitol, and 250 mm sucrose. The extract was then subjected to centrifugation: first 1000 × g for 5 min and then 100,000 × g for 30 min. The supernatant was taken and subjected to ultracentrifugation for 16 h at 100,000 × g. The pellet was resuspended and loaded on a nondenaturing polyacrylamide gel using the protocol previously described (42).
Immunoblot Analysis
The protein mix was mixed with Laemmli sample buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol and 0.125 m Tris-HCl), heated at 95 °C for 5 min, and loaded on a 10% polyacrylamide-SDS gel. Following electrophoresis, proteins were transferred to cellulose nitrate 0.45-mm membranes (Schleicher & Schuell). The antibodies used were: monoclonal DO-1, 1801, 421 anti-HA (Sigma), and anti-actin (Santa Cruz Biotechnology). Secondary antibodies were horseradish peroxidase-linked Goat anti-mouse (Jackson ImmunoResearch). Signals were detected using the Ez-ECL kit (Biological Industries).
Cells and Transfections
The cell lines used were as follows: HCT116, p53 null HCT116 cells, A31N-ts20 BALB/c mouse cell line that harbors a temperature-sensitive E1 ubiquitin-activating enzyme (43), MEFs p53−/− Mdm2−/− (kindly provided by Prof. Oren from the Weizmann Institute of Science), and NIH3T3 cells. The cells were grown as described previously (24). Transfection was performed when cells reached 80–90% confluence. p53 null HCT116 were transfected with JetPEI (PolyPlus Transfection). A31N-ts20 and MEFs p53−/− Mdm2−/− cells were transfected with Lipofectamine 2000 (Invitrogen).
20 S Proteasomal Degradation Assay
20 S proteasomes were purified from mice livers as described previously (7). Purified proteins were incubated with the 20 S proteasomes in degradation buffer (50 mm Tris, pH 7.5, 150 mm NaCl) at 37 °C for up to 60 min. The degradation reaction was stopped with the addition of Laemmli sample buffer and heating at 95 °C for 5 min.
Purified p53 Proteins
p53 lacking amino acids 363–393 (HA-p53 C30), his-p53C-terminus domain of p53 (amino acids 311–393) and wild-type p53 were previously described (44, 45). The p53ΔN22 and p53ΔN97 proteins used were constructed and expressed by similar means (46). Infection and purification of p53 proteins from insect cells was performed as before (45). In vitro translation was performed using the TnT® quick coupled transcription/translation system (Promega, WI) and plasmids mentioned above.
Protein Half-life Determination
24 h after seeding or transfection, cells were starved in methionine-free Dulbecco's modified Eagle's medium with dialyzed serum (Met−) (Biological industries) for 30 min. Cells were then collected, pelleted, and resuspended in Met− and then labeled with prewarmed [35S]Met (10 mCi/ml; Amersham Biosciences), reaching a final concentration of 0.2 mCi/ml for different times as mentioned above (1–60 min) (in the NIH 3T3 experiments, cells were not starved for methionine before labeling with [35S]Met). 50 μm MG132 (Calbiochem) was added 20 min before labeling where indicated. The cells were then pelleted, washed with 1× phosphate-buffered saline, and resuspended in prewarmed medium containing unlabeled Met (2%) Met+. The cells were then aliquoted into 0.5 ml of prewarmed Met+ and incubated at 37 °C for the indicated times. Cells were pelleted and lysed in radioimmune precipitation buffer (7), and samples were subjected to immunoprecipitation with anti-HA, anti-p53 (1801), or anti-p53 (421) antibodies. Immunoprecipitates were washed four times in radioimmune precipitation buffer and run on SDS-PAGE, and radioactivity present in individual bands was determined using the Fuji Bas2500 phosphorimaging device. Cycloheximide experiments were done as described previously (47). The data analysis and the mathematical fits were done utilizing the cftool in MATLAB program.
Protein Accumulation Assay
NIH 3T3 cells were UV- (50 J/m2) or γ-irradiated (6 grays). After 30 min, cells were collected, pelleted, and resuspended in M− containing [35S]Met (10 mCi/ml; Amersham Biosciences) and incubated at 37 °C for the indicated times. Cells were then collected and treated as described above.
RESULTS
The N Terminus of p53 Is Required for p53 Degradation by the 20 S Proteasome in Vitro
The 20 S proteasome preferentially degrades IUPs (2, 3). p53 is intrinsically unstructured at both the N terminus and the C terminus (38). This led us to explore whether these regions of p53 are responsible for its susceptibility to 20 S proteasomal degradation. To address this question, we utilized in vitro translated or baculovirus-expressed and -purified WT and mutant p53 proteins that lacked sequences at either the N terminus or the C terminus of the protein and used 20 S proteasomes purified from mouse livers as described previously (7).
p53 was efficiently degraded by the 20 S proteasome in vitro, exhibiting almost complete degradation within 30 min (Fig. 1A). The ΔC30 p53 truncation mutant, missing the last 30 amino acids, was degraded to the same extent as the WT. The C terminus region in isolation CT-(311–393) was completely degraded by the 20 S proteasome, confirming its unstructured nature (Fig. 1B). In vitro synthesized [35S]methionine-labeled C terminus deletions of p53 were also susceptible to 20 S proteasomal degradation just as the wild-type protein (Fig. 1G), suggesting that the C terminus is not responsible for the susceptibility of p53 to 20 S proteasomal degradation.
FIGURE 1.

The N terminus of p53 is crucial for susceptibility to 20 S proteasomal degradation. Various truncated forms of p53 that were either purified from baculovirus-infected cells as described (45) (A–D) or in vitro translated and [35S]methionine-labeled (E–H) were incubated with purified 20 S proteasomes for different time points (A–C and E) or for 1 h (D and F–H) at 37 °C. The degradation reaction was stopped with the addition of Laemmli sample buffer, and the mixture was heated at 95 °C for 5 min and electrophoresed on SDS-PAGE. Following electrophoresis, proteins were transferred to cellulose nitrate membranes, and proteins were detected by immunoblot (IB) analysis with the indicated monoclonal anti-p53 antibodies (A–D) or by autoradiography (E–H). H, graphic representation of at least three 20 S proteasome degradation experiments with various p53 truncation forms. CT, C terminus.
In contrast, the ΔN97p53-(97–393) was resistant to 20 S proteasomal degradation (Fig. 1C). To further narrow down the region, we utilized N-terminally deleted ΔN22p53-(22–393). In contrast to ΔN97, the WT and ΔN22p53 were completely degraded (Fig. 1D). Similar results were obtained with in vitro synthesized [35S]methionine-labeled p53 truncations (Fig. 1, E and F). Both ΔN40p53-(40–393) and ΔNPRp53-(Δ60–102) were degraded by the 20 S proteasome (Fig. 1F), whereas the ΔN97p53 was resistant to this degradation (Fig. 1, E and F). The degradation experiments were performed at least three times, and the results indicated that only the ΔN97 is resistant to degradation by the 20 S proteasome (Fig. 1H), suggesting that an unstructured domain at the N terminus of p53 is crucial for its susceptibility to 20 S proteasomal degradation.
p53 Is Degraded by the 20 S Proteasome via Its N Terminus
The role of the N terminus in the susceptibility of p53 to 20 S proteasomal degradation may suggest that p53 degradation is initiated at the N terminus. To test this possibility, we examined the p53 partial degradation products by the 20 S proteasome using antibodies specific for different epitopes of p53 (Fig. 2A). When probing a partial p53 digest using an antibody recognizing the N terminus (DO-1, recognizing amino acids 21–25), no new fragments were detected after the degradation. Only one fragment was detected by the 1801 antibody (amino acids 46–55), whereas several fragments were detected by the C-terminal 421 antibody (amino acids 371–380) (Fig. 2B). This pattern of fragments is best explained by the assumption that the degradation of p53 by the 20 S proteasome is initiated from the N terminus.
FIGURE 2.

p53 degradation by the 20 S proteasome is initiated only at the N terminus (N). A, schematic representation of the p53 protein and the recognition map of the various antibodies used in this work. C, C terminus. B, baculovirus-purified p53 was incubated with the 20 S proteasome 30 min, the reactions were analyzed by SDS-PAGE, and the detection of degradation products was done by immunoblotting (IB) with antibodies specific for different epitopes of p53. C, purified p53 was incubated with 20 S proteasomes for 1 h in the presence or absence of either DO-1 or 421 antibodies. The level of degradation was monitored by immunoblotting, and the amount of antibodies added was monitored by the levels of heavy chain (HC). D, in vitro translated [35S]methionine-labeled p53 was incubated for 1 h with 20 S proteasomes in the presence or absence of increasing amounts of either in vitro translated Hdmx or in vitro translated Mdm2. Protein levels were detected by autoradiography.
The Binding of the N Terminus of p53 Can Protect It from 20 S Proteasomal Degradation
We have previously shown that binding of a protein can protect a client substrate protein from 20 S proteasomal degradation (7, 16). To see whether this also holds for p53, we subjected p53 to 20 S degradation in the presence of specific antibodies. The binding of DO-1 antibody to the N terminus prevented p53 degradation, whereas the binding of 421 to the C terminus had no effect (Fig. 2C). These results suggest that binding of proteins to the N terminus of p53 may protect p53 in this degradation pathway.
Mdm2 and Hdmx (Hdm4) share high sequence resemblance and have been both shown to bind p53 at the unstructured N terminus, inducing a best fit structure (48, 49). We incubated p53 with and without Mdm2/Hdmx and subjected these proteins to 20 S degradation. Both of these proteins could protect p53 from degradation in this in vitro system (Fig. 2D), further strengthening the possibility that binding to the unstructured N terminus of p53 rescues p53 from 20 S degradation.
p53 Degradation in Vivo Is Biphasic
Previous studies have shown that p53 is susceptible to degradation by both ubiquitin-dependent and ubiquitin-independent mechanisms. However, it was not clear how each mechanism contributes to the overall degradation of p53 and to the kinetics of this degradation. To address this problem, we used a mathematical approach to analyze the decay kinetics of p53. The basic model of p53 degradation is that p53 is synthesized (S) and then degraded by the ubiquitin system (Ks) (Fig. 3A). In this case, the equation for p53 levels would be as shown in Equation 1, and the solution of this equation when synthesis is stopped (S = 0 and p53[0] = A0) is described in Equation 2
If applying Ln to Equation 2, a linear monophasic decay curve is expected with the slope of −Ks from which the half-life of the protein can be calculated (t½ = Ln(2)/Ks). On the other hand, our in vitro data have shown that p53 is susceptible to degradation by the 20 S proteasome, but binding to another protein can prevent this degradation. These results lead to a model in which free p53, such as newly synthesized p53 that has not yet formed a complex with a partner, can undergo fast degradation by the 20 S proteasome (Kf) or form a functional complex at the rate of B (Fig. 3B). Once in a complex, p53 can only be degraded by the ubiquitin system at the rate of Ks. Under these assumptions, the equations that would describe the behavior of the two forms of p53, the free form (f(t)) and the form in a complex (c(t)), are described in Equations 3 and 4 respectively.
FIGURE 3.

p53 degradation is biphasic. A and B, schematic representation of the two proposed mechanisms of p53 degradation. Pulse-chase experiments on endogenous p53 in NIH3T3 cells were performed at least three times for the two pulse times (5 and 30 min). N, N terminus; C, C terminus; S, synthesis; E2, ubiquitin carrier protein. C, the 5-min pulse results were plotted (experimental), and a one-exponent (Mono-phasic) and two-exponent fit (Bi-phasic) was performed utilizing cftool of MATLAB. D, the graphs described in C were plotted after Ln was applied. E and F, experimental data of p53 pulse-chase experiments for the two different pulse times (5 and 30 min).
The solution of these equations combined ([p53] = c(t)+f(t)) when synthesis is stopped (S = 0, f(0) = F0, and c(0) = C0) is described in Equation 5
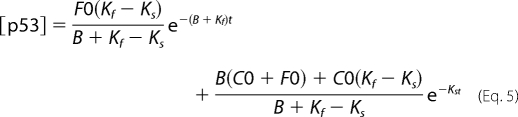 |
With these two models at hand, we looked at the decay curve of endogenous p53 after a pulse-chase experiment with 5-min labeling with [35S]methionine. We fitted to the experimental data the best fit for a single exponent function (monophasic) and double exponent function (biphasic) (supplemental Table 1). The two-exponent fit was much better, as can be visualized (Fig. 3C). A monophasic curve cannot encompass the initial fast decay and the slower later decay to such an extent as the two-exponent function. To emphasize the better fit of the two-exponent function, we applied Ln to the experimental data and the two fits (Fig. 3D). A biphasic degradation mode better explains the experimental data. To further validate that indeed there is biphasic degradation in cells, we utilized another approach whereby the decay curve of endogenous p53 was examined following different labeling times. If indeed the first phase of degradation is fast, longer labeling of a protein will result in the minimization of the relative contribution of the fast phase in the overall decay kinetics, whereas the second slow phase should not be affected. As expected, with long 30-min labeling, the relative contribution of the fast phase in overall decay kinetics became minor, whereas the second phase remained the same (Fig. 3E). Specifically, when looking only at the first phase of degradation, in the 5-min pulse, the kinetics is k = −0.034 (R2 = 0.99 t½ = 20 min), and with the 30-min pulse, the k = −0.024 (R2 = 0.99 t½ = 29 min) (Fig. 3F). This kinetics is consistent with the biphasic degradation of p53 in these cells with the first phase being faster than the second.
The First Phase of p53 Degradation Is Mediated by the 20 S Proteasome
To quantify p53 decay, we performed a pulse-chase experiment with a minimal pulse of 1 min and a longer pulse of 30 min (Fig. 4A). Analyzing the decay of p53 after applying Ln revealed that p53 exhibited biphasic degradation in HCT116 cells and that the first phase decreased when the pulse was longer (Fig. 4A). This degradation is proteasomal and was completely inhibited by MG132, an inhibitor of the proteasome (Fig. 4B).
FIGURE 4.

The rapid phase of p53 degradation is mediated by the 20 S proteasome. HCT116 cells were pulsed with 35S[methionine] for 1 and 30 min (A) or for 10 min (B and C) in the absence (A and C) or presence (B) of 50 μm MG132. Cells were collected at the indicated time points, and p53 was immunoprecipitated (IP). The levels of labeled p53 were detected by autoradiography. IB, immunoblotting. C–E, HCT116 cells were transduced with control or Rpn2 shRNAmir. After 96 h after infection, pulse-chase experiments were performed on p53 in the transduced cells (C). The ratio of the 20 S/26 S proteasomes was determined after their separation on native gels (D), and the total protein levels of 20 S and 26 S proteasome subunits were analyzed (E).
Given the susceptibility of the free p53 N terminus to 20 S proteasomal degradation, we reasoned that in the cells, the newly synthesized p53 is at higher risk of degradation by this pathway, accounting for the first fast decay phase. To test our hypothesis, we knocked down the Rpn2 subunit of the 19 S proteasome cap to prevent the assembly of 26 S proteasomes, whereas not affecting 20 S proteasomes. If our hypothesis is correct, under this condition, the first decay phase should remain intact, whereas the second phase should disappear (as it is ubiquitin- and 26 S proteasome-dependent). HCT116 cells were infected with a viral vector encoding either scrambled control shRNAmir or a specific target for Rpn2. Under these conditions, we performed a pulse-chase experiment. In the control knockdown, p53 was degraded throughout the 2-h chase (Fig. 4C) in a biphasic degradation pattern. In the Rpn2 knockdown cells, the second phase was completely inhibited, whereas the first remained intact (Fig. 4C). To verify that the knockdown of Rpn2 indeed changed the 20 S/26 S ratio, we examined this ratio on a native gel. We observed that following the knockdown of Rpn2, the 20 S/26 S ratio significantly increased (Fig. 4D) and that there was a significant decrease in the protein level of Rpn2 and an increase in the p53 levels (Fig. 3E). These results strongly suggest that the first fast phase of p53 degradation is mediated by the 20 S proteasomes.
The First Phase of p53 Degradation Is UI
Newly synthesized p53 can be degraded by the 20 S proteasome, a process that is expected to be UI. To validate its UI nature, we utilized TS20 cells in which under 39 °C the ubiquitin system is inactivated (43). Pulse-chase experiments revealed that endogenous p53 was indeed susceptible to degradation at 39 °C (Fig. 5A). Consistent with the results in the NIH3T3 and HCT116 cells (Figs. 3 and 4), we observed that in the TS20 cells, p53 had a biphasic degradation curve as well (Fig. 5B). Remarkably, at 39 °C, only the first phase of degradation was evident, demonstrating that this phase is UI. The results are not due to an altered 20 S/26 S ratio that remained the same at both temperatures (supplemental Fig. 1). Due to the rapid kinetics of the first phase of degradation, no p53 degradation was detectable with a 60-min-long pulse (Fig. 5C). In addition, under 39 °C and in the presence of the proteasome inhibitor MG132, the fast degradation of the newly synthesized p53 vanished (Fig. 5D), confirming that this process is proteasome-dependent but ubiquitin-independent.
FIGURE 5.

Newly synthesized p53 is degraded by the 20 S proteasome in a UI manner. A–E, a pulse-chase experiment was performed as described under ‘Experimental Procedures.‘ After incubation of TS20 cells for 16 h at 32 °C or 39 °C, endogenous p53 (A–D) or transfected p53 constructs (E) were immunoprecipitated (IP). 50 μm MG132 was added where specified, 30 min before labeling (D). The decay of p53 was monitored by autoradiography, and the levels of immunoprecipitated p53 were detected by immunoblotting with p53 antibodies (421 and 248 for endogenous and HA for transfected).
In the in vitro degradation assay, we have shown that the ΔN97 p53 mutant lacking the N-terminal disordered region was resistant to 20 S proteasomal degradation. To validate that the degradation in vivo at 39 °C is mediated by the 20 S proteasome, we followed ΔN97 p53 degradation in TS20 cells. In agreement with the in vitro data, WT p53 was degraded, whereas the ΔN97 mutant was resistant at 39 °C (Fig. 5E). These results infer that newly synthesized p53 undergoes UI degradation by the 20 S proteasome, whereas the second phase of degradation is the ubiquitin- and 26 S proteasome-mediated degradation.
p53 N-terminal Binding Proteins Can Stabilize p53 in the UI Degradation Pathway
Unstructured domains such as the N terminus of p53 have been shown to be associated with binding diversity (50). Our model predicts that when p53 is bound to other proteins (specifically via its N terminus), it is protected from degradation by default (Fig. 3B). To this end, we utilized Hdmx as a model p53-binding protein. It was shown that the unstructured N terminus of p53 can acquire structure when bound to Mdm2/Hdmx (48, 49) and the 70-kDa subunit of human replication protein A (hRPA70) (51). Moreover, in vitro, p53 can be protected from 20 S UI degradation by p53 N-terminal binding proteins (Hdmx, Mdm2, and DO-1 antibody) (Fig. 2).
Next we asked whether this principle is followed in cells. Hdmx stabilized p53 on the protein level (Fig. 6A) and did not affect p53 translation (Fig. 6B). Furthermore, Hdmx stabilization of p53 could be achieved in the UI degradation pathway of p53, suggesting that Hdmx could protect newly synthesized p53 from UI degradation (Fig. 6C). Moreover, the inability of Hdmx to stabilize only the ΔN40 form of p53 suggested that the binding to the N terminus of p53 is crucial for the stabilization of p53 in the UI pathway (Fig. 6E). Hdmx stabilization can be achieved in the absence of mdm2 as it could stabilize p53 and inhibit the first phase degradation of p53 in an Mdm2 null background (Fig. 6G).
FIGURE 6.

p53 is stabilized by N-terminal binding proteins in the UI pathway. p53-HA was transfected into HCT−/− p53 cells in the presence or absence of Hdmx. After 24 h, 25 μg/ml cycloheximide (CHX) was added, and cells were collected at the indicated time points (A) or 50 μm of MG132 was added for 30 min and then the cells were labeled with [35S]methionine for 30 min (B). Cells were then collected, extracted, and subjected to immunoprecipitation (IP). TS20 cells were transfected with p53 with increasing amounts of Hdmx (C) or increasing amounts of Mdm2 and Hdmx (D) and were incubated for 16 h at either 32 °C or 39 °C. E, WT and ΔN40 p53 were transfected into TS20 cells with or without increasing amounts of Hdmx and incubated for 16 h at 39 °C. GFP, green fluorescent protein. F–G, MEF p53−/− Mdm2−/− cells were transfected with p53 in the presence or absence of increasing amounts of Hdmx and ΔRING Mdm2-(1–441) (F). G, p53 was transfected with or without Hdmx, and a pulse-chase experiment was performed as described above.
Mdm2 is another model protein that can prevent p53 UI degradation in vitro (Fig. 2). As expected and unlike Hdmx, in the TS20 cells, Mdm2 at 32 °C caused p53 degradation (Fig. 6D, left panel). Remarkably, under 39 °C, Mdm2 played an opposite role and stabilized p53 (Fig. 6D, right panel). Inactive Mdm2 (1–441 mutant, lacking E3 ligase activity), like Hdmx, could stabilize p53 in the p53−/− and Mdm2−/− MEFs (Fig. 6F). Overall, the data suggest that p53 can be stabilized in the presence of Hdmx or inactive Mdm2, both p53 N-terminal binding proteins. Thus, binding to p53 prevents its degradation by default, a process that is both ubiquitin-independent and Mdm2-independent.
Biphasic Degradation Enables Differential Accumulation and Altered Steady State Levels of p53 Following Different Stresses
A double mode of degradation of p53 can enable a differential accumulation pattern of p53 following different stressors. We set out to examine whether indeed this is the case. For example, p53 is differentially accumulated following UV as compared with IR (52), but the underlying mechanism remained elusive. When NIH3T3 cells were exposed to either UV (50 J/m2) or IR (6 grays), the accumulation kinetics behaved differently. Following UV, p53 accumulation was delayed as compared with the IR-induced accumulation. Mdm2 level was not increased by UV, resulting in higher levels of p53 at later times (Fig. 7A). IR induced a rapid accumulation of p53 that was followed by a decline after 90 min, probably due to an increase in the Mdm2 level. Next we wanted to quantify the levels of p53 accumulation and degradation following UV and IR by labeling the cells with [35S]methionine. For both assays, cells were labeled starting 30 min after stress induction. Looking at the accumulation of labeled p53, as expected, the control steady state level was the lowest but increased in response to UV and to a much higher level with IR (Fig. 7B). The difference in the accumulation cannot be explained by changes in p53 translation (supplemental Fig. 2). To understand the accumulation, we followed the degradation decay at the same experimental conditions as in the accumulation experiment. Biphasic degradation was observed in the control samples. Remarkably, UV blocked the UD second phase but seemed to have little effect on the UI degradation (Fig. 7C). IR, on the other hand, blocked the first phase and attenuated the second phase of degradation (Fig. 7C). Similar results were obtained in HCT116 cells (supplemental Fig. 3). Overall, these results indicate that discriminative inhibition of the two phases of degradation enables discrete accumulation kinetics of p53 programmed to cope with unique types of stress.
FIGURE 7.

Differential accumulation and degradation of p53 following UV and IR. A, NIH 3T3 cells were exposed to either UV (50 J/m2) or IR (6 grays (6Gy)), and either total levels of proteins (A) or the accumulation of [35S]methionine-labeled p53 following incubation of the cells with [35S]methionine for the indicted time points (B) or the decay of labeled p53 after a pulse with [35S]methionine for 10 min (C) 30 min after exposure of cells were detected as described above. IB, immunoblot; Ip, immunoprecipitation; A.U., absorbance units.
DISCUSSION
p53, a key regulator of cell fate, has unstructured domains at both termini. We show that the lack of structure of the N terminus of p53 is crucial for its UI degradation enabling vectorial processing with an N to C terminus direction by the 20 S proteasome. It has been demonstrated that an unstructured domain at the N terminus of a protein may facilitate degradation by the proteasome (53). On the other hand, as shown for IκB (20) and ornithine decarboxylase (54), the unstructured domain at the C terminus initiates degradation, ruling out an N terminus rule in proteasomal degradation. Given the fact that the C terminus of p53 is also unstructured (38), the question remains as to what traits of an unstructured segment make it a good initiator of degradation by the 20 S proteasome.
The two distinct degradation pathways of p53, one UD and the other UI, lead us to bring about a working model that is described in Fig. 3B. In this model, the newly synthesized p53 is susceptible to rapid UI degradation via the free unstructured N terminus. Once the N terminus is occupied by a second protein, p53 degradation requires ubiquitination. We challenged this model by examining the decay of endogenous p53 in cells, and we show that p53 undergoes biphasic decay kinetics. The immediate and fast decay kinetics represents the newly synthesized p53 that was degraded by the 20 S proteasome in a UI manner, whereas p53 that escapes the first phase of degradation continues to be subjected to the second phase of degradation, that is UD, with much slower kinetics.
It has been previously shown that as much as a third of newly synthesized total proteins are degraded within the first half hour exhibiting biphasic behavior with a fast degradation phase within the first hour of synthesis and slower degradation that follows (55–57). Moreover, at least some of the newly synthesized proteins were shown to be degraded by the proteasome in a UI manner (55). It has been suggested that this process is important to get rid of the misfolded proteins, which are referred to as defective ribosomal products (DRIPs), as a quality control mechanism (56). Despite the similarity between these findings and ours on p53, the underlying mechanisms appear to be completely different. In our case, the behavior of p53 is the result of an inherent property of the functional protein that is intrinsically unstructured. Although DRIPs need chaperones to acquire proper folding and escape degradation (55), the intrinsically unstructured proteins, although having a positive correlation with binding to proteins, have a negative correlation with binding to chaperones (58). In contrast to the DRIP model, we found that the first decay phase is under regulation and that its turning off is programmed. Therefore, the biphasic behavior of p53 degradation is not the result of a quality control mechanism but rather an inherent feature.
Our model predicts that an IUP is susceptible for default degradation as long as the unstructured segment is free. IUPs may escape 20 S proteasomal degradation by binding to a protein partner (16, 59). Hdmx that binds the N terminus of p53 was used as a model protein. We demonstrate here that the N-terminal binding proteins of p53 cannot only prevent the degradation of p53 by the 20 S proteasome in vitro but also protect newly synthesized p53 from being degraded by the UI degradation pathway. This novel degradation pathway is important as p53 protein levels are largely regulated at the level of escaping degradation. The unstructured N terminus of p53 is modified mostly by phosphorylation in response to stress-induced kinases (40). These modifications sharply reduce p53 affinity to Mdm2 and therefore protect p53 from degradation. This simple model does not explain how the kinetics of p53 accumulation differs in response to the first rapid decay kinetics following IR and the lack of inhibition by UV. Apart from allowing discrete steady states, the biphasic kinetics allows reaching a maximal level of p53 much beyond what a monophasic mechanism permits. The differences between the two may change with the weight and the kinetics of the rapid phase. Also, the dual mode of the p53 degradation mechanism (each with distinct kinetics), in theory, may program at least four discrete steady state levels. In this study, we demonstrated the possibility of separately blocking each of the degradation phases. Finally, it has been suggested that the p53-Mdm2 feedback loop generates a ‘digital‘ clock that releases scheduled quanta of p53 in response to DNA damage (60, 61). Given that under DNA damage p53 is at least partially refractory to Mdm2, it is possible that such discrete pulses are generated by regulating the first UI decay kinetics. This can be observed when comparing p53 accumulation following UV and IR.
The effect of dual degradation modes on the outcome of stress response was also recently demonstrated in the case of NFκB. NFκB can bind and stabilize IκBα from UI degradation, a process that is crucial for NFκB activation (22). Furthermore, the dual degradation mode was shown to be crucial in NFκB response to UV (21). Thus, the combination of a double mode protein degradation mechanism and biphasic decay kinetics might be a more general phenomenon. We propose that this molecular scenario permitting programmable discrete steady state levels is a hallmark of many stress-responding effectors. The fact that a large number of effector proteins contain inherently unstructured regions fits well with this emerging principle.
Finally, the stabilizing role of the p53 N-terminal binding proteins described here is distinct from the chaperone-like activity described in other contexts (62). There are many N-terminal binding proteins of p53 (41, 63) that induce different p53 activities In this study, we used Hdmx as a model protein to hold ‘nanny‘ activity, namely protecting newly synthesized p53 from degradation, allowing it to mature to function. Whether other proteins with disordered regions have to have their specific ‘nannies‘ is an important prediction of our model that can be experimentally challenged.
Acknowledgments
We thank Drs. C. Kahana and M. Oren (Weizmann Institute of Science) for the materials and help and Drs. L. Pismen (Technion) and D. Holchman (Weizmann Institute of Science) for the guidance and help in the mathematical aspect of the work.
This work was supported by grants from the Samuel Waxman Cancer Research Foundation, from the Israel Science Foundation, and from the Minerva Foundation with funding from the Federal German Ministry for Education and Research and Journal of Cell Science student travel fellowship.
This article was selected as a Paper of the Week.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3 and a table.
- UD
- ubiquitin-dependent
- UI
- ubiquitin-independent
- IR
- irradiation
- IUP
- intrinsically unstructured protein
- E1
- ubiquitin-activating enzyme
- E3
- ubiquitin-protein isopeptide ligase
- HA
- hemagglutinin
- MEF
- mouse embryonic fibroblast
- DRIP
- defective ribosomal product
- WT
- wild type.
REFERENCES
- 1.Jariel-Encontre I., Bossis G., Piechaczyk M. (2008) Biochim. Biophys. Acta 1786, 153–177 [DOI] [PubMed] [Google Scholar]
- 2.Liu C. W., Corboy M. J., DeMartino G. N., Thomas P. J. (2003) Science 299, 408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsvetkov P., Asher G., Reiss V., Shaul Y., Sachs L., Lotem J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5535–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunker A. K., Obradovic Z., Romero P., Garner E. C., Brown C. J. (2000) Genome Inform. Ser. Workshop Genome Inform. 11, 161–171 [PubMed] [Google Scholar]
- 5.Gsponer J., Futschik M. E., Teichmann S. A., Babu M. M. (2008) Science 322, 1365–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baugh J. M., Viktorova E. G., Pilipenko E. V. (2009) J. Mol. Biol. 386, 814–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asher G., Tsvetkov P., Kahana C., Shaul Y. (2005) Genes Dev. 19, 316–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X., Barton L. F., Chi Y., Clurman B. E., Roberts J. M. (2007) Mol. Cell 26, 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheaff R. J., Singer J. D., Swanger J., Smitherman M., Roberts J. M., Clurman B. E. (2000) Mol. Cell 5, 403–410 [DOI] [PubMed] [Google Scholar]
- 10.Tofaris G. K., Layfield R., Spillantini M. G. (2001) FEBS Lett. 509, 22–26 [DOI] [PubMed] [Google Scholar]
- 11.Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 12.Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E. (2009) Annu. Rev. Biochem. 78, 959–991 [DOI] [PubMed] [Google Scholar]
- 13.Dyson H. J., Wright P. E. (2005) Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 14.Elliott E., Tsvetkov P., Ginzburg I. (2007) J. Biol. Chem. 282, 37276–37284 [DOI] [PubMed] [Google Scholar]
- 15.Touitou R., Richardson J., Bose S., Nakanishi M., Rivett J., Allday M. J. (2001) EMBO J. 20, 2367–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsvetkov P., Asher G., Paz A., Reuven N., Sussman J. L., Silman I., Shaul Y. (2008) Proteins 70, 1357–1366 [DOI] [PubMed] [Google Scholar]
- 17.Sickmeier M., Hamilton J. A., LeGall T., Vacic V., Cortese M. S., Tantos A., Szabo B., Tompa P., Chen J., Uversky V. N., Obradovic Z., Dunker A. K. (2007) Nucleic Acids Res. 35, D786–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krappmann D., Wulczyn F. G., Scheidereit C. (1996) EMBO J. 15, 6716–6726 [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez-Castelao B., Castaño J. G. (2005) FEBS Lett. 579, 4797–4802 [DOI] [PubMed] [Google Scholar]
- 20.Kroll M., Conconi M., Desterro M. J., Marin A., Thomas D., Friguet B., Hay R. T., Virelizier J. L., Arenzana-Seisdedos F., Rodriguez M. S. (1997) Oncogene 15, 1841–1850 [DOI] [PubMed] [Google Scholar]
- 21.O'Dea E. L., Kearns J. D., Hoffmann A. (2008) Mol. Cell 30, 632–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathes E., O'Dea E. L., Hoffmann A., Ghosh G. (2008) EMBO J. 27, 1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogelstein B., Lane D., Levine A. J. (2000) Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 24.Asher G., Lotem J., Sachs L., Kahana C., Shaul Y. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 13125–13130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camus S., Menéndez S., Cheok C. F., Stevenson L. F., Laín S., Lane D. P. (2007) Oncogene 26, 4059–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haupt Y., Maya R., Kazaz A., Oren M. (1997) Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 27.Kubbutat M. H., Jones S. N., Vousden K. H. (1997) Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 28.Leng R. P., Lin Y., Ma W., Wu H., Lemmers B., Chung S., Parant J. M., Lozano G., Hakem R., Benchimol S. (2003) Cell 112, 779–791 [DOI] [PubMed] [Google Scholar]
- 29.Dornan D., Wertz I., Shimizu H., Arnott D., Frantz G. D., Dowd P., O'Rourke K., Koeppen H., Dixit V. M. (2004) Nature 429, 86–92 [DOI] [PubMed] [Google Scholar]
- 30.Sharp D. A., Kratowicz S. A., Sank M. J., George D. L. (1999) J. Biol. Chem. 274, 38189–38196 [DOI] [PubMed] [Google Scholar]
- 31.Stad R., Little N. A., Xirodimas D. P., Frenk R., van der Eb A. J., Lane D. P., Saville M. K., Jochemsen A. G. (2001) EMBO Rep. 2, 1029–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stad R., Ramos Y. F., Little N., Grivell S., Attema J., van Der Eb A. J., Jochemsen A. G. (2000) J. Biol. Chem. 275, 28039–28044 [DOI] [PubMed] [Google Scholar]
- 33.Vega F. M., Sevilla A., Lazo P. A. (2004) Mol. Cell Biol. 24, 10366–10380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finch R. A., Donoviel D. B., Potter D., Shi M., Fan A., Freed D. D., Wang C. Y., Zambrowicz B. P., Ramirez-Solis R., Sands A. T., Zhang N. (2002) Cancer Res. 62, 3221–3225 [PubMed] [Google Scholar]
- 35.Gilkes D. M., Chen L., Chen J. (2006) EMBO J. 25, 5614–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu B., Gilkes D. M., Farooqi B., Sebti S. M., Chen J. (2006) J. Biol. Chem. 281, 33030–33035 [DOI] [PubMed] [Google Scholar]
- 37.Shvarts A., Steegenga W. T., Riteco N., van Laar T., Dekker P., Bazuine M., van Ham R. C., van der Houven van Oordt W., Hateboer G., van der Eb A. J., Jochemsen A. G. (1996) EMBO J. 15, 5349–5357 [PMC free article] [PubMed] [Google Scholar]
- 38.Bell S., Klein C., Müller L., Hansen S., Buchner J. (2002) J. Mol. Biol. 322, 917–927 [DOI] [PubMed] [Google Scholar]
- 39.Joerger A. C., Fersht A. R. (2008) Annu. Rev. Biochem. 77, 557–582 [DOI] [PubMed] [Google Scholar]
- 40.Brooks C. L., Gu W. (2003) Curr. Opin. Cell Biol. 15, 164–171 [DOI] [PubMed] [Google Scholar]
- 41.Braithwaite A. W., Del Sal G., Lu X. (2006) Cell Death Differ. 13, 984–993 [DOI] [PubMed] [Google Scholar]
- 42.Glickman M. H., Rubin D. M., Fried V. A., Finley D. (1998) Mol. Cell Biol. 18, 3149–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chowdary D. R., Dermody J. J., Jha K. K., Ozer H. L. (1994) Mol. Cell Biol. 14, 1997–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jayaraman J., Prives C. (1995) Cell 81, 1021–1029 [DOI] [PubMed] [Google Scholar]
- 45.Jayaraman L., Freulich E., Prives C. (1997) Methods Enzymol. 283, 245–256 [DOI] [PubMed] [Google Scholar]
- 46.Cain C., Miller S., Ahn J., Prives C. (2000) J. Biol. Chem. 275, 39944–39953 [DOI] [PubMed] [Google Scholar]
- 47.Levy D., Adamovich Y., Reuven N., Shaul Y. (2008) Mol. Cell 29, 350–361 [DOI] [PubMed] [Google Scholar]
- 48.Popowicz G. M., Czarna A., Rothweiler U., Szwagierczak A., Krajewski M., Weber L., Holak T. A. (2007) Cell Cycle 6, 2386–2392 [DOI] [PubMed] [Google Scholar]
- 49.Kussie P. H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A. J., Pavletich N. P. (1996) Science 274, 948–953 [DOI] [PubMed] [Google Scholar]
- 50.Dunker A. K., Cortese M. S., Romero P., Iakoucheva L. M., Uversky V. N. (2005) Febs. J. 272, 5129–5148 [DOI] [PubMed] [Google Scholar]
- 51.Vise P. D., Baral B., Latos A. J., Daughdrill G. W. (2005) Nucleic Acids Res. 33, 2061–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu X., Lane D. P. (1993) Cell 75, 765–778 [DOI] [PubMed] [Google Scholar]
- 53.Prakash S., Tian L., Ratliff K. S., Lehotzky R. E., Matouschek A. (2004) Nat. Struct. Mol. Biol. 11, 830–837 [DOI] [PubMed] [Google Scholar]
- 54.Zhang M., MacDonald A. I., Hoyt M. A., Coffino P. (2004) J. Biol. Chem. 279, 20959–20965 [DOI] [PubMed] [Google Scholar]
- 55.Qian S. B., Princiotta M. F., Bennink J. R., Yewdell J. W. (2006) J. Biol. Chem. 281, 392–400 [DOI] [PubMed] [Google Scholar]
- 56.Schubert U., Antón L. C., Gibbs J., Norbury C. C., Yewdell J. W., Bennink J. R. (2000) Nature 404, 770–774 [DOI] [PubMed] [Google Scholar]
- 57.Wheatley D. N., Giddings M. R., Inglis M. S. (1980) Cell Biol. Int. Rep. 4, 1081–1090 [DOI] [PubMed] [Google Scholar]
- 58.Hegyi H., Tompa P. (2008) PLoS Comput. Biol. 4, e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Asher G., Reuven N., Shaul Y. (2006) Bioessays 28, 844–849 [DOI] [PubMed] [Google Scholar]
- 60.Geva-Zatorsky N., Rosenfeld N., Itzkovitz S., Milo R., Sigal A., Dekel E., Yarnitzky T., Liron Y., Polak P., Lahav G., Alon U. (2006) Mol. Syst. Biol. 2, 2006 0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lahav G., Rosenfeld N., Sigal A., Geva-Zatorsky N., Levine A. J., Elowitz M. B., Alon U. (2004) Nat. Genet. 36, 147–150 [DOI] [PubMed] [Google Scholar]
- 62.Marques C., Guo W., Pereira P., Taylor A., Patterson C., Evans P. C., Shang F. (2006) Faseb. J. 20, 741–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prives C., Hall P. A. (1999) J. Pathol. 187, 112–126 [DOI] [PubMed] [Google Scholar]