

Abstract
Peroxisome proliferator-activated receptor α (PPARα) is an important transcription factor in liver that can be activated physiologically by fasting or pharmacologically by using high-affinity synthetic agonists. Here we initially set out to elucidate the similarities in gene induction between Wy14643 and fasting. Numerous genes were commonly regulated in liver between the two treatments, including many classical PPARα target genes, such as Aldh3a2 and Cpt2. Remarkably, several genes induced by Wy14643 were upregulated by fasting independently of PPARα, including Lpin2 and St3gal5, suggesting involvement of another transcription factor. Using chromatin immunoprecipitation, Lpin2 and St3gal5 were shown to be direct targets of PPARβ/δ during fasting, whereas Aldh3a2 and Cpt2 were exclusive targets of PPARα. Binding of PPARβ/δ to the Lpin2 and St3gal5 genes followed the plasma free fatty acid (FFA) concentration, consistent with activation of PPARβ/δ by plasma FFAs. Subsequent experiments using transgenic and knockout mice for Angptl4, a potent stimulant of adipose tissue lipolysis, confirmed the stimulatory effect of plasma FFAs on Lpin2 and St3gal5 expression levels via PPARβ/δ. In contrast, the data did not support activation of PPARα by plasma FFAs. The results identify Lpin2 and St3gal5 as novel PPARβ/δ target genes and show that upregulation of gene expression by PPARβ/δ is sensitive to plasma FFA levels. In contrast, this is not the case for PPARα, revealing a novel mechanism for functional differentiation between PPARs.
Hepatic lipid metabolism is governed by a complex interplay between hormones, transcription factors, and energy substrates, allowing for rapid adaptations to changes in metabolic needs (21). According to the traditional view, energy substrates such as fatty acids influence lipid metabolism by promoting flux through a particular pathway via mass action. However, it has become clear that energy substrates can also directly govern the transcription of enzymes involved in lipid metabolism via mechanisms analogous to those of many hormones. Indeed, it is now evident that glucose and fatty acids play a major regulatory role in hepatic lipid metabolism via direct activation or inhibition of specific transcription factors, including carbohydrate response element binding protein (6, 63), sterol response element binding protein 1 (SREBP1) (2, 41, 58, 61, 62), and peroxisome proliferator-activated receptor α (PPARα) (38).
Although numerous transcription factors have been shown to be activated by fatty acids in vitro, recent data suggest that PPARα is dominant in mediating the effects of dietary fatty acids on gene expression in liver (48). PPARα is a member of the superfamily of nuclear receptors and is closely related to the other PPAR isoforms, β/δ and γ (32). Similar to several other nuclear receptors, PPARs function as heterodimers with the retinoid X receptor and bind to specific sequences on the DNA referred to as PPAR response elements (PPREs) (8, 11, 26). Numerous studies have shown that fatty acids can directly bind to PPARs and activate DNA transcription (12, 17, 24, 28, 31, 50). Binding of fatty acids changes the conformation of the PPAR protein (13, 23, 37, 60) and leads to recruitment of coactivator proteins (31, 48). Besides fatty acids and their derivatives, PPARs bind synthetic agonists, including the thiazolidionediones, which serve as agonists for PPARγ, and the fibrates, which are PPARα agonists (51).
Most of the information about the function of PPARα in liver and its impact on target genes is based on studies that have used high-affinity synthetic PPARα agonists. These pharmacological studies have shown that PPARα regulates a remarkably large number of genes, many of which are involved in hepatic lipid metabolism, thereby explaining the positive effect of synthetic PPARα agonists on plasma lipid parameters (9, 38). However, PPARα did not evolve as a receptor for fibrates but rather as a fatty acid sensor. Accordingly, the question arises to what extent results from pharmacological studies reflect the physiological function of PPARα.
Physiological experiments using PPARα−/− mice have shown that PPARα is especially important for the adaptive response to fasting. During fasting, the absence of PPARα elicits a complex phenotype characterized by fatty liver, hypoketonemia, hypoglycemia, hypothermia, and elevated plasma free fatty acid (FFA) levels (1, 19, 27, 34). Furthermore, the hepatic induction of numerous metabolic genes during fasting is abolished in PPARα−/− mice. While both pharmacological and physiological studies thus support a major role for PPARα in hepatic lipid metabolism, evidence suggests that there is only partial overlap between genes upregulated by PPARα during fasting and genes upregulated by synthetic PPARα agonists (45). One possible explanation is that PPARα responds differently to pharmacological compared to physiological activation. Additionally, there may be a role for other PPAR subtypes. Besides PPARα, PPARβ/δ has been shown to be well expressed in hepatocytes (10, 22). However, the functional role of PPARβ/δ in hepatocytes and its physiological mechanisms of activation remain unknown.
Here we initially set out to elucidate the similarities and discrepancies in gene regulation in liver between pharmacological PPARα activation by Wy14643 and physiological PPARα activation by fasting. While our data reveal major overlap between the effects of Wy14643 and fasting, the data also indicate that a number of pharmacological PPARα target genes are induced by fasting independently of PPARα. Subsequent analysis uncovered a role for PPARβ/δ in hepatic gene regulation and revealed different mechanisms of activation of PPARα versus PPARβ/δ in mouse liver. Specifically, we found that upregulation of gene expression by PPARβ/δ is sensitive to plasma FFAs, while this is not the case for PPARα.
MATERIALS AND METHODS
Materials.
Tridocosahexaenoin was obtained from Nu-Chek-Prep, Inc. (Elysian, MN). SYBR green was purchased from Eurogentec (Seraing, Belgium). Protease inhibitor cocktail was purchased from Roche Diagnostics (Almere, The Netherlands), sonicated salmon sperm DNA was from Invitrogen (Breda, The Netherlands), and proteinase K was from Fermentas (St. Leon-Rot, Germany). All other chemicals were from Sigma (Zwijndrecht, The Netherlands).
Animals.
Purebred Sv129 PPARα−/− mice (129S4/SvJae) and corresponding wild-type mice (129S1/SvImJ) were purchased from Jackson Laboratory (Bar Harbor, ME). The PPARβ/δ−/− mice were on a mixed background (Sv129/C57BL/6) and have been previously described (42). The Angptl4−/−, Angptl4+/−, and transgenic mice were on a C57BL/6 background and have been previously described (30, 36, 39). Angptl4-transgenic mice overexpress Angptl4 in numerous tissues, including adipose tissue (36, 39), while the Angptl4−/− mice lack Angptl4 expression in all tissues (30). Only male mice were used, with 4 to 10 mice per group. For the fasting experiment, food was withdrawn for 24 h starting at the onset of the light cycle.
PPARα ligand testing.
Wild-type and PPARα−/− mice were fasted for 4 h and thereafter given an intragastric gavage of 400 μl Wy14643 (10 mg/ml in 0.5% carboxymethyl cellulose; 160 mg/kg of body weight). The control treatment was 400 μl of 0.5% carboxymethyl cellulose. Livers were collected after 6 h.
Oral lipid load testing.
Wild-type and PPARα−/− mice were given an intragastric gavage of 400 μl synthetic triglyceride (tridocosahexaenoin) after a 4-hour fast. The control treatment was 0.5% carboxymethyl cellulose (400 μl). Livers were collected 6 h after gavage.
PPARβ/δ ligand testing.
Wild-type mice were given a single oral gavage of 150 μg GW501516 (6 mg/kg). Alternatively, PPARα−/− mice were fed 0.025% (wt/wt) L165041 mixed in food for 5 days (40 mg/kg).
Mice were anesthetized with a mixture of isoflurane (1.5%), nitrous oxide (70%), and oxygen (30%). Blood was collected by orbital puncture, after which the mice were sacrificed by cervical dislocation. Livers were dissected and directly frozen in liquid nitrogen. For RNA analyses, tissue from the same part of the liver lobe was used.
The animal studies were approved by the Local Committee for Care and Use of Laboratory Animals at Wageningen University, The Netherlands, and the University of Lausanne, Switzerland.
Affymetrix microarray.
Total RNA from mouse liver was extracted with TRIzol reagent, purified, and DNase treated using the SV total RNA isolation system (Promega, Leiden, The Netherlands). RNA quality measurements were performed on an Agilent 2100 bioanalyzer (Agilent Technologies, Amsterdam, The Netherlands) using 6000 Nano Chips in combination with the eukaryote total RNA Nano assay. RNA was judged as suitable for array hybridization only if samples showed intact bands corresponding to the 18S and 28S rRNA subunits, displayed no chromosomal peaks or RNA degradation products, and had an RNA integrity number above 8.0. Five micrograms of RNA was used for the cRNA synthesis mixture for one cycle (Affymetrix, Santa Clara, CA). Hybridization, washing, and scanning of Affymetrix mouse genome 430 2.0 arrays/Affymetrix Nugo mouse arrays were carried out according to standard Affymetrix protocols.
Packages from the Bioconductor Project were used for analyzing the scanned arrays (14). Arrays were normalized using quantile normalization, and expression estimates were compiled using GC-RMA (GeneChip robust multiarray average), applying the empirical Bayes approach (59). A nonspecific filtering step was applied to remove genes with low variation and included only those genes that had an interquartile range across the samples of at least 0.25 on a log2 scale (55).
Gene set enrichment analysis (GSEA) was used to relate changes in gene expression to functional changes between mice treated with the PPARα agonist Wy14643 for 6 h and mice fasted for 24 h. GSEA takes into account a broad context of physically interacting networks in which gene products function, including biochemical, metabolic, and signal transduction routes (53). Gene sets with a false discovery rate P value of <0.1 were considered significantly overrepresented.
Plasma metabolites.
Plasma was obtained from blood by centrifugation for 10 min at 10,000 × g. Plasma triglycerides and the glycerol concentration in cell culture medium were determined using kits from Instruchemie (Delfzijl, The Netherlands). Plasma free fatty acids were determined using a kit from WAKO Chemicals (Instruchemie, Delfzijl, The Netherlands).
Fat explants.
Epididymal adipose tissue was excised and cut into 0.2- to 0.3-cm3 pieces. The explants were subsequently incubated for 15 min at 37°C in Dulbecco's modified Eagle's medium (DMEM) containing 1% lipid-free bovine serum albumin (BSA) and 1 mg/ml collagenase type 1. Fat cells were liberated by gentle stirring followed by centrifugation of the cell suspension for 1 min at 400 × g. Fat cells were isolated from the surface and washed once in phosphate-buffered saline (PBS). Subsequently, fat cells were incubated in DMEM containing 1% lipid-free BSA and 1 μM isoproterenol at 37°C with 5% CO2. Medium was collected at various time points and frozen for measurement of free fatty acids and glycerol (kits from Instruchemie, Delfzijl, The Netherlands).
RNA isolation and quantitative reverse transcription-PCR.
Total liver RNA was isolated with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. A NanoDrop ND-1000 spectrophotometer (Isogen Life Science, Ijsselstein, The Netherlands) was used to determine RNA concentrations. One microgram of total RNA was reverse transcribed using iScript (Bio-Rad, Veenendaal, The Netherlands). cDNA was amplified on a Bio-Rad MyIQ or iCycler PCR machine using Platinum Taq DNA polymerase (Invitrogen). PCR primer sequences were taken from the PrimerBank (56) and ordered from Eurogentec. Sequences of the primers used are available upon request.
Transactivation assay.
Conserved PPREs were identified at 1,291 or 23,333 nucleotides downstream of the transcriptional start site (TSS) of the mouse Lpin2 or St3gal5 gene, respectively, using a published algorithm (20). A 201-nucleotide and 183-nucleotide fragment surrounding the putative PPRE within the Lpin2 and St3gal5 genes, respectively, was PCR amplified from mouse genomic DNA (strain C57BL/6) and subcloned into the KpnI and BglII sites of the pGL3 promoter vector (Promega, Leiden, The Netherlands). The reporter vector (PPRE)3-TK-luciferase was included as a positive control. Reporter vectors were transfected into human hepatoma HepG2 cells together with an expression vector (pSG5) for mouse PPARβ/δ, in the presence or absence of GW501516 (1 μM). A β-galactosidase reporter vector was cotransfected to normalize for differences in transfection efficiencies. Transfections were carried out using Nanojuice (Novagen, Nottingham, United Kingdom). Luciferase activity was measured 24 h posttransfection using the Promega luciferase assay kit on a Fluoroskan Ascent FL apparatus (Thermo Labsystems, Breda, The Netherlands). β-Galactosidase activity was measured in the cell lysate by a standard assay using 2-nitrophenyl-β-d-galactopyranoside as a substrate.
For the GAL4 transactivation assay, the human osteosarcoma cell line U2OS was maintained in DMEM Glutamax containing 10% fetal calf serum (Invitrogen), 100 μg of penicillin/ml, and 100 μg streptomycin/ml (Invitrogen). For luciferase reporter assays, cells were seeded in 24-well plates and transiently transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Each well was cotransfected with 1 μg of the 5×Gal4-E1BTATA-pGL3 reporter, 10 ng of pcDNA3-GAL4-hPPARδDBD, and 2 ng of pCMV-Renilla (Promega, Madison, WI). The next day, medium was replaced with medium containing ligands. At 24 h after the incubation, cells were harvested in passive lysis buffer (Promega) and assayed for luciferase activity according to the manufacturer's protocol (Promega dual-luciferase reporter assay system) and for Renilla luciferase activity to correct for transfection efficiency. The relative light units were measured by a Centro LB 960 luminometer (Berthold Technologies, Bad Wildbad, Germany). Experiments were performed in triplicate.
Chromatin immunoprecipitation (ChIP) assay.
It is becoming increasingly apparent that most nuclear receptor binding sites, including PPREs, are not found in proximity of the annotated TSS of a gene but are often located quite distant (4, 33, 43). Nuclear receptors bound to such distal sites likely contact the basal transcription machinery via DNA looping. Binding of PPAR to distant PPREs can thus be demonstrated by showing cross-linking of PPAR to the TSS (5, 49).
Wild-type and PPARα−/− mice on an Sv129 background were fed or fasted for 24 h (n = 3). Transgenic mice overexpressing Angptl4 (Angptl4-Tg), wild-type mice (Angptl4+/+), and homozygous knockout mice (Angptl4−/−) (n = 3/group) were fasted for 24 h. At the end of the fasting period, mice were killed by cervical dislocation and livers were extracted. Livers were cut in smaller pieces and directly put in PBS containing 1% formaldehyde. Cross-linking was stopped after 15 min by adding glycine to a final concentration of 0.125 M for 5 min at room temperature. After a short centrifugation to collect the liver pieces, two washing steps with ice-cold PBS were carried out. Livers were homogenized and thereafter centrifuged. After supernatant was removed, liver homogenate was resuspended in lysis buffer (1% sodium dodecyl sulfate [SDS], 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, protease inhibitors), and the lysates were sonicated with a Bioruptor (Diagenode, Liège, Belgium) to achieve a DNA length of 300 to 1,000 bp. After removal of cellular debris by centrifugation, supernatants were diluted 1:10 in ChIP dilution buffer (150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 7.5, protease inhibitors). Chromatin was incubated overnight at 4°C with 2 μg antibody, 25 μl BSA (10 mg/ml), and 2.4 μl sonicated salmon sperm (10 mg/ml). Antibodies used were anti-PPARα (sc-9000), anti-PPARGC1α (sc-13067), and anti-PPARβ/δ (sc-7197), all of which were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Immunocomplexes were collected with 25 μl MagaCell protein A magnetic beads (Isogen Life Science) for 1 h at room temperature and subsequently washed sequentially with 700 μl of the following buffers: twice with ChIP wash buffer 1 (150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8, protease inhibitors), once with ChIP wash buffer 2 (500 mM NaCl, 1% Triton X-100, 2 mM EDTA, 0.1% SDS, 20 mM Tris-HCl, pH 8, protease inhibitors), once with ChIP wash buffer 3 (250 mM LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8), and two times with TE buffer (1 mM EDTA, 10 mM Tris-HCl, pH 8). Elution of immunocomplexes was carried out in 250 μl of elution buffer (10 mM EDTA, 0.5% SDS, 25 mM Tris-HCl, pH 7.5) at 64°C for 30 min. After collection of supernatant, elution was repeated with 250 μl elution buffer at room temperature for 2 min. After combining the supernatants, cross-linking was reversed at 64°C overnight with 2.5 μl proteinase K (20 mg/ml) for digestion of any remaining proteins. Genomic DNA fragments were recovered by phenol-chloroform extraction with phase lock gel (Eppendorf, Wesseling-Berzdorf, Germany), followed by salt-ethanol precipitation. Samples were diluted in sterile H2O and analyzed by quantitative PCR.
Primers were from Eurogentec and designed to cover the TSS of the following genes: Aldh3a2 (forward [F], 5′-CAGGTGAGGGAGCACAGTAC-3′; reverse [R], 5′-CGCTTGGCTCTTTTCTGAAG-3′); Cpt2 (F, 5′-GCCAGTCACGCAACAGCAG-3′; R, 5′-TAGTTTAGAGACCGCTTCCG-3′); Lpin2 (F, 5′-CCGTCTTGTGATTGGGCAGG-3′; R, 5′-GAAGGAAACTCACCAGAATCC-3′); St3gal5 (F, 5′-GCCTTCCACTATCTAATCACG-3′; R, 5′-GTGTCCGCTCTGCCGACTG-3′); Rplp0 (F, 5′-CGAGGACCGCCTGGTTCTC-3′; R, 5′-GTCACTGGGGAGAGAGAGG-3′).
FAO cell culture.
Rat hepatoma FaO cells were grown in DMEM containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were incubated with albumin only (control), albumin-bound oleic or linoleic acid (100 μM; molar ratio for FA:BSA of 3.5:1), or GW501516 (100 nM) for 4 h, followed by RNA isolation and quantitative reverse transcription-PCR.
Cofactor recruitment assay.
Nuclear receptor PamChip arrays (PamGene, s'Hertogenbosch, The Netherlands) were used as described previously (29). Upon binding a ligand, PPARβ/δ undergoes a conformational change which promotes the formation of a cofactor binding pocket, subsequently allowing interaction with the so-called LxxLL motif within some coregulators. The PamChip arrays consist of 53 peptides encompassing the LxxLL motifs of 22 different coregulator proteins. Briefly, the arrays were incubated with glutathione S-transferase-tagged PPARβ/δ ligand binding domain (LBD) (Invitrogen, Breda, The Netherlands) in the presence and absence of ligand (GW501516 at 400 nM; oleic and linoleic acids at 125 μM). Quantification of the interaction between PPARβ/δ and coregulators was made using Alexa 488-conjugated anti-glutathione S-transferase rabbit polyclonal antibody (Invitrogen).
Microarray accession numbers.
All microarray experimental results have been deposited into the Gene Expression Omnibus database under accession number GSE 8396 and GSE 17863.
RESULTS
Overlap in gene regulation between pharmacological and physiological PPARα activation.
PPARα in liver can be activated pharmacologically by using synthetic agonists such as Wy14643 or physiologically by fasting. To assess the similarities and discrepancies in gene regulation between these two stimuli, we compared microarray data from livers of mice treated with the synthetic PPARα agonist Wy14643 for 6 h and livers of mice subjected to 24 h of fasting. GSEA showed great similarity and overlap in top-regulated pathways between fasting and Wy14643 treatment, almost all of which corresponded to pathways of lipid metabolism (Fig. 1A). Much less overlap was observed at the individual gene level (Fig. 1B). Nevertheless, a substantial number of genes upregulated by Wy14643 were also induced by fasting. Many of these genes represent classical PPARα target genes involved in fatty acid catabolism, such as Acox1, Cpt2, Aldh3a2, Acot8, Ehhadh, and Hmgcs2. Consistent with an important role of PPARα, induction of classical PPARα target genes by fasting was abolished in PPARα−/− mice (Fig. 1C and D). In contrast, a number of Wy14643-responsive genes could be identified that were more significantly upregulated by fasting in PPARα−/− mice than in wild-type mice, suggesting PPARα-independent regulation during fasting (Fig. 1C and D). Overall, these data indicate that targets of pharmacological PPARα activation exhibit diverse responses following physiological PPARα activation by fasting, being either up- or downregulated and showing a variable dependence on PPARα.
FIG. 1.

Hepatic genes activated by Wy14643 and fasting show a variable dependence on PPARα. Livers from wild-type and PPARα−/− mice treated with the PPARα agonist Wy14643 for 6 h or fasted for 24 h were used for gene expression profiling (n = 4 to 5 mice per group). (A) Overlap in top-regulated pathways between Wy14643 treatment and fasting according to gene set enrichment analysis. Gene sets with a false discovery rate P value of <0.1 were considered significant. (B) Overlap of upregulated genes between Wy14643 treatment and fasting (criteria for inclusion: P < 0.01 and a change of >1.5-fold). (C) Scatter plot showing the effects of fasting in genes significantly upregulated by Wy14643. The y axis and x axis show the effects of fasting in wild-type and PPARα−/− mice, respectively. Red dots represent classical PPARα target genes, while blue dots are Wy14643-responsive genes that are more significantly upregulated by fasting in the PPARα−/− mouse compared to wild type. (D) Heatmap showing the changes (n-fold) of genes compared to the wild-type control/fed state. (Upper panel) Classical PPARα target genes, showing a PPARα-dependent increase in gene expression upon Wy14643 treatment as well as fasting. Genes in the lower panel exhibit a PPARα-dependent induction upon Wy14643 treatment but are induced independently of PPARα upon fasting.
To explore the possible mechanism underlying the more significant induction by fasting of a number of pharmacological PPARα targets in PPARα−/− mice compared to wild-type mice, two representative genes were investigated in more detail: Lpin2 and St3gal5. Remarkably, in contrast to classical PPARα targets Cpt2 and Aldh3a2 (Fig. 2A and C), induction of Lpin2 and St3gal5 by fasting and dietary fatty acids was largely maintained in PPARα−/− mice (Fig. 2B and D). These results imply that the effects of fasting and dietary fatty acids on hepatic expression of Lpin2 and St3gal5 may be partially mediated by a transcription factor other than PPARα. On the contrary, effects of fasting and dietary fatty acids on Cpt2 and Aldh3a2 are entirely mediated by PPARα. It should be mentioned that the expression profiles of Cpt2 and Aldh3a2 are representative of a large set of classical PPARα targets (Fig. 1D).
FIG. 2.

Lpin2 and St3gal5 are induced during fasting independently of PPARα. Livers from wild-type and PPARα−/− mice fasted for 24 h or treated with tridocosahexaenoin (DHA) for 6 h were used for gene expression profiling (n = 4 to 5 mice per group). (A and B) Gene expression of classical PPARα targets Aldh3a2 and Cpt2 (A) and Lpin2 and St3gal5 (B) in fed and fasted wild-type and PPARα−/− mice. (C and D) Gene expression of Aldh3a2 and Cpt2 (C) and Lpin2 and St3gal5 (D) after treatment with the dietary fatty acid tridocosahexaenoin. Error bars represent standard errors of the means. *, significantly different according to Student's t test (P < 0.05).
PPARβ/δ as an alternative transcription factor to PPARα in mouse liver during fasting.
One obvious candidate alternative transcription factor is PPARβ/δ, which is well expressed in liver (10, 16). Supporting regulation of Lpin2 and St3gal5 by PPARβ/δ, the PPARβ/δ agonists GW501516 and L165041 significantly induced Lpin2 and St3gal5 mRNA (Fig. 3A). To establish whether Lpin2 and St3gal5 are direct PPAR target genes, we identified a conserved PPRE within the Lpin2 and St3gal5 genes (Fig. 3B) and cloned a genomic region encompassing the PPRE in front of a luciferase reporter to perform transactivation assays. GW501516 significantly increased reporter activity for the Lpin2 and St3gal5 genomic regions, which was further enhanced by cotransfection with PPARβ/δ (Fig. 3C). In subsequent ChIP experiments, PPARβ/δ as well as PPARα could be cross-linked to the TSS of the Lpin2 and St3gal5 genes, at least in the fasted state, which provided evidence for the presence of a distant functional PPRE (Fig. 3D). These data suggest that Lpin2 and St3gal5 genes represent direct PPAR target genes. Interestingly, while fasting increased binding of both PPARα and PPARβ/δ to the Lpin2 and St3gal5 genes, fasting increased binding of only PPARα to the Aldh3a2 and Cpt2 genes (Fig. 3D). No binding of PPARα and PPARβ/δ to the negative control gene Rplp0 was observed. All together these data suggest that Lpin2 and St3gal5 are dual targets of PPARα and PPARβ/δ, whereas Aldh3a2 and Cpt2 are exclusive targets of PPARα. In agreement with this notion, induction of Lpin2 and St3gal5 by fasting was partially abolished in PPARβ/δ−/− mice (Fig. 3E).
FIG. 3.

PPARβ/δ as alternative transcription factor to PPARα in mouse liver. (A) Lpin2 and St3gal5 expression in livers of wild-type mice (n = 5) treated with the PPARβ/δ agonist GW501516 for 6 h or PPARα−/− mice (n = 5) treated with the PPARβ/δ agonist L165041 for 5 days. Error bars represent standard errors of the means. (B) PPREs conserved between mouse and human were identified 1,291 bp and 23,333 bp downstream of the TSS of the Lpin2 and St3gal5 genes. (C) HepG2 cells were transfected with a PPARβ/δ expression vector and a simian virus 40 reporter vector containing 201-nucleotide and 183-nucleotide fragments with the putative PPREs within the Lpin2 and St3gal5 genes, respectively. The reporter vector (PPRE)3-TK-luciferase served as a positive control. Luciferase and β-galactosidase activities were determined 24 h after exposure of the cells to 1 μM GW501516. Error bars represent standard errors of the means. (D) Chromatin was extracted from livers of fed or 24-h-fasted wild-type and PPARα−/− mice (n = 3 per group). ChIP was performed with antibodies against PPARα and PPARβ/δ on the TSS of Lpin2, St3gal5, Aldh3a2, Cpt2, and Rplp0. Rabbit IgG was used as a specificity control. Gray bars, fed state; black bars, 24-h fasted state. Error bars represent standard deviations. (E) Expression of Lpin2 and St3gal5 in livers of fed and 24-h-fasted wild-type and PPARβ/δ−/− mice (n = 4 to 5 per group). Relative induction by fasting is indicated. Error bars represent standard errors of the means. (F and G) Plasma FFA levels in wild-type and PPARα−/− mice (F) or wild-type and PPARβ/δ−/− mice (G) sacrificed in a fed or 24-h-fasted state. Error bars represent standard errors of the means. *, significantly different according to Student's t test (P < 0.05).
Given the more pronounced induction of Lpin2 and St3gal5 by fasting in PPARα−/− versus wild-type mice, we speculated that either expression of PPARβ/δ may be upregulated in PPARα−/− mice as a compensatory mechanism or that ligand activation of PPARβ/δ is enhanced in PPARα−/− mice. While we could not detect a change in PPARβ/δ mRNA in PPARα−/− mice (data not shown), consistent with the second scenario, plasma FFA levels were markedly elevated in fasted PPARα−/− mice (Fig. 3F), which was associated with marked induction of PPARβ/δ binding to the Lpin2 and St3gal5 promoter (Fig. 3D). These data suggest that in the absence of PPARα plasma FFAs can induce Lpin2 and St3gal5 expression via PPARβ/δ. It should be noted that levels of plasma FFAs during fasting are unaltered in PPARβ/δ−/− mice (Fig. 3G).
Circulating FFAs activate PPARβ/δ but not PPARα in mouse liver.
Importantly, recent evidence suggests that PPARα in liver cannot be (ligand) activated by plasma FFAs, while it can be activated by fatty acids synthesized de novo (3). To study the activation of PPARα and PPARβ/δ by plasma FFAs, we modulated fasting plasma FFA levels by taking advantage of a unique transgenic model system based on whole-body overexpression or inactivation of the mouse Angptl4 gene, which encodes a prolipolytic factor involved in lipid metabolism (39, 52). Previously, intravenous injection of Angptl4 was shown to cause an immediate increase in plasma FFA (65). In support, Angptl4 increased release of glycerol from 3T3-L1 adipocytes (Fig. 4A).
FIG. 4.

Angptl4 stimulates adipose tissue lipolysis. (A) Glycerol concentration in medium of 3T3-L1 cells treated for 30 min with isoproterenol or with concentrated conditioned medium of HEK293 cells transfected with mAngptl4. Control cells were treated with conditioned medium of nontransfected HEK293 cells. Error bars represent standard errors of the means. *, significantly different according to Student's t test (P < 0.05). (B) Increase in fatty acid and glycerol concentrations in medium of adipose tissue explants from transgenic mice overexpressing Angptl4 (Tg), wild-type (+/+), and homozygous knockout (−/−) mice. Values are corrected for weight of explants. (C and D) Plasma FFAs (C) and triglycerides (D) in transgenic mice overexpressing Angptl4 (Angptl4-Tg) and wild-type (Angptl4+/+), heterozygous (Angptl4+/−), and homozygous (Angptl4−/−) mice fed or in a 24-h-fasted state (n = 5). Gray bars, fed state; black bars, 24-h-fasted state. Error bars represent standard errors of the means. Different letters indicate statistically significant differences (Student's t test; P < 0.05). (E) Eosin and hematoxylin staining of epididymal adipose tissue.
Consistent with a prolipolytic effect of Angptl4, Angptl4 overexpression in mice was associated with a significant increase in release of fatty acids and glycerol from adipose tissue, whereas the opposite was observed in Angptl4−/− mice (Fig. 4B). In agreement with these data, fasting plasma FFA levels were increased or decreased upon Angptl4 overexpression or inactivation, respectively (Fig. 4C). In fact, the fasting-induced increase in plasma FFA was entirely blunted in Angptl4−/− mice. Plasma triglyceride levels were increased or decreased upon Angptl4 overexpression or inactivation (Fig. 4D), respectively, reflecting the well-documented inhibitory effect of Angptl4 on lipoprotein lipase activity (25). Finally, the defective lipolysis in Angptl4+/− and Angptl4−/− mice was supported by the absence of changes in adipocyte cell size upon fasting, in contrast to Angptl4-Tg and wild-type mice (Fig. 4E). These results corroborate the stimulatory effect of Angptl4 on adipose tissue lipolysis, which we exploited to study the effect of plasma FFAs on hepatic gene expression.
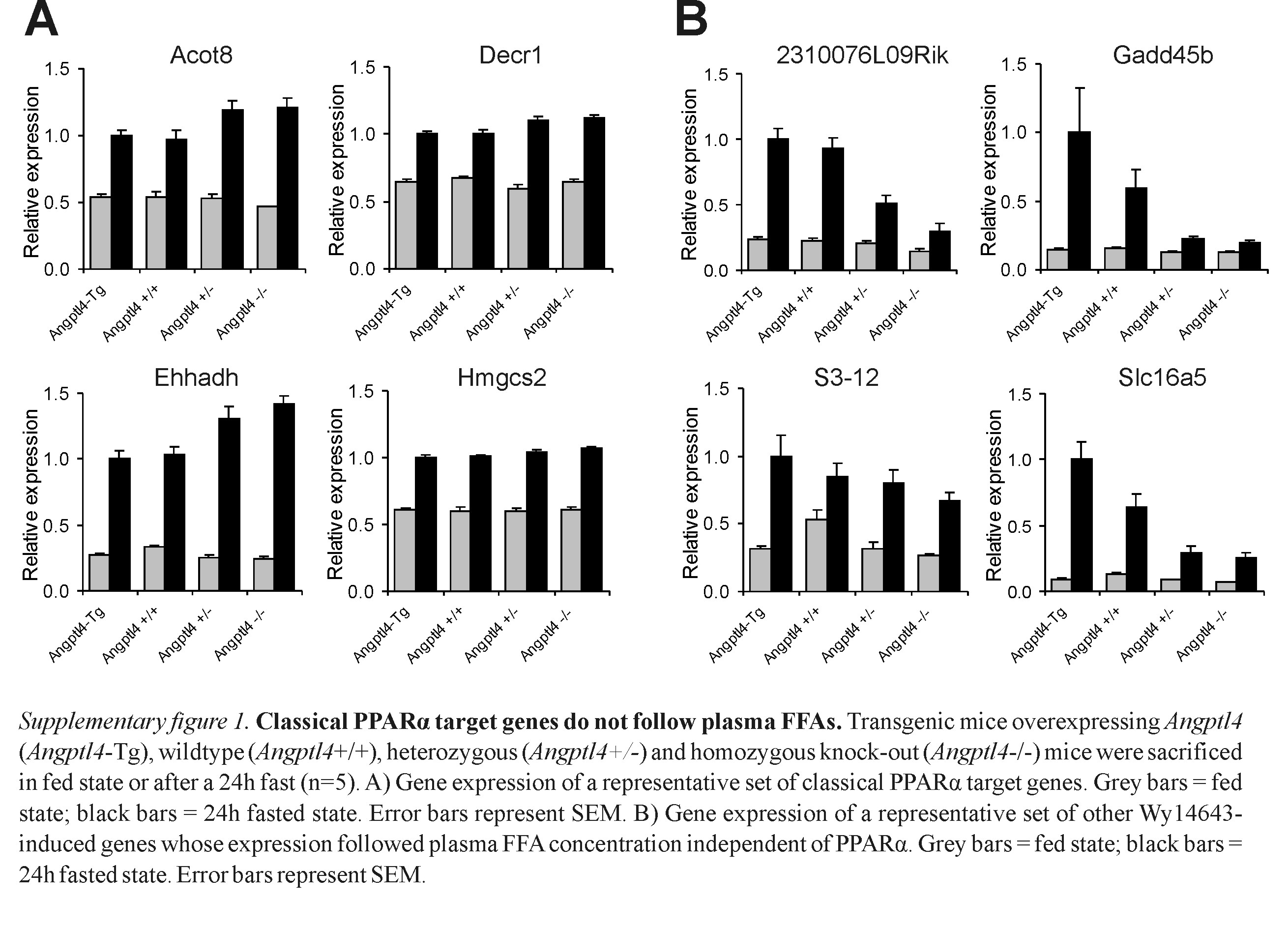
If hepatic PPARα is activated by plasma FFAs, expression of classical PPARα target genes during fasting would be expected to be proportional to the plasma FFA level throughout the various Angptl4 mouse models. Remarkably, rather than going down, gene expression of classical PPARα targets Aldh3a2, Cpt2, and others was stable or went up as plasma FFAs declined (Fig. 5A; see also Fig. S1A in the supplemental material). Expression of Pparα itself, which is auto-regulated, followed a very similar pattern (Fig. 5B), suggesting that regulation of classical PPARα targets is determined by PPARα expression level. Supporting the use of the Angptl4 mouse models to study hepatic gene regulation by FFA, hepatic expression of Srebp1, which is known to be suppressed by fatty acids, negatively correlated with plasma FFA concentration (data not shown). In combination with previously published data (3), these data strongly suggest that PPARα is not activated by plasma FFA in mouse liver.
FIG. 5.

Plasma FFAs do not activate hepatic PPARα. Transgenic mice overexpressing Angptl4 (Angptl4-Tg) or wild-type (Angptl4+/+), heterozygous (Angptl4+/−), and homozygous knockout (Angptl4−/−) mice were sacrificed in the fed state or after a 24-h fast (n = 5). Results show hepatic gene expression of classical PPARα targets Aldh3a2 and Cpt2 (A), Pparα (B), and Pparβ/δ (F). Gray bars, fed state; black bars, 24-h-fasted state. Error bars represent standard errors of the means. *, P < 0.05. (C) Fatty acids activate PPARβ/δ in a transactivation assay using a GAL4-LBDPPARβ/δ fusion. Fatty acids were used at 125 μM or 250 μM (normal or bold plus sign). (D) A nuclear receptor PamChipH assay was used to measure the interaction between PPARβ/δ and immobilized peptides corresponding to specific coregulator-nuclear receptor binding regions in the presence and absence of fatty acids (125 μM). (E) Fatty acids (100 μM) upregulated expression of PPARβ/δ target Adfp in rat FaO hepatoma cells. (G) Chromatin was extracted from livers of 24-h-fasted transgenic mice overexpressing Angptl4 (Tg) and wild-type (+/+) and homozygous Angptl4 knockout (−/−) mice (n = 3 per group). ChIP was performed with antibodies against PPARα and PPARβ/δ on the TSS of Lpin2, St3gal5, and Rplp0. Rabbit IgG was used as a specificity control. Error bars represent standard deviations. Different lowercase letters indicate statistically significant differences (Student's t test; P < 0.05). (H) Hepatic gene expression of Lpin2 and St3gal5. Gray bars, fed state; black bars, 24-h-fasted state. Error bars represent standard errors of the means. Different lowercase letters indicate statistically significant differences (Student's t test; P < 0.05).
While plasma FFAs seemingly do not activate hepatic PPARα, data presented above suggested that plasma FFAs induce Lpin2 and St3gal5 expression by activating PPARβ/δ, at least when PPARα is absent. In order to explore activation of hepatic PPARβ/δ by plasma FFA, we first determined that fatty acids are able to bind and activate PPARβ/δ in vitro. In a transactivation assay using the GAL4-LBDPPARβ/δ fusion, oleic and linoleic acids markedly induced luciferase activity, indicating activation of PPARβ/δ (Fig. 5C). Next we studied the effects of oleic and linoleic acids on the interaction between PPARβ/δ and peptides corresponding to specific coregulator-nuclear receptor binding regions (29). Both fatty acids and GW501516 promoted the interaction between PPARβ/δ and numerous coregulator peptides, which demonstrated their binding to PPARβ/δ (Fig. 5D). Finally, oleic and linoleic acids significantly induced expression of the PPARβ/δ marker gene Adfp in hepatoma cells (Fig. 5E). These data demonstrate that fatty acids directly activate PPARβ/δ. To assess activation of PPARβ/δ in vivo by circulating FFAs in the presence of PPARα, we determined binding of PPARβ/δ to the Lpin2 and St3gal5 genes in the Angptl4 mouse models by using ChIP. Importantly, independent of PPARβ/δ gene expression levels, which remained constant (Fig. 5F), binding of PPARβ/δ to the Lpin2 and St3gal5 genes was proportional to the plasma FFA concentration and mimicked fasting Lpin2 and St3gal5 expression levels (Fig. 5G and H). In contrast, binding of PPARα to the Lpin2 and St3gal5 genes was minimal and did not follow the plasma FFA concentration (Fig. 5G). Again, no binding of PPARα and PPARβ/δ to the negative control gene Rplp0 was observed. These data suggest that PPARβ/δ can be activated by plasma FFAs. Other Wy14643-induced genes whose expression followed the plasma FFA concentration independent of PPARα included lipid droplet proteins 2310076L09Rik (MLDP), and S3-12, as well as Slc16a5 and Gadd45b, suggesting they might represent targets of PPARβ/δ as well (see Fig. S1B in the supplemental material).
PPARα target genes may be upregulated during fasting via induction of PGC1α.
If elevated plasma FFAs cannot account for the induction of classical PPARα activation during fasting, the question arises what other mechanism may be responsible. One possibility is increased coactivator expression. The coactivator PPAR coactivator 1α (PGC1α) plays a major role in the liver during fasting by upregulating genes involved in gluconeogenesis and fatty acid oxidation/ketogenesis, mediating activation by several transcription factors, including PPARα (47, 54, 64). In agreement with previous data (64), expression of Pgc1a went up significantly during fasting (Fig. 6A). Importantly, fasting markedly enhanced binding of PGC1α to the TSS of the PPARα target genes Aldh3a2 and Cpt2, which was abolished in PPARα−/− mice (Fig. 6B). No binding of PGC1α to Rplp0 was observed. These data suggest that upregulation of Pgc1α mRNA may contribute to induction of classical PPARα target genes during fasting via increased PPARα-dependent binding of PGC1α to gene promoters.
FIG. 6.

PPARα activation during fasting may be mediated by PGC1α upregulation. (A) Expression of Pgc1a in livers of fed or fasted wild-type mice (n = 5). Fasting statistically significantly induced gene expression of Pgc1α (P < 0.05). Error bars represent standard errors of the means. (B) Chromatin was extracted from livers of fed and 24-h-fasted wild-type and PPARα−/− mice (n = 3 per group). ChIP was performed with antibodies against PGC1α on the TSS of Aldh3a2 and Cpt2 and the negative control gene Rplp0. Rabbit IgG was used as a specificity control. Gray bars, fed state; black bars, 24-h-fasted state. Error bars represent standard deviations. Fasting significantly induced binding of PGC1α in wild-type but not PPARα−/− mice (P < 0.05).
DISCUSSION
It has been clearly established that PPARα governs the fasting-induced upregulation of numerous genes involved in hepatic fatty acid oxidation, many of which are direct PPARα target genes (19, 27, 34). However, it has remained unclear whether elevated plasma FFAs themselves are responsible for the induction of hepatic fatty acid catabolism via enhanced ligand activation of PPARα (19, 27, 34). Recently, using mice with liver-specific inactivation of the Fasn gene, Chakravarthy et al. showed that unlike dietary fatty acids and de novo-synthesized fatty acids, circulating FFAs fail to activate hepatic PPARα (3). In the present study, by using mice differentially expressing Angptl4 we arrived at essentially the same conclusion.
Importantly, our data also suggest that in contrast to PPARα, hepatic PPARβ/δ can be activated by plasma FFA, which accounts for the plasma FFA- and fasting-dependent upregulation of several genes in wild-type and PPARα−/− mice, including Lpin2 and St3gal5. Indeed, we demonstrated that Lpin2 and St3gal5 expression and binding of PPARβ/δ to the Lpin2 and St3gal5 promoter closely mirror plasma FFA levels. The role of PPARβ/δ in gene regulation by plasma FFA during fasting was substantiated by the observation that induction of Lpin2 and St3gal5 by fasting is reduced in PPARβ/δ−/− mice.
In contrast to plasma FFAs, evidence abounds indicating that dietary fatty acids are able to activate PPARα (40, 44, 46). Recently, it was shown that the effects of dietary fatty acids on hepatic gene expression are quantitatively almost entirely mediated by PPARα (48). Additionally, the present data suggest that dietary fatty acids can also activate PPARβ/δ, as induction of Lpin2 and St3gal5 by dietary fat was entirely or partially maintained in PPARα−/− mice.
It may be argued that the lack of effect of declining plasma FFAs on hepatic PPARα activation may be because PPARα, in contrast to PPARβ/δ, is already saturated with fatty acids at low plasma FFA levels, thus allowing no further activation. Previously, it has been shown that fatty acids bind to PPARβ/δ with an about 5- to10-fold-lower affinity than to PPARα (60). However, as treatment with synthetic agonists clearly results in more pronounced PPARα activation compared to fasting (44), the argument of PPARα saturation is only tenable if we assume fatty acids act as partial agonists that do not elicit full PPARα activity compared to synthetic agonists. Saturation of PPARα is also not supported by the findings reported by Chakravarthy et al. (3).
Alternatively, it is conceivable that fatty acids are present in hepatocytes in distinct pools which have different activities toward PPARα and PPARβ/δ. In this context, it should be realized that dietary fatty acids present in chylomicron remnants are internalized differently compared to plasma FFAs. Whereas the former are liberated after endosomal and lysosomal degradation of cholesteryl esters and triglycerides, plasma FFAs are likely internalized via diffusion as well as via specific fatty acid transport proteins, including CD36. The third contributor to the hepatic fatty acid pool is de novo lipogenesis, a process which occurs in the cytosol. It is presently unclear to what extent these three sources of fatty acids undergo similar metabolic fates. Recent studies support the existence of distinct hepatic fatty acid pools that are differentially shuttled into various metabolic pathways, including oxidation and incorporation into very-low-density lipoprotein (VLDL) triglycerides (67). For example, there is evidence that fatty acids generated by de novo lipogenesis only marginally contribute to VLDL triglycerides, in contrast to plasma FFAs (15). Our present and previous data suggest that in terms of gene regulation, a similar type of segregation occurs between the three sources of fatty acids (3). The mechanism underlying the differential activity of fatty acids from distinct pools toward PPARα and PPARβ/δ remains unknown. One could hypothesize a role for fatty acid binding proteins (FABPs). It can be speculated that FABP1, which has been shown to interact with PPARα (57), picks up lipoprotein-derived fatty acids and shuttles them to PPARα, whereas another FABP expressed in liver such as FABP2 may selectively bind free fatty acids coming from plasma and shuttle them to PPARβ/δ.
An important lingering question is that if plasma FFAs do not activate hepatic PPARα during fasting, what mechanism accounts for activation of PPARα-dependent gene regulation during fasting? Previously, a role for PGC1α in fasting-dependent upregulation of hepatic mitochondrial fatty acid oxidation and ketogenesis was shown (47, 54). Our ChIP analysis indicates enhanced recruitment of PGC1α, which itself is upregulated by fasting, to classical PPARα target genes during fasting. Accordingly, activation of PPARα by fasting may be driven by the increase in PGC1α expression, although an important role for other coactivators cannot be excluded. Recently, it was shown that PGC1α cooperates with BAF60a (SMARCD1) to activate transcription of PPARα target genes involved in peroxisomal and mitochondrial fatty acid oxidation genes (35).
While our data suggest that PPARβ/δ mediates the effects of plasma FFAs on a small set of genes in the liver, the overall importance of PPARβ/δ in hepatic gene regulation by FFAs remains unclear. The same is true for the actual functional role of PPARβ/δ in liver. Presently, combined transcriptomics and metabolomics analyses of livers of PPARα−/− and PPARβ/δ−/− mice are under way to gain more understanding about the role of PPARβ/δ in the liver and to determine the extent to which PPARα and PPARβ/δ regulate distinct sets of genes and govern distinct metabolic pathways, especially under physiological circumstances.
In this study we have used variable expression of the Angptl4 gene to create variations in fasting plasma FFA levels in mice. Angptl4 is a potent inhibitor of lipoprotein lipase and hepatic lipase and decreases uptake of lipoprotein remnants by the liver, thereby decreasing hepatic uptake of dietary fatty acids (36). In addition, it stimulates adipose tissue lipolysis, as shown by the acute increase in plasma FFA upon injection of recombinant Angptl4 (65) and by elevated plasma FFAs and glycerol levels in mice overexpressing Angptl4 (39). The prolipolytic effect of Angptl4 is supported by recent data in humans (52). In the present paper, Angptl4 markedly induced glycerol release from 3T3-L1 adipocytes. Activation of lipolysis by Angptl4 was further substantiated by the altered release of fatty acids and glycerol from adipose tissue explants from Angptl4-Tg and Angptl4−/− mice, as well as by the lack of an increase in FFA during fasting in Angptl4−/− mice. As a consequence, hepatic VLDL production is reduced in Angptl4−/− mice (7). By inhibiting lipoprotein lipase and stimulating adipose tissue lipolysis, Angptl4 promotes switching of hepatic fatty acid uptake from remnant-derived fatty acids toward plasma FFAs (39). Importantly, the variations in plasma FFAs in the Angptl4 mouse models are specifically elicited by fasting, permitting study of the impact of differential plasma FFAs on hepatic gene expression during fasting.
While in vivo and in vitro studies using synthetic PPARα agonists are extremely relevant to assess the toxicological and pharmacological impacts and significance of PPARα, it is unclear to what extent they report on the physiological role of PPARα in liver. Our results reveal that several genes upregulated following pharmacological PPARα activation are not induced by PPARα under physiological conditions such as fasting, or are induced by fasting independently of PPARα but are dependent on PPARβ/δ. It is well known that in reporter assays PPARα and PPARβ/δ (and PPARγ) can activate the same genes, suggesting that all PPARs share an intrinsic ability to transactivate any given PPAR target gene. The present data on Lpin2 and St3gal5 are consistent with the notion that in vivo the dominant receptor in the regulation of a particular PPAR target is context dependent and importantly may differ between pharmacological and physiological stimuli. Genes other than Lpin2 and St3gal5 that have been shown to be activated by both PPARα and PPARβ/δ include Adfp (18), G0s2 (66), and Pdk4 (5). Overall, the data imply that studies using high-affinity synthetic PPAR agonists are not perfectly suited to assess the functions of PPARs during normal physiology.
Supplementary Material
Acknowledgments
We thank Laeticia Lichtenstein, Janna van Diepen, Karin Mudde, Bianca Knoch, Shohreh Keshtkar, José van den Heuvel, Jenny Jansen, and Mechteld Grootte-Bromhaar for laboratory analysis. We also thank Anja Köster (Eli Lilly) for the gift of Angptl4−/− animals.
Footnotes
Published ahead of print on 5 October 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bandsma, R. H., T. H. Van Dijk, A. Harmsel At, T. Kok, D. J. Reijngoud, B. Staels, and F. Kuipers. 2004. Hepatic de novo synthesis of glucose 6-phosphate is not affected in peroxisome proliferator-activated receptor alpha-deficient mice but is preferentially directed toward hepatic glycogen stores after a short term fast. J. Biol. Chem. 279:8930-8937. [DOI] [PubMed] [Google Scholar]
- 2.Botolin, D., Y. Wang, B. Christian, and D. B. Jump. 2006. Docosahexaneoic acid (22:6,n-3) regulates rat hepatocyte SREBP-1 nuclear abundance by Erk- and 26S proteasome-dependent pathways. J. Lipid Res. 47:181-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakravarthy, M. V., Z. Pan, Y. Zhu, K. Tordjman, J. G. Schneider, T. Coleman, J. Turk, and C. F. Semenkovich. 2005. “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1:309-322. [DOI] [PubMed] [Google Scholar]
- 4.Deblois, G., and V. Giguere. 2008. Nuclear receptor location analyses in mammalian genomes: from gene regulation to regulatory networks. Mol. Endocrinol. 22:1999-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Degenhardt, T., A. Saramaki, M. Malinen, M. Rieck, S. Vaisanen, A. Huotari, K. H. Herzig, R. Muller, and C. Carlberg. 2007. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J. Mol. Biol. 372:341-355. [DOI] [PubMed] [Google Scholar]
- 6.Dentin, R., F. Benhamed, J. P. Pegorier, F. Foufelle, B. Viollet, S. Vaulont, J. Girard, and C. Postic. 2005. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Investig. 115:2843-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desai, U., E. C. Lee, K. Chung, C. Gao, J. Gay, B. Key, G. Hansen, D. Machajewski, K. A. Platt, A. T. Sands, M. Schneider, I. Van Sligtenhorst, A. Suwanichkul, P. Vogel, N. Wilganowski, J. Wingert, B. P. Zambrowicz, G. Landes, and D. R. Powell. 2007. Lipid-lowering effects of anti-angiopoietin-like 4 antibody recapitulate the lipid phenotype found in angiopoietin-like 4 knockout mice. Proc. Natl. Acad. Sci. USA 104:11766-11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desvergne, B., and W. Wahli. 1999. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 20:649-688. [DOI] [PubMed] [Google Scholar]
- 9.Duval, C., M. Muller, and S. Kersten. 2007. PPARalpha and dyslipidemia. Biochim. Biophys. Acta 1771:961-971. [DOI] [PubMed] [Google Scholar]
- 10.Escher, P., O. Braissant, S. Basu-Modak, L. Michalik, W. Wahli, and B. Desvergne. 2001. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 142:4195-4202. [DOI] [PubMed] [Google Scholar]
- 11.Evans, R. M., G. D. Barish, and Y. X. Wang. 2004. PPARs and the complex journey to obesity. Nat. Med. 10:355-361. [DOI] [PubMed] [Google Scholar]
- 12.Forman, B. M., J. Chen, and R. M. Evans. 1997. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 94:4312-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fyffe, S. A., M. S. Alphey, L. Buetow, T. K. Smith, M. A. Ferguson, M. D. Sorensen, F. Bjorkling, and W. N. Hunter. 2006. Recombinant human PPAR-beta/delta ligand-binding domain is locked in an activated conformation by endogenous fatty acids. J. Mol. Biol. 356:1005-1013. [DOI] [PubMed] [Google Scholar]
- 14.Gentleman, R. C., V. J. Carey, D. M. Bates, B. Bolstad, M. Dettling, S. Dudoit, B. Ellis, L. Gautier, Y. C. Ge, J. Gentry, K. Hornik, T. Hothorn, W. Huber, S. Iacus, R. Irizarry, F. Leisch, C. Li, M. Maechler, A. J. Rossini, G. Sawitzki, C. Smith, G. Smyth, L. Tierney, J. Y. H. Yang, and J. H. Zhang. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbons, G. F., D. Wiggins, A. M. Brown, and A. M. Hebbachi. 2004. Synthesis and function of hepatic very-low-density lipoprotein. Biochem. Soc. Trans. 32:59-64. [DOI] [PubMed] [Google Scholar]
- 16.Girroir, E. E., H. E. Hollingshead, P. He, B. Zhu, G. H. Perdew, and J. M. Peters. 2008. Quantitative expression patterns of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) protein in mice. Biochem. Biophys. Res. Commun. 371:456-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottlicher, M., E. Widmark, Q. Li, and J. A. Gustafsson. 1992. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 89:4653-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta, R. A., J. A. Brockman, P. Sarraf, T. M. Willson, and R. N. DuBois. 2001. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J. Biol. Chem. 276:29681-29687. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto, T., W. S. Cook, C. Qi, A. V. Yeldandi, J. K. Reddy, and M. S. Rao. 2000. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 275:28918-28928. [DOI] [PubMed] [Google Scholar]
- 20.Heinaniemi, M., J. O. Uski, T. Degenhardt, and C. Carlberg. 2007. Meta-analysis of primary target genes of peroxisome proliferator-activated receptors. Genome Biol. 8:R147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herzig, S., S. Hedrick, I. Morantte, S. H. Koo, F. Galimi, and M. Montminy. 2003. CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 426:190-193. [DOI] [PubMed] [Google Scholar]
- 22.Hoekstra, M., J. K. Kruijt, M. Van Eck, and T. J. Van Berkel. 2003. Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells. J. Biol. Chem. 278:25448-25453. [DOI] [PubMed] [Google Scholar]
- 23.Hostetler, H. A., A. D. Petrescu, A. B. Kier, and F. Schroeder. 2005. Peroxisome proliferator-activated receptor alpha interacts with high affinity and is conformationally responsive to endogenous ligands. J. Biol. Chem. 280:18667-18682. [DOI] [PubMed] [Google Scholar]
- 24.Keller, H., C. Dreyer, J. Medin, A. Mahfoudi, K. Ozato, and W. Wahli. 1993. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. USA 90:2160-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kersten, S. 2005. Regulation of lipid metabolism via angiopoietin-like proteins. Biochem. Soc. Trans. 33:1059-1062. [DOI] [PubMed] [Google Scholar]
- 26.Kersten, S., B. Desvergne, and W. Wahli. 2000. Roles of PPARs in health and disease. Nature 405:421-424. [DOI] [PubMed] [Google Scholar]
- 27.Kersten, S., J. Seydoux, J. M. Peters, F. J. Gonzalez, B. Desvergne, and W. Wahli. 1999. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 103:1489-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kliewer, S. A., S. S. Sundseth, S. A. Jones, P. J. Brown, G. B. Wisely, C. S. Koble, P. Devchand, W. Wahli, T. M. Willson, J. M. Lenhard, and J. M. Lehmann. 1997. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 94:4318-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koppen, A., R. Houtman, D. Pijnenburg, E. H. Jeninga, R. Ruijtenbeek, and E. Kalkhoven. 20 July 2009. Nuclear receptor-coregulator interaction profiling identifies TRIP3 as a novel PPARγ cofactor. Mol. Cell Proteomics [Epub ahead of print.] [DOI] [PMC free article] [PubMed]
- 30.Koster, A., Y. B. Chao, M. Mosior, A. Ford, P. A. Gonzalez-DeWhitt, J. E. Hale, D. Li, Y. Qiu, C. C. Fraser, D. D. Yang, J. G. Heuer, S. R. Jaskunas, and P. Eacho. 2005. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of Angptl4 and Angptl3: regulation of triglyceride metabolism. Endocrinology 146:4943-4950. [DOI] [PubMed] [Google Scholar]
- 31.Krey, G., O. Braissant, F. L'Horset, E. Kalkhoven, M. Perroud, M. G. Parker, and W. Wahli. 1997. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 11:779-791. [DOI] [PubMed] [Google Scholar]
- 32.Lefebvre, P., G. Chinetti, J. C. Fruchart, and B. Staels. 2006. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 116:571-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefterova, M. I., Y. Zhang, D. J. Steger, M. Schupp, J. Schug, A. Cristancho, D. Feng, D. Zhuo, C. J. Stoeckert, Jr., X. S. Liu, and M. A. Lazar. 2008. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 22:2941-2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leone, T. C., C. J. Weinheimer, and D. P. Kelly. 1999. A critical role for the peroxisome proliferator-activated receptor alpha (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 96:7473-7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li, S., C. Liu, N. Li, T. Hao, T. Han, D. E. Hill, M. Vidal, and J. D. Lin. 2008. Genome-wide coactivation analysis of PGC-1α identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab. 8:105-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lichtenstein, L., J. F. Berbee, S. J. van Dijk, K. W. van Dijk, A. Bensadoun, I. P. Kema, P. J. Voshol, M. Muller, P. C. Rensen, and S. Kersten. 2007. Angptl4 upregulates cholesterol synthesis in liver via inhibition of LPL- and HL-dependent hepatic cholesterol uptake. Arterioscler. Thromb. Vasc. Biol. 27:2420-2427. [DOI] [PubMed] [Google Scholar]
- 37.Lin, Q., S. E. Ruuska, N. S. Shaw, D. Dong, and N. Noy. 1999. Ligand selectivity of the peroxisome proliferator-activated receptor alpha. Biochemistry 38:185-190. [DOI] [PubMed] [Google Scholar]
- 38.Mandard, S., M. Muller, and S. Kersten. 2004. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol. Life Sci. 61:393-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandard, S., F. Zandbergen, E. van Straten, W. Wahli, F. Kuipers, M. Muller, and S. Kersten. 2006. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J. Biol. Chem. 281:934-944. [DOI] [PubMed] [Google Scholar]
- 40.Martin, P. G., H. Guillou, F. Lasserre, S. Dejean, A. Lan, J. M. Pascussi, M. Sancristobal, P. Legrand, P. Besse, and T. Pineau. 2007. Novel aspects of PPARα-mediated regulation of lipid and xenobiotic metabolism revealed through a nutrigenomic study. Hepatology 45:767-777. [DOI] [PubMed] [Google Scholar]
- 41.Mater, M. K., A. P. Thelen, D. A. Pan, and D. B. Jump. 1999. Sterol response element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J. Biol. Chem. 274:32725-32732. [DOI] [PubMed] [Google Scholar]
- 42.Nadra, K., S. I. Anghel, E. Joye, N. S. Tan, S. Basu-Modak, D. Trono, W. Wahli, and B. Desvergne. 2006. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor beta/delta. Mol. Cell. Biol. 26:3266-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen, R., T. A. Pedersen, D. Hagenbeek, P. Moulos, R. Siersbaek, E. Megens, S. Denissov, M. Borgesen, K. J. Francoijs, S. Mandrup, and H. G. Stunnenberg. 2008. Genome-wide profiling of PPARγ: RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 22:2953-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patsouris, D., J. K. Reddy, M. Muller, and S. Kersten. 2006. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 147:1508-1516. [DOI] [PubMed] [Google Scholar]
- 45.Rakhshandehroo, M., L. M. Sanderson, M. Matilainen, R. Stienstra, C. Carlberg, P. J. de Groot, M. Muller, and S. Kersten. 2007. Comprehensive analysis of PPARα-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res. 2007:26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren, B., A. P. Thelen, J. M. Peters, F. J. Gonzalez, and D. B. Jump. 1997. Polyunsaturated fatty acid suppression of hepatic fatty acid synthase and S14 gene expression does not require peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 272:26827-26832. [DOI] [PubMed] [Google Scholar]
- 47.Rhee, J., Y. Inoue, J. C. Yoon, P. Puigserver, M. Fan, F. J. Gonzalez, and B. M. Spiegelman. 2003. Regulation of hepatic fasting response by PPARgamma coactivator-1α (PGC-1α): requirement for hepatocyte nuclear factor 4α in gluconeogenesis. Proc. Natl. Acad. Sci. USA 100:4012-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanderson, L. M., P. J. de Groot, G. J. Hooiveld, A. Koppen, E. Kalkhoven, M. Muller, and S. Kersten. 2008. Effect of synthetic dietary triglycerides: a novel research paradigm for nutrigenomics. PLoS ONE 3:e1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saramaki, A., S. Diermeier, R. Kellner, H. Laitinen, S. Vaisanen, and C. Carlberg. 2009. Cyclical chromatin looping and transcription factor association on the regulatory regions of the p21 (CDKN1A) gene in response to 1α,25-dihydroxyvitamin D3. J. Biol. Chem. 284:8073-8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt, A., N. Endo, S. J. Rutledge, R. Vogel, D. Shinar, and G. A. Rodan. 1992. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol. Endocrinol. 6:1634-1641. [DOI] [PubMed] [Google Scholar]
- 51.Staels, B., M. Maes, and A. Zambon. 2008. Fibrates and future PPARα agonists in the treatment of cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 5:542-553. [DOI] [PubMed] [Google Scholar]
- 52.Staiger, H., C. Haas, J. Machann, R. Werner, M. Weisser, F. Schick, F. Machicao, N. Stefan, A. Fritsche, and H. U. Haring. 2008. Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via PPARδ and is of metabolic relevance in humans. Diabetes 58:579-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramanian, A., P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander, and J. P. Mesirov. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102:15545-15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vega, R. B., J. M. Huss, and D. P. Kelly. 2000. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 20:1868-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Heydebreck, A., W. Huber, and R. Gentleman. 2004. Differential expression with the Bioconductor Project. Bioconductor Project working papers, no. 7. bepress, Berkeley, CA. http://www.bepress.com/bioconductor/paper7.
- 56.Wang, X. W., and B. Seed. 2003. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 31:e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolfrum, C., C. M. Borrmann, T. Borchers, and F. Spener. 2001. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha- and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc. Natl. Acad. Sci. USA 98:2323-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Worgall, T. S., S. L. Sturley, T. Seo, T. F. Osborne, and R. J. Deckelbaum. 1998. Polyunsaturated fatty acids decrease expression of promoters with sterol regulatory elements by decreasing levels of mature sterol regulatory element-binding protein. J. Biol. Chem. 273:25537-25540. [DOI] [PubMed] [Google Scholar]
- 59.Wu, Z. J., R. A. Irizarry, R. Gentleman, F. Martinez-Murillo, and F. Spencer. 2004. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 99:909-917. [Google Scholar]
- 60.Xu, H. E., M. H. Lambert, V. G. Montana, D. J. Parks, S. G. Blanchard, P. J. Brown, D. D. Sternbach, J. M. Lehmann, G. B. Wisely, T. M. Willson, S. A. Kliewer, and M. V. Milburn. 1999. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 3:397-403. [DOI] [PubMed] [Google Scholar]
- 61.Xu, J., M. T. Nakamura, H. P. Cho, and S. D. Clarke. 1999. Sterol regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. A mechanism for the coordinate suppression of lipogenic genes by polyunsaturated fats. J. Biol. Chem. 274:23577-23583. [DOI] [PubMed] [Google Scholar]
- 62.Yahagi, N., H. Shimano, A. H. Hasty, M. Amemiya-Kudo, H. Okazaki, Y. Tamura, Y. Iizuka, F. Shionoiri, K. Ohashi, J. Osuga, K. Harada, T. Gotoda, R. Nagai, S. Ishibashi, and N. Yamada. 1999. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J. Biol. Chem. 274:35840-35844. [DOI] [PubMed] [Google Scholar]
- 63.Yamashita, H., M. Takenoshita, M. Sakurai, R. K. Bruick, W. J. Henzel, W. Shillinglaw, D. Arnot, and K. Uyeda. 2001. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 98:9116-9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoon, J. C., P. Puigserver, G. Chen, J. Donovan, Z. Wu, J. Rhee, G. Adelmant, J. Stafford, C. R. Kahn, D. K. Granner, C. B. Newgard, and B. M. Spiegelman. 2001. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413:131-138. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida, K., T. Shimizugawa, M. Ono, and H. Furukawa. 2002. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J. Lipid Res. 43:1770-1772. [DOI] [PubMed] [Google Scholar]
- 66.Zandbergen, F., S. Mandard, P. Escher, N. S. Tan, D. Patsouris, T. Jatkoe, S. Rojas-Caro, S. Madore, W. Wahli, S. Tafuri, M. Muller, and S. Kersten. 2005. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem. J. 392:313-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang, Y. L., A. Hernandez-Ono, C. Ko, K. Yasunaga, L. S. Huang, and H. N. Ginsberg. 2004. Regulation of hepatic apolipoprotein B-lipoprotein assembly and secretion by the availability of fatty acids. I. Differential response to the delivery of fatty acids via albumin or remnant-like emulsion particles. J. Biol. Chem. 279:19362-19374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}