

Abstract
Inhibition of the TOR signalling pathway by genetic or pharmacological intervention extends lifespan in invertebrates, including yeast, nematodes and fruit flies1–5. However, whether inhibition of mTOR signalling can extend life in a mammalian species was unknown. We report here that rapamycin, an inhibitor of the mTOR pathway, extends median and maximal lifespan of both male and female mice when fed beginning at 600 days of age. Based on age at 90% mortality, rapamycin led to an increase of 14% for females and 9% for males. The effect was seen at three independent test sites in genetically heterogeneous mice, chosen to avoid genotype-specific effects on disease susceptibility. Disease patterns of rapamycin-treated mice did not differ from those of control mice. In a separate study, rapamycin fed to mice beginning at 270 days of age also increased survival in both males and females, based on an interim analysis conducted near the median survival point. Rapamycin may extend lifespan by postponing death from cancer, by retarding mechanisms of ageing, or both. These are the first results to demonstrate a role for mTOR signalling in the regulation of mammalian lifespan, as well as pharmacological extension of lifespan in both genders. These findings have implications for further development of interventions targeting mTOR for the treatment and prevention of age-related diseases.
Because incidences of most diseases rise rapidly with age6, interventions that delay ageing would greatly benefit health7–8. To date, dietary additives that delay ageing and increase lifespan in rodent models have shown only weak effects9–11. Before clinical studies are considered, anti-ageing interventions must be repeatable and effective in many mouse genotypes, and not merely postpone strain-specific diseases12–14.
The National Institute on Aging Interventions Testing Program (ITP) evaluates agents that may delay ageing and increase lifespan in genetically heterogeneous mice15–17. Agents are chosen as summarized at www.nia.nih.gov/ResearchInformation/ScientificResources/InterventionsTestingProgram.htm. Studies are simultaneously replicated at three test sites: The Jackson Laboratory (TJL), the University of Michigan (UM), and the University of Texas Health Science Center (UT). BALB/cByJ × C57BL/6J F1 (CB6F1) females and C3H/HeJ × DBA/2J F1 (C3D2F1) males are supplied to each site by The Jackson Laboratory, and mated to produce genetically heterogeneous populations in which each animal is genetically unique, but a full sibling of all other mice in the population18. Sufficient mice are used to provide 80% power to detect a 10% increase (or decrease) in mean lifespan with respect to unmanipulated controls of the same sex, even if data from one of the three test sites were to be unavailable. Here we report that dietary encapsulated rapamycin increases mouse survival, including survival to the last decile, a measure of maximal lifespan.
Rapamycin reduces function of the rapamycin target kinase TOR and has anti-neoplastic activities, and genetic inhibition of TOR extends lifespan in short-lived model organisms. In male and female mice at each of three collaborating research sites, median and maximum lifespan were extended by feeding encapsulated rapamycin starting at 600 days of age (Figure 1). We analyzed the dataset as of February 1, 2009, with 2% (38 of 1901) of mice still alive. For data pooled across sites, a log-rank test rejected the null hypothesis that treatment and control groups did not differ (p < 0.0001); mice fed rapamycin were longer lived than controls (p < 0.0001) in both males and females. Expressed as mean lifespan, the effect sizes were 9% for males and 13% for females in the pooled dataset. Expressed as life expectancy at 600 days (the age of first exposure to rapamycin), the effect sizes were 28% for males and 38% for females. Mice treated with other agents (enalapril and CAPE) evaluated in parallel did not differ from controls at the doses used (Supplemental Figure 1).
Figure 1.
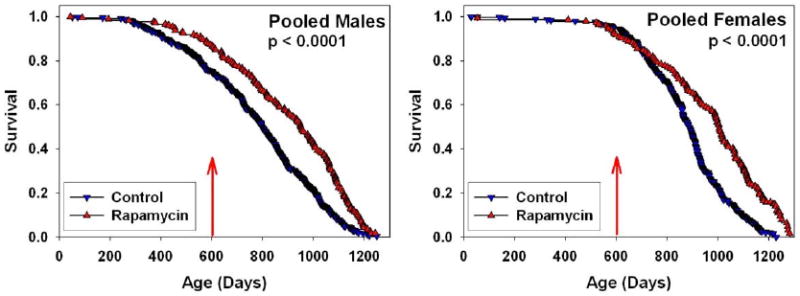
Survival plots for male (left) and female (right) mice, comparing control mice to those fed rapamycin in the diet starting at 600 days of age, pooling across the three test sites. P-values were calculated by the log-rank test. 4% of the control mice, and 3% of rapamycin-assigned mice were removed from the experiment for technical reasons. Only 5 animals (3 controls, 2 rapamycin) were removed after the start of rapamycin treatment at 600 days. Thus there were no significant differences between groups in censoring.
Rapamycin-fed and control mice were then compared separately for each combination of site and gender. Rapamycin had a consistent benefit, compared to controls, with p-values ranging from 0.03 to 0.0001 (Figure 2).
Figure 2.
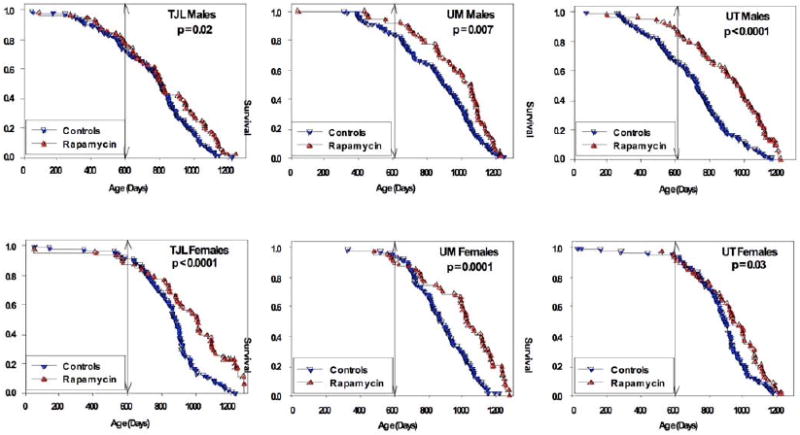
Survival of control and rapamycin-treated mice for males (top) and females (bottom) for each of the three test sites separately. P-values represent results of log-rank calculations. Vertical lines at age 600 days indicate the age at which the mice were first exposed to rapamycin.
Female mice at all three sites had improved survival after rapamycin feeding (Figure 2). Mean lifespan increases for females were 15%, 16%, and 7% (TJL, UM, UT), and life expectancy at 600 days increased by 45%, 48%, and 22% for females at the three sites. Median lifespan estimates of control females were consistent across sites (881–895 days), and were similar to values noted in Cohort 2004, which ranged from 858 to 909 days15. Thus the improvement in survival seen in the rapamycin-fed females is not an artefact of low survival for the control females.
Male mice at all three sites also had improved survival after rapamycin feeding (Figure 2). Mean lifespan increases for males were 5%, 8%, and 15% (TJL, UM, UT), and male life expectancy at 600 days increased by 16%, 23% and 52%. Interpretation is somewhat complicated by differences among sites in survival of control males, and because mice assigned to the rapamycin-fed group at UT and perhaps at UM had lower mortality prior to 600 days than controls. Control mice at UT and UM differed from those fed rapamycin, not only in exposure to rapamycin from 600 days of age, but also in specific formulation of the mouse chows (all based on the NIH-31 standard) used between weaning and 600 days. We thus cannot rule out the possibility that improved survival among males in the rapamycin group, at UT and at UM, might reflect differences in nutritional or health status between control and rapamycin groups prior to 600 days, rather than solely the effects of rapamycin. Importantly, the significant benefits of rapamycin on male (and female) survival at TJL could not have been affected by diet prior to drug administration, because at TJL both control and rapamycin-fed mice received the same chow (Purina 5LG6) throughout this period.
Maximum lifespan was increased by rapamycin feeding. Table 1 shows the ages at the 90th percentile for control and rapamycin-treated mice, along with the 95% upper confidence bound for the controls. For each site and sex, the 90th percentile age for rapamycin-treated mice is higher than the upper limit for the corresponding control group, showing that rapamycin increases the age for 90th percentile survival.
Table 1. The effect of rapamycin on maximum lifespan.
| Comparison | Sites | Age at 90th Percentile for Controls (UL*) | Age at 90th Percentile for Rapa | Percent Increase |
|---|---|---|---|---|
| Females, Rapa vs Control | All sites | 1094 (1136) | 1245 | 14% |
| Females, Rapa vs Control | TJL | 1100 (1165) | 1282 | 17% |
| Females, Rapa vs Control | UM | 1094 (1149) | 1250 | 14% |
| Females, Rapa vs Control | UT | 1089 (1159) | 1179 | 8% |
| Males, Rapa vs Control | All sites | 1078 (1111) | 1179 | 9% |
| Males, Rapa vs Control | TJL | 1035 (1091) | 1142 | 10% |
| Males, Rapa vs Control | UM | 1141 (1177) | 1188 | 4% |
| Males, Rapa vs Control | UT | 1020 (1101) | 1179 | 16% |
Note: “UL” = upper limit of the 95% confidence interval for control mice. For example, top row, for females pooled across sites, the 95% confidence interval for controls goes up to 1136 days, and the estimate for 90th percentile survival for the rapamycin-treated mice is 1245 days. This gives good evidence that the 90th percentile survival for Rapa (1245) is substantially above that for Controls (1094).
To determine if increases in maximal lifespan due to rapamycin feeding are statistically significant, we compared the proportion of living mice in each group when 90% had died in the joint life table19 (Details in Supplementary Table S1). Summing across the three sites, 4.8% of the female control mice were alive at these ages, compared to 21.5% of the rapamycin-treated females (p < 0.0001). For males, the corresponding values were 5.9% of controls and 20.2% of rapamycin mice (p < 0.0001). The site-specific calculations documented a significant effect on females at both TJL (p = 0.0006) and UM (p = 0.0001); for males we noted a significant effect at both TJL (p = 0.008) and UT (p = 0.0001), with a marginal effect at UM (p = 0.07). Rapamycin initiated at 600 days of age thus leads to a significant increase in maximal lifespan.
To test if the spectrum of lesions was altered by dietary rapamycin, complete necropsies were conducted on 31 control and 40 rapamycin-fed mice that were either found dead or euthanized when moribund (Details in Supplemental Table S2). Although rapamycin postpones death, it did not change the distribution of presumptive causes of death.
A separate group of mice was used to evaluate the effects of encapsulated rapamycin initiated at 270 days of age (Figure 3A). At the time of analysis, 51% of the females and 68% of the males had died, and a stratified log-rank test showed significantly lower mortality risk in the rapamycin-treated mice compared to controls, pooling across the three test sites (p = 0.0002 for males and p < 0.0001 for females). When each site was evaluated separately, the beneficial effect of rapamycin for females was significant at each site (p < 0.005); for males, the effect was significant (p < 0.025) at UM and UT, but not at TJL. Rapamycin appears to reduce mid-life mortality risk when started at 270 days of age, but additional data are needed to provide an accurate estimate of effect size, and to evaluate effects on maximal longevity.
Figure 3.
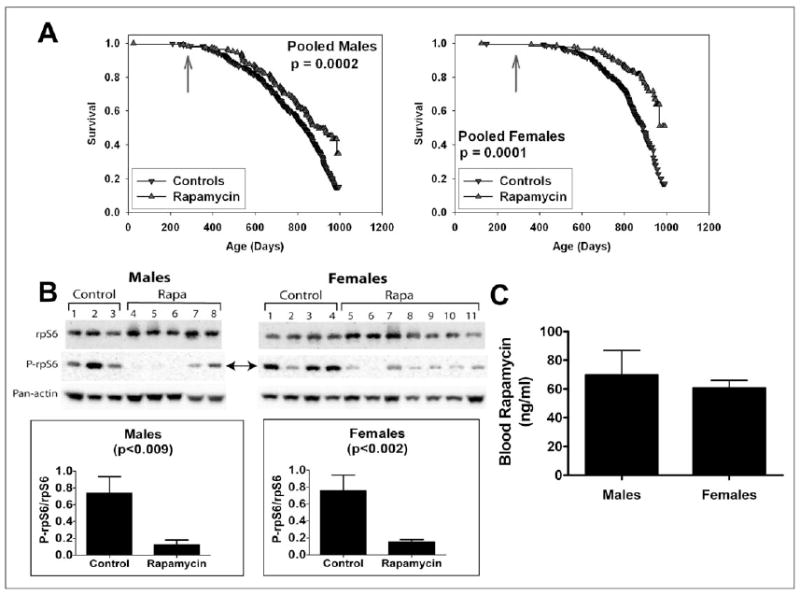
A. Survival plots for male (left) and female (right) mice, comparing control mice to rapamycin-treated mice of a separate (“Cohort 2006”) population, in which mice were treated with rapamycin from 270 days of age. Because at the time of the interim analysis all live mice were between 800 and 995 days of age, we have only limited information about the shape of the survival curve at ages above 900 days, and the apparent change in slope at the oldest ages (> 990 days) reflects this experimental uncertainty. P-values calculated by the log-rank test. B. Effects of dietary rapamycin on an mTORC1 effector in the visceral fat pads from 750- to 880-day-old male and female mice. Ribosomal subunit protein S6 (rpS6) and its phosphorylation status (P-rpS6 - double arrow) were immunoassayed in tissue lysates prepared from mice consuming microencapsulated rapamycin-containing (Rapa) or control diets. Antibodies used are shown to the left. The ratio of intensity values for P-rpS6: rpS6 are shown in the graphs for females and males. Pan-actin was also immunoassayed in the blots to provide an indication of protein loading for each lane. C. Whole blood rapamycin content in 750- to 880-day-old male and female mice.
To document biochemical effects of rapamycin at the dose used for the lifespan studies, we evaluated the phosphorylation status of ribosomal protein subunit S6 (rpS6), a target substrate of S6 kinase 1 in the mTOR signalling pathway20, in visceral white adipose tissue (a sensitive indicator of mTOR inhibition by rapamycin treatment in vivo). Figure 3B shows that rapamycin reduced phosphorylated rpS6 4–5 fold when fed from 270 to about 800 days of age. Blood levels of rapamycin in the treated mice were equivalent in males and females, between 60 and 70 ng/ml.
Initial evidence that reduced TOR function can extend longevity came primarily from studies in yeast1,2 and invertebrates3–5. Beneficial effects of diet restriction (DR)21 and dwarf mutations, both of which extend lifespan in rodents, may to some degree result from repression of the mTOR complex 1 (mTORC1) pathway22,23. It is not yet known to what extent inhibition of mTOR will recapitulate other aspects of the phenotypes associated with DR or dwarf mutations. Our demonstration that rapamycin feeding increases lifespan even when started late in life, as well as the absence of changes in body weight (data not shown), distinguish our results from studies using DR; in all cases DR reduces body weight, and in most reports21, though not all24, DR produces little if any benefit if started after about 550 days of age.
Rapamycin may extend lifespan in old genetically heterogeneous mice through a combination of anti-neoplastic effects25,26 and effects on cellular stress resistance and response to nutrient dynamics27–29. The increase in both median and maximum lifespan seen in rapamycin-fed mice is consistent with the hypothesis that inhibiting the mTORC1 pathway retards mammalian ageing, but is not compelling proof that ageing rates are altered, which would require testing whether the intervention decelerates age-dependent changes in multiple organs, cell types, and intra-cellular and extra-cellular processes14. Comparing effects of rapamycin treatment and other models of decelerated ageing will help narrow the list of possible mechanisms for longevity extension.
At the cellular level, mTORC1 helps to coordinate growth and survival responses induced by alterations in nutrient availability, energy status, growth factor stimuli and exposure to potentially lethal cell stresses27–29; this strategic position at the nexus of nutrient/stress sensing pathways may contribute to the importance of TOR function in regulating lifespan in invertebrates and in mammals as well. It is especially noteworthy that rapamycin feeding can extend mouse lifespan even when started late in life; in terms of the percentage of the maximal lifespan, a 600-day-old mouse is roughly the equivalent of a 60-year-old person14. An effective anti-ageing intervention that could be initiated later than the midpoint of the lifespan could prove to be especially relevant to clinical situations, in which the efficacy of anti-ageing interventions would be particularly difficult to test in younger volunteers. Our data justify special attention to the role of the TOR pathway in control of ageing in mammals and in the pathogenesis of late-life illnesses.
Methods Summary
Mice
Specific pathogen free (SPF) mice were produced at each of the 3 test sites by mating CB6F1 females with C3D2F1 males to produce a genetically heterogeneous population15. Weanlings at the 3 sites were fed similar diets (but not identical; see supplementary material for details) until they were started on food containing rapamycin, in Purina 5LG6, at 600 days of age. A second independent study was begun one year later, with rapamycin initiated at 270 days rather than at 600 days of age. The principal endpoint was age at death (for mice found dead at daily inspections) or age at euthanasia (for mice deemed unlikely to survive for more than an additional 48 hrs).
Diet preparation
Rapamycin (from LC Labs, Woburn, MA) was microencapsulated by Southwest Research Institute (San Antonio, TX), using a spinning disk atomization coating process with the enteric coating material Eudragit S100 (Röhm Pharma, Germany). This coating increased the fraction of rapamycin that survived the food preparation process by 3- to 4-fold, and protected the agent from digestion in the stomach16. Encapsulated rapamycin was then incorporated into 5LG6 mouse chow and distributed to all 3 test sites. Rapamycin in blood and diet was measured by HPLC with UV detection as detailed in supplementary materials.
Rapamycin effectiveness
To test effects of rapamycin on mTORC1 targets, we measured phosphorylation of ribosomal protein S6 (Ser240/244), a substrate of S6 kinase 1, in visceral adipose tissue. Adipose tissue was dissected from mice that had been fed rapamycin diet for 420 days; lysates were loaded on a 4–12% gradient PAGE and electrophoresed overnight at 5V. After transfer to membranes, blocking and incubation with appropriate antibodies, total amounts of rpS6 and amounts of phosphorylated rpS6 were quantitated by chemiluminescence. Techniques are detailed in supplementary materials.
Supplementary Material
Supplementary Table S1: Details of calculation for comparison of surviving proportion of mice at the 90th percentile age.
Supplementary Table S2: Lesions in rapamycin-treated mice and in controls at the time of death.
Supplemental Figure 1 legend. Survival plots for male (left) and female (right) mice, comparing control mice to those fed enalapril, CAPE or rapamycin pooling across the three test sites. Enalapril and CAPE were added to the diet at 4 months of age, and rapamycin at 20 months. P-values were calculated by the log-rank test.
Acknowledgments
This work was supported by NIA grants AG022303 (RAM), AG025707 and AG022308 (DEH), and AG022307 and AG13319 (RS), and the Department of Veterans Affairs (RAM and RS). We wish to thank Pamala J. Krason, Patricia J. Harrison, Elizabeth Adler, Vivian Diaz, Jessica Sewald, Lisa Burmeister, Bill Kohler, Melissa Han, and Maggie Lauderdale for reliable technical assistance, Scott Pletcher and Andrzej Galecki for statistical assistance, and Huber Warner for scientific counsel.
Footnotes
Supplementary Information accompanies the paper on www.nature.com/nature.
References
- 1.Kaeberlein M, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 2.Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 4.Kapahi P, et al. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vellai T, et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 6.Kohn RR. Principles of Mammalian Aging. 2nd. Prentice-Hall; NJ: 1978. p. 151. 1978 Fig. 6.6. [Google Scholar]
- 7.Miller RA. Extending life: scientific prospects and political obstacles. Milbank Quarterly. 2002;80:155–174. doi: 10.1111/1468-0009.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olshansky SJ, Perry D, Miller RA, Butler RN. In pursuit of the longevity dividend. The Scientist. 2006;20:28–35. [Google Scholar]
- 9.Schneider EL, Miller RA. Anti-Aging Interventions. In: Tallis R, Fillit H, Brockelhurst JC, editors. Brockelhurst's Textbook of Geriatric Medicine. Churchill Livingstone; New York: 1998. pp. 193–199. [Google Scholar]
- 10.Archer JR, Harrison DE. L-deprenyl treatment in aged mice slightly increases lifespans, and greatly reduces fecundity by aged males. J Gerontol Biol Sci. 1996;51A:B448–453. doi: 10.1093/gerona/51a.6.b448. [DOI] [PubMed] [Google Scholar]
- 11.Schneider EL, Reed JD., Jr Life extension. New Engl J Med. 1985;312:1159–1168. doi: 10.1056/NEJM198505023121805. [DOI] [PubMed] [Google Scholar]
- 12.Phelan JP, Austad SN. Selecting animal models of human aging. Inbred strains often exhibit less biological uniformity than F1 hybrids. J Gerontol. 1994;49:B1–11. doi: 10.1093/geronj/49.1.b1. [DOI] [PubMed] [Google Scholar]
- 13.Klebanov SE, et al. Maximum life spans in mice are extended by wild strain alleles. Exp Biol Med. 2001;226(9):854–859. doi: 10.1177/153537020122600908. [DOI] [PubMed] [Google Scholar]
- 14.Flurkey K, Currer JM, Harrison DE. The Mouse in Aging Research. In: Fox JG, et al., editors. The Mouse in Biomedical Research. Second. III. Academic Press; 2007. pp. 637–672. [Google Scholar]
- 15.Miller RA, et al. An Aging Interventions Testing Program: study design and interim report. Aging Cell. 2007;6:565–575. doi: 10.1111/j.1474-9726.2007.00311.x. [DOI] [PubMed] [Google Scholar]
- 16.Nadon NL, et al. Design of Aging Intervention Studies: the NIA Interventions Testing Program. 2008. AGE online - http://dx.doi.org/10.1007/s11357-008-9048-1. [DOI] [PMC free article] [PubMed]
- 17.Strong R, et al. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell. 2008;7:641–650. doi: 10.1111/j.1474-9726.2008.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roderick TH. Selection for radiation resistance in mice. Genetics. 1963;48:205–216. doi: 10.1093/genetics/48.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, Li Q, Redden DT, Weindruch RD, Allison B. Statistical methods for testing effects on “maximum lifespan”. Mech Ageing Dev. 2004;125(9):629–632. doi: 10.1016/j.mad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N. mTOR signaling: implications for cancer and anticancer therapy. Br J Cancer. 2007;96(Suppl):R11–15. [PubMed] [Google Scholar]
- 21.Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev. 2005;126:913–922. doi: 10.1016/j.mad.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Sharp ZD, Bartke A. Evidence for down-regulation of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-dependent translation regulatory signaling pathways in Ames dwarf mice. J Gerontol A Biol Sci Med Sci. 2005;60:293–300. doi: 10.1093/gerona/60.3.293. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh CC, Papaconstantinou J. Akt/PKB and p38 MAPK signaling, translational initiation and longevity in Snell dwarf mouse livers. Mech Ageing Dev. 2004;125:785–798. doi: 10.1016/j.mad.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Dhahbi JM, et al. Temporal linkage between the phenotypic and genomic responses to caloric restriction. PNAS. 2004;101:5524–5529. doi: 10.1073/pnas.0305300101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garber K. Rapamycin's resurrection: a new way to target the cancer cell cycle. J Natl Cancer Inst. 2001;93:1517–1519. doi: 10.1093/jnci/93.20.1517. [DOI] [PubMed] [Google Scholar]
- 26.Lorberg A, Hall MN. TOR: the first 10 years. Curr Top Microbiol Immunol. 2004;279:1–18. doi: 10.1007/978-3-642-18930-2_1. [DOI] [PubMed] [Google Scholar]
- 27.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene. 2006;25:6373–6383. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- 29.Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell. 2007;28:721–729. doi: 10.1016/j.molcel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 30.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: Details of calculation for comparison of surviving proportion of mice at the 90th percentile age.
Supplementary Table S2: Lesions in rapamycin-treated mice and in controls at the time of death.
Supplemental Figure 1 legend. Survival plots for male (left) and female (right) mice, comparing control mice to those fed enalapril, CAPE or rapamycin pooling across the three test sites. Enalapril and CAPE were added to the diet at 4 months of age, and rapamycin at 20 months. P-values were calculated by the log-rank test.