

Abstract
The T4 bacteriophage helicase loader (gp59) is one of the main eight proteins that play an active role in the replisome. gp59 is a small protein (26 kDa) that exists as a monomer in solution and in the crystal. It binds preferentially to forked DNA and interacts directly with the T4 helicase (gp41), single-stranded DNA-binding protein (gp32), and polymerase (gp43). However, the stoichiometry and structure of the functional form are not very well understood. There is experimental evidence for a hexameric structure for the helicase (gp41) and the primase (gp61), inferring that the gp59 structure might also be hexameric. Various experimental approaches, including gel shift, fluorescence anisotropy, light scattering, and fluorescence correlation spectroscopy, have not provided a clearer understanding of the stoichiometry. In this study, we employed single-molecule photobleaching (smPB) experiments to elucidate the stoichiometry of gp59 on a forked DNA and to investigate its interaction with other proteins forming the primosome complex. smPB studies were performed with Alexa 555-labeled gp59 proteins and a forked DNA substrate. Co-localization experiments were performed using Cy5-labeled forked DNA and Alexa 555-labeled gp59 in the presence and absence of gp32 and gp41 proteins. A systematic study of smPB experiments and subsequent data analysis using a simple model indicated that gp59 on the forked DNA forms a hexamer. In addition, the presence of gp32 and gp41 proteins increases the stability of the gp59 complex, emphasizing their functional role in T4 DNA replication machinery.
T4 bacteriophage is one of the major model systems for acquiring information on DNA replication and repair processes. The T4 replisome is composed of eight major proteins that are subgrouped into the holoenzyme (gp43 polymerase, gp45 clamp, and gp44/62 clamp loader) and the primosome (gp41 helicase, gp59 helicase loader, gp32 single-stranded DNA-binding protein, and gp41 primase). Replication takes place through the coordinated function of all these proteins (1).
The helicase loader (gp59) functions to load the helicase onto a DNA strand that is coated with gp32. In addition, gp59 inhibits the initiation of polymerization by gp43 (2, 3). Furthermore, it acts in recombination and recombination-dependent DNA replication (4, 5) and functions as a gatekeeper in origin-dependent replication in vivo (6). The gp59 protein is known to exist as a monomer in solution and in an x-ray crystal structure (7, 8). It binds to several types of DNA substrates, preferring a replication fork (i.e. forked DNA) (9). Based on its similarity to the members of the high mobility group (HMG) family of proteins, a model structure has been proposed by Mueser et al. (8) for gp59 bound to forked DNA in which the N-terminal domain of gp59 binds to the duplex DNA, whereas the lagging strand passes through a narrow groove that lies between the N- and C-terminal domains and the leading strand binds to the bottom surface of the C-terminal domain.
An interaction between gp59 and gp32 is required for loading the helicase on gp32-coated DNA. The dissociation constant (Kd) observed for gp59 binding to a gp32-DNA complex is ∼2 nm (10). The N terminus of gp59 is involved in the gp32 interaction, and the C terminus interacts with gp41 (11, 12). On a forked DNA, gp59 prefers to co-localize with gp32 rather than the DNA (13). In the absence of gp32, gp59 has a binding preference for DNA in the order forked DNA > single-stranded DNA > double-stranded DNA (9). Electron microscopic studies of the active replisome revealed bobbin-like structures on the lagging strand that may contain gp59, suggesting that the protein remains as part of the replisome during replication, although its role is unclear (14, 15).
Functional assays using single-turnover and steady-state kinetics showed that DNA unwinding by the helicase is enhanced by 200-fold in the presence of gp59, where maximum unwinding is observed when the proteins exist in equal stoichiometry (16). In the analogous Escherichia coli proteins, DnaC (helicase loader) binds to the DnaB (helicase) hexamer likewise at a 1:1 stoichiometry (17). Cross-linking experiments observed complexes of gp59 up to a pentamer in the presence of gp32, gp41, and DNA.3 Isothermal titration calorimetry experiments were consistent with a stoichiometry of 0.75 gp59 subunit to 1 gp41 subunit with a dissociation constant of ∼150 nm.3 The electron microscopic studies on the structure of the gp41-gp61 complex revealed two hexamers in a 1:1 stoichiometry (19). Based on the collective data, it could be postulated that gp59 should form a functional higher order complex ranging from tetramer to hexamer.
In this study, we have experimentally investigated the stoichiometry and interaction of gp59 on its primary forked DNA substrate in the presence of gp32 and gp41 proteins using the single-molecule photobleaching (smPB)4 method. The smPB technique has been successfully used to elucidate the stoichiometry of fluorescently labeled complexes of membrane proteins (20) and packaging RNA in bacteriophage Φ29 (21). In this method, each subunit in a protein complex is labeled with a fluorescent dye and imaged using laser illumination. The dye molecules undergo photobleaching; thus, counting the number of photobleaching steps gives us an estimate of the number of subunits present. We used gp59 monomer labeled with Alexa 555 fluorophore and forked DNA substrates with a biotin tag. In one experiment, we used unlabeled DNA, whereas in another, we used DNA labeled with Cy5 dye to study co-localization of the forked DNA and proteins.
The experimental results give convincing evidence that gp59 forms a hexameric complex on the forked DNA because of a protein-DNA interaction. A simple model was used to analyze the part played by other primosome proteins (gp32 and gp41) in the gp59-forked DNA interaction.
MATERIALS AND METHODS
Purification and Labeling of T4 Proteins
Primosome proteins gp41 (11), gp32 (22), and gp59 (23) and two mutants of gp59 (C42A and C215A) (24) were expressed in E. coli and purified as described previously. Mutant gp59 proteins were labeled by cysteine modification. In brief, the protein was dialyzed in labeling buffer (20 mm Tris (pH 7.5), 150 mm NaCl, and 10% glycerol) for 8 h and incubated with a 5-fold excess of Alexa 555 (Alexa Fluor 555 C2 maleimide, Invitrogen) for ∼4 h. The unreacted dye was removed by chromatography using a cation-exchange resin and a salt gradient. Dithiothreitol (∼2 mm) was added to the gp59-Alexa 555 proteins and frozen in aliquots at −70 °C. Protein concentration was determined by Bradford assay, and the extent of labeling was calculated by using absorbance of the fluorescent dye as described previously (25). gp59 mutants with one accessible cysteine were found to have 1 Alexa 555/protein, which implies that protein is 100% labeled. An ATPase assay was carried out to check the activity of the gp59-Alexa 555 proteins in the presence of gp32 and gp41. The labeled proteins were found to be as active as the wild-type protein.
Preparation of Forked DNA Substrates
Various high pressure liquid chromatography-purified DNA primers were bought from Integrated DNA Technologies (Coralville, IA) for smPB (with biotin and Cy5 dye attachments). The structures of the forked DNAs are shown in Fig. 1. The sequences of the primers are given in Table 1. These primers were mixed at a 1:1 molar ratio in complex buffer (20 mm Tris acetate, 150 mm potassium acetate, and 10 mm magnesium acetate (pH 7.8)) to a final concentration of 5 μm. The mixture was heated to 95 °C for 5 min, followed by slow cooling to room temperature.
FIGURE 1.

Structures of the forked DNA substrates used in this study. 1, forked DNA (10/40/40-mer) with biotin at the 5′-end of the leading strand for the smPB study using unlabeled substrate (unlabeled forked DNA); 2, forked DNA (34/62/50-mer) with Cy5 dye at the 5′-end of the primer and biotin at the 3′-end of the lagging strand (Cy5-labeled forked DNA) for the smPB study with labeled substrate and gp59-Alexa 555.
TABLE 1.
Primer sequences of forked DNA substrates used in this study
Various forked DNA substrates were made by annealing the indicated primers. The primers were mixed at a 1:1 molar ratio and heated to 95 °C for 5 min, followed by slow cooling to room temperature. Unlabeled forked DNA (FkD-U) was used for the smPB experiment done with forked DNA and the various proteins (gp59, gp32, and gp41). Cy5-labeled forked DNA (FkD-Cy5) was used for the smPB co-localizing experiments done with proteins.
| Name | Sequence |
|---|---|
| FkD-U | 5′-Biotin-ACACAGACGTACTATCATGAGGTGAGGGAGGGTGGGTGGT-3′ (40-mer) |
| 5′-TGTGTCTGCATGATAGTACGGGTGAGGGAGGGTGGGTGGT-3′ (40-mer) | |
| 5′-ACCCCCACCC-3′ (10-mer) | |
| FkD-Cy5 | 5′-TATGAATCAGAGTGTAAGTTCCGAGTGATACAATGATAGTACGTCTGTGT-biotin-3′ (50-mer) |
| 5′-ACACAGACGTACTATCATGACGCATCAGACAACGTGCGTCAAAAATTACGTGCGGAAGGAGT-3′ (62-mer) | |
| 5′-Cy5-ACTCCTTCCGCACGTAATTTTTGACGCACGTTGT-3′ (34-mer) | |
smPB Experiments
Quartz microscope slides and glass coverslips were prepared according to a published protocol (26). A flow cell with channels was constructed from a quartz slide and glass coverslip. Each channel was washed with buffer and then with a solution of bovine serum albumin as a surface blocker, followed by streptavidin solution. The biotinylated forked DNA substrate was attached on the quartz slide through streptavidin-biotin linkage. The protein solution (either gp59(C42A)-Alexa 555 or gp59(C215A)-Alexa 555) was filtered using a 0.1-μm syringe filter, introduced into the channel containing forked DNA, allowed to equilibrate for 1 min, and then washed with complex buffer. For the smPB experiment with two dyes, the slide was coated with forked DNA containing Cy5 through a streptavidin-biotin linkage, and then the protein solution was introduced, equilibrated, and washed as described above.
The smPB experiments were done using a homebuilt prism-based total internal reflection fluorescence microscope and imaging system. The sample was excited using a green laser (532 nm, 300 milliwatts; Ventus, Laser Quantum). The fluorescence from the sample was collected by a 1.2-numerical aperture/×60 water immersion objective (Plan Apo, Nikon, Tokyo) on a Nikon microscope (inverted TE-2000U) with a 550-nm long-pass filter (Chroma, Rockingham, VT) and a 640-nm dichroic mirror (Chroma) and imaged onto an EMCCD camera (Cascade 512B, Roper Scientific, Tucson, AZ) with 3 × 3-pixel binning at a 100-ms exposure time. For experiments with Alexa 555 and Cy5, a low intensity red diode laser (645 nm, 25 milliwatts; Coherent Inc.) was used to excite Cy5. An automated shutter was used to switch between the red and green lasers after seven frames of the movie. The movies were collected until the bright fluorescent spots completely photobleached or disappeared. The fluorescence images were converted into fluorescence traces using mathematical codes written in IDL (Interface Definition Language software, ITT Visual Information Solutions, Inc.). Traces with very high photon intensities (>105) were discarded, and others were counted manually. Histograms were built from the number of photobleaching events, and the data were fit to a described model with Equation 4 (see below) employing code written in MATLAB.
Single-molecule Data Analysis
The smPB data were fit using a simple monomer-binding model to study the relative binding and affinity of protein-DNA and protein-protein interactions, respectively. In this case, we assumed a hexameric complex (A6) was formed from stepwise interaction of monomers (A) in a sequential manner.
The overall pathway of the hexamer formation is written as in Equation 1.
The total concentration of various oligomer species formed from an initial concentration of monomer ([A0]) is written as in Equation 2.
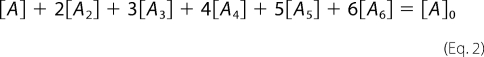 |
The steady-state equilibrium relation at each oligomerization step could be written as follows.
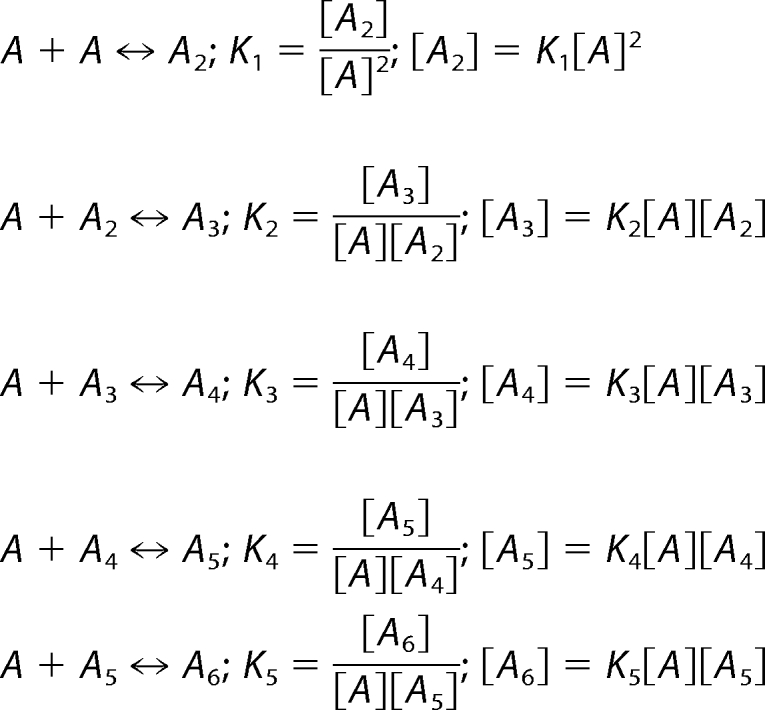 |
Now, substituting the concentration of each species ([A2] to [A6]) in terms of [A] into Equation 2 and introducing an affinity parameter (σ) associated with higher order oligomerization (e.g. Kn = σKn−1 where n = 2∼6 and K1 = K), we get the following equations for normalization (Equation 3) and the oligomer concentration distribution as a function of n, [A], K, and σ (Equation 4).
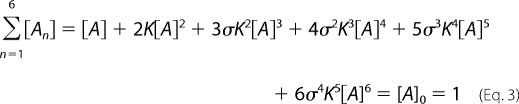 |
The dimerization of gp59 is the first step in complex formation. This has only a single binding parameter associated with the process (i.e. K1 = K). As the oligomer becomes bigger (for example, n > 2), the tendency for initiating or inhibiting a protein-protein interaction increases; this warrants the need for affinity parameter σ in the binding equilibrium.
A histogram is constructed by analyzing the time traces of the fluorescent spots from the recorded sequence of video frames. The number of photobleaching steps indicates the number of monomers in the complex. The events are grouped and normalized so that their number represents the population of the respective species. The experimental data were fit to Equation 4. The discrete nature of photobleaching steps requires the equation to be valid only for integer values of n (from 1 to 6). The normalized number of events and their corresponding photobleaching steps were provided as the input, whereas [A], K, and σ were used as fitting parameters. We employed a nonlinear least-square fitting method based on the Levenberg-Marquardt algorithm using MATLAB. After several rounds of iterations, the values of K and σ, as well as the residuals and a measure of the goodness of fit, were obtained.
RESULTS
smPB Studies
The experiments involving gp59, forked DNA, and other proteins were carried out as described under “Materials and Methods.” The fluorescence traces were also extracted from the recorded video frames as mentioned above. The time traces of the fluorescence signal were analyzed, and the photobleaching steps were counted manually by visual inspection. A representative view of the traces with various photobleaching steps is given in Fig. 2 (a–c). After analyzing the traces, a histogram was built as shown in Fig. 3. The numbers of events observed are normalized with respect to the total number of frames counted.
FIGURE 2.

Analysis of photobleaching steps of protein-forked DNA complexes. a–c, the smPB fluorescence images were collected at a 100-ms exposure time with 3 × 3-pixel binning with an EMCCD camera. The fluorescence images were converted into fluorescence traces using mathematical codes written in IDL. Representative plots of fluorescence intensity versus time (ms) show photobleaching steps 1, 2, and 6. a.u., arbitrary units.
FIGURE 3.

Histogram plot of smPB events. The histogram represents the smPB experiment done with Cy5-labeled forked DNA (FkD-Cy5) with gp59-Alexa 555 and other proteins. Histograms show the normalized events versus number of photobleaching steps. Similar histograms were built for smPB experiments with unlabeled forked DNA with the same proteins (data not shown). The experiments were done in triplicate, and the errors assigned. The population of the hexamer seen is indicated by the circle.
Forked DNA with Primosome Proteins gp59, gp32, and gp41
A compilation of histograms obtained upon addition of various proteins that are bound to forked DNA is given in Fig. 3. The data were fit to a monomer model using Equation 4 in MATLAB. Initially, a model was applied such that the constant Keq was involved in the formation of all the species (i.e. monomer, trimer, etc.) with the same affinity, but the experimental data did not fit properly for such an assumption. Hence, we introduced the affinity parameter σ to account for higher order oligomerization. The value of σ could vary depending on the nature of the binding event. Thus, the resulting value of K does not give an absolute value of the binding association constant in terms of molar concentration but represents a relative parameter for the magnitude and strength of the actual binding equilibrium constant (Keq). The obtained K and σ values are given in Table 2. The root mean square error obtained for values of K and σ was less than ±0.02. smPB experiments were also performed with gp59 in the presence of forked DNA, gp32, and gp41. All data obtained were analyzed as described above and are shown in Fig. 4 (A–D), and the obtained values of the parameters K and σ are provided in Table 2 (Nos. 1–4).
TABLE 2.
smPB data results
The K and σ values obtained from fitting the experimental data (Fig. 4) to Equation 4 are tabulated below. The K and σ values measure the magnitude of the association constant and relative strength of the binding events. The values are subgrouped and given as proteins with unlabeled forked DNA (FkD-U) in Nos. 1–4, with no DNA in Nos. 5–8, and with Cy5-labeled forked DNA (FkD-Cy5) in Nos. 9–12. Along with K and σ values and the residuals, a measure of the goodness of fit was obtained. The root mean square error obtained for all the fittings was less than ± 0.02. gp59*, Alexa 555-labeled gp59.
| No. | System | K | σ |
|---|---|---|---|
| 1 | FkD-U + gp59* | 0.90 | 0.86 |
| 2 | FkD-U + gp59* + gp32 | 0.38 | 0.78 |
| 3 | FkD-U + gp59* + gp41 | 0.64 | 0.97 |
| 4 | FkD-U + gp59* + gp32 +gp41 | 0.68 | 1.2 |
| 5 | gp59* | 0.43 | 0.06 |
| 6 | gp59* + gp32 | 0.27 | 0.03 |
| 7 | gp59* + gp41 | 0.40 | 0.07 |
| 8 | gp59* + gp32 + gp41 | 0.43 | 0.06 |
| 9 | FkD-Cy5 + gp59* | 0.71 | 0.73 |
| 10 | FkD-Cy5 + gp59* + gp32 | 0.32 | 0.39 |
| 11 | FkD-Cy5 + gp59* + gp41 | 0.93 | 0.87 |
FIGURE 4.

Plots of smPB experiments. The following plots show the experimental and fitted data for various smPB experiments. The data were fitted to a monomer model using Equation 4 as described under “Materials and Methods.” The normalized number of events and their corresponding photobleaching steps were provided as the input, whereas [A], K, and σ were used as fitting parameters. A–D, unlabeled forked DNA + gp59 and other proteins (i.e. gp32 and gp41); E–H, gp59 and other proteins alone; I–L, Cy5-labeled forked DNA and gp59 and other proteins.
Protein-Protein Interaction
The control experiments were performed in the absence of forked DNA. After introduction of the protein followed by repeated washing, some fluorescent spots remained on the slide. Hence, we performed similar experiments in the absence of forked DNA. The recorded data were analyzed as described above and are shown in Fig. 4 (E–H), and values of the fit parameters are given in Table 2 (Nos. 5–8). The values of K and σ were significantly different in the presence and absence of forked DNA.
Co-localization of Forked DNA and gp59
Co-localization experiments were performed using Cy5-labeled forked DNA and gp59-Alexa 555 as described above. These co-localization experiments helped us to consider only the protein-DNA interaction and to eliminate any nonspecific adsorption at the surface. For this purpose, we designed a forked DNA substrate with a Cy5 dye such that any fluorescence resonance energy transfer (FRET) between Alexa 555 (on gp59) and Cy5 (on forked DNA) would be minimal. The experiments were performed with a red and a green laser as described under “Materials and Methods.” Traces were picked in which both dyes were seen, indicating that the protein was bound to the forked DNA. As expected, the Cy5 dye on the forked DNA photobleached in a single step. Multiple photobleaching steps of Alexa 555 were observed in the presence of various combinations of primosome proteins (gp59, gp32, and gp41). The data analysis was performed as discussed above, and the results are shown in Fig. 4 (I–L) and Table 2 (Nos. 9–12).
The results of co-localization experiments are consistent with the experiments performed in the presence of the forked DNA without label. The control experiment inferred that proteins can interact off the DNA. The most important observation is the hexamer seen in the presence of forked DNA. The population of the hexameric complex varied depending on the presence of other proteins. Because the labeling efficiency is ∼1, we did not need to carry out any statistical analysis based on a binomial distribution of the gp59 oligomers. We did not see higher oligomers (n > 7), which led us to frame a model of depicting monomer interacting stepwise to form a hexameric complex.
DISCUSSION
The smPB results unambiguously demonstrate the formation of a hexameric gp59 complex on the forked DNA. A pictorial view of gp59 hexamer is given in Fig. 5a. We observed a distribution of smaller complexes (n = 1–6) seen by counting the number of photobleaching steps. It is known from the ensemble experiments that the value of Kd obtained for gp59 dissociation is in the order of nm. Single-molecule fluorescence imaging experiments are usually performed at very low protein concentrations, in the range of about pm to sub-nm. Thus, the data obtained from single-molecule studies include a condition in which the protein complexes would be partially dissociated (27). In our smPB experiments, we observed ∼3% of the population to be hexameric; this lower yield is most likely the consequence of the gp59 complex being partially dissociated into oligomers. Also the intermediate oligomers observed could be due to the linear binding of gp59 on the single-strand and forked junction of the DNA, as shown in Fig. 5b.
FIGURE 5.

Schematic representation of oligomers of gp59 on a replication fork. a, hexamer of gp59; b, random oligomer of gp59; c, gp59 at the forked DNA and co-localized on gp32; d, ternary complex between a hexamer of gp59 and gp32 and gp41 in the presence of forked DNA.
The data analysis done by fitting to a simple model involving stepwise monomer association provided a reasonable estimate of the interaction of protein-protein and protein-DNA interactions. However, we cannot rule out other pathways for the formation of the hexamer, i.e. association of three dimers, two trimers, or their combinations.
The results of 12 sets of smPB experiments involving gp59 are tabulated in Table 2. The values of K and σ given in Table 2 measure the magnitude of the association constant and relative strength of the binding events. The experiments can be grouped into three sections. The three sections are (a) experiments in which unlabeled forked DNA was used (Table 2, Nos. 1–4), (b) studies involving protein-protein interactions without forked DNA (Nos. 5–8), and (c) experiments using Cy5-labeled forked DNA and gp59-Alexa 555 interactions along with other proteins (Nos. 9–12).
To rule out contributions from any nonspecific binding of proteins to the surface, we performed co-localization experiments. Only Cy5-labeled forked DNA whose fluorescence co-localized with gp59-Alexa 555 spots was used for analysis. All experiments using unlabeled forked DNA (Table 2, Nos. 1–4) were also repeated with co-localization experiments, and the results are tabulated in Table 2 (Nos. 9–12).
At the onset, it is evident from Fig. 4 (E–H) that, in experiments involving only proteins, no oligomeric complexes higher than n > 4 were found. The comparison of K, the association values, observed in protein interaction experiments involving unlabeled forked DNA is in the order gp59 > gp59 + gp32 + gp41 > gp59 + gp41 > gp59 + gp32; their binding affinity (σ) is in the order gp59 + gp32 + gp41 > gp59 + gp41 > gp59 > gp59 + gp32. The K value in the case of Cy5-labeled forked DNA is in the order gp59 + gp41 > gp59 + gp32 + gp41 > gp59 > gp59 + gp32; their binding affinity (σ) in the order gp59 + gp32 + gp41 > gp59 + gp41 > gp59 > gp59 + gp32. The trend observed for the K values for forked DNA with and without the label varies to a slight extent; however, the trends in σ are the same. Because co-localization results are restricted to the protein-forked DNA interaction and not any spurious nonspecific binding, those values are considered to reflect the interactions of interest.
To understand the actual protein-protein and protein-DNA interactions, we need to consider both the binding parameter K and the affinity parameter σ. Detailed comparison among these sets of experiments and the numerical values of these parameters allow us to draw a complete picture of the nature of the protein-DNA interaction for the primosome proteins.
First, considering the experiment with gp59 in the absence of DNA (Table 2, No. 5), the protein gp59 seems to interact with a weak association (K = 0.43). The formations of small oligomeric complexes arise from this weak protein-protein interaction and are reflected in the low affinity binding parameter (σ = 0.06). This possibly means that, in the absence of DNA, gp59 forms small oligomers that may not have defined stoichiometry or functional significance.
Now, comparing these results with the experiment in which the gp59 protein was added to both labeled and unlabeled forked DNAs (Table 2, Nos. 1 and 9), we see a significant increase in the value of K = 0.90 and 0.71. This ∼2-fold increase in the presence of forked DNA is undoubtedly due to the protein-DNA interaction. This is also accompanied by a 15-fold increase in the affinity binding parameter (σ = 0.86). This is an indication that formation of higher oligomeric complexes is favored in the presence of forked DNA. It is evident from the results of smPB studies that gp59 exists in oligomers up to a hexamer only in the presence of forked DNA (Fig. 4, A, E, and I). Considering these observations, binding of one monomer to the DNA facilitates the binding of other monomers to form a hexameric ring, which is reflected in the higher K and σ values.
Table 2 (No. 10) shows the results when gp32 is added in addition to gp59 on the forked DNA. The marked decrease in the value of K (0.32) in this case relative to No. 9 is an indication that, in the presence of gp32, gp59 prefers to co-localize with gp32 and not with the forked DNA, as shown in Fig. 5c. The K value is very similar to the result in the absence of forked DNA (K = 0.27) (Table 2, No. 6). In the presence of DNA, a significant increase of ∼ 13-fold in the binding affinity parameter was observed (Table 2, No. 10). This implies that the higher oligomerization of gp59 is facilitated by the presence of a forked DNA + gp32 combination.
Upon adding the gp41 helicase to the above-mentioned proteins (gp59 and gp32) in the presence of forked DNA, we noted an abrupt increase in the value of the binding parameter from K = 0.32 to K = 0.83 (Table 2, No. 12). This >2-fold increase indicates that binding of gp59 to the forked DNA is further stabilized by gp41 in the presence of gp32. Likewise, the affinity parameter increases by ∼2-fold to a maximum value (σ = 0.93). This is consistent with the higher stability of the ternary complex of forked DNA -gp59-gp32-gp41 in the replisome assembly as modeled in Fig. 5d. These results provide evidence for gp59 forming a hexameric complex on the replication fork and loading the gp41 hexamer at a 1:1 stoichiometry (16, 17).
The case in which we have all necessary proteins (gp59 + gp32 + gp41) with forked DNA (Table 2, No. 12) can be compared with that without forked DNA (No. 8). The results show a significant decrease in the value of K from 0.83 to ∼0.43. It is known that gp59 interacts with gp32 via the N terminus and that the C terminus is involved in binding to gp41 (11, 13). It is interesting to note that the value of σ decreases by ∼15-fold in the absence of forked DNA. This clearly indicates that the proteins interact off the forked DNA but do not favor higher oligomerization. From these collective results, we can conclude that hexameric complexes of gp59 are favored in the presence of forked DNA and that the complex is stabilized by gp32 and gp41. Further comparison of the experimental sets (Table 2, No. 7 versus 8 and No. 11 versus 12) allows us to deduce that the presence of gp32 is, however, not a major contributor to either the equilibrium association constant or the affinity parameter.
In summary, our results strongly support the formation of a gp59 hexameric complex in the T4 replisome. This is the first unambiguous experimental observation of the hexameric form of gp59 in the presence of forked DNA, gp32, and gp41. With this result, the understanding of the stoichiometry of a functional pre-primosome complex is more complete. The smPB experiments provide a significant distinction between protein-protein and protein-DNA interactions centered on gp59. The observation of a gp59 hexamer supports the hypothesis that this functional form assists in loading the hexameric helicase gp41 and has a strong interaction with the forked DNA arm and gp32. Our results also support the initiation of replication at a D-loop, where gp59 first binds and then recruits gp32 and gp41 so that the fork arm becomes a lagging strand template (18).
Supplementary Material
Acknowledgments
We thank Dr. Michelle M. Spiering for protein samples and Dr. Padmaja P. Misra for help with smPB experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant GM13306 (to S. J. B.).
We dedicate this work to the late Prof. Nancy G. Nossal for her seminal work on T4 DNA replication.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
F. T. Ishmael, unpublished data.
- smPB
- single-molecule photobleaching.
REFERENCES
- 1.Benkovic S. J., Valentine A. M., Salinas F. (2001) Annu. Rev. Biochem. 70, 181–208 [DOI] [PubMed] [Google Scholar]
- 2.Xi J., Zhang Z., Zhuang Z., Yang J., Spiering M. M., Hammes G. G., Benkovic S. J. (2005) Biochemistry 44, 7747–7756 [DOI] [PubMed] [Google Scholar]
- 3.Nelson S. W., Yang J., Benkovic S. J. (2006) J. Biol. Chem. 281, 8697–8706 [DOI] [PubMed] [Google Scholar]
- 4.Kreuzer K. N., Yap W. Y., Menkens A. E., Engman H. W. (1988) J. Biol. Chem. 263, 11366–11373 [PubMed] [Google Scholar]
- 5.Kreuzer K. N. (2000) Trends Biochem. Sci. 25, 165–173 [DOI] [PubMed] [Google Scholar]
- 6.Dudas K. C., Kreuzer K. N. (2005) J. Biol. Chem. 280, 21561–21569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yonesaki T. (1994) J. Biol. Chem. 269, 1284–1289 [PubMed] [Google Scholar]
- 8.Mueser T. C., Jones C. E., Nossal N. G., Hyde C. C. (2000) J. Mol. Biol. 296, 597–612 [DOI] [PubMed] [Google Scholar]
- 9.Jones C. E., Mueser T. C., Nossal N. G. (2000) J. Biol. Chem. 275, 27145–27154 [DOI] [PubMed] [Google Scholar]
- 10.BiocheXu H., Wang Y., Bleuit J. S., Morrical S. W. (2001) Biochemistry 40, 7651–7661 [DOI] [PubMed] [Google Scholar]
- 11.Ishmael F. T., Alley S. C., Benkovic S. J. (2002) J. Biol. Chem. 277, 20555–20562 [DOI] [PubMed] [Google Scholar]
- 12.Delagoutte E., von Hippel P. H. (2005) J. Mol. Biol. 347, 257–275 [DOI] [PubMed] [Google Scholar]
- 13.Lefebvre S. D., Wong M. L., Morrical S. W. (1999) J. Biol. Chem. 274, 22830–22838 [DOI] [PubMed] [Google Scholar]
- 14.Chastain P. D., 2nd, Makhov A. M., Nossal N. G., Griffith J. (2003) J. Biol. Chem. 278, 21276–21285 [DOI] [PubMed] [Google Scholar]
- 15.Nossal N. G., Makhov A. M., Chastain P. D., 2nd, Jones C. E., Griffith J. D. (2007) J. Biol. Chem. 282, 1098–1108 [DOI] [PubMed] [Google Scholar]
- 16.Raney K. D., Carver T. E., Benkovic S. J. (1996) J. Biol. Chem. 271, 14074–14081 [DOI] [PubMed] [Google Scholar]
- 17.San Martin C., Radermacher M., Wolpensinger B., Engel A., Miles C. S., Dixon N. E., Carazo J. M. (1998) Structure 6, 501–509 [DOI] [PubMed] [Google Scholar]
- 18.Jones C. E., Mueser T. C., Dudas K. C., Kreuzer K. N., Nossal N. G. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8312–8318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norcum M. T., Norcum M. T., Warrington J. A., Spiering M. M., Ishmael F. T., Trakselis M. A., Benkovic S. J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3623–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das S. K., Darshi M., Cheley S., Wallace M. I., Bayley H. (2007) ChemBioChem 8, 994–999 [DOI] [PubMed] [Google Scholar]
- 21.Shu D., Zhang H., Jin J., Guo P. (2007) EMBO J. 26, 527–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J., Trakselis M. A., Roccasecca R. M., Benkovic S. J. (2003) J. Biol. Chem. 278, 49828–49838 [DOI] [PubMed] [Google Scholar]
- 23.Ishmael F. T., Alley S. C., Benkovic S. J. (2001) J. Biol. Chem. 276, 25236–25242 [DOI] [PubMed] [Google Scholar]
- 24.Xi J., Zhuang Z., Zhang Z., Selzer T., Spiering M. M., Hammes G. G., Benkovic S. J. (2005) Biochemistry 44, 2305–2318 [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z., Spiering M. M., Trakselis M. A., Ishmael F. T., Xi J., Benkovic S. J., Hammes G. G. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3254–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha T., Rasnik I., Cheng W., Babcock H. P., Gauss G. H., Lohman T. M., Chu S. (2002) Nature 419, 638–641 [DOI] [PubMed] [Google Scholar]
- 27.van Oijen A. M. (2008) Nat. Chem. Biol. 4, 440–443 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.