

Abstract
Although human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) has been extensively studied, there are still significant questions about the effects of mutations on the maturation and stability of RT. We show here that a significant fraction (>80%) of the single point mutations we generated in the thumb subdomain of HIV-1 (RT) affect the stability of RT in virions. Fragments of the unstable mutant RTs can be detected in Western blots of virion proteins; however, the degree of degradation varies. The titers of the mutants whose virions contain degraded RTs are reduced. Some, but not all, of the unstable RT thumb subdomain mutants we analyzed have a temperature-sensitive phenotype. A preliminary survey of mutations in other subdomains of RT shows that some of these mutations also destabilize RT. The stability of the RT mutants is enhanced by the addition of a protease inhibitor, suggesting that the viral protease plays an important role in the degradation of the mutant RTs. These results confirm and extend earlier reports of mutations that affect the stability of RT in virions. The data suggest that the stability of a mutant RT in virions could be a major factor in determining the virus titer and, by extension, viral fitness, which could affect whether a mutation in RT is acceptable to the virus.
Although mutations arise frequently during human immunodeficiency virus type 1 (HIV-1) replication, most mutations are rapidly lost because they have a negative effect on viral replication. Mutations can be retained if their effect on viral function is minimal or fixed if there is selective pressure such as, for example, drug treatment that confers a selective advantage for the mutation. However, the extent to which most mutations affect viral replication is not known. In the case of the HIV-1 reverse transcriptase (RT), understanding the effects of mutations is complicated by both the pathway used to make the mature protein and its structure. RT is synthesized as part of the Gag-Pol polyprotein and is produced from Gag-Pol by cleavage with the protease (PR). Mature HIV-1 RT is a heterodimer composed of two related subunits, p66 and p51. The two subunits share a common amino terminus but differ at the carboxy terminus. In the Gag-Pol polyprotein, the carboxy terminus of PR is linked to the amino terminus of both of the RT subunits, and the amino terminus of integrase (IN) is linked to the carboxy terminus of the larger subunit of RT, p66 (26). Because HIV-1 is a heterodimer, most RT mutations cause changes in both the p66 and the p51 subunits. Changes in either subunit, or in both subunits, can affect the behavior of RT.
We generated single point mutations in the thumb subdomain of RT. More than 80% of the mutations tested affected the stability of RT in virions. Even in virions in which RT was shown to be extensively degraded in Western blots of virion proteins, there were normal, or near normal, amounts of both IN and PR, showing that processing of the mutant Gag-Pol is reasonably normal. This phenotype is similar to the phenotype reported by Huang et al. for their triple RT mutant in the fingers subdomain (15). The Huang et al. triple mutant was temperature sensitive, as were some of the single point mutants we analyzed. There is a report that a point mutation in the thumb subdomain caused the complete and selective loss of RT in virions (24), and there is a mutation in the connection subdomain that destabilizes RT (28). There are additional reports of mutations that appear to destabilize RT, but these mutations were reported to have more global effects on the processing of Gag-Pol and/or Gag (1, 16, 21).
Viral fitness is a major consideration in HIV-infected patients. There is an inverse relationship between the ability of the virus to replicate (fitness) and the long-term health of patients. Drug treatments are successful because they dramatically reduce the fitness of the virus. Unfortunately, HIV can become resistant to all of the approved drugs. The development of drug resistance is always accompanied by a loss of fitness in the absence of drugs. The loss of fitness can be small; however, a simple thought experiment shows that drug resistance mutations reduce viral fitness. At any given time, the major strain that is present, the wild type (WT), is the most fit. Any variant that arises and is more fit than the WT will become the new WT. This process occurs all of the time, as the virus continually adapts to the changing environment in the patient. In patients undergoing drug treatment, resistant virus is selected because it is more fit in the presence of the drug(s) than the WT. However, any resistant virus must have been less fit than WT in the absence of drugs, or it would have been the predominant (WT) virus before drug therapy was initiated.
Unfortunately, in most cases, the reduction in fitness that accompanies the development of resistance is too slight to have much benefit for the patient. There is evidence that mutations that confer resistance to nucleoside analogs reduce viral fitness in patients (9), and preliminary data suggest that some of the primary mutations that cause resistance to the new IN inhibitor raltegravir can have a significant impact on viral fitness (10). In thinking about the mutations that are selected by various drug therapies, the question arises: what causes the loss of fitness? In some cases, resistance mutations have a negative impact on the enzymatic activity of the target protein. For example, the M184V mutation in RT, which confers resistance to lamivudine and emtricitabine, reduces polymerase activity (2, 5, 12, 13, 20, 23).
The fact that a majority of the RT mutations we tested affect the stability of RT suggests that there could be a number of mutations that would otherwise be selected, either in response to immune selection or drug therapy, that are not selected because the mutations also reduce RT stability. This idea is supported by the effects of mutations at G190, which cause non-nucleoside RT inhibitor resistance but have a negative impact on viral fitness (16). The data, taken together, suggest that the virus may not have as much latitude in the mutations that can be selected in RT as was previously thought, suggesting that the quest to find new drugs that will select mutants with significantly reduced fitness may not be as difficult a task as we might otherwise think.
MATERIALS AND METHODS
Construction of the HIV-1 mutants.
The one-round HIV-1 viral vector, pNLNgoMIVR-E-HSA, has been described (19). Mutations were made by using the QuikChange Multi site-directed mutagenesis kit (Stratagene). DNA oligonucleotides (Invitrogen) containing the desired mutations were used in the mutagenesis protocol. The plasmids were analyzed by restriction endonuclease digestion and DNA sequencing to confirm that only the desired mutations were present.
Cells.
The human embryonal kidney cell line 293 was obtained from the American Type Culture Collection; the 293T cell line was obtained from GenHunter Corp. The human osteosarcoma cell line HOS was obtained from Richard Schwartz (Michigan State University, Lansing, MI). 293, 293T, and HOS cells were maintained in Dulbecco modified Eagle medium (DMEM; Invitrogen) supplemented with 5% fetal bovine serum, 5% newborn calf serum, and penicillin (50 U/ml) plus streptomycin (50 μg/ml) (Quality Biologicals).
Transfection, infection, and phenotyping.
293 cells were transfected with 3 μg of pNLNgoMIVR-E-HSA and 1.5 μg of pHCMV-g (obtained from Jane Burns, University of California at San Diego) by using the calcium phosphate method. pHCMV-g expresses the vesicular stomatitis virus glycoprotein (3, 30). 293 cells were plated in 100-mm-diameter dishes at a density of 1.5 × 106 cells per plate on the day prior to transfection. At 6 to 7 h after the calcium phosphate precipitate was added, the plates were washed twice with phosphate-buffered saline (PBS), and fresh medium was added. The 48-h supernatants were clarified by low-speed centrifugation, and an aliquot was used to infect HOS cells. The amount of p24 in the supernatant was determined by using a Perkin-Elmer Alliance HIV-1 p24 enzyme-linked immunosorbent assay kit; the p24 concentration was used to control the amount of virus in the samples. HOS cells were plated in 60-mm-diameter dishes at a density of 1.5 × 105 cells per plate on the day prior to infection. The virus was allowed to absorb to the cells for 4 h, and then fresh medium was added. At 48 h after infection, cells were harvested from the plate with 1.0 ml of versene (Gibco), an additional 3 ml of PBS without Ca2+/Mg2+ was added, and the cells were collected by centrifugation, washed, and resuspended in 100 μl of PBS. The cells were labeled with phycoerythrin-conjugated rat anti-mouse CD24 monoclonal antibody (MAb; Pharmingen) using standard procedures and fixed with paraformaldehyde, and fluorescence-activated cell sorting was used to determine the virus titer. The relative virus titers were calculated from the linear range of the titer assay.
Real-time PCR.
293 cells were transfected with 5 μg of the HIV vector DNA and 3 μg of pHCMV-g DNA by using the calcium phosphate method. Forty-eight-hour supernatants (9 ml) were harvested, clarified by slow-speed centrifugation, filtered through a 45-μm-pore-size syringe filter (Millipore), and treated for 30 min with Turbo DNase (Ambion). The supernatants were concentrated to 0.5 ml using 20-ml 300,000-MWCO concentrators (Vivascience). Medium was added to give a final volume of 7.25 ml per sample. Then, 1.75 ml of each virus sample was added to four 60-mm plates of HOS cells for 2 h. HOS cells were washed with 2 ml of PBS, fresh medium was added, and DNA was isolated at 2-, 4-, 6-, and 24-h time points using the Biorobot EZ-1 (Qiagen). The elution volume was 100 μl for each sample. Real-time PCRs were performed using the 2× Universal TaqMan Master Mix (Applied Biosystems) in 50-μl reaction volumes using previously described reagents and conditions (19). Viral DNA copy numbers were determined by using the ABI 7700 (Applied Biosystems) and normalized for the amount of p24 antigen in the supernatant.
Ritonavir treatment of virions.
293 cells were seeded at 2 × 105/well on a six-well dish in DMEM with 5% fetal calf/newborn calf serum and then incubated at 37°C for 2 h. Each well was transfected with 3 μg of HIV DNA and 1 μg of pHCMV-g for 4 to 7 h by using the calcium phosphate method. Cells were gently washed twice with PBS without Ca2+/Mg2+ and refed with DMEM that contained 2% fetal calf/newborn calf sera. Ritonavir was added at 1, 0.l, and 0 μM final concentrations for 48 h. Virions were harvested, filtered, and collected by ultracentrifugation as described in the Western blot protocol. The viral pellet was resuspended in 20 μl of 1× NuPage LDS sample buffer (Invitrogen catalog no. NP0007), H2O, and 10 mM dithiothreitol and run on a gel for Western blots.
Western blots.
Virus stocks were generated by transfecting 293 (or 293T in the temperature-sensitive assay) cells with 15 μg of HIV vector DNA and 4 μg of pHCMV-g DNA by using the calcium phosphate method. Cells were washed twice with PBS without Ca2+/Mg2+ and refed with DMEM that contained 2% fetal calf/newborn calf serum. The cells were incubated at either 37 or 32°C. Low serum was used to reduce the amount of immunoglobulin G in the Western blots. After 16 h, the supernatants were clarified by low-speed centrifugation and filtered through a 0.45-μm-pore-size filter. The virus was concentrated by using Sartorius Vivaspin concentrators, and the p24 levels were determined by ELISA (Perkin-Elmer). Supernatants containing 500 ng of p24 were underlaid with 1 ml of 20% sucrose-PBS and centrifuged at 25,000 rpm at 4°C for at least 35 min. Viral pellets were resuspended in 25 μl of 1× NuPage LDS sample buffer, H2O, and 10 mM dithiothreitol; heated to 95°C for 5 min; fractionated on a NuPage 4 to 12% Bis-Tris gel (Invitrogen); and wet blot transferred onto Hybond-ECL nitrocellulose (Amersham). Blots were incubated in block solution (5% dry milk in Tris-buffered saline-Tween) for 4 h. Primary antibody was diluted in block solution, and blots were incubated for 4 to 24 h. Blots were washed 10 times for 6 min each (1 h total). Secondary antibody was diluted in block solution, and the blots were incubated for 2 to 4 h. The blots were washed five times for 6 min each time (30 min total), and the antibody was detected with SuperSignal West Pico chemiluminescent substrate (Pierce). The individual mouse anti-RT antibodies have been described (11); the MAb mix was prepared using equal amounts of MAbs 19, 21, 42, 48, and 50. The MAbs used individually were 28 and 51. The mouse MAbs were diluted 1:500; rabbit anti-PR antibody (kindly provided by Ron Swanstrom and Janera Harris) was diluted 1:2,000, and goat anti-CA antibody (kindly provided by Dave Ott) was diluted 1:10,000. The rabbit anti-CA antibody (kindly provided by Rob Gorelick) was diluted 1:10,000. The rabbit anti-IN antibody (kindly provided by Duane Grandgenett) was diluted 1:10,000.
RESULTS
Mutations in the thumb subdomain can affect the stability of RT.
We created mutations in the HIV-1 vector pNLNgoMIVR-E-HSA (Table 1). Most of the mutations were in the thumb subdomain of RT. A number of the mutations were near points of contact between the p66 subunit and nucleic acid (14, 17, 22). Some of the mutants had been tested for polymerase activity using a recombinant form of RT (4, 12). We measured the effects of the mutations on titer, the biosynthesis of RT in cultured cells, and the ability of the mutant RTs to carry out viral DNA synthesis in cells. All titer data were corrected for the amount of virus and are given as the percentage of WT (5.4 × 103/pg of p24).
TABLE 1.
HIV-1 RT mutants and their propertiesa
| Mutant | Location | Mean relative titer (%) ± SD |
p66/p51 (37°C) | |
|---|---|---|---|---|
| 37°C | 32°Cb | |||
| R78K | Fingers | 10 ± 4 | 45 ± 10 | Some degradation |
| V148S | Boundary/fingers-palm | 18 ± 1 | 52 ± 9 | Some degradation |
| P150G | Boundary/fingers-palm | 0 ± 1 | (0) | Very low/absent |
| G152A | Boundary/fingers-palm | 0 ± 2 | (0) | Some degradation |
| M230A | Boundary/palm-thumb | 0 ± 0 | (4) | Normal |
| D256N | Thumb | 30 ± 5 | 100 ± 34 | Some degradation |
| I257T/W229G | Thumb | 0 ± 0 | (3) | Very low/absent |
| I257T | Thumb | 0 ± 1 | ND | Very low/absent |
| G262A | Thumb | 0 ± 4 | (6) | Normal |
| L264S | Thumb | 0 ± 1 | 1 ± 0 | Very low/absent |
| N265D | Thumb | 16 ± 17 | ND | Normal |
| W266T | Thumb | 0 ± 0 | (0) | Some degradation |
| I274T | Thumb | 4 ± 6 | 59 ± 1 | Very low/absent |
| L279S | Thumb | 1 ± 3 | 43 ± 0 | Very low/absent |
| G285A | Thumb | 100 ± 0 | ND | Some degradation |
| L289A | Thumb | 86 ± 2 | ND | Some degradation |
| T290I | Thumb | 28 ± 4 | ND | Some degradation |
| A299L | Thumb | 1 ± 1 | (1) | Very low/absent |
| E302Q | Thumb | 16 ± 4 | ND | Very low/absent |
| L310S | Thumb | 0 ± 4 | 2 ± 1 | Very low/absent |
The table shows the locations of the mutations and the relative virus titers at 37 and 32°C and describes the degree of degradation of p66/p51 at 37°C. All of the 37°C relative titers were measured, in independent experiments, at least three times. The data in the table are the averages of three measurements. If the relative titer at 32°C was very low and matched the relative titer at 37°C, the experiment was not repeated. For mutants that had a significantly higher relative titer at 32°C than at 37°C, the titer was measured at 32°C in at least two independent experiments. The data are the average results of the experiments.
ND, not determined. Titers in parentheses were determined once at 32°C and were found to be not significantly different from those at 37°C.
Previous analysis with vectors that contained mixtures of fully active RT and of RT with no polymerase activity (19) suggested that virions that contain 50% of the normal amount of polymerase activity have titers that are ca. 40% of the WT; a further reduction in the level of polymerase activity to 25% of WT dramatically reduced the virus titer and caused a significant reduction in viral DNA synthesis.
Viral DNA synthesis proceeds in well-defined steps. Viral RNA is plus strand, the first DNA made is a minus-strand copy of RU5. After the first strand transfer, minus-strand DNA is synthesized progressively along the RNA genome, proceeding from U3 to Gag. Although plus-strand DNA is initiated relatively early, the second (plus-strand) jump cannot take place until all of Gag is copied into minus-strand DNA. We measured the levels of RU5, Gag, and the plus-strand transfer product at various times after infection (Fig. 1). The RU5, Gag, and plus-strand transfer probes have been previously described (19). The 5′ ends of the three probes correspond, respectively, to positions 493, 9682, and 919 in the sequence of PNL4-3.
FIG. 1.

Viral DNA synthesis. Real-time PCR was used to measure the amounts of viral DNA at various times for three important stages of viral DNA synthesis: the probes were designed to measure the amount of viral DNA made early (RU5), near the completion of minus-strand synthesis (Gag) and at the second transfer between templates (plus-strand transfer). Mutants that were analyzed at the same time are shown together. (A) Levels of viral DNA present at various times after infections with the D256N and N265D mutants. (B) Levels of viral DNA present at various times after infections with the I257T, G262A, and W266T. (C) Levels of viral DNA present at various times after infections with the I274T, T290I, and E302Q mutants. The plus-strand transfer and RU5 experiments were done at least twice, while the Gag experiments were done once.
There are some mutants in which there was very little, if any, intact p66 or p51 in the virions, judged by Western blot analysis (P150G, I257T, L264S, L279S, A299L, and L310S; see Fig. 2). The titers of all of these mutants are low (≤1% of the WT titer, see Table 1). When I257T mutant virions were used to infect cells, the initiation of viral DNA synthesis appears to be delayed (Fig. 1). Low amounts of RU5 DNA were detected at 2 h; however, this mutant synthesized some viral DNA (perhaps 10% of the WT amount) after 24 h. We were somewhat surprised that a mutants such as I257T, whose virions have little if any intact RT in Western blots, can still carry out a significant amount of DNA synthesis. The last step in viral DNA synthesis that can accurately be measured using real-time PCR is plus-strand transfer. This step is about halfway though the process of viral DNA synthesis, and it is possible that the I257T mutant was not able to synthesize significant mounts of full-length viral DNA. As mentioned in the introduction, it appears that in the unstable RT mutants, p66 is cleaved from Gag-Pol relatively normally, suggesting that some intact RT is produced, at least transiently, in the mutant virions. It is also possible that some of the cleavages in RT could allow the heterodimer to remain intact, and some of this cleaved RT could retain a portion of its enzymatic activity.
FIG. 2.

Virions of some of the RT mutants contain degraded RT; IN and PR are processed normally and are present in normal amounts. The blots are from the same filter probed sequentially with four antisera. (A) Western blots of RT mutants probed with anti-RT sera. (B) The same blots probed with anti-IN. (C) Probed with anti-PR. (D) Probed with anti-CA.
The I274T mutation had a relative titer ca. 4% of WT, the E302Q had a titer ca. 16% of WT, and the D256N mutant had a titer ca. 30% of WT. For all three of these mutants, DNA synthesis appeared to be delayed relative to WT. At the earlier time points, there was an obvious reduction in the amount of viral DNA synthesized; the reduction was particularly marked for the viral DNA segments synthesized later in the reverse transcription process. However, at later time points, there was an increase in the amount of viral DNA, although the amount of DNA made at late times was still reduced, which presumably explains the reduced titer.
Not all of the thumb mutants showed a reduction in the amounts of intact p66 and p51 RT in the virions. The G262A and N265D mutant virions showed no sign of RT degradation, nor did M230A, which is in the palm near the base of the thumb. The D256N, W266T, G285A, L289A, and T290I mutant virions, which contained some degraded RT, also contained significant amounts of intact p66 and p51 (Fig. 2). Although the E302Q mutant virions contained a relatively small amount of intact p66 and p51 in the experiment shown in Fig. 2, there appears to be more intact E302Q RT in the experiments shown in Fig. 3 and 4. Two of the mutants, N265D and T290I, had a DNA synthesis phenotype that was similar to the mutants whose virions contained reduced amount of intact RT. The N265D and T290I mutants showed a delay in viral DNA synthesis, but these two mutants synthesized almost as much Gag DNA and plus-strand transfer product as WT at 24 h. At 4 and 6 h the G262A mutant produced less Gag and plus-strand transfer DNA than the N265D and T290I mutants; however, G262A had largely caught up at 24 h. In contrast, the W266T mutant showed a strong delay in the synthesis of RU5 DNA and synthesized only small amounts of either Gag DNA or the plus-strand transfer product, even after 24 h. This is consistent with the in vitro properties of this mutant, which has significant defects in the activities of both polymerase and RNase H (12).
FIG. 3.
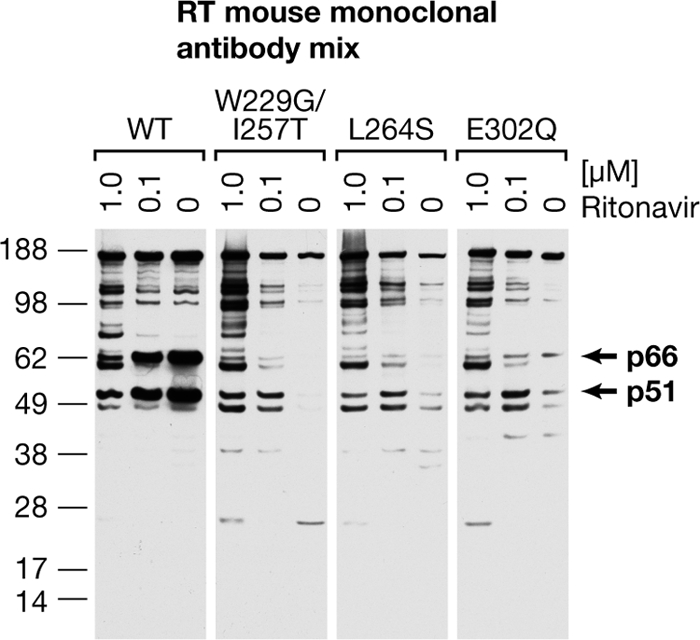
The PR inhibitor, ritonavir, reduces the degradation of the mutant RTs. No ritonavir or 1.0 or 0.1 μM ritonavir was added to cells transfected with RT mutant expression plasmids. Virions were collected and analyzed by Western blotting using a mixture of MAbs to RT.
FIG. 4.

Pattern of degradation of the RT mutants in blots with specific MAbs. (A) No ritonavir or 0.1 or 1.0 μM ritonavir was added to cells transfected with DNA encoding vectors with mutant RTs. Virions were collected and analyzed by Western blotting using two MAbs that recognize different epitopes in RT. MAb 51 reacts with an epitope very near the C terminus of the p51 subunit of RT. (B) An identical blot was probed with MAb 28, which reacts with an epitope in the middle of the Pol domain of RT. (11).
Mutations outside the thumb subdomain can affect the stability of RT.
We analyzed a small number of mutations in the other subdomains in RT, some of which affected the stability of RT. One mutation in the fingers (R78K), had a titer ca. 10% of WT. Three mutations are near the boundary of the fingers and the palm (V148S, P150G, and G152A). V148S had a titer ca. 18% of WT; the other two mutants both had very low titers. Neither the P150G nor the G152A mutants made significant amounts of RU5, Gag, or plus-strand transfer DNA, even at 24 h (data not shown). The P150G mutant virions contained very low levels of intact p66 or p51 RT; the other mutations have a less dramatic effect on RT stability. The double mutant W229G/I257T, which has one change in the thumb and one change in the palm, had very low levels of intact p66 and p51 in virions. The I257T mutation alone can dramatically alter the stability of RT, and this mutation appears to be sufficient to account for the phenotype of the double mutant.
PR and IN are present in normal, or nearly normal, amounts in the mutant virions.
Some or all of the unstable RT mutations could have affected the folding of Gag-Pol in a way that would disrupt the processing of the Gag-Pol polyprotein, which could affect the maturation of PR, leading to a defect in Gag processing. However, Western blots showed that the levels and apparent sizes of IN, PR, and CA were normal, or nearly normal, in all of the mutant virions (Fig. 2). It is possible that the level of PR and IN is modestly reduced in the P150G mutant; however, CA is processed normally, suggesting that PR is fully functional. Some of the mutant virions (V148S, D256N, G262A, L264S, I274T, and L279S) appeared to contain low levels of partially processed Gag-Pol products, suggesting that these mutations have a modest effect on the folding/processing of Gag-Pol. However, because these mutants contain normal amounts of mature IN and PR, the majority of Gag-Pol must be properly processed.
PR participates in the breakdown of the mutant RTs.
To test whether PR plays a significant role in the breakdown of RT, two different concentrations of ritonavir, a potent inhibitor of PR, were added to cells producing virions that contain some of the unstable RTs. Ritonavir caused an increase in the amount of RT polypeptides in the virions (Fig. 3). Wapling et al. reported a similar result with their connection subdomain mutant (28). At the higher concentration of ritonavir, both the WT and the mutant virions contained a number of proteins that reacted with the mixture of MAbs used to detect RT. The pattern of RT polypeptides in the different mutants appeared to be similar, suggesting that the cleavages were similar. This raises the question of whether the mutations are creating new PR cleavage sites in RT or are exposing sites in RT that PR could cleave if RT was not properly folded and/or dimerized.
PR cleaves the mutant RTs at similar positions.
The first set of Western blots were performed with a mixture of MAbs (Fig. 3). The blots were repeated with individual MAbs (Fig. 4). Although the exact patterns of the RT fragments and the extent of breakdown of the RT mutants were not identical, both the sizes of the RT fragments, and which fragments reacted with individual MAbs were similar. This suggests that the primary effect of the RT mutations is to increase the exposure of preexisting PR cleavage sites rather than creating new sites. Examination of the sequence of WT RT shows that there are a number of sites where PR might be expected to cleave if it had access (7, 27); moreover, the mutations in the thumb that increased the susceptibility of RT to PR cleavage do not create sites where PR would be expected to cleave (data not shown).
Some of the unstable RT mutants are temperature sensitive.
293T cells were transfected at 37°C for 6 h, washed, and then transferred to 32°C to produce the viral stock. After 38 h, virus was harvested and used to infect cells at 37°C or cells at 32° for the first 6 h. Similar results were obtained when the infections were done at either temperature; the data we report here were from infections done at 37°. Cells produce much less virus at 32°C than at 37°C; the titers were normalized for the amount of CA (p24). Some of the mutants (M230A, G262A, L310S) and the double mutant (W229G/I257T) showed what may have been a modest increase in the relative titer at the lower temperature (from undetectable to 2 to 6% of WT). However, five of the mutants (R78K, V148S, D256N, I274T, and L279S) showed large increases in the relative titer (Table 1). All of the mutants that had a strong temperature-sensitive phenotype also had, at 37°C, reduced amounts of intact RT. For all of the temperature-sensitive mutants there was an obvious increase in the amount of p66 and p51 at 32°C (Fig. 5). Even the WT showed a small amount of RT degradation at 37°C, but none at 32°C. We also tested some of the unstable mutants that did not show an increased titer at 32°C. For two of the unstable mutants (A264S and L310S) that had very low titers at 32°C there was a significant increase in p66 and p51 at 32°C. Presumably, these two mutations affect the activity of RT as well as its stability. With the possible exception of the G262A mutant, which had a titer of 6% of the WT at 32°C, none of the mutants we tested that had normal amounts of RT at 37°C showed any evidence that they were temperature sensitive.
FIG. 5.

Some of the RT mutants show increased stability at 32°C. Virions were grown at either 37 or 32°C, the virions were harvested, and RT was analyzed by Western blotting. Western blots were probed first with a mixture of MAbs to RT, and then the blots were stripped and reprobed with a polyclonal antibody against CA.
DISCUSSION
A majority of the point mutations we made in the thumb subdomain of HIV-1 RT affected the stability of RT in virions. Because there are reports of mutations in RT that affect the processing of Gag-Pol and/or Gag (1, 16, 21), we considered the possibility that the point mutations affected the folding of Gag-Pol. However, the amount of PR and IN is normal or near normal in our mutants, showing that the primary effect is on the stability of RT. Figure 6 shows the locations of the mutations on the HIV-1 RT heterodimer. If the mutant RTs form less-stable dimers, the individual subunits of RT could be susceptible to degradation by PR. Wapling et al. (28) proposed that the mutations at W401 that allowed RT degradation affected the ability of RT to dimerize. Although we have no evidence that speaks directly to this possibility, most of the positions where there are temperature-sensitive mutations are not part of the dimer interface (Fig. 6). Some of mutations in the thumb subdomain that cause instability in RT are near the interface between the thumb of p51 and RNase H; however, none of the mutations are at the dimer interface. Because the thumb of the p51 subunit makes extensive interactions with the RNase H domain, it is possible that these mutations could indirectly destabilize the dimer. Similarly, none of the three mutations in the fingers subdomain, which when present together cause a temperature-sensitive phenotype (15), are at the interface between p66 and p51. There are mutations (for example, L310S) that are in the thumb subdomain and mutations elsewhere (such as the P150G mutation, which is near the boundary between the fingers and palm) that destabilize RT and are far from the interface between the subunits.
FIG. 6.

Positions of the thumb subdomain mutations in RT and their effects on RT stability. (A) High-resolution structure of HIV-1 RT heterodimer (8) with the p66 subunit in tan and the p51 subunit in gray. The thumb subdomain of both subunits is shown in green. (B) Close-up view of the isolated thumb subdomain from p66. In both panels, mutations that cause the most severe degradation phenotype (little or no intact p66 and p51) are shown in pink; these mutations are likely to destabilize the hydrophobic core that is responsible for the folding of thumb subdomains. Mutations for which there is either no effect on RT stability or a modest effect on RT stability (significant amounts of p66 and p51 remaining in the virion) are shown in blue.
Many of the mutations that reduce the stability of RT appear to be in the hydrophobic core of the thumb subdomain. Many, but not all, of the mutations that cause RT instability involve the substitution of a hydrophobic amino acid for a hydrophilic amino acid; these include the three mutants for which the lower temperature had the greatest effect on stability (I274T, L279S, and L310S). This suggests that these mutations allow the thumb to partially unfold at 37°C, rendering RT susceptible to cleavage by PR. If it is the thumb of p66 that is primarily responsible for the phenotype, the increase in the susceptibility of RT to PR would be a direct effect; if it is the thumb of p51, a partial unfolding of the thumb could either directly enhance PR cleavage or destabilize the heterodimer. It is possible that the thumbs of both subunits are involved in the instability phenotype. The fact that a number of the mutants are temperature sensitive supports the underlying hypothesis: the lower temperature helps stabilize the hydrophobic core, thus reducing proteolytic degradation. This interpretation is supported by the results of Wrobel et al., who showed, using an HIV-1 Pol bacterial expression system, that mutations in the fingers and palm subdomains that affected the production of mature, properly processed RT usually involved either buried hydrophobic residues or residues involved in hydrogen bonds (29).
How general is this phenotype? A surprising percentage (>80%) of the point mutations we tested in the thumb subdomain make RT more susceptible to degradation by PR. Our data and the data of others show that mutations in other subdomains can have a similar phenotype (15). There are also reports of mutations in the fingers (21), palm (16), and RNase H (1) that appear to affect both RT and Gag-Pol and/or Gag, and there are mutations in capsid (CA) that cause it to be degraded in virions. Although there is no direct evidence, the simplest interpretation is that the CA mutants are degraded by PR (18, 25).
Mutations can also affect the stability of cellular proteins. All of the mutations that caused a loss of hypoxanthine-guanine phosphoribosyltransferase activity in cultured cells also decreased the half-life of the mutant proteins (6). It seems likely that many proteins (both host and viral) have been selected to be folded into compact structures that are resistant to degradation by PRs. There is complex proteolytic degradative machinery host proteins must be able to avoid, and the host's degradative machinery is carefully regulated. In the HIV-1 virion, the situation is much simpler. There is only one PR inside the virion. HIV-1 PR is able to recognize and cleave a fairly broad array of peptide substrates, including sequences in the mature Gag and Pol proteins that are not normally cleaved to any appreciable extent in virions.
It appears to be relatively easy to find mutations that allow PR to degrade RT. This suggests that the stability of proteins in the virion is an important factor that helps determine the fitness of the virus. The observation that a mutation in PR partially compensates for a mutation that destabilizes RT supports this idea (21). Taken together, the data suggest that the effects of a mutation on the enzymatic activity of RT is not the only factor that determines whether a given mutation can be tolerated and/or selected. Although it may be possible to select compensatory mutations that will stabilize a mutant RT, the data suggest that, for RT, what are acceptable mutations may be more constrained than we previously thought. From the point of view of developing new and more broadly effective drugs, this is a good result. Resistance mutations must allow the target enzyme to evade the drug and retain most of its enzymatic activity but not significantly increase the susceptibility of RT to PR. It is also possible that HIV-1 RT may be particularly constrained, relative to other HIV proteins, in terms of which mutations are acceptable. Although the two subunits of RT are folded into similar subdomains, the relationship of the subdomains differs in the two subunits, and any mutation in the fingers, palm, or connection subdomains actually causes two changes in the heterodimeric protein. If the change in either subunit leads to a significant increase in the degradation of RT, the mutation will cause a corresponding decrease in fitness of the virus.
Acknowledgments
This study was supported, in part, by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and by NIH grant AI 27690 to E.A.
We are grateful to Eric Freed for helpful discussions. We thank Michael Abram for assistance with the ritonavir experiments, George Kassey for fluorescence-activated cell sorting analysis, Jiro Wada and Tammy Schroyer for assistance on figures, and Teresa Burdette for help with the manuscript.
Footnotes
Published ahead of print on 16 September 2009.
REFERENCES
- 1.Abram, M. E., and M. A. Parniak. 2005. Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and the RT RNase H domain. J. Virol. 79:11952-11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Back, N. K., and B. Berkhout. 1997. Limiting deoxynucleoside triphosphate concentrations emphasize the processivity defect of lamivudine-resistant variants of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 41:2484-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartz, S. R., and M. A. Vodicka. 1997. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods 12:337-342. [DOI] [PubMed] [Google Scholar]
- 4.Boyer, P. L., A. L. Ferris, P. Clark, J. Whitmer, P. Frank, C. Tantillo, E. Arnold, and S. H. Hughes. 1994. Mutational analysis of the fingers and palm subdomains of human immunodeficiency virus type-1 (HIV-1) reverse transcriptase. J. Mol. Biol. 243:472-483. [DOI] [PubMed] [Google Scholar]
- 5.Boyer, P. L., and S. H. Hughes. 1995. Analysis of mutations at position 184 in reverse transcriptase of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 39:1624-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capecchi, M. R., N. E. Capecchi, S. H. Hughes, and G. M. Wahl. 1974. Selective degradation of abnormal proteins in mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA 71:4732-4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou, K. C., A. G. Tomasselli, I. M. Reardon, and R. L. Heinrikson. 1996. Predicting human immunodeficiency virus protease cleavage sites in proteins by a discriminant function method. Proteins 24:51-72. [DOI] [PubMed] [Google Scholar]
- 8.Das, K., J. D. Bauman, A. D. Clark, Jr., Y. V. Frenkel, P. J. Lewi, A. J. Shatkin, S. H. Hughes, and E. Arnold. 2008. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. USA 105:1466-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deeks, S. G., R. Hoh, T. B. Neilands, T. Liegler, F. Aweeka, C. J. Petropoulos, R. M. Grant, and J. N. Martin. 2005. Interruption of treatment with individual therapeutic drug classes in adults with multidrug-resistant HIV-1 infection. J. Infect. Dis. 192:1537-1544. [DOI] [PubMed] [Google Scholar]
- 10.Delelis, O., I. Malet, L. Na, L. Tchertanov, V. Calvez, A. G. Marcelin, F. Subra, E. Deprez, and J. F. Mouscadet. 2009. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res., in press. [DOI] [PMC free article] [PubMed]
- 11.Ferris, A. L., A. Hizi, S. D. Showalter, S. Pichuantes, L. Babe, C. S. Craik, and S. H. Hughes. 1990. Immunologic and proteolytic analysis of HIV-1 reverse transcriptase structure. Virology 175:456-464. [DOI] [PubMed] [Google Scholar]
- 12.Gao, H. Q., P. L. Boyer, E. Arnold, and S. H. Hughes. 1998. Effects of mutations in the polymerase domain on the polymerase, RNase H and strand transfer activities of human immunodeficiency virus type 1 reverse transcriptase. J. Mol. Biol. 277:559-572. [DOI] [PubMed] [Google Scholar]
- 13.Gao, L., M. N. Hanson, M. Balakrishnan, P. L. Boyer, B. P. Roques, S. H. Hughes, B. Kim, and R. A. Bambara. 2008. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse transcriptase derive solely from a dNTP utilization defect. J. Biol. Chem. 283:9196-9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang, H., R. Chopra, G. L. Verdine, and S. C. Harrison. 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282:1669-1675. [DOI] [PubMed] [Google Scholar]
- 15.Huang, M., R. Zensen, M. Cho, and M. A. Martin. 1998. Construction and characterization of a temperature-sensitive human immunodeficiency virus type 1 reverse transcriptase mutant. J. Virol. 72:2047-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang, W., A. Gamarnik, K. Limoli, C. J. Petropoulos, and J. M. Whitcomb. 2003. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J. Virol. 77:1512-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobo-Molina, A., A. D. Clark, Jr., R. L. Williams, R. G. Nanni, P. Clark, A. L. Ferris, S. H. Hughes, and E. Arnold. 1991. Crystals of a ternary complex of human immunodeficiency virus type 1 reverse transcriptase with a monoclonal antibody Fab fragment and double-stranded DNA diffract X-rays to 3.5-Å resolution. Proc. Natl. Acad. Sci. USA 88:10895-10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi, A., K. Nagashima, and E. O. Freed. 2006. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, gag-gag interactions, gag-membrane binding, and virion maturation. J. Virol. 80:7939-7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Julias, J. G., A. L. Ferris, P. L. Boyer, and S. H. Hughes. 2001. Replication of phenotypically mixed human immunodeficiency virus type 1 virions containing catalytically active and catalytically inactive reverse transcriptase. J. Virol. 75:6537-6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krebs, R., U. Immendorfer, S. H. Thrall, B. M. Wohrl, and R. S. Goody. 1997. Single-step kinetics of HIV-1 reverse transcriptase mutants responsible for virus resistance to nucleoside inhibitors zidovudine and 3-TC. Biochemistry 36:10292-10300. [DOI] [PubMed] [Google Scholar]
- 21.Olivares, I., A. Mulky, P. I. Boross, J. Tozser, J. C. Kappes, C. Lopez-Galindez, and L. Menendez-Arias. 2007. HIV-1 protease dimer interface mutations that compensate for viral reverse transcriptase instability in infectious virions. J. Mol. Biol. 372:369-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarafianos, S. G., K. Das, C. Tantillo, A. D. Clark, Jr., J. Ding, J. M. Whitcomb, P. L. Boyer, S. H. Hughes, and E. Arnold. 2001. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 20:1449-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma, P. L., and C. S. Crumpacker. 1999. Decreased processivity of human immunodeficiency virus type 1 reverse transcriptase (RT) containing didanosine-selected mutation Leu74Val: a comparative analysis of RT variants Leu74Val and lamivudine-selected Met184Val. J. Virol. 73:8448-8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takehisa, J., M. H. Kraus, J. M. Decker, Y. Li, B. F. Keele, F. Bibollet-Ruche, K. P. Zammit, Z. Weng, M. L. Santiago, S. Kamenya, M. L. Wilson, A. E. Pusey, E. Bailes, P. M. Sharp, G. M. Shaw, and B. H. Hahn. 2007. Generation of infectious molecular clones of simian immunodeficiency virus from fecal consensus sequences of wild chimpanzees. J. Virol. 81:7463-7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang, S., T. Murakami, B. E. Agresta, S. Campbell, E. O. Freed, and J. G. Levin. 2001. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J. Virol. 75:9357-9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Telesnitsky, A., and G. P. Goff. 1997. Reverse transcriptase and the generation of retroviral DNA, p. 121-160. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- 27.Tomasselli, A. G., J. L. Sarcich, L. J. Barrett, I. M. Reardon, W. J. Howe, D. B. Evans, S. K. Sharma, and R. L. Heinrikson. 1993. Human immunodeficiency virus type-1 reverse transcriptase and ribonuclease H as substrates of the viral protease. Protein Sci. 2:2167-2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wapling, J., K. L. Moore, S. Sonza, J. Mak, and G. Tachedjian. 2005. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J. Virol. 79:10247-10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wrobel, J. A., S. F. Chao, M. J. Conrad, J. D. Merker, R. Swanstrom, G. J. Pielak, and C. A. Hutchison III. 1998. A genetic approach for identifying critical residues in the fingers and palm subdomains of HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 95:638-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yee, J. K., T. Friedmann, and J. C. Burns. 1994. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 43(Pt. A):99-112. [DOI] [PubMed] [Google Scholar]