

Abstract
Helicobacter pylori strains harboring the cag pathogenicity island (PAI) have been associated with more severe gastric disease in infected humans. The cag PAI encodes a type IV secretion (T4S) system required for CagA translocation into host cells as well as induction of proinflammatory cytokines, such as interleukin-8 (IL-8). cag PAI genes sharing sequence similarity with T4S components from other bacteria are essential for Cag T4S function. Other cag PAI-encoded genes are also essential for Cag T4S, but lack of sequence-based or structural similarity with genes in existing databases has precluded a functional assignment for the encoded proteins. We have studied the role of one such protein, Cag3 (HP0522), in Cag T4S and determined Cag3 subcellular localization and protein interactions. Cag3 is membrane associated and copurifies with predicted inner and outer membrane Cag T4S components that are essential for Cag T4S as well as putative accessory factors. Coimmunoprecipitation and cross-linking experiments revealed specific interactions with HpVirB7 and CagM, suggesting Cag3 is a new component of the Cag T4S outer membrane subcomplex. Finally, lack of Cag3 lowers HpVirB7 steady-state levels, further indicating Cag3 makes a subcomplex with this protein.
Helicobacter pylori infects 50% of the world population. Stomach infection with this bacterium is associated with the development of several gastric diseases, including chronic active gastritis, peptic ulcers, gastric cancer, and mucosa-associated lymphoid tissue lymphoma. Factors influencing disease outcomes are not completely understood, but bacterial, host, and environmental factors have been identified that affect the dynamics of this bacterium-host interaction (30). A hallmark of H. pylori infection is the induction of mucosal inflammation, which is a risk factor for developing more severe pathology (27).
Epidemiological studies have established that infection with strains harboring the cag pathogenicity island (PAI) leads to a higher risk for development of severe disease (27). The cag PAI size varies between 35 and 40 kb and encodes 27 putative proteins (1, 13). Several of the encoded proteins share sequence similarities with components of the prototypical type IV secretion (T4S) system VirB/D4 of Agrobacterium tumefaciens (15, 16). Based on research done in A. tumefaciens, the components of the molecular machinery have been divided into channel or core complex components (VirB6, VirB7, VirB8, VirB9, and VirB10), energetic components (VirB11, VirB4, and VirD4), and extracellular appendage components (VirB2 and VirB5). VirB6, VirB8, and VirB10 are components anchored at the inner membrane with domains spanning the periplasm, while VirB7 and VirB9 are located at the outer membrane. Energetic components are located at the inner membrane, and pilus components include the main subunit VirB2 and accessory components, such as VirB5, which functions as an adhesin (15, 16). The VirB/D4 T4S is thought to be energized by the inner membrane ATPases, and this energy is transduced to VirB10 and the outer membrane complex for protein translocation (11). The lipoprotein VirB7 is critical for the stability of HpVirB9 at the outer membrane (19).
While the extent of homology of the H. pylori cag T4S components is often limited, sequence analysis has allowed the identification of the VirB11 (HP0525 and HpVirB11), VirB10 (HP0527 and HpVirB10), VirB9 (HP0528 and HpVirB9), and VirD4 (HP0524 and HpVirD4) homologues as summarized in Table S1 of the supplemental material (1, 13, 28). HpVirB9 and HpVirB10 homologies are not distributed along the entire length of the protein. For example, HpVirB10 is a very large protein with only a short domain similar to VirB10. HpVirB10 is also reported to localize on the external surface of the pilus (31), while VirB10 is tethered in the inner membrane. HP0529 (HpVirB6) and HP0530 (HpVirB8) have been assigned as homologs of VirB6 and VirB8, respectively (28). HP0523 (HpVirB1) has lytic transglycosylase activity, supporting its designation as a VirB1 homolog (38). HP0532 (HpVirB7) has a lipoprotein attachment site, suggesting a role as a VirB7 homolog (1, 28), and has been suggested to stabilize a Cag T4S outer membrane subcomplex containing CagM, HpVirB9, and HpVirB10 (28).
The activity of the cag PAI-encoded T4S system is responsible for the translocation of the effector protein CagA and induction of proinflammatory chemokine and cytokine secretion, including the chemokine interleukin-8 (IL-8) (7). CagA T4S-mediated translocation into host cells is followed by tyrosine phosphorylation on specific tyrosine phosphorylation motifs (EPIYA motifs) at the C-terminal region of the protein and both phosphorylation-dependent and -independent interference with host cellular pathways. The induction of proinflammatory chemokine production is mediated by a still-uncharacterized Cag T4S-mediated delivery of peptidoglycan into host cells and subsequent activation of Nod receptors (37), and it has also been reported that CagA itself has proinflammatory properties (9). The molecular mechanisms responsible for Cag T4S system assembly and activity remain unclear.
Null alleles of the genes with homology to T4S components (HpVirB11, HpVirB4, HpVirB6, HpVirB7, HpVirB8, HpVirB9, and HpVirB10) abolish both CagA translocation and IL-8 induction, with the exception of HpVirD4, which affects CagA translocation but not IL-8 induction (20). Other genes of the island also essential for Cag T4S function do not share sequence or structural homology with known T4S components. More detailed analysis of these Cag T4S essential genes allowed the recent assignment of several proteins as functional homologs of additional VirB components. HP0546 was suggested as a VirB2 homolog, the main subunit of other T4S system pili (3). Ultrastructural work suggested that HpVirB10 is also a major subunit of the Cag T4S system pilus (31, 35), but clear evidence that either HpVirB2 or HpVirB10 is the main pilus subunit is still lacking. CagL (HP0539) has been identified (29) as an adhesin (functionally similar to VirB5) whose binding to host cell receptors is required for activation of the secretion process, and CagF (HP0543) has been characterized as a CagA chaperone (17). CagD (HP0545) has been recently reported as a multifunctional Cag T4S component essential for CagA translocation and full IL-8 secretion induction (12).
We have characterized the biochemical role of an additional essential H. pylori-specific gene, HP0522/cag3, in Cag T4S. A previous yeast two-hybrid screen that investigated interactions among cag PAI proteins suggested Cag3 could interact with HpVirB8, HpVirB7, CagM (HP0537), and CagG (HP0542) (10). To begin to understand the molecular basis of Cag3 function in T4S we investigated the subcellular localization of the Cag3 protein and the protein-protein interactions this protein establishes in H. pylori cells. We found evidence suggesting that Cag3 is an integral part of the Cag T4S outer membrane subcomplex required to maintain HpVirB7 levels.
MATERIALS AND METHODS
Bacterial strains and growth media.
All H. pylori strains used in this study are listed in Table 1. NSH57 (5) is a mouse-adapted derivative of strain G27 (18). Liquid medium for growth of H. pylori consisted of brucella broth (Difco) supplemented with 10% fetal bovine serum (Invitrogen). H. pylori was maintained on blood agar (HB) plates consisting of 4% Columbia agar base (Oxoid) and 5% defibrillated horse blood (Hemostat Laboratories) supplemented with 0.2% β-cyclodextrin (Sigma), vancomycin (10 μg ml−1; Sigma), cefsulodin (5 μg ml−1; Sigma), polymyxin B (2.5 U ml−1; Sigma), trimethoprim (5 μg ml−1; Sigma), and amphotericin B (8 μg ml−1; Sigma) at 37°C either under a microaerobic atmosphere generated using a CampyGen sachet (Oxoid) in a GasPak jar or in an incubator equilibrated with 14% CO2 and 86% air. AGS gastric epithelial cells (ATCC CRL 1739) were cultured in Dulbecco modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum and grown in a humidified 5% CO2 incubator. For resistance marker selection, bacterial medium was additionally supplemented with kanamycin (50 μg ml−1), chloramphenicol (15 μg ml−1), or sucrose (6%).
TABLE 1.
H. pylori strains used in this study
| Strain or genotype | Description | Source or reference |
|---|---|---|
| NSH57 | Wild type, mouse adapted | 5 |
| G27 | 13 | |
| G27 rdxA::aphA3-sacB | G27 rdxA::aphA3-sacB | 8 |
| NSH57 rdxA::aphA3-sacB | NSH57 rdxA::aphA3-sacB | This study |
| cag3 | NSH57 cag3::cat | This study |
| cag3 rdxA::cag3 | NSH57 cag3::cat rdxA::cag3 | This study |
| cag3 | NSH57 cag3::aphA3 | This study |
| ΔHpvirB4 | NSH57 ΔHpvirB4::cat | This study |
| ΔHpvirB6 | NSH57 ΔHpvirB6::cat | This study |
| ΔHpvirB7 | NSH57 ΔHpvirB7::cat | This study |
| ΔHpvirB8 | NSH57 ΔHpvirB8::cat | This study |
| ΔHpvirB9 | NSH57 ΔHpvirB9::cat | This study |
| ΔHpvirB10 | NSH57 ΔHpvirB10::cat | This study |
| ΔHpvirB11 | NSH57 ΔHpvirB11::cat | This study |
| cagM | NSH57 cagM::Tn3 | This study |
| HpvirB7-3XFLAG | NSH57 HpvirB7::3XFLAG::cat | This study |
| 07KS-B7FLAG | G27 cag2::aphA3-sacB HpvirB7::3XFLAG::cat | This study |
| ΔPAI | NSH57 ΔPAI::aphA3 | This study |
| NSH57 rdxA::cag3 | NSH57 rdxA::cag3 | This study |
| ΔPAI cag3 | NSH5 ΔPAI::aphA3 rdxA::cag3 | This study |
| cag3 HpvirB7-3XFLAG | NSH57 cag3::aphA3 HpvirB7::3XFLAG::cat | This study |
| cagM HpvirB7-3XFLAG | NSH57 cagM::Tn3 HpvirB7::3XFLAG::cat | This study |
Molecular biology.
DNA manipulations (restriction digests, PCR, and agarose gel electrophoresis) were performed according to standard procedures (4). Genomic DNA was prepared from H. pylori by using a Wizard genomic DNA preparation kit (Promega). A list of primers used for PCR and sequencing is given in Table S2 of the supplemental material.
H. pylori isogenic knockout mutants and complemented strains.
All null alleles were constructed in the H. pylori G27 strain background. Mutations were moved to the NSH57 strain background by natural transformation with 1 μg genomic DNA from G27 clones and selection for the appropriate resistance cassette. All experiments in this study used null alleles in the NSH57 background unless otherwise indicated. Null alleles of HpVirB4 (HP0544), HpVirB6 (HP0529), HpVirB7 (HP0532), HpVirB9 (HP0528), HpVirB10 (HP0527), HpVirB11 (HP0525), and HpVirD4 (HP0524) were constructed using a vector-free allelic replacement strategy, as described previously (14), to generate insertion/deletion mutants in which 80 to 90% of the coding sequence of the gene is replaced with a chloramphenicol resistance cassette while preserving the start and stop codons. A similar strategy was used to generate the cag3 mutants, except that the resistance cassette (cat or aphA3) was inserted in the cag3 gene at position 39 of the coding sequence without removal of gene sequence. Additionally, for cag3, the oligo 2 used to produce the N-terminal fragment was designed to introduce a point mutation encoding a premature stop codon at position 29. Primers used for this procedure are listed in Table S2 of the supplemental material. Three clones were evaluated by PCR to confirm replacement of the wild-type allele with the null allele. Urease activity and flagellum-based motility were also confirmed. Strains derived from verified single clones were used for experiments in this study. The chloramphenicol and kanamycin resistance cassettes contain their own promoter but lack transcriptional terminators, and in all cases they were inserted in the same direction of transcription as that of the native gene.
The complemented strain NSH57 (cag3::cat rdxA::cag3) was constructed in two steps. In the first step, genomic DNA from a G27 clone (rdxA::aphA3_sacB; a kind gift from Karen Guillemin [8]) was used to transform NSH57 and produce NSH57 rdxA::aphA3_sacB. This strain was transformed with 10 μg of the pLC292-Cag3 plasmid, and integration into the rdxA locus was selected on sucrose plates. Sucrose-resistant and kanamycin-sensitive clones were selected for further analysis. To generate pLC292-Cag3, cag3 coding sequence plus 250 bp of upstream sequence were cloned into the pLC292 vector (36) by using EcoRI/SalI sites. The cag3 gene was amplified using primers Ucag3Eco and Cag3CSalI. The NSH57 cagM::Tn3 strain was constructed by transformation with genomic DNA derived from G27 cagM::Tn3, a gift from Antonello Covacci (13). The Tn3 insertion is located at position 296 of the cagM coding sequence. The transposon insertion was verified by using CagMF and CagMR primers in combination with primers Kan5′ and Kan3′, which are complementary to the aphA3 gene (conferring kanamycin resistance) encoded in the transposon. The generation of the strain expressing HpVirB7-3XFLAG utilized the same PCR strategy described above for creation of null alleles, adding the FLAG sequences and a stop codon right before the endogenous stop codon in the oligos used to generate stitching product (see Table S2 in the supplemental material). All strains expressing HpVirB7-3XFLAG were constructed by natural transformation with genomic DNA isolated from NSH57: HpvirB7::3XFLAG::cat, including 07KS-B7FLAG (Table 1). 07KS is a cag2 insertion mutant whose defect in Cag T4S can be rescued by Cag3 expression from the rdxA locus (27) (N. R. Salama, unpublished data). NSH57 ΔPAI::aphA3 rdxA::cag3 was constructed by transformation of NSH57 rdxA::cag3 with genomic DNA from G27 ΔPAI::aphA3 (a gift from Manuel Amieva [34]). Clones were selected on kanamycin-supplemented HB plates.
Antibody production.
The predicted mature Cag3 coding sequence (mCag3), lacking the first 23-amino-acid N-terminal signal peptide, was amplified from H. pylori G27 genomic DNA using oligos 5′Cag3Bam5-23 and 3′Cag3EcoR1. The amplified product was directionally cloned into pGEX-2T (Pharmacia) using BamH1 and EcoRI sites to produce pGEX-2T-mCag3. For protein expression pGEX-2T-mCag3 was transferred into the protease-deficient strain E. coli BL21 by electroporation. An overnight culture derived from a single E. coli BL21 colony was used to inoculate 100 ml of Luria broth liquid medium, and purification of the glutathione S-transferase-mCag3 fusion protein was performed as follows: bacterial cells were grown to mid-log phase at 37°C and then transferred to 30°C for induction of protein expression with 0.1 mM isopropyl-β-d-thiogalactopyranoside for 5 h. Cell pellets were collected by centrifugation and resuspended in lysis buffer (50 mM Tris, pH 7.5, 2 mM MgCl2, 0.2% sodium dodecyl sulfate [SDS], 1 M NaCl) plus 15 mM dithiothreitol, 75 μg/ml lysozyme, and 1× Complete (Roche) protease inhibitors. Three freeze-thaw cycles followed by sonication in a Branson sonifier cell disruptor 185 (50% duty, 80% power settings) was performed to break the cells apart. Unbroken cells and unsoluble material were removed by 30 min of centrifugation at 16,000 rpm. Whole-cell extract was added to a 20-ml gluthatione-Sepharose 4B column (Pharmacia) previously reconstituted by washing with lysis buffer. After binding, washes were performed as follows: wash 1, 25 mM Tris, pH 7.5, 1 mM MgCl2, 0.1% SDS, 0.5 M NaCl; washes 2 and 3, thrombin digestion buffer (100 mM NaCl, 20 mM Tris, pH 8.4, 2 mM CaCl2, 0.1% Triton X-100). Glutathione S-transferase was removed by in-column digestion with 0.5 U thrombin/mg of protein (Novagen) for 3 h at room temperature (RT), and Cag3 protein was eluted with 1 M NaCl, 0.1% Triton X-100, 100 mM Tris, pH 8.0, 2 mM MgCl2. The purified recombinant protein (5 mg/ml) was used for antibody production in rabbits by a commercial vendor (RR Rabbitry).
Immunoblotting.
Proteins separated by SDS-polyacryamide gel electrophoresis (SDS-PAGE) were transferred onto polyvinylidene difluoride membranes (Amersham) using a semidry transfer system (Semiphor transphor unit; Amersham) for 2 h at 40 mA. Membranes were blocked for 1 h at RT or overnight at 4°C with 3% nonfat milk-Tris-buffered saline with Tween 20 (TBS-T; 0.5 M Tris, 1.5 M NaCl, pH 7.4, plus 0.05% Tween 20), followed by incubation for 1 h at RT with primary antibody at a 1:10,000 dilution in TBS-T (anti-Cag3 and anti-CagA [2]). Three 15-min washes with TBS-T were followed by a 1-h incubation at RT with horseradish peroxidase-conjugated rabbit immunoglobulin G (Santa Cruz Biotechnology) at a 1:10,000 dilution in TBS-T. After three more TBS-T washes, antibody detection was performed with an ECL Plus immunoblotting detection kit following the manufacturer's protocol (GE Healthcare). The same procedure was used for the antibodies against HSP, a mouse monoclonal antibody directed against an H. pylori-specific heat shock protein, and OMP, a mouse monoclonal antibody directed against an H. pylori outer membrane protein. Both antibodies (US Biological) were used at a 1:100 dilution. Horseradish peroxidase-conjugated mouse immunoglobulin G at a 1:10,000 dilution was used as the secondary antibody (Santa Cruz Biotechnology). For detection of phospho-tyrosine residues the same procedure was used except membranes were blocked for 1 h at RT using 3% bovine serum albumin-TBS-T, followed by incubation for 1 h at RT with primary antibody at 1:1,000 (4G10; Santa Cruz Biotechnology) in TBS-T. Monoclonal anti-FLAG M2 (Sigma) antibody for HpVirB7-FLAG detection was used at a 1:1,000 dilution.
H. pylori cell fractionation.
A total of 1.2 × 1010 H. pylori cells growing on HB plates were harvested and washed twice with phosphate-buffered saline (PBS; 0.14 M NaCl, 2.68 mM KCl, 10.14 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.4). After resuspension in lysis buffer (20 mM Tris, pH 7.9), cells were sonicated three times for 20 s each using a Branson sonifier cell disruptor 185 with a small probe at 60% power and output control 7. After removal of unbroken cells by centrifugation at 4,000 × g for 5 min, the supernatant was recovered and centrifuged at 100,000 × g (SW41 rotor) for 1 h in a Beckman coulter Optima L-90K ultracentrifuge. The supernatant fraction was considered to be a mixture of cytoplasm and periplasm (HSP fraction), and pellets were considered to be the membrane fraction (OMP fraction). Equivalent volumes of the fractions were loaded on 4 to 12% bis-Tris gradient gels (Invitrogen) with buffers following the manufacturer's instructions.
AGS cell infection experiments.
A total of 5 × 104 AGS cells were seeded on 24-well plates for 12 to 16 h before infection. A total of 5 × 105 H. pylori cells from mid-log-phase liquid cultures were added to seeded AGS cells for a multiplicity of infection of 10:1, and infection was allowed to proceed for the indicated time intervals. Supernatants were taken at 3, 6, and 24 h after infection for determination of IL-8 by using the human IL-8 enzyme-linked immunosorbent assay Biotrak system (GE Healthcare) following the manufacturer's protocol. At 24 h, infected cells were washed twice with PBS plus 10 mM sodium orthovanadate and resuspended in 100 μl of 2.5× SDS loading buffer (25% glycerol, 0.125 M Tris-HCl, pH 6.8, 5% SDS, 0.1% bromophenol blue, with 100 mM dithiothreitol added fresh each time). Ten-microliter aliquots of the extracts were used for determination of CagA tyrosine phosphorylation.
Gel filtration chromatography.
A whole-cell lysate was prepared from 7.5 × 109 cells of the appropriate H. pylori strain. Cells were collected from freshly growing solid cultures, washed twice in PBS, and resuspended in 1 ml lysis buffer (50 mM Tris, pH 7.9, 5 mM MgCl2, 150 mM NaCl, 10% glycerol, 5 U DNase [Roche], 0.05 mg/ml RNase A [Roche], and 1× Complete [Roche]). Cells were lysed as for H. pylori cell fractionation. 3-[(3-Cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) detergent was added to a 0.3% final concentration for 20 min at RT. Removal of unbroken cells and unsoluble material was done by centrifugation at 12,000 rpm for 15 min. Supernatant was clarified by filtration through a 0.4-μm filter, and 100 μl of final extract was loaded in a Superdex 200 column. A 100-μl aliquot of whole-cell extract was used to determine the apparent sizes of Cag3 and HpVirB7 complexes. Experiments were performed at a flow rate of 0.3 ml/min on an Akta prime plus chromatography system (Amersham GE) with a small SuperDex 200 HR 10/30 column (Amersham GE). The void volume was determined by running dextran blue 200 resuspended in lysis buffer. Calibration of the column was performed by running a molecular calibration mix (Amersham HMW and LMW gel filtration calibration kits) resuspended in lysis buffer. Fractions (1 ml) were precipitated with 10% trichloroacetic acid. Proteins were resuspended in 50 μl of 2.5× SDS loading buffer, and 10-μl aliquots were loaded in 10% acrylamide gels for determination of Cag3 and HpVirB7-3XFLAG elution volume by immunoblotting.
Coimmunoprecipitation.
A total of 1.2 × 1010 H. pylori cells grown overnight on HB plates were washed twice with PBS and then resuspended in 5 ml NP-40 lysis buffer (50 mM Tris, pH 7.9, 150 mM NaCl, 0.1 mM EDTA, 1% NP-40) plus 1× Complete protease inhibitors (Roche). Cells were broken as described above. Twenty-five units of DNase (Roche) and 0.05 mg/ml RNase A (Roche) were added to the lysate, which was rocked for 20 min at RT. Centrifugation at 4,000 rpm for 5 min removed unbroken cells. The lysate was then passed through a 0.2-μm filter. One milliliter of whole-cell lysate was incubated with 50 μl of protein A-coupled Sepharose CL-4B (Zymed) beads for 1 h at 4°C for preclearing. Beads were pelleted by centrifugation at maximal speed for 30 s. Supernatant (precleared lysate) was incubated with 30 μl of anti-Cag3 protein A-Sepharose beads (prepared by incubation of 3 μl of anti-Cag3 with 30 μl of protein A-Sepharose CL-4B in PBS for 1 h at 4°C) plus 1× Complete protease inhibitors (Roche) for 1 h at 4°C. Beads were pelleted and washed three times with lysis buffer. After the final wash, beads were resuspended in 30 μl of loading buffer, and 10 μl of boiled supernatant was loaded in 10% acrylamide gels. For M2 coimmunoprecipitations, 30 μl of anti-FLAG M2 affinity gel (Sigma) was used after reconstitution in lysis buffer.
Expression of Cag3 in different type IV secretion system backgrounds.
A total of 3 × 108 fresh-growing H. pylori cells were taken from plates, washed twice with PBS, and resuspended in 50 μl of 2.5× loading buffer (0.02 optical density units [OD]/μl). Threefold serial dilutions (18×, 9×, 3×, 1× [1 μl], 1/3×, and 1/9×) were loaded on 10% acrylamide gels for subsequent immunoblot analysis. Membranes were probed with anti-Cag3 antibody (1:10,000) for Cag3 detection. As a loading control blots were reprobed with anti-HSP antibody (1:100) after removal of anti-Cag3 by incubation with stripping buffer (62.5 mM Tris, pH 6.8, 2% SDS) prewarmed to 55°C for 30 min at RT.
Cross-linking experiments.
Cells freshly growing on HB plates were resuspended in PBS buffer and washed twice. A total of 6 × 108 cells were resuspended in 200 μl of PBS and formaldehyde was added to a 1% final concentration. The cross-linking reaction mixture was incubated for 30 min on ice and stopped by addition of 0.125 M glycine, pH 2.0, and a 10-min incubation on ice. After one wash with PBS the cells were pelleted and resuspended in 50 μl 2.5× SDS loading buffer. Ten microliters of cross-linked extract was loaded in 7.5% (for Cag3 cross-linked species) or 10% (for HpVirB7 cross-linked species) bisacrylamide gels. Cross-linked species were identified by immunoblotting as described above.
Affinity chromatography followed by mass spectrometry.
An anti-Cag3 affinity chromatography column was constructed by incubating protein A-Sepharose CL-4B (Zymed) with the anti-Cag3 antibody. Antibody was covalently linked to protein A beads by using the cross-linker dimethyl pimelimidate. Two separate columns were constructed: one for the wild-type extract and another one for the cag3 extract. A whole-cell lysate was prepared from 3 × 1010 cells of the appropriate H. pylori strain, grown for 12 to 16 h in HB plates. Cells were harvested, washed twice in PBS, and resuspended in 5 ml of lysis buffer (50 mM Tris, pH 7.9, 5 mM MgCl2, 150 mM NaCl, 1% NP-40) with DNase, RNase, and 1× Complete protease inhibitors added to the mix. (The detergent NP-40 was used because it is a nondenaturant [protein-protein interactions are thus maintained], and we observed Cag3 was very soluble when cells were broken in its presence.) Cells were broken by three freeze-thaw cycles, and unbroken cells were removed by centrifugation at 8,000 rpm. Lysates were applied in batch to a 1-ml column previously equilibrated with lysis buffer, for 2 h at 4°C. The columns were washed as follows: wash 1, 10 bed volumes of lysis buffer; wash 2, 10 bed volumes of urea buffer (2 M urea, 0.2 M NaCl, 0.1 M Tris, pH 7.5) and 10 bed volumes of high-salt buffer (0.5 M NaCl, 20 mM Tris, pH 8.0); wash 3, 10 bed volumes of low-salt buffer (50 mM NaCl, 10 mM Tris, pH 8.0) (32). Elution was performed with 5 bed volumes of 0.1 M glycine pH 3.0 (to disrupt the anti-Cag3 antibody-Cag3 protein interaction and release all proteins bound to Cag3) into 50 μl of 1 M Tris, pH 7.9, and beads were immediately reconstituted by washing with 10 bed volumes of 1 M Tris, pH 7.9 (22). The eluates were concentrated using Amicon ultra-15 centrifugal filters (molecular weight cutoff, 10,000). The final mix was analyzed at the Mass Spectrometry Facility of the Fred Hutchinson Cancer Research Center (FHCRC) Shared Resource, and data were transferred through automated pipelines to systems supported by the FHCRC Computing Support Shared Resource. Data were analyzed using the Computational Proteomics Analysis System, a database repository and analysis tool developed at the FHCRC. The peptide population was analyzed using Computational Proteomics Analysis System software against the G27 predicted peptide population. Peptides were filtered by using a PeptideProphet score of ≥0.9 and percent ionization of ≥30. The PeptideProphet score provides an estimate of the accuracy of peptide identification made by tandem mass spectrometry (25). The more stringent criteria for protein identification required the detection of more than one peptide representing the respective protein. Proteins present in the wild-type eluates and absent from cag3 eluates were considered to be Cag3-specific interactors. cag PAI-encoded proteins were analyzed independently and were reported even if identified by only one peptide (Table 2). Non-cag PAI-encoded proteins identified by the more stringent criteria are reported in Table S3 of the supplemental material.
TABLE 2.
Proteins of the cag PAI identified by anti-Cag3 affinity purification followed by mass spectrometrya
| Annotated gene | Protein name | No. of WT peptides detected |
Prophet score | No. of cag3 peptides detected |
||
|---|---|---|---|---|---|---|
| Total | Unique | Total | Unique | |||
| HP0520 | Cag1 | 5 | 4 | 1 | 0 | 0 |
| HP0521 | Cag2 | 0 | 0 | 0 | 0 | |
| HP0522 | Cag3 | 66 | 22 | 1 | 0 | 0 |
| HP0523 | HpVirB1 | 0 | 0 | 0 | 0 | |
| HP0524 | HpVirD4 | 3 | 3 | 0.99 | 0 | 0 |
| HP0525 | HpVirB11 | 1 | 1 | 0.98 | 0 | 0 |
| HP0526 | 1 | 1 | 0 | 1 | 1 | |
| HP0527 | HpVirB10 | 1 | 1 | 1* | 1* | |
| HP0528 | HpVirB9 | 1 | 1 | 0.99 | 0 | 0 |
| HP0529 | HpVirB6 | 0 | 0 | 0 | 0 | |
| HP0530 | HpVirB8 | 0 | 0 | 1 | 1 | |
| HP0531 | CagU | 0 | 0 | 0 | 0 | |
| HP0532 | HpVirB7 | 8 | 7 | 1 | 0 | 0 |
| HP0533 | 0 | 0 | 0 | 0 | ||
| HP0534 | CagS | 0 | 0 | 0 | 0 | |
| HP0535 | 1 | 1 | 0.35 | 0 | 0 | |
| HP0536 | 0 | 0 | 0 | 0 | ||
| HP0537 | CagM | 2 | 2 | 0.55 | 0 | 0 |
| HP0538 | CagN | 1 | 1 | 0.13 | 0 | 0 |
| HP0539 | CagL, HpVirB5 | 0 | 0 | 0 | 0 | |
| HP0540 | CagI | 0 | 0 | 0 | 0 | |
| HP0541 | CagH | 0 | 0 | 0 | 0 | |
| HP0542 | CagG | 0 | 0 | 0 | 0 | |
| HP0543 | CagF | 2 | 2 | 0.99 | 1 | 1 |
| HP0544 | HpVirB4 | 1 | 1 | 0.75 | 0 | 0 |
| HP0545 | CagD | 6 | 5 | 1 | 1 | 1 |
| HP0546 | HpVirB2 | 1 | 1 | 0.93 | 0 | 0 |
| HP0547 | CagA | 38 | 28 | 1 | 22 | 19 |
Results shown in bold indicate protein peptides with Prophet scores of ≥0.9, indicating high-confidence protein identification. An asterisk indicates the identified peptide in cag3 extract is different from the identified peptide in wild-type extract.
Pulse-chase assay.
For each strain, cells were grown overnight in DMEM plus 10% PBS dialyzed serum to a 0.8 OD600/ml. After growth, bacterial cells were harvested by centrifugation and washed once with DMEM-Cys-Met (plus 10% dialyzed serum and 1× glutamine [Invitrogen]). A 20-ml culture at 0.4 OD600/ml was started in DMEM-Cys-Met (plus 10% dialyzed serum) and allowed to grow for 1 h. Cells were pelleted and resuspended in 5 ml of prewarmed medium, supplemented with 50 μl of Tran35S-label (Cys-Met) (10 mCi/ml; 500 μCi; MP Biomedicals) for 30 min. After the pulse, excess cold amino acids (2 mM Cys, 2 mM Met) were added and aliquots were taken at 0, 10, 30, and 60 min. At each time point, a 750-μl aliquot was taken and added to the same volume of 0.04% sodium azide solution. Cells were pelleted and then frozen in liquid nitrogen. After the chase, all samples were resuspended in lysis buffer (50 mM Tris, pH 7.9, 150 mM NaCl, 0.1 mM EDTA, 2% Triton X-100, 1× Complete protease inhibitors) and incubated for 20 min on ice. After incubation, cells were disrupted in a Diagenode Bioruptor at high setting, five times for 5 min each on ice. The lysate was centrifuged for 20 min at maximal speed in an Eppendorf microcentrifuge. The supernatant was precleared by incubation with 100 μl of protein A-Sepharose for 1 h at room temperature. Precleared lysate was incubated with the 40 μl of the respective antibody-bound beads for 1 h at 4°C. Beads were pelleted and washed three times with wash buffer (lysis buffer plus 0.1% Triton X-100). Beads were resuspended in 40 μl of loading buffer and boiled 5 min. Samples were fractionated on SDS-PAGE gels. Gels were dried and subsequently analyzed with a phosphorimager. Protein half-life was determined as described previously (6). Briefly, phosphorimager scans were used to quantify protein intensity at each time point, and the natural log of the intensity was plotted versus time to determine the first-order decay degradation constant (k). The half-life was determined using the equation t1/2 = ln2/k.
RESULTS
cag3 is essential for Cag T4S function.
Previous work had suggested that cag3 (HP0522) was essential for Cag T4S, although the absence of complementation left open the possibility that neighboring genes contributed to the observed phenotype (20). We constructed a null cag3 mutant by insertion of a chloramphenicol acetyltransferase cassette in this locus and performed complementation by expression of the gene at the unrelated rdxA locus (33). The functionality of the Cag T4S system was evaluated by incubation of the strain with AGS cells and determination of host cell-catalyzed CagA phosphorylation and induction of IL-8 secretion, two of the in vitro phenotypes associated with Cag T4S activity. The cag3 strain was unable to translocate CagA, as seen by lack of CagA phosphorylation (Fig. 1A), and unable to induce IL-8 (Fig. 1B) when incubated with AGS cells. We confirmed that expression of Cag3 from the rdxA locus was able to rescue these two phenotypes. These results confirm and definitively establish the essential role of the Cag3 protein in Cag T4S function.
FIG. 1.
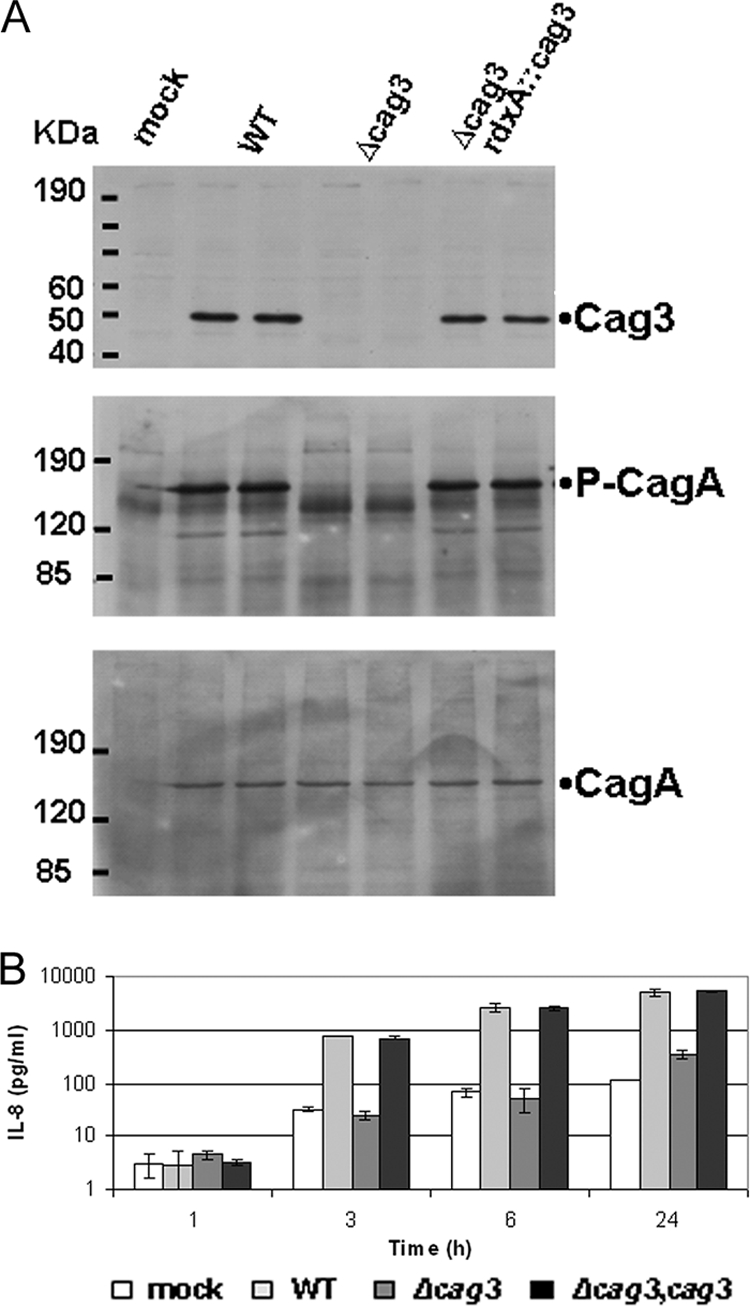
Cag3 is essential for cag PAI-encoded IV secretion. (A) Immunoblot analysis of total protein extracts prepared from AGS cells cocultured with the respective H. pylori strains for 24 h at a multiplicity of infection of 10:1 (in duplicate). Protein extracts were separated by 10% (upper panel) or 6% (middle and lower panels) SDS-PAGE and probed with anti-rabbit Cag3 antibody (upper) or with anti-mouse 4G10 antibody to detect host cell-catalyzed CagA phosphorylation (middle panel) and subsequently probed with anti-rabbit CagA antibody to detect total CagA (lower panel). Molecular mass marker sizes in kDa are indicated. (B) Induction of IL-8 secretion. Medium harvested from the same coculture experiment as shown in panel A was used for determination of secreted IL-8 by enzyme-linked immunosorbent assay. Means from triplicate cultures from a representative experiment are shown. Error bars indicate 1 standard deviation.
Cag3 is a membrane-associated protein.
While Cag3 is required for full activity of the Cag T4S system, the protein does not show sequence homology to any previously described T4S component (or other characterized protein motif). Cag3 does not contain predicted transmembrane helices but does have a predicted N-terminal signal sequence, suggesting the protein is translocated into the periplasmic space and thus may interact directly with other T4S core components. To investigate Cag3 subcellular localization, we fractionated H. pylori cells by selective solubilization and differential centrifugation followed by immunoblotting of each fraction. As seen in Fig. 2, Cag3 was found enriched in the membrane-associated fraction, also enriched for a control OMP, but not the cytoplasmic HSP. While the majority of Cag3 protein is associated with membranes, a soluble pool also appears to exist.
FIG. 2.
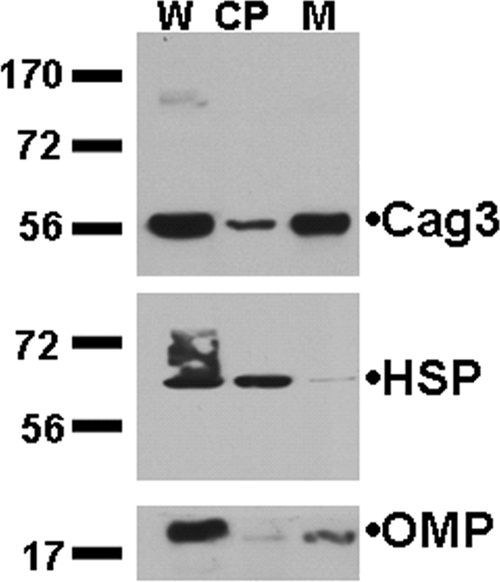
Cag3 fractionates with H. pylori membranes. Equivalent volumes of whole-cell extracts (W), cytoplasmic-periplasmic (CP), and membrane fractions (M) from strain NSH57 were loaded, and immunoblots were probed with antibodies to Cag3, a cytoplasmic 58-kDa H. pylori HSP, or a 19-kDa H. pylori OMP as indicated. Molecular mass marker sizes (in kDa) are indicated. Data shown are representative of three independent experiments.
Cag3 copurifies with predicted T4S components.
To identify Cag3-interacting partners we used affinity purification followed by mass spectrometry. Total extracts from wild-type strains and cag3 strains were loaded onto anti-Cag3 affinity columns, and peptide profiles from eluates were compared. A number of proteins copurified specifically with Cag3 (Fig. 3). Mass spectrometry analysis of the liquid mixtures identified several cag PAI-encoded proteins (Table 2). HpVirB7, HpVirD4, CagD, and Cag1 were identified by more than one peptide and a PeptideProphet score higher than 0.9. Additionally, peptides for these proteins were absent from the column loaded with the cag3 strain. Thus, these four proteins represent the most specific interactors identified based on the abundance of representative peptides with high sequence quality (PeptideProphet) scores. We also identified HpVirB4, HpVirB11, HpVirB10, HpVirB9, CagM, and HpVirB2 by unique peptides present in wild-type columns but absent from cag3 columns. Some of these proteins were identified by Prophet scores lower than 0.9 or identification of only one representative protein peptide. HpVirB2, HpVirB4, HpVirB11, and HpVirB9 were all present in wild-type samples and absent in cag3 samples, and only one representative peptide was identified for these proteins. Prophet scores differed among these proteins. Two unique peptides were found for CagM. HpVirB10 was a special case, since we identified only one peptide in the wild-type mix and a different unique peptide in the cag3 mix. HpVirB10 has two features, a large size (>150-kDa protein) and multiple cysteines that may preclude good detection by the mass spectrometry methods used, but we predicted that the band of highest molecular weight in silver-stained gels (Fig. 3) might correspond to this protein. Excision and mass spectrometry analysis confirmed that this protein band corresponded to HpVirB10, which was thus present in the eluate of wild-type but not cag3 extracts.
FIG. 3.
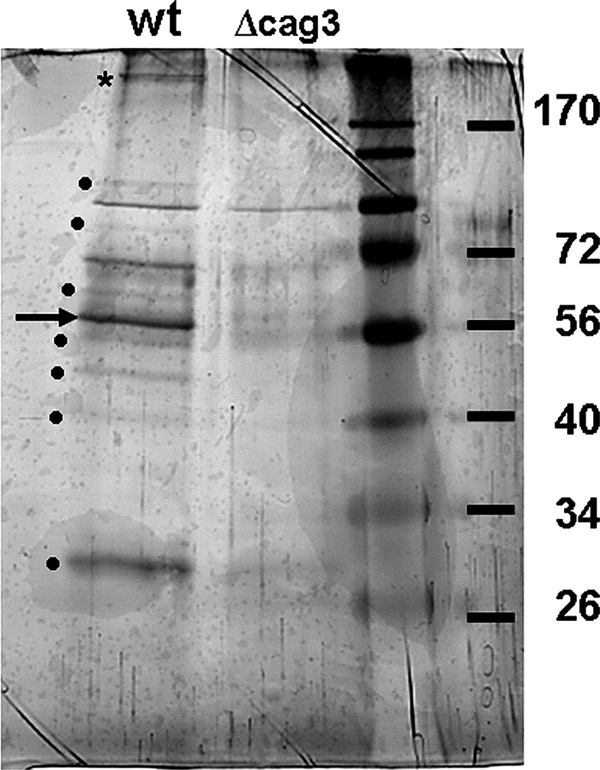
Cag3 copurifies with predicted Cag T4S components. Anti-Cag3 antibody purified a number of proteins from wild-type (wt) extracts but not from cag3 extracts. Dots indicate protein bands present in wild-type extracts that were not present in the cag3::cat extract. The arrow indicates a protein band predicted to be Cag3 protein (55 kDa), and the asterisk indicates the HpVirB10-containing band excised for mass spectrometry analysis (see text). Molecular mass marker sizes in kDa are indicated.
The substrate CagA and its chaperone CagF were identified from both wild-type and cag3 mixes. However, enrichment for CagA and CagF peptides was found in the wild-type column. Thus, CagF and CagA appear to specifically interact with Cag3 under these conditions, although confirmation of this interaction will require an independent approach.
We also identified non-cag PAI proteins in the eluates from wild-type extract that were not detected by cag3 extract affinity purification that included highly abundant proteins and proteins that localize to the cell envelope, such as flagellin subunits (see Table S3 in the supplemental material). Although these proteins may represent Cag3-specific interactions, they may represent false positives from nonspecific binding to Cag3 and associated proteins detected because of the mild purification conditions used. Further experiments are required to address the significance of these interactions.
Cag3 interacts with the predicted lipoprotein HpVirB7 independently of CagM.
We were interested in the possible interaction of Cag3 with HpVirB7, since HpVirB7 has been shown to be part of the outer membrane subcomplex (28), and previous results from a yeast two-hybrid screen (10) had suggested that the two proteins could interact. We fused three FLAG epitopes to the C terminus of HpVirB7 and expressed this protein from its endogenous locus. The HpVirB7-3XFLAG protein retained T4S activity as determined by the ability of strains expressing this protein to induce IL-8 and translocate CagA into host cells (see Fig. S1 in the supplemental material). The interaction of HpVirB7-3XFLAG with Cag3 was confirmed by coimmunoprecipitation (Fig. 4A). Anti-FLAG antibodies were able to coimmunoprecipitate Cag3 in a wild-type strain expressing HpVirB7-3XFLAG. No protein of a size corresponding to Cag3 was observed when HpVirB7-3XFLAG was immunoprecipitated from the cag3 strain. We also confirmed that anti-Cag3 antibodies could pull down HpVirB7-3XFLAG from the wild-type strain but not from the cag3 strain (Fig. 4B). Since it was suggested previously that HpVirB7 interacts with CagM and that this interaction is required for subsequent interaction with HpVirB9 and HpVirB10 (10, 28), we asked whether the Cag3 and HpVirB7 interaction was dependent on CagM. In the absence of CagM, HpVirB7-3XFLAG levels were somewhat reduced (Fig. 4B, ΔcagM versus wild type). However, the interaction between Cag3 and HpVirB7 was still detected in the absence of CagM, as demonstrated by the ability of the anti-Cag3 antibody to pull down HpVirB7-3XFLAG in a ΔcagM mutant (Fig. 4B). This result suggests that Cag3 can interact with HpVirB7 independently of CagM. A more dramatic reduction in HpVirB7 steady-state levels was observed in the absence of Cag3 (Fig. 4B, cag3 input versus wild-type input), suggesting that the Cag3-HpVirB7 interaction stabilizes HpVirB7.
FIG. 4.
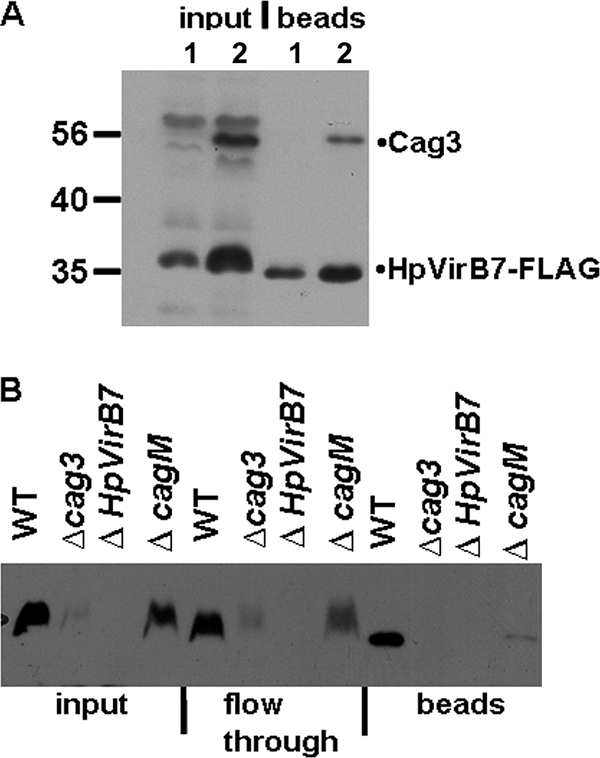
Cag3 interacts with HpVirB7, and this interaction does not require CagM. (A) Whole-cell lysates from a cag3 mutant strain (lanes 1) or wild type (WT; lanes 2) expressing HpVirB7-3XFLAG protein were immunoprecipitated with anti-FLAG antibody, and the input and bead fractions were probed with anti-FLAG and anti-Cag3 antibodies. (B) Anti-Cag3 serum-immunoprecipitated material from the indicated strains containing HpVirB7-3XFLAG was probed with anti-FLAG antibody to detect HpVirB7-3XFLAG. Equivalent volumes of input and flowthrough and one-third of the precipitated bead fractions were separated by SDS-PAGE. HpVirB7-3XFLAG steady-state levels were reduced compared to the wild type in the cagM (cagM::Tn3) strain and further reduced in the cag3 (cag3::aphA3) strain. Molecular mass marker sizes in kDa are indicated. WT, HpvirB7::3XFLAG::cat; cag3, cag3::aphA3 HpvirB7::3XFLAG::cat; ΔHpVirB7, HpvirB7::cat; cagM, cagM::Tn3 HpvirB7::3XFLAG::cat. Data shown are representative of two independent experiments.
Cag3 and HpVirB7 fractionate in a high-molecular-weight complex.
To determine the size of the Cag3 complexes in the cell and to gain further evidence for interactions between Cag3 and HpVirB7, we performed gel filtration chromatography. Whole-cell extracts solubilized in 0.3% CHAPS were loaded in a SuperDex 200 gel filtration column (Fig. 5). Cag3 eluted in two peaks: one peak overlapped with the void volume and the other one corresponded to a 150-kDa peak, both larger than the size predicted for a Cag3 monomer (55 kDa). These results suggest that Cag3 exists in the cell as part of two pools. The lower-molecular-mass form may represent a soluble pool (perhaps the cytoplasmic-periplasmic pool seen in cell fractionation), while the higher-molecular-mass form may represent the Cag3 membrane-associated complex. Interestingly, HpVirB7 comigrated with Cag3 in the void volume but was not seen in any other pool, indicating HpVirB7 assembles into a very large complex which might also contain Cag3, although large Cag3- and HpVirB7-independent complexes may also exist.
FIG. 5.

Cag3 and HpVirB7-3XFLAG fractionate as a high-molecular-mass complex. A whole-cell extract of NSH57, virB7::3XFLAG::cat (WT), was fractionated on a Superdex 200 gel filtration column, and the presence of Cag3 and HpVirB7-3XFLAG in each fraction was determined by immunoblotting with anti-Cag3 or anti-FLAG antibody. Molecular mass marker sizes in kDa are indicated. Fraction 1 is the void volume at which dextran blue eluted, the 443-kDa marker eluted in fraction 3, and the 150-kDa marker eluted in fraction 6. WT, HpvirB7::3XFLAG::cat; cag3, cag3::aphA3. Data shown are representative of two independent experiments.
In a strain lacking the cag PAI but expressing cag3 from the rdxA locus, Cag3 fractionated primarily in a single peak corresponding to the same size of the low-molecular-mass peak observed in wild-type extracts. This result indicates that in the absence of products of the cag PAI, Cag3 does not assemble into the large complex (see Fig. S2 in the supplemental material) but may form a multimeric complex with itself or other proteins not encoded in the cag PAI.
Protein cross-linking reveals HpVirB7-CagM and Cag3-CagM interactions.
We explored the proximity of the Cag3-HpVirB7 interaction by a cross-linking approach. H. pylori cells were cross-linked in vivo with formaldehyde, and cross-linked extracts were loaded on 7.5% acrylamide gels for detection of Cag3 cross-linked species or on 10% acrylamide gels for detection of HpVirB7-3XFLAG cross-linked species. In addition to a monomer size band, several high-molecular-mass Cag3-containing complexes were observed (Fig. 6A). None of these species appeared to contain HpVirB7, since they were still evident in the ΔHpvirB7 mutant. We did, however, detect a Cag3 form that may represent a Cag3-CagM interaction, because it is absent in the cagM mutant and corresponds to the expected size of a heterodimer of these proteins (99 kDa). The Cag3 high-molecular-mass forms were seen in a strain lacking the entire cag PAI and expressing cag3 from the rdxA locus (ΔPAI; rdxA::cag3), suggesting that they represent a Cag3 interaction with itself.
FIG. 6.
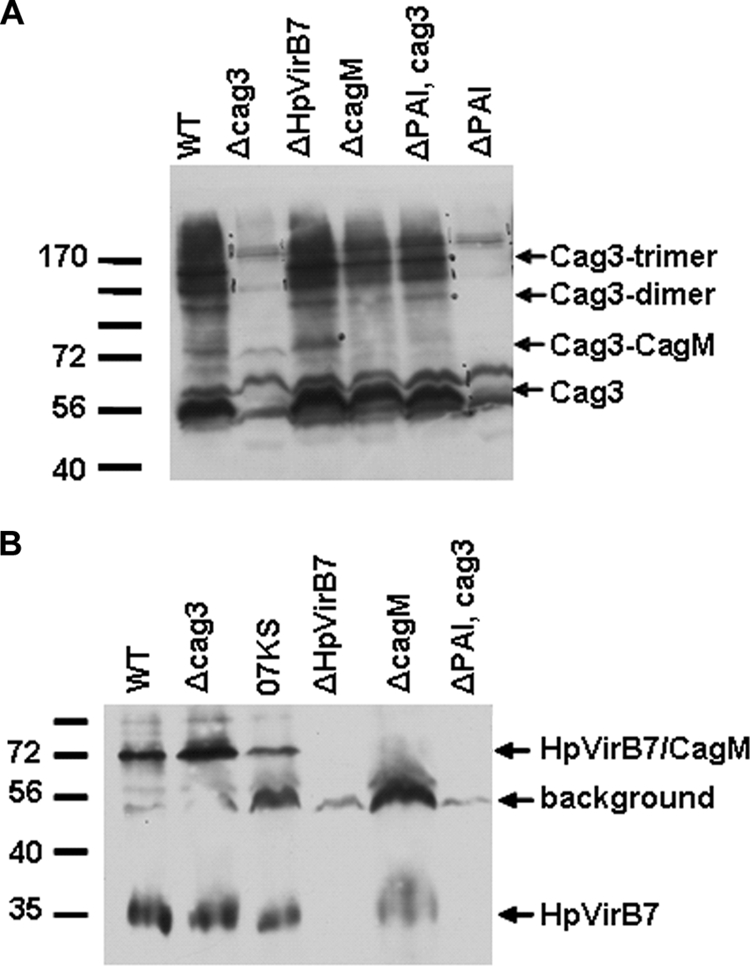
Cross-linking reveals both Cag3 and HpVirB7 interact with CagM, and Cag3 interacts with itself. Formaldehyde cross-linked whole-cell extracts expressing HpVirB7-3XFLAG were fractionated by SDS-PAGE and immunoblotted with anti-Cag3 or anti-FLAG to detect Cag3 (A) or HpVirB7-3XFLAG (B). Molecular mass marker sizes in kDa are indicated. WT, HpvirB7::3XFLAG::cat; cag3, cag3::aphA3 HpvirB7::3XFLAG::cat; 07KS, G27 Δcag2::aphA-sacB HpvirB7::3XFLAG::cat; ΔHpVirB7, HpvirB7::cat; cagM, cagM::Tn3 HpvirB7::3XFLAG::cat; ΔPAI Cag3, cagPAI::aphA3 rdxA::cag3; ΔPAI, cagPAI::aphA3. Data shown are representative of two independent experiments.
In contrast to the large number of cross-linked bands observed with the Cag3 antibody, we detected two main species with the anti-FLAG antibody: a band corresponding to the HpVirB7-3XFLAG monomer size of 35 kDa and a band of approximately 72 kDa (Fig. 6B). This 72-kDa band likely represents an HpVirB7-CagM interaction, because this band was not observed in a cagM strain and is the predicted mass of one monomer each of HpVirB7-3XFLAG (35 kDa) and CagM (44 kDa). This HpVirB7-CagM interaction does not depend on Cag3, because this band was still observed in a cag3 strain.
Cag3 and HpVirB7 promote each other's stability.
Since loss of Cag3 had an effect on HpVirB7 steady-state levels (Fig. 4B), we further explored the possibility that lower HpVirB7 levels were the result of increased protein turnover due to the lack of its Cag3 partner. As shown in Fig. 7, during a 35S metabolic labeling pulse-chase experiment HpVirB7-3XFLAG appeared less stable in the cag3 mutant strain background compared to the wild type. Quantification of the immunoprecipitated band intensities revealed a half-life of 150 min for HpVirB7-3XFLAG in wild-type cells that was reduced to 39 min after mutation of cag3. As a control, we also measured the half-life of CagA in this experiment. The half-life of CagA was 130 min in wild-type cells and 71 min after mutation of cag3.
FIG. 7.
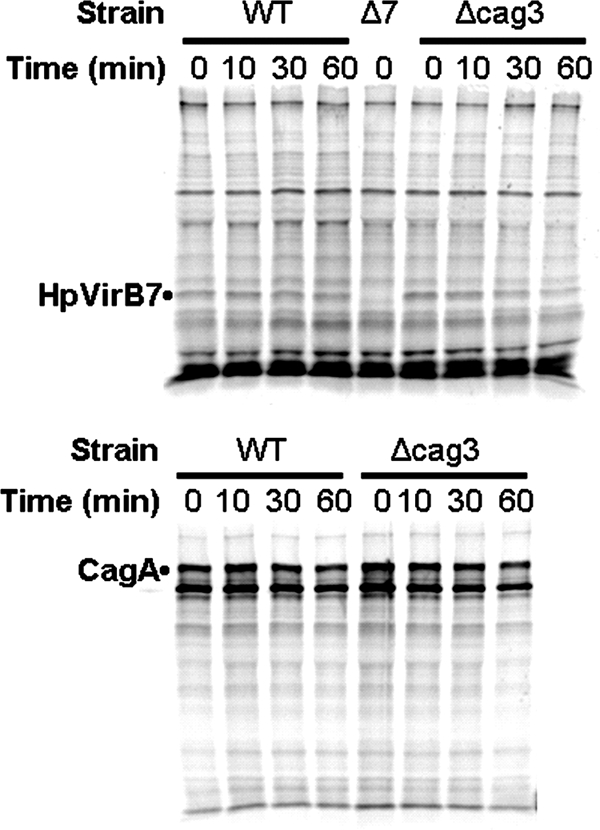
HpVirB7 is unstable in a cag3 strain. The indicated strains were metabolically labeled with Tran35S-label Cys-Met during liquid culture growth for a 30-min pulse. After the pulse, two aliquots were taken from each culture for determination of HpVirB7-3XFLAG and CagA labeling by immunoprecipitation with anti-FLAG (top) or anti-CagA (bottom), respectively, after the indicated time of chase in the presence of unlabeled amino acids. WT, HpvirB7::3XFLAG::cat; Δcag3, cag3::aphA3 HpvirB7::3XFLAG::cat; Δ7, HpvirB7::cat.
Several T4S periplasmic subassemblies have been described in the A. tumefaciens VirB/D4 T4S system, where the loss of one component leads to protein instability of the other subunits (19, 26). We thus examined the steady-state levels of Cag3 in the absence of other predicted T4S components. Cag3 steady-state levels were lower in the absence of HpVirB7 (HP0532), HpVirB9 (HP0528), or HpVirB10 (HP0527) but normal in the absence of HpVirB6 (HP0529), HpVirB4 (HP0544), HpVirB11 (HP0525), or HpVirD4 (HP0524). A representative experiment is shown in Fig. S3 of the supplemental material, demonstrating severely reduced Cag3 levels in a ΔHpvirB7 strain while Cag3 steady-state levels were not affected in a ΔHpvirB4 strain. Taken together with the pulse-chase analysis of HpVirB7 protein levels, these results suggest that Cag3 and HpVirB7 mutually stabilize each other in an outer membrane protein subcomplex that likely also includes HpVirB9 and HpVirB10.
DISCUSSION
T4S systems are macromolecular machines spanning the bacterial cell walls of gram-negative bacteria which allow the translocation of DNA-protein complexes and proteins into other cells, into the extracellular milieu, or even to import DNA, as is the case for the Com apparatus of H. pylori (23, 24). Much of the biochemical work done to understand T4S has used the prototypical VirB/D4 T4S system encoded in the T-DNA of the plant pathogen A. tumefaciens. Based on research done in this system the components of the molecular machinery have been divided into channel or core complex components (VirB6, VirB7, VirB8, VirB9, and VirB10), energetic components (VirB11, VirB4, and VirD4), and extracellular appendage components (VirB2 and VirB5).
While sequence or functional homologs of most VirB/D components have been identified, H. pylori has additional proteins that are essential to Cag T4S and thus represent additional functional Vir homologs or H. pylori-specific factors. We have studied one such protein, Cag3. Our Cag3 affinity purification experiment suggests that Cag3 interacts with an assembled Cag T4S complex that includes the CagA substrate (Fig. 8). HpVirB7, HpVirB9, HpVirB10, and CagM are thought to form an outer membrane subcomplex (10, 28, 31, 35). The inner membrane ATPases HpVirD4, HpVirB4, and HpVirB11, but not other inner membrane components, such as HpVirB8 and HpVirB6, also copurified with Cag3. Additional proteins that copurified with Cag3 were CagD and Cag1, as well as the Cag T4S substrate CagA and its chaperone, CagF. Since we found interactions of Cag3 with all these proteins, our data indicate that Cag3 might bridge inner and outer membrane complexes or that the entire complex consisting of both inner and outer membrane components is stable under the conditions used for immunoprecipitation. In support of this hypothesis, we observed interactions with CagD (HP0545) and Cag1 (HP0520). CagD has been suggested to be an accessory protein located in the periplasm and extracellularly that is necessary for CagA translocation but only partially required for pilus assembly (12). Cag1 is not essential for Cag T4S function and is predicted to have a single transmembrane domain. The lack of a predicted signal peptide suggests that Cag1 might localize to the inner membrane. Collectively, these results confirm that Cag3 is able to interact with inner membrane, outer membrane, and periplasmic proteins. The fact that we found interactions with Cag T4S components that are both essential and nonessential (such as Cag1) for IL-8 induction and CagA translocation indicates that some components may have other still-unknown functions during the infection process.
FIG. 8.

Cag3 function in H. pylori cag T4S. A model is presented for Cag3 function as a component of a cag T4S outer membrane (OM) subcomplex containing Cag3 (3), CagM (M), the lipoprotein HpVirB7 (B7), HpVirB9 (B9), and HpVirB10 (B10) (shown in dark gray). Black arrows indicate proteins that copurified with Cag3 by anti-Cag3 affinity purification, and double arrows indicate interactions confirmed by coimmunoprecipitation or cross-linking experiments. Cag3-interacting proteins include the pilus monomer homolog HpVirB2 (B2; light gray); the accessory proteins CagD (D) and Cag1 (1) (square checkerboard); the inner membrane ATPases HpVirD4 (D4), HpVirB4 (B4), and HpVirB11 (B11) (diamond checkerboard); and the effector substrate and its chaperone CagA (A) and CagF (F) (vertical stripes). No interactions were observed with the lytic transglycosylase HpVirB1 (B1), HpVirB8 (B8), or HpVirB6 (B6) (white). Cell fractionation, gel filtration, and cross-linking experiments suggested Cag3 may also oligomerize as a trimer with itself or other proteins not encoded within the cag PAI. The double-pointed white arrow suggests a hypothetical equilibrium between unassembled Cag3 trimer and assembled Cag3-containing cag T4S complex. OM, outer membrane; PG, peptidoglycan; IM, inner membrane.
While the Cag T4S components are expressed in broth culture, full elaboration of Cag T4S-associated pili appears to require host cell contact (29, 31). HpVirB2 copurification with Cag3 suggests that the pilus monomer may contact the machinery before signaling from the host cells induces pilus extension. Interestingly, we did not pull down HpVirB1 in our experiment. In the A. tumefaciens VirB/D4 T4S, VirB1 has been reported to interact with core components VirB8 and VirB9 as well as the ATPase VirB11 (16). HpVirB1 may have been missed because we failed to pull down HpVirB8. If HpVirB1 associates with HpVirB9 and HpVirB11, it is possible that the lower abundance of these proteins limited the ability to detect HpVirB1 with this approach. Alternatively, HpVirB1 may associate with the Cag T4S only upon contact with host cells.
Previous results suggested interactions of Cag3 with HpVirB7, CagM (HP0537), and CagG (HP0542), as well as with itself, using a yeast two-hybrid approach (10). We confirmed interactions of Cag3 with HpVirB7 and CagM in a more biologically relevant context (Fig. 4 and 6). We also found evidence that Cag3, which has a predicted molecular mass of 55 kDa, multimerizes with itself. Cag3 migration with a relative size of 150 kDa during gel filtration chromatography and the observation of high-molecular-mass forms observed in our formaldehyde cross-linking experiments even when Cag3 was expressed in the absence of other cag PAI genes suggest that Cag3 may exist as a trimer (Fig. 5 and 6). The possibility that Cag3 interacts with additional non-T4S proteins, however, cannot be excluded.
Kutter et al. suggested on the bases of mutual stabilization observations and copurification experiments an assembly order for the outer membrane subcomplex consisting of HpVirB7, CagM, HpVirB9, and HpVirB10. In contrast to the A. tumefaciens VirB/D4 T4S system, HpVirB7 does not interact directly with HpVirB9, but the interaction occurs through the H. pylori-specific protein CagM (28). To place Cag3 in this interaction network, we performed coimmunoprecipitation and cross-linking experiments. Our cross-linking methodology detected interactions between HpVirB7 and CagM, also reported previously (10, 28), and this interaction was not dependent on Cag3. Although cross-linking did not confirm that Cag3 can directly interact with HpVirB7, we detected an interaction between Cag3 and CagM that was not dependent on the presence of HpVirB7. Coimmunoprecipitation experiments demonstrated that the interaction between Cag3 and HpVirB7 does not require CagM, although the amount of Cag3-HpVirB7 complex detected was reduced in a cagM strain. Taken together these results confirm interactions among Cag3, CagM, and HpVirB7, placing Cag3 as a new component of this outer membrane subcomplex (Fig. 8). These results suggest Cag3 may interact with HpVirB7 indirectly through another protein, possibly CagM. Alternatively, Cag3 and HpVirB7 might physically interact in the complex, but the lack of close reactive residues available for formaldehyde cross-linking prevented detection of this interaction using this methodology.
Our purification of Cag3-interacting proteins was performed with the uninduced state of the secretion complex, in the absence of host cell contact. It is likely that we have identified the most stable associations among Cag T4S proteins under these conditions, as less stable protein interactions may have been lost during our purification. Based on our results, we hypothesize that the outer membrane subcomplex connects strongly with the inner membrane ATPases in the uninduced state, while engagement of the remaining inner membrane components, such as HpVirB6 and HpVirB8, occurs upon contact with host cells. Purification of Cag3-interacting partners in the induced state of the secretion apparatus, upon host cell contact, might reveal interactions with these components. Lack of Cag3 expression lowered HpVirB7 steady-state levels as demonstrated by immunoblotting and pulse-chase experiments (Fig. 7; see also Fig. S3 in the supplemental material). CagM loss also affected HpVirB7 levels, although to a lesser extent (Fig. 4). It is intriguing that lack of CagM does not have an effect on Cag3 steady-state levels but that lack of HpVirB7 does. Further experiments are required to understand the mechanisms by which HpVirB7 and Cag3 mutual stabilization occurs. In A. tumefaciens, a covalent interaction between VirB7 and VirB9 is required for its stabilization, but these cysteine residues are not conserved in the H. pylori proteins (19). Interestingly, a recently reported structure of a plasmid-encoded T4S subcomplex demonstrated a core channel consisting of VirB7, -9, and -10 that requires no additional proteins for assembly when expressed in E. coli (21). Thus, the mechanisms of assembly and stabilization of the T4S core appear to be system specific.
Our purification methodology provides a good starting point for isolation of the complete Cag T4S complex, which could facilitate further study of its structure and function. Information accumulated regarding the individual components and the ability to integrate this information with systems biology approaches could provide a better understanding on how the components work together in concerted action to accomplish Cag T4S functions.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Pew Charitable Trusts and grant AI054423 from the NIH to N.R.S.
The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the Pew Charitable Trusts.
We thank Jason Hogan and Deepa Hedge in the FHCRC Proteomics Shared Resource for help with mass spectrometry analysis and Maralice Conacci-Sorrell, Daniel Diolati, and Robert Eisenman for help with metabolic labeling. We thank Antonello Covacci, Karen Guillemin, Manuel Amieva, and Karen Ottemann for strains, antibodies, and plasmids. We thank members of the Salama lab, Hector Rincon, Ferric Fang, Beth Traxler, and Colin Manoil, for helpful suggestions and discussions.
Footnotes
Published ahead of print on 2 October 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Akopyants, N. S., S. W. Clifton, D. Kersulyte, J. E. Crabtree, B. E. Youree, C. A. Reece, N. O. Bukanov, E. S. Drazek, B. A. Roe, and D. E. Berg. 1998. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol. Microbiol. 28:37-53. [DOI] [PubMed] [Google Scholar]
- 2.Amieva, M. R., N. R. Salama, L. S. Tompkins, and S. Falkow. 2002. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell Microbiol. 4:677-690. [DOI] [PubMed] [Google Scholar]
- 3.Andrzejewska, J., S. K. Lee, P. Olbermann, N. Lotzing, E. Katzowitsch, B. Linz, M. Achtman, C. I. Kado, S. Suerbaum, and C. Josenhans. 2006. Characterization of the pilin ortholog of the Helicobacter pylori type IV cag pathogenicity apparatus, a surface-associated protein expressed during infection. J. Bacteriol. 188:5865-5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ausubel, F., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1997. Short protocols in molecular biology, 3rd ed. John Wiley & Sons, New York, NY.
- 5.Baldwin, D. N., B. Shepherd, P. Kraemer, M. K. Hall, L. K. Sycuro, D. M. Pinto-Santini, and N. R. Salama. 2007. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect. Immun. 75:1005-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belle, A., A. Tanay, L. Bitincka, R. Shamir, and E. K. O'Shea. 2006. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. USA 103:13004-13009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourzac, K. M., and K. Guillemin. 2005. Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol. 7:911-919. [DOI] [PubMed] [Google Scholar]
- 8.Bourzac, K. M., L. A. Satkamp, and K. Guillemin. 2006. The Helicobacter pylori cag pathogenicity island protein CagN is a bacterial membrane-associated protein that is processed at its C terminus. Infect. Immun. 74:2537-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandt, S., T. Kwok, R. Hartig, W. Konig, and S. Backert. 2005. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA 102:9300-9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busler, V. J., V. J. Torres, M. S. McClain, O. Tirado, D. B. Friedman, and T. L. Cover. 2006. Protein-protein interactions among Helicobacter pylori cag proteins. J. Bacteriol. 188:4787-4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cascales, E., and P. J. Christie. 2004. Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proc. Natl. Acad. Sci. USA 101:17228-17233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cendron, L., M. Couturier, A. Angelini, N. Barison, M. Stein, and G. Zanotti. 2009. The Helicobacter pylori CagD (HP0545, Cag24) protein is essential for CagA translocation and maximal induction of interleukin-8 secretion. J. Mol. Biol. 386:204-217. [DOI] [PubMed] [Google Scholar]
- 13.Censini, S., C. Lange, Z. Xiang, J. E. Crabtree, P. Ghiara, M. Borodovsky, R. Rappuoli, and A. Covacci. 1996. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 93:14648-14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalker, A. F., H. W. Minehart, N. J. Hughes, K. K. Koretke, M. A. Lonetto, K. K. Brinkman, P. V. Warren, A. Lupas, M. J. Stanhope, J. R. Brown, and P. S. Hoffman. 2001. Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 183:1259-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christie, P. J. 2004. Type IV secretion: the Agrobacterium VirB/D4 and related conjugation systems. Biochim. Biophys. Acta 1694:219-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christie, P. J., K. Atmakuri, V. Krishnamoorthy, S. Jakubowski, and E. Cascales. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59:451-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Couturier, M. R., E. Tasca, C. Montecucco, and M. Stein. 2006. Interaction with CagF is required for translocation of CagA into the host via the Helicobacter pylori type IV secretion system. Infect. Immun. 74:273-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Covacci, A., S. Censini, M. Bugnoli, R. Petracca, D. Burroni, G. Macchia, A. Massone, E. Papini, Z. Xiang, N. Figura, et al. 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 90:5791-5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez, D., G. M. Spudich, X. R. Zhou, and P. J. Christie. 1996. The Agrobacterium tumefaciens VirB7 lipoprotein is required for stabilization of VirB proteins during assembly of the T-complex transport apparatus. J. Bacteriol. 178:3168-3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer, W., J. Puls, R. Buhrdorf, B. Gebert, S. Odenbreit, and R. Haas. 2001. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 42:1337-1348. [DOI] [PubMed] [Google Scholar]
- 21.Fronzes, R., E. Schafer, L. Wang, H. R. Saibil, E. V. Orlova, and G. Waksman. 2009. Structure of a type IV secretion system core complex. Science 323:266-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harlow, E., and D. Lane. 1988. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Hofreuter, D., S. Odenbreit, G. Henke, and R. Haas. 1998. Natural competence for DNA transformation in Helicobacter pylori: identification and genetic characterization of the comB locus. Mol. Microbiol. 28:1027-1038. [DOI] [PubMed] [Google Scholar]
- 24.Karnholz, A., C. Hoefler, S. Odenbreit, W. Fischer, D. Hofreuter, and R. Haas. 2006. Functional and topological characterization of novel components of the comB DNA transformation competence system in Helicobacter pylori. J. Bacteriol. 188:882-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keller, A., A. I. Nesvizhskii, E. Kolker, and R. Aebersold. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383-5392. [DOI] [PubMed] [Google Scholar]
- 26.Krall, L., U. Wiedemann, G. Unsin, S. Weiss, N. Domke, and C. Baron. 2002. Detergent extraction identifies different VirB protein subassemblies of the type IV secretion machinery in the membranes of Agrobacterium tumefaciens. Proc. Natl. Acad. Sci. USA 99:11405-11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusters, J. G., A. H. van Vliet, and E. J. Kuipers. 2006. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19:449-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kutter, S., R. Buhrdorf, J. Haas, W. Schneider-Brachert, R. Haas, and W. Fischer. 2008. Protein subassemblies of the Helicobacter pylori Cag type IV secretion system revealed by localization and interaction studies. J. Bacteriol. 190:2161-2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwok, T., D. Zabler, S. Urman, M. Rohde, R. Hartig, S. Wessler, R. Misselwitz, J. Berger, N. Sewald, W. Konig, and S. Backert. 2007. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449:862-866. [DOI] [PubMed] [Google Scholar]
- 30.McNamara, D., and E. El-Omar. 2008. Helicobacter pylori infection and the pathogenesis of gastric cancer: a paradigm for host-bacterial interactions. Dig. Liver Dis. 40:504-509. [DOI] [PubMed] [Google Scholar]
- 31.Rohde, M., J. Puls, R. Buhrdorf, W. Fischer, and R. Haas. 2003. A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol. Microbiol. 49:219-234. [DOI] [PubMed] [Google Scholar]
- 32.Salama, N. R., J. S. Chuang, and R. W. Schekman. 1997. Sec31 encodes an essential component of the COPII coat required for transport vesicle budding from the endoplasmic reticulum. Mol. Biol. Cell 8:205-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smeets, L. C., J. J. Bijlsma, S. Y. Boomkens, C. M. Vandenbroucke-Grauls, and J. G. Kusters. 2000. comH, a novel gene essential for natural transformation of Helicobacter pylori. J. Bacteriol. 182:3948-3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan, S., L. S. Tompkins, and M. R. Amieva. 2009. Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog. 5:e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka, J., T. Suzuki, H. Mimuro, and C. Sasakawa. 2003. Structural definition on the surface of Helicobacter pylori type IV secretion apparatus. Cell Microbiol. 5:395-404. [DOI] [PubMed] [Google Scholar]
- 36.Terry, K., S. M. Williams, L. Connolly, and K. M. Ottemann. 2005. Chemotaxis plays multiple roles during Helicobacter pylori animal infection. Infect. Immun. 73:803-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viala, J., C. Chaput, I. G. Boneca, A. Cardona, S. E. Girardin, A. P. Moran, R. Athman, S. Memet, M. R. Huerre, A. J. Coyle, P. S. DiStefano, P. J. Sansonetti, A. Labigne, J. Bertin, D. J. Philpott, and R. L. Ferrero. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5:1166-1174. [DOI] [PubMed] [Google Scholar]
- 38.Zahrl, D., M. Wagner, K. Bischof, M. Bayer, B. Zavecz, A. Beranek, C. Ruckenstuhl, G. E. Zarfel, and G. Koraimann. 2005. Peptidoglycan degradation by specialized lytic transglycosylases associated with type III and type IV secretion systems. Microbiology 151:3455-3467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.