

Abstract
The DH-PH domain tandems of Dbl-homology guanine nucleotide exchange factors catalyze the exchange of GTP for GDP in Rho-family GTPases, and thus initiate a wide variety of cellular signaling cascades. Although several crystal structures of complexes of DH-PH tandems with cognate, nucleotide free Rho GTPases are known, they provide limited information about the dynamics of the complex and it is not clear how accurately they represent the structures in solution. We used a complementary combination of nuclear magnetic resonance (NMR), small-angle X-ray scattering (SAXS), and hydrogen-deuterium exchange mass spectrometry (DXMS) to study the solution structure and dynamics of the DH-PH tandem of RhoA-specific exchange factor PDZRhoGEF, both in isolation and in complex with nucleotide free RhoA. We show that in solution the DH-PH tandem behaves as a rigid entity and that the mutual disposition of the DH and PH domains remains identical within experimental error to that seen in the crystal structure of the complex, thus validating the latter as an accurate model of the complex in vivo. We also show that the nucleotide-free RhoA exhibits elevated dynamics when in complex with DH-PH, a phenomenon not observed in the crystal structure, presumably due to the restraining effects of crystal contacts. The complex is readily and rapidly dissociated in the presence of both GDP and GTP nucleotides, with no evidence of intermediate ternary complexes.
Keywords: RhoA, guanine nucleotide exchange factors, protein structure, protein dynamics, nuclear magnetic resonance, small angle X-ray scattering, deuterium-hydrogen exchange mass spectrometry
Introduction
Rho (Ras homology) GTPases are a homologous family of 22 proteins that act in pathways regulating various aspects of cell physiology, including remodeling of the cytoskeleton and gene transcription. The most ubiquitous and consequently the most studied among them are RhoA, Cdc42, and Rac1.1–3 All are poor enzymes with very low kcat values, so that the GTP-bound form (i.e., enzyme-substrate complex) has a long half-life. The conformation of the protein in this complex is distinct from that in the enzyme-product complex (Rho•GDP), so that the bound GTP nucleotide confers biological activity on RhoA enabling it to bind to effectors. The stability of the substrate-bound and product-bound states of Rho GTPases allows them to serve as cellular molecular switches, controlled by accessory proteins that can load GTP at the beginning of the cycle and accelerate its hydrolysis to GDP to terminate the biological signal.
The loading of GTP is catalyzed by guanine nucleotide exchange factors, or GEFs.4–6 These large, multidomain cytosolic proteins contain a tandem of DH (Dbl-homology) and PH (plekstrin-homology) domains, which together constitute the minimal functional module responsible for catalysis. It is generally accepted that the tandem functions by binding to and stabilizing nucleotide-free Rho, which upon dissociation is more likely to bind GTP than GDP because the concentration of the former in the cell is an order of magnitude higher. Other types of protein domains found in many GEFs serve additional regulatory purposes, including targeting to specific cellular sites or inhibition via intramolecular interactions. Given the biological importance of Rho GTPases and their role in various diseases, including cancer,7 there is considerable interest in the elucidation of molecular mechanisms that govern the DH-PH catalytic activity and specificity.
The RGSL family of GEFs, comprises p115,8 the leukemia associated RhoA-specific guanine exchange factor (LARG),9,10 and the PDZ-containing RhoA exchange factor (PDZRhoGEF).11 All three proteins show high specificity for RhoA and are activated by select G-protein coupled receptors.12 Their common feature is the presence of the RGSL (regulator of G-protein signaling-like) domain upstream of the DH-PH tandem. This domain interacts with the α-subunit of trimeric G-proteins, allowing for a unique cross-talk between these receptor-coupled GTP-ases and the cytosolic Rho proteins. In addition, LARG and PDZRhoGEF contain a PDZ domain at the N-terminus.
The RGSL GEFs exert their biological function in a variety of tissues, including smooth muscle, where they regulate contractility through Ca2+ independent pathways, a phenomenon known as Ca2+-sensitization.13–15 The DH-PH tandems of LARG and PDZRhoGEF were characterized by X-ray crystallography: the former has been studied both in isolation and in complex with RhoA,16 while the PDZRhoGEF was reported in complex with RhoA.17 Although in both complexes the mutual disposition of the DH and PH domains is similar, it is different from the structures of homologous tandems in other GEFs.18–22 This structural variation, along with the limited interactions typically seen in complexes between the PH domain and the GTPases, raises the possibility of distortion of the DH-PH/RhoA complex structures by crystal packing forces. Although single domain proteins are rarely distorted in any significant way by packing interactions, multidomain systems in which supramodular architecture is dependent on flexible linkers may crystallize in a number of low energy conformational states, not necessarily representing the one in solution.23 Thus, it is not clear if the DH and PH domains have fixed relative orientations in solution, or if they assume this tertiary structure only in a complex with the GTPase. Further, the constraints of the crystal lattice critically limit insight into structure dynamics in solution.
To address these questions we investigated the structure of the DH-PH tandem of the PDZRhoGEF in solution, both in isolation and in complex with RhoA, using high field-heteronuclear NMR, small angle X-ray scattering (SAXS), and H/D exchange mass spectrometry (DXMS). We find that in solution the DH-PH tandem assumes a tertiary architecture that is observed in the crystal structure of the complex, thus enabling it to readily bind the GTPase. However, we also show that in solution the DH-PH/RhoA complex displays sophisticated dynamics that are not revealed by the crystal structure. The nucleotide-free RhoA is significantly disordered when bound to the DH-PH tandem, a phenomenon that cannot be inferred from crystallographic data. Finally, we provide evidence that the complex dissociates rapidly in the presence of either GTP or GDP, virtually ruling out the presence of stable intermediates.
Results
The solution structure of the free DH-PH tandem
As in other structurally characterized GEFs,16,17,19,20,24 the DH and PH domains of PDZRhoGEF are connected by a long α-helix, that is, helix α6 of the DH domain.20 Thus, the relative disposition of the two domains depends on the stability of the connecting α-helix, which in the crystal structure is well resolved with low displacement parameters (B factors).17 To assess if this helix is rigid in the absence of stabilizing intermolecular packing forces in the crystal, we investigated its dynamics in solution.
We used secondary chemical shifts of backbone atoms25 to probe the dynamics of isolated DH and PH domains, and of the intact DH-PH tandem. The backbone order parameters (S2) were computed using the RCI server25 based on our recently determined assignment of individual domains and DH-PH tandem26 (Fig. 1). As expected, the order parameters in individual domains are very similar to those of the tandem. Lower S2 values in the tandem indicate enhanced backbone flexibility and correlate well with the location of surface loops. The PH domain exhibits significantly higher intrinsic dynamics than the DH domain, with several surface loops showing particularly high mobility and in particular the loop encompassing residues 1008–1021, which is completely disordered in the crystal. With respect to the α6-helix, which connects the DH and PH domains, the chemical shifts do not indicate any increase in dynamics, although the assignment could not be extended beyond Ala923 except for shifts assigned to residues 935–937 within the PH-domain portion of the helix (Fig. 1). We have additionally measured 15N{1H} NOE spectra and found that Gly935 does not show any decrease in the heteronuclear NOE effect, consistent with a well ordered conformation (Supporting Information Figure 1). These results strongly suggest that the α6-helix remains intact in the DH-PH tandem in solution, with the concomitant fixed relative disposition of DH and PH domains. To obtain independent experimental confirmation, we used two complementary approaches: residual dipolar couplings (RDCs) and small angle SAXS.
Figure 1.
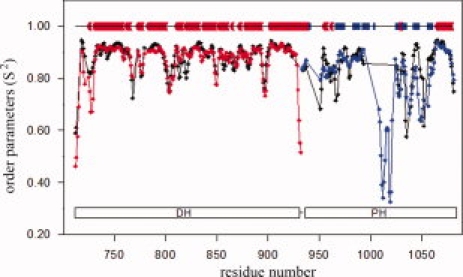
Order parameters (S2) derived from backbone chemical shifts of DH (red), PH (blue), and DH-PH (black) using RCI server.25 The diagram above the plot shows the location of secondary structure elements in the crystal structure of DH-PH in complex with RhoA; α-helices are shown in red and β-strands are blue.
Residual dipolar couplings (RDCs) provide accurate measure of the orientation of backbone N—H groups (1DHN) and allow for direct comparison with X-ray coordinates.27–29 We measured 1DHN RDCs for the DH-PH tandem aligned in a positively charged polyacrylamide gel (Fig. 2). Despite considerable broadening of the signals, we obtained a set of 69 1DHN values, that is, ∼30 % of all assigned backbone amides, including 35 within the DH domain and 34 within the PH domain (see Table I). First, we assessed if the 1DHN values of the individual domains are consistent with the DH-PH/RhoA crystal structure, using the Q quality factors.30 Fitting the experimental RDCs to the two crystallographically independent DH domains yielded Q-factors of 36.1% and 37.1%, a good agreement assuming an experimental error of 4 Hz. For the two PH domains the Q-factors were 32.6% and 29.3%, respectively. Next, to establish the mutual orientation of the DH and PH domains we calculated their respective alignment tensors (Da) from the fit of experimental RDCs to the crystal structure. Their magnitudes are very similar and the Euler angles representing the average orientation of the molecules relative to the alignment tensor frame are also similar (Fig. 2, Table I). Simultaneous fitting of the full set of 1DHN RDCs to the DH-PH tandem in the crystal structure yields a Q-factor of 37.2%, comparable to the fit for the individual domains. Thus, the NMR data confirm that the two domains in the tandem are mutually oriented in the same way as in the crystal structure. However, these data do not exclude a change in relative translation.
Figure 2.
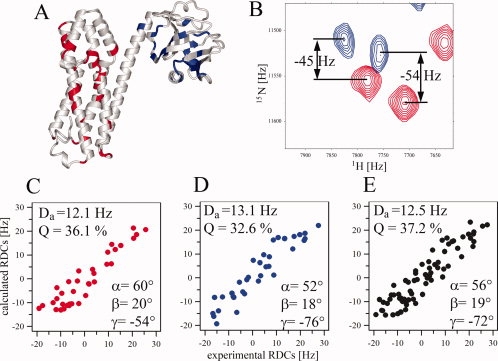
Domain orientation in DH-PH studied by RDCs. (A) DH-PH from the crystal structure of the complex showing the locations of 1DHN RDCs measured for residues in DH (red) and PH (blue); (B) an example of 1H-15N TROSY-HSQC (red); and 1H-15N HSQC (blue) for 2H,13C,15N-DH-PH aligned in positively charged polyacrylamide gel used to extract 1DHN RDCs. (C–E) Fitting of experimental RDCs for DH, PH, and DH-PH, respectively, to the crystal structure. Magnitude of the alignment tensor (Da), Q-factors and Euler angles are shown for each analysis.
Table I.
Analysis of Alignment Tensor Parameters Based on RDCs Measured for DH-PH and DH-PH/RhoA Complex
| Q [%] | Da [Hz] | α | β | γ | N | |
|---|---|---|---|---|---|---|
| DHa | 36.1 ± 2.6 | 12.1 ± 0.4 | 59.6 ± 1.1 | 20.5 ± 1.7 | −54.5 ± 16.5 | 35 |
| PHa | 32.6 ± 2.1 | 13.1 ± 0.5 | 52.4 ± 1.3 | 18.5 ± 1.2 | −75.8 ± 2.6 | 34 |
| DH-PHa | 37.2 ± 1.7 | 12.5 ± 0.3 | 56.0 ± 0.8 | 19.0 ± 0.9 | −71.7 ± 3.9 | 69 |
| DH-PH/RhoAb | 39.0 ± 2.3 | 16.5 ± 0.6 | 57.5 ± 2.3 | 19.3 ± 1.2 | −78.5 ± 3.2 | 37 |
Calculations of Q-factors (Q), magnitude of the alignment (Da) and Euler angles (α, β, γ) defining rotations of molecular coordinates about x, y, and z axes, relative to principal axes frame of alignment tensor were carried out employing program PALES31 based on the crystal structure of DH-PH/RhoA complex (PDB code 1XCG, molecule A) and using N experimental 1DHN RDCs (see Materials and Methods for details).
The RDCs measured for DH-PH were fit to coordinates of separate DH, PH domains, and DH-PH tandem.
The analysis of RDCs measured for DH-PH in the complex with RhoA.
We used small angle SAXS to determine the overall shape of the molecule in solution. The composite experimental scattering pattern from DH-PH is presented in Figure 3, and the overall structural parameters computed from the SAXS data are given in Table II. Several independent runs of the ab initio program DAMMIN32 yielded reproducible molecular shapes. The average model generated by DAMAVER33 is consistent with the crystal structure of the DH-PH tandem in complex with RhoA (Fig. 3). The value of normalized spatial discrepancy (NSD) between the two is 1.0, that is, the two molecular envelopes are virtually identical at low resolution. Furthermore, a simulated scattering curve computed by CRYSOL34 using the atomic coordinates of DH-PH fits very well to the experimental data (Table II).
Figure 3.
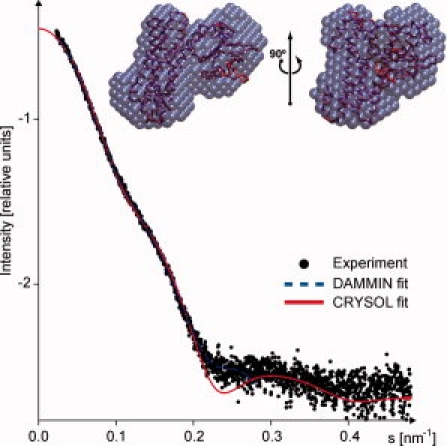
SAXS profiles of DH-PH (see text for details). Dots denote the experimental data, the fits obtained by DAMMIN32 and CRYSOL are shown as blue dashed and red solid lines, respectively. Inset: The averaged ab initio shape obtained from SAXS data is presented by blue beads, the crystallographic model is shown as red Cα-trace.
Table II.
Overall Structural Parameters of DH-PH Obtained by SAXS
| Rg (nm) | Dmax (nm) | Vp (nm3) | MM (kDa) | χs | χcryst |
|---|---|---|---|---|---|
| 2.9 ± 0.1 | 9.5 ± 0.5 | 60 ± 15 | 40 ± 10 | 1.6 | 1.6 |
Rg, Dmax, Vp, and MM are radius of gyration, maximum size, excluded volume, and molecular mass, respectively.
χs and χcryst are discrepancy between the experimental data and computed scattering curves from ab initio shape and atomic model, respectively.
The structure of the DH-PH/RhoA complex in solution: Overview of the NMR strategy
To gain further insight into the structure and dynamics of the DH-PH/RhoA complex in solution, we analyzed two samples: 2H,13C,15N-labeled DH-PH bound to unlabeled RhoA; and 2H,13C,15N-labeled RhoA bound to an unlabeled DH-PH tandem. This approach allowed for independent analyses of the DH-PH tandem and RhoA in the complex. Despite the size of the complex (64 kDa), the 1H-15N TROSY-HSQC spectra were of high quality [Fig. 4(A,C)]. When either of the two labeled proteins was in excess, the corresponding spectra showed two sets of signals (not shown) consistent with slow exchange kinetics, typical of strong complexes with dissociation constants in the nM range. As expected, the formation of the complex results in strong chemical shift perturbations for both RhoA and DH-PH (Fig. 4). Importantly, changes in the spectra of RhoA upon binding to the DH-PH tandem are more pronounced compared to those occurring in the tandem, consistent with significant structural perturbations of RhoA
Figure 4.
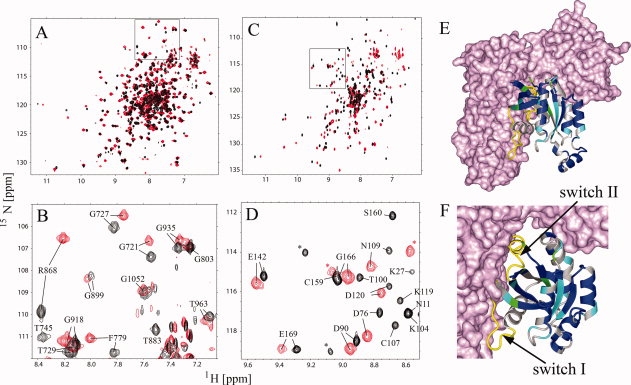
Solution NMR of the DH-PH/RhoA complex. (A) Comparison of the 1H-15N TROSY-HSQC spectra of DH-PH (black) and DH-PH/RhoA (red); (C) comparison of the 1H-15N TROSY-HSQC spectra of RhoA (black) and RhoA-DHPH (red); (B,D) assigned fragments of the squared regions in (A) and (C), respectively; unassigned residues are labeled with asterisk; (E,F) mapping of the assigned resonances onto the model of RhoA (ribbon) in the complex with DH-PH (surface representation); residues exhibiting small chemical shift perturbations (Δσ < 0.06 ppm) are blue, medium (0.06 ppm < Δσ < 0.15 ppm) are cyan and strongest (Δσ > 0.15 ppm) are green; residues that were not assigned are gray; chemical shift perturbations were calculated using the following formula sqrt(ΔσHN2+ 0.1 ΔσN2). Switch I and II regions of RhoA which were not assigned are shown in yellow and labeled.
To obtain chemical shift assignments for quantitative analysis of the two proteins in the complex, we conducted TROSY-based HNCO, and HNCA experiments and 15N-edited NOESY at 900 MHz, for samples in which either the DH-PH tandem or RhoA were triple-labeled. The assignment was based on sequential information derived from HNCA and NOESY experiments and comparison of backbone chemical shifts to those of isolated proteins. Using this strategy, we assigned 184 of the 258 residues previously assigned for free DH-PH. This was primarily due to peak overlap although a number of resonances were strongly perturbed and difficult to assign unambiguously. With respect to RhoA, we were only able to assign 89 residues in the complex, although in this case the analysis was complicated by strong broadening of numerous resonances (Figure 4). In particular, we could not assign any of the residues from Switch I and Switch II that are in direct contact with DH domain in the crystal structure.
The structure of the DH-PH tandem in complex with RhoA
To assess how the two proteins are affected by complex formation, we mapped the chemical shift perturbations onto the crystal structure of the DH-PH/RhoA complex [Fig. 5(A)]. In the DH-PH tandem, the most pronounced changes affect, as expected, those residues within the DH domain which are in the direct contact with RhoA in the crystal structure. A total of 43 amides within the DH domain showed resonances that were shifted to such degree that they could not be assigned. In the PH domain, the chemical shift perturbations were much smaller than those in the DH domain and were limited to two surface patches: one involving Met966, and Glu969 and some neighboring residues, in proximity of both RhoA and the DH domain, and the second, involving Ser1065 and its neighbors, in contact with Glu97 of RhoA. The small magnitude of the chemical shift changes observed for PH amides in the latter patch indicate that the contact is weak.
Figure 5.
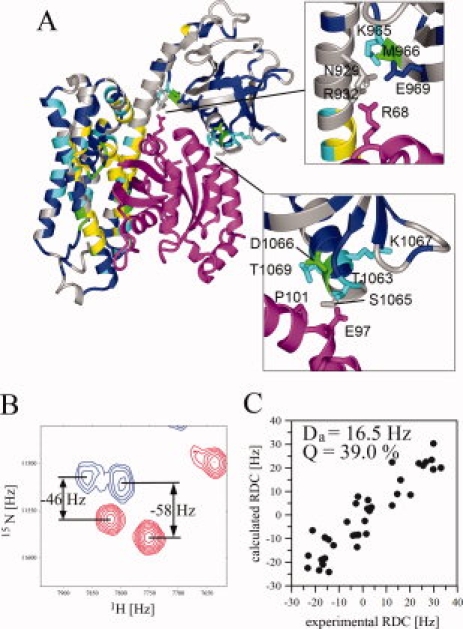
Characterization of the structure of DH-PH/RhoA complex in solution; (A) mapping of chemical shift perturbations within DH-PH upon binding to RhoA (magenta); two regions showing contacts involving RhoA and PH domain are shown in insets; residues exhibiting small chemical shift perturbations (Δσ < 0.05) are blue, medium (0.05 < Δσ < 0.10) are cyan and strongest (Δσ > 0.10) are green; residues that could be assigned only for free DH-PH and experience very strong perturbations in the complex are yellow; residues without assignment for free DH-PH are gray; chemical shift perturbations were calculated using the following formula sqrt(ΔσHN2+0.1ΔσN2). (B) An example of 1H-15N TROSY-HSQC (red) and 1H-15N HSQC (blue) for 2H,13C,15N-DH-PH/RhoA aligned in positively charged polyacrylamide gel, used to extract 1DHN RDCs; (C) fitting of RDCs measured for the DH-PH in the complex with RhoA to the crystal structure.
We also conducted cross-saturation experiments,35 with saturation of the protonated RhoA and monitoring of deuterated DH-PH (not shown). We found that the only residues exhibiting significant cross-saturation effect were within the DH domain. No cross saturation was detected between RhoA and PH, confirming that these contacts are weak.
To determine experimentally the orientation the two domains in DH-PH module in the complex, we used residual dipolar couplings (RDCs), measured for the complex aligned under conditions similar to those used for the free DH-PH tandem [Fig. 5(B,C)]. Because of the strong broadening and overlap of the signals, we were able to measure accurately only 37 1DHN RDCs including 16 and 21 RDCs for DH and PH, respectively. Fitting of these RDCs to the crystal structure yielded the Q-factor of 39%. Although the number of RDCs included in this calculation is relatively small, this result strongly suggests that the relative orientation of the DH and PH domains in the complex in solution is accurately represented by the crystal structure.
Structure and dynamics of RhoA in complex with DH-PH
Close examination of assigned chemical shifts for RhoA in complex with DH-PH reveals significant structural perturbations throughout the protein, including sites distant from DH-PH binding surface [Fig. 4(C)]. We find that in solution the subset of RhoA resonances in the DH-PH bound state exhibit significant degree of broadening, consistent with a much higher level of dynamic disorder, with ∼20% of amides, including all of those in Switch I and Switch II, broadened beyond detection in the 1H-15N TROSY-HSQC spectrum. Such resonance broadening is not caused by overall increase in molecular weight of RhoA in the complex but most likely it results from backbone motions occurring on the microsecond to millisecond timescale. In contrast, the crystal structure of RhoA in complex with DH-PH of PDZRhoGEF36 shows relatively little disorder, with temperature (B) factors elevated slightly for residues in the RhoA's Switch I region.
To further probe the dynamics in the DH-PH/RhoA complex, we used DXMS. This technique is useful for monitoring the dynamics of interacting surfaces in protein–protein complexes on a significantly longer timescale than that probed by NMR.37,38 When the quenched complex is proteolysed, the peptides with the comparatively lower deuterium content map the interacting surfaces or regions with increased stability. In our case, proteolytic digestion yielded 44 and 24 reporter peptides, covering 94% and 96% of the DH-PH tandem and RhoA sequences, respectively (Supporting Information Figure 2). The H/D experiments were carried out for the isolated DH-PH tandem, isolated RhoA•GDP, and for the DH-PH/RhoA complex. The results were mapped on the crystal structure of the complex [Fig. 6(A)]. For both proteins there were several overlapping reporter peptides that provided a consistency test.
Figure 6.
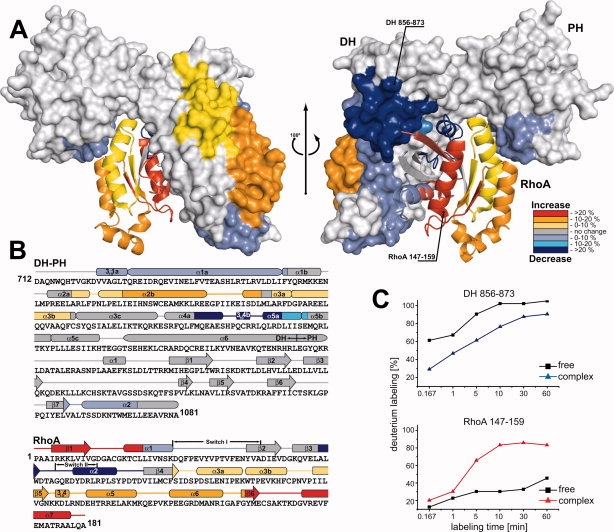
The deuterium exchange map of the DH-PH/RhoA complex. (A) Deuteration level of the DH-PH/RhoA complex shown after 60 min of ion-exchange reaction. Regions of DH-PH (surface) and RhoA (ribbon) affected by the formation of complex are color-coded, based on the change in the deuteration level, suggested by H/D exchange. Areas of the protein with significant decrease in exchange upon complex formation are colored dark blue (deuteration reduction of >20%), blue (reduction of 10–20%), and slate (reduction of 0–10 %). Regions of increased solvent accessibility are shown in red (deuteration increase of >20%), orange (10–20%), and yellow (0–10%). Parts of the protein with no significant changes in deuteration level are shown in grey (PDB ID: 1XCG). (B) The secondary structure elements of DH-PH and RhoA are shown above the protein sequences, and the color-coding represents changes in the levels of deuteration. (C) Two examples of the differences in deuteration levels as a function of time in free DH-PH and RhoA, and after formation of the binary complex. Reduction in deuteration was observed for peptide 856–873 from DH-PH tandem, whereas H/D exchange level increased for peptide 147–159 from RhoA.
We identified three sets of peptides with H/D exchange rates that are lower in the complex than in the free proteins (Fig. 6). The first set maps to the contact between helices 3101a and α1a of the DH-PH tandem, and the Switch I region of RhoA; these peptides show a modest, but measurable ∼5 % reduction in deuterium labeling compared to free proteins. A second set of reporter peptides corresponds to the contact between fragment comprising helices α4a, 3104b, and α5a of DH-PH, and Switch II region of RhoA; in this case we observe a much more dramatic, ∼20 % decrease in the H/D exchange. These results are in apparent contrast to the NMR spectra which show conspicuous absence of both Switch I and Switch II amide resonances in the 1H-15N TROSY-HSQC spectra (see above). However, broadening of NMR resonances results from dynamics on microsecond to millisecond timescale, which is significantly faster than the time required to obtain DXMS data. The interaction involving Switch I of RhoA appears to be generic for a variety of RhoGEFs,19,20,24,36 while the Switch II interaction was suggested to be largely responsible for RhoA selectivity in the p115 family of GEFs.17
Finally, the third area of contact between DH-PH and RhoA involves Ser1065 and Asn1068 from the helix α2 of the PH domain (residues 1060–1073), which form hydrogen bonds with Glu97 of RhoA.17 The DXMS results show modest (∼5%) reduction in H/D exchange in the α2 helix. Unexpectedly, large portions of both DH-PH and RhoA exhibited higher H/D exchange in the complex than in isolation. In the DH domain, helices α2a, α2b, α3a, and α3b, comprising residues 776–832 and distal to the RhoA-binding surfaces, all show ∼10 % increase in the H/D exchange, suggesting that they are destabilized as a result of complex formation with RhoA. The GTPase, on the other hand, exhibits marked overall 5–35 % increase in H/D exchange compared to its GDP-bound form, and in agreement with the enhanced dynamics suggested by the NMR data (above). The only exceptions are the two Switch regions, both of which interact with the DH domain.
A comparison of the DXMS data for the DH-PH/RhoA complex with temperature (B) factors from the crystal structure reveals poor correlation (Supporting Information Figure 3). This is particularly evident in the DH domain helices α2a, α2b, α3a, and α3b (residues 776–832). The ∼10 % increase in H/D exchange in this region is inconsistent with relatively low crystallographic B-factor values. However, the most dramatic contrast between the DXMS and X-ray crystallographic data, relates to Switch I and II regions of RhoA. DXMS indicates that both of these regions become significantly shielded from H/D exchange upon formation of a complex with DH-PH. On the other hand, temperature (B) factors show increase in the Switch I region and only mild decrease for Switch II compared to the RhoA•GDP structure, while the NMR data is consistent with very strong conformational fluctuations of both Switches on a short timescale.
The interpretation of the data was not straightforward because we were unable to obtain NMR data for the nucleotide-free RhoA. The differences between RhoA in complex with DH-PH and RhoA•GDP are not really representative of the simple transition from nucleotide-free RhoA to GEF-bound form, but rather of a two-step transition from GDP-bound RhoA to nucleotide-free RhoA and then to complex with DH-PH. The overall destabilization of RhoA observed by DXMS in complex with DH-PH is most probably a direct consequence of the loss of nucleotide. Both Switch regions remain shielded in the complex from solvent, and the complex is quite stable on a long timescale, but within the complex the Switches undergo rapid conformational exchange. The loss of conformational entropy in RhoA as a result of complex formation may be in part compensated by the partial destabilization of the DH domain (helices α2 and α3), resulting in an “entropy transfer” effect. Overall, the temperature (B) factors of the complex in the crystal structure are not representative of true complex dynamics, because they are constrained by extensive crystal contacts, particularly between the DH domain and RhoA from a symmetry related complex.
Dissociation of the DH-PH/RhoA complex by guanine nucleotides
The dynamic behavior of RhoA in the complex may be an important functional feature which facilitates nucleotide exchange. To directly observe whether guanine nucleotides are able to bind to and dissociate from the complex, we titrated the DH-PH/2H,15N-RhoA complex with GDP and GTP (Fig. 7). The addition of even substoichiometric amounts of nucleotides, in the presence of 1 mM Mg2+ gave rise to a second set of signals characteristic of free RhoA. Thus, in the presence of the guanine nucleotide, either GDP or GTP, the complex undergoes rapid dissociation and regeneration of the nucleotide-bound RhoA. We see no evidence of any transition complex involving DH-PH and nucleotide-bound RhoA, as was suggested by others,39 indicating that even if such a transition state exists transiently, it is unstable and undergoes very rapid dissociation.
Figure 7.
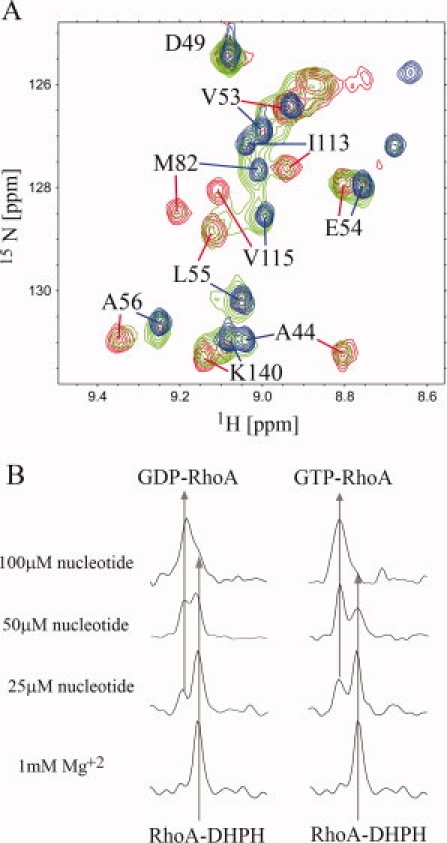
Nucleotide induced dissociation of the DH-PH/RhoA complex. (A) fragment of the superimposed 1H-15N TROSY-HSQC spectra showing the comparison of the of 80 μM nucleotide free DH-PH/2H,15N-RhoA (red), the 80 μM DH-PH/2H,15N-RhoA complex in the presence of 50 μM GDP (green) and 100 μM 2H,15N RhoA•GDP (blue). (B) extraction of 1D traces from 1H-15N TROSY-HSQC spectra used to follow the dissociation of the complex by guanine nucleotides; the arrows show chemical shifts of the C-terminal Ala181 amide proton of RhoA bound to DH-PH (upfield signal) and nucleotide (downfield signal); left and right panels shows the titration of DH-PH/RhoA complex with GDP and GTP, respectively.
We next used NMR spectroscopy to compare the relative abilities of GDP and GTP to dissociate RhoA from the complex with DH-PH. The advantage of this approach was that we monitored the behavior of the proteins in the presence of unmodified nucleotides. For this purpose, we quantified the amount of dissociated RhoA based on the intensities of the amide proton resonance of C-terminal Ala181. As shown in Figure 7(B), in the presence of GTP the dissociation of RhoA is somewhat more efficient than in the presence of GDP. We estimated that GTP has 1.5 times higher affinity towards the complex than GDP. We have also obtained similar results in the independent fluorescence-based assay titrating DH-PH/RhoA complex with N-methylanthraniloyl (mant) analogues of GTP and GDP (data not shown). The slightly higher affinity of GTP to the complex is not sufficient to drive the formation of the active RhoA•GTP complex in the cell. Therefore, the major factor is the ten times higher cellular level of GTP compared to GDP.
Discussion
Using heteronuclear NMR and SAXS we showed that the two domains within the isolated DH-PH tandem retain fixed relative position in solution, so that the protein tumbles as a monomeric and rigid body. Although we were not able to fully assign all the amide resonances within the connecting α6-helix of the free tandem, we see no indication of disorder or noticeably increased dynamics within this helix that would be consistent with independent motions of the two domains. It is not immediately obvious if the rigid architecture is a product of cooperative effects, or if the α6-helix provides an intrinsically stable scaffold, but the data in Figure 1 suggest that in the absence of the PH domain the C-terminus of the helix unfolds.
The rigid architecture of the DH-PH tandem sets it apart from a number of other multidomain proteins, such as calmodulin in which the two globular domains are connected through a linker which adopts a well defined α-helical conformation in the crystal structure but is clearly very flexible in solution.40 Further, we show that the mutual disposition of the two modules in the isolated tandem is identical, within the experimental error, to that observed in the crystal structure of the DH-PH/RhoA complex.17
The stable mutual disposition of the DH and PH domains raises the question of its biological significance. Other structural studies of DH-PH tandems in various GEFs, both in isolation and in complexes with cognate GTPases, seem to support the notion that this moiety functions as a rigid unit. In some cases, this architecture primes the tandem for binding of GTPase in a cooperative fashion, so that both the DH and PH domains engage the GTPase. This appears to be the case for Dbs and Trio.18,41 Recently, two independent structural investigations have shown that in Vav1, a cysteine-rich domain (CRD) located downstream of PH, packs together with the latter against the C-terminal helix of the DH domain to stabilize its conformation.42,43 However, the impact of this architecture on the nucleotide exchange is indirect, because neither the PH domain nor the CRD domain engages the GTPase.
It has been shown previously that in the absence of the PH domain, the catalytic efficiency of the isolated DH domain of PDZRhoGEF is impaired, and the rate of nucleotide exchange reaction is lowered 3-fold.36 However, the mutations of the PH residues Ser1065 and Asn1068 to alanines, both of which are in the contact with RhoA, have no impact on the nucleotide exchange kinetics.36 The modest PH-RhoA interface and small perturbations of PH chemical shifts upon RhoA binding suggest that the interaction is weak and transient. The second site within the PH domain which exhibits chemical shift changes upon RhoA binding is found in the region bridging DH, PH, and RhoA. In the crystal structure, Asn929 (DH domain) forms hydrogen bonds with Arg68 (RhoA) and with the backbone carbonyl of Met966 (PH domain). Mutation of Asn929 to Ala results in significant reduction of nucleotide exchange rate.36 Thus, the PH domain seems to have no direct role in the nucleotide exchange process, but indirectly affects the DH interaction with RhoA. Also, neither the PH domain of PDZRhoGEF nor the homologous PH domains of the related LARG and p115 exchange factors, bind phospholipids, ruling out their involvement in membrane targeting of the GEFs. However, we note that the electrostatic potential surface of the rigid DH-PH tandem may be of importance in orienting the DH domain in a favorable position to interact with membrane anchored RhoA, assuming that the translocation of the GTPase to the membrane from the RhoGDI/RhoA complex precedes the nucleotide exchange event.
It is noteworthy, that the DH-PH tandem of LARG shows a significant difference in the position of its PH domain between the crystal structures of the isolated tandem and of the complex with RhoA: the PH domain is laterally translated by more than 10 Å16. Assuming that the results of our solution studies extend to LARG, this can only be rationalized by a distortion of the LARG tandem due to crystal contacts. Indeed, we find a unique set of crystal contacts in the structure of the isolated DH-PH tandem, so that each DH domain contacts six symmetry related DH domains, while each PH domain contacts only one other PH domain. While crystal packing forces rarely have a significant impact on the conformation of single domain globular proteins, their energy is sufficient to distort the mutual disposition of domain connected by relatively flexible secondary structure elements.23
Finally, using NMR and DXMS, we show that RhoA shows significantly increased dynamics in solution in complex with DH-PH, specifically within the Switch I and Switch II regions, a phenomenon not evident from the crystal structure which is effectively stabilized by both the crystal contacts and the low temperature of the experiment. Molecular dynamics play an important role in protein-protein interactions in solution, but are not well represented by the crystallographic models. Destabilization of one of the partners within a complex may be a relatively common occurrence. In the case of the nucleotide exchange reaction, this may be particularly important, as exemplified by the study of Rab8, which undergoes partial unfolding during such exchange catalyzed by its exchange factor MSS4.44 Similar effect of conformational dynamics was observed for the AN 1–4 ankyrin repeats in iκB, which become destabilized upon interaction with NF-κB.45 In the case of the DH-PH/RhoA complex, the GTPase shows increased dynamics on the microseconds to milliseconds scale, particularly within the Switch I and Switch II regions. However, both these regions are protected from H/D exchange in the complex on a longer time scale, indicating that the integrity of the complex is not compromised by the dynamics of the GTP-ase. It is also interesting to note that portions of the DH domain distal to the RhoA-binding site seem to be destabilized in the complex, compared to isolated state, as judged by the DXMS analysis. One possible explanation is that unfavorable loss of entropy by the GTPase on binding the DH-PH tandem is compensated by increase in the entropy in the DH domain. This suggests that specific interactions of other domains within intact GEFs, with the distal face of the DH domain, may provide additional regulatory mechanism, effectively lowering the affinity of the DH domain for RhoA, and inhibiting the nucleotide exchange rate.
Our experiments underscore the power of solution structural studies as a complement to crystallographic investigations of regulatory complexes.
Materials and Methods
Protein expression and purification
The DH-PH tandem of human PDZ-RhoGEF (residues 712–1081) was expressed as fusion protein with GST in E. coli BL21(DE3)-RIL (Stratagene) and purified as described previously.46 The purified protein was concentrated in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1 mM DTT. Human truncated RhoA (residues 1–181), was expressed in pETUni vector47 in fusion with a His6-tag. RhoA was purified by Ni-NTA-agarose affinity chromatography in 50 mM Tris-HCl, 400 mM NaCl, 1 mM MgCl2, 5 mM imidazole, pH 8.0, and eluted with 150 mM imidazole buffer, before final purification using gel filtration. Proteins labeled with stable isotopes (2H, 13C, 15N) were expressed in minimal media with (15NH4)2SO4 as sole source of nitrogen and 13C- or 2H,13C-glucose as a source of carbon. The media were enhanced by the addition of labeled BioExpress (Cambridge Isotope Labs). Four types of labeling scheme were used: 15N; 13C, 15N; 2H, 15N; and 2H, 13C, 15N.
Preparation of protein samples for NMR experiments
For assignment experiments a sample of 0.9 mM13C,15N RhoA was prepared in 25 mM TRIS buffer, pH 7.5 with 50 mM NaCl, 5 mM MgCl2, 1 mM DTT and saturated with GDP. Samples of DH-PH for NMR experiments contained between 0.2 and 0.35 mM protein exchanged to 200 mM MOPS/TRIS, pH 7.5 buffer.26 The DH-PH/RhoA complex was prepared by mixing approximately equimolar amounts of the DH-PH tandem and RhoA, with 10% excess of the unlabelled component and subsequent dialysis of the sample to remove GDP and Mg+2 ions. We found that long term solubility of DH-PH/RhoA complex in 200 mM MOPS/TRIS, pH 7.5 is slightly lower than that of DH-PH tandem, but the samples of the complex were nevertheless stable at 0.25 mM for the period of a week at 25°C.
NMR experiments
Measurements for 13C,15N-RhoA were carried out using Varian Inova 500 MHz at 25°C. To assign backbone chemical shifts we recorded standard triple resonance experiments [HNCO, HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH and 3D 15N-edited NOESY with 200 ms mixing time] and we have been able to assign 148 of 168 (88 %) backbone amides. To assign the DH-PH and RhoA in the complex we prepared two 0.35 mM samples of 2H,13C,15N-labeled RhoA complexed to unlabeled DH-PH and 2H,13C,15N-labeled DH-PH in complex with unlabeled RhoA. The spectra (TROSY-HNCA, TROSY-HNCO, 3D 15N-edited TROSY-NOESY with 250 ms mixing time) were measured using Varian Inova 900 MHz spectrometer at 28°C. Assignment of the binary DH-PH/RhoA complex was based on comparison of HN, N, C′, Cα backbone chemical shifts to the chemical shifts of free proteins.26 The sequential connectivities were additionally verified using correlations from TROSY-HNCA and TROSY-NOESY experiments. Overall, we assigned the total of 50% and 53% of backbone amides for the complexed DH-PH and RhoA, respectively.
The TROSY-based 15N{1H} heteronuclear NOE experiment with 3 s saturation was measured for free DH-PH at 28°C using a Bruker Avance III 800 MHz spectrometer.
To measure cross-saturation effects we modified the 1H-15N TROSY-HSQC experiments to introduce saturation of aliphatic resonances.35 The cross-saturation spectra for 2H,13C,15N-RhoA bound to unlabeled DH-PH were measured at 28°C with 0.6 and 1.2 s saturation times using Varian Inova 600 MHz. Experiments for the 2H,13C,15N-DH-PH complexed with unlabeled RhoA were carried out at 28°C with saturation times of 0.3, 0.6, and 1.0 s using Varian Inova 900 MHz.
Measurement and analysis of RDCs
The alignment of the DH-PH and DH-PH/RhoA complex was successfully obtained using a positively charged gel: 50% (3-acrylamidopropyl)-trimethylammonium chloride/50% acrylamide (referred to hereafter as 50+M).48 The RDCs were measured for 0.5 and 0.35 mM samples of 2H,13C,15N-labeled DH-PH and 0.35 mM2H,13C,15N-labeled DH-PH/RhoA, respectively. Introduction of weak alignment resulted in signal broadening and decreased the quality of the spectra, making it impossible to measure J–couplings by HNCO-based triple resonance experiments. Instead, we measured 1H-15N HSQC and 1H-15N TROSY-HSQC in interleaved manner and extracted the anisotropic 1JHN coupling from the peak positions in 15N dimension. All measurements were carried out using Varian Inova 900 MHz spectrometer at 30°C. To calculate the 1DHN RDCs we assumed the isotropic 1JHN value of −94 Hz.49 The calculated 1DHN ranged from −19 to +28 Hz and −23 to +33 Hz for DH-PH and DH-PH/RhoA, respectively. On the basis of the repeated experiments, we estimated 4 Hz error in 1DHN determination.
The determination of the alignment tensor parameters (Da and R) and of the Euler angles (α, β, γ) defining rotations of molecular coordinates about x, y, and z axes, relative to principal axes frame of alignment tensor, was carried out using the PALES program31 and the crystal structure of DH-PH/RhoA complex (PDB code 1XCG). Compatibility between experimental and calculated RDC was evaluated based on quality factors Q calculated from the formula: Q = rms (Dcalc−Dobs)/rms(Dobs).30 Evaluation of the error in determination of alignment tensor and domain orientation has been done using jack-knife procedure by performing 100 cycles of calculation with random elimination of 10% of the data.23
Titration of DH-PH/RhoA with nucleotides
For the titration of the DH-PH/RhoA complex with nucleotides, we prepared a sample of unlabeled DH-PH in complex with 2H,15N-labeled RhoA, at 100 μM concentration, in 200 mM MOPS/TRIS buffer, pH 7.5 with 1 mM MgCl2. NMR samples were prepared by mixing the complex with stock solutions of GDP and GTP in the same buffer. Final concentration of the protein complex in NMR samples was 80 μM and nucleotides were at 25, 50, and 100 μM. To minimize the hydrolysis of GTP all samples were prepared and stored on ice. The 1H-15N TROSY-HSQC spectra were measured at 28°C for 30 min.
Small angle X-ray scattering
SAXS measurements were performed at the X33 beamline (DESY, EMBL Outstation Hamburg, Germany)50 using MAR345 area detector. The sample–detector distance was 2.7 m. Data were collected at protein concentrations of 1–10 mg/mL with exposure times of 120 s, at 5°C. SAXS intensity I is represented as a function of the momentum transfer modulus q [q = 4π/λ sin(θ), where λ is the radiation wavelength(1.5 Å), and 2θ is the scattering angle. Scattering from the buffer was collected before and after that of the sample, then averaged and subtracted from the sample scattering curve to generate the scattering profile of the protein [I(q)]. Data at different concentrations were scaled and merged using the program PRIMUS.51 A Guinier approximation [I(q) = I(0)exp(−q2Rg2/3)] describing scattering intensity at a low angle region of the scattering profile was used to calculate the radius of gyration (Rg) and zero-angle scattering [I(0)] from a linear fit to the Guinier plot using the program AutoRg.52 The molecular mass (MM) of the solute was evaluated by comparison with the reference solutions of bovine serum albumin using the proportion MM ∼I(0)/c. In addition, the excluded (Porod) volumes Vp53 of the solute was also analyzed taking advantage of the fact that this parameter does not depend on concentration. The maximum size Dmax and real-space distance distribution function [P(r)] was determined with the program GNOM,54 which implements the indirect Fourier transform method applied to the experimental scattering plot I(q). Multiple independent reconstructions of three-dimensional shapes with the best fit to the experimental scattering curves were modeled ab initio using the DAMMIN program,32 and subsequently aligned and averaged in the program DAMAVER.33 The averaged low-resolution SAXS envelope was aligned with the high-resolution atomic structure of the DH-PH tandem (PDB ID: 1XCG) using the program SUPCOMB55 which minimizes normalized spatial deviation (NSD) between the models. The scattering from the atomic model was calculated using the program CRYSOL,34 which either predicts theoretical scattering patterns or fits the experimental data by adjusting the excluded volume and the contrast of the hydration layer.
Hydrogen-deuterium exchange mass spectrometry
Samples of the DH-PH and RhoA were prepared as described above. The DH-PH/RhoA complex was prepared by mixing DH-PH tandem and RhoA in 1:1.5 molar ratio and subsequent dialysis of the sample to 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, and 1 mM DTT. To remove the excess unbound RhoA, the complex was subsequently purified using gel filtration chromatography.
Hydrogen/deuterium (H/D) isotopic exchange in the DH-PH fragment, RhoA, or their complex was initiated by diluting 10 μL of the stock solution (∼0.8 mg/mL) into 90 μL D2O solvent, pH 7.4 at room temperature. The H/D exchange reaction was performed for varying times (10, 60, 300, 600, 1800, and 3600 s) before quenching. Quenching was accomplished using an equal volume of cold 0.1M NaH2PO3 buffer with 4M GuanHCl, pH 2.5. The deuterium-labeled proteins were then cleaved into reporter peptides on an immobilized pepsin column (∼100 μL in bed volume). The resulting peptide mixture was desalted for 6 min in a C8 peptide trapping column (Michrom Bioresources, Auburn, CA), then eluted by acetonitrile gradient from 15 to 40% over 15 min at flow rate of 5 μL/min (after splitting). The eluted peptide mixture was then separated by a Vydac C18 column (0.3 × 50 mm, Grace Vydac, Hesperia, CA), and detected by an ion trap mass spectrometer. To minimize artifactual exchange during the analysis time, all the columns, loops, and lines were immersed in an ice bath during all the experiments.
The deuteration level for each reporter peptide was calculated by the following equation:
where m is the measured centroid mass of the deuterated peptide after a particular labeling time; m0 and m100 are the two centroid mass limits of a reporter peptide from zero-deuteration and full-deuteration control experiments, respectively. The factor of 0.9 in the denominator accounts for the 90% deuterium in the labeling solution. Mass spectra were acquired by a linear ion trap mass spectrometer with a standard electrospray ionization source (LTQ, Thermo Electron, San Jose, CA). The capillary temperature was 250°C, spray voltage—4.3 kV, sheath gas flow—40 unit, capillary voltage—15 V, and tube lens voltage—105 V.
Acknowledgments
The authors thank Natalya Olekhnovich for help in the preparation of protein samples and outstanding technical assistance. This research benefited from access to the Southeast Collaboratory for High-Field Biomolecular NMR, a resource at the University of Georgia, funded by the National Institute of General Medical Sciences (GM66340) and the Georgia Research Alliance. We thank Dr. John Glushka and Dr. Yizhou Liu for assistance in setting up NMR experiments.
References
- 1.Jaffe AB, Hall A. RHO GTPASES: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 2.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 3.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. (Part 2) [PMC free article] [PubMed] [Google Scholar]
- 4.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 5.Erickson JW, Cerione RA. Structural elements, mechanism, and evolutionary convergence of Rho protein-guanine nucleotide exchange factor complexes. Biochemistry. 2004;43:837–842. doi: 10.1021/bi036026v. [DOI] [PubMed] [Google Scholar]
- 6.Whitehead IP, Campbell S, Rossman KL, Der CJ. Dbl family proteins. Biochim Biophys Acta. 1997;1332:F1–F23. doi: 10.1016/s0304-419x(96)00040-6. [DOI] [PubMed] [Google Scholar]
- 7.Kandpal RP. Rho GTPase activating proteins in cancer phenotypes. Curr Protein Pept Sci. 2006;7:355–365. doi: 10.2174/138920306778018025. [DOI] [PubMed] [Google Scholar]
- 8.Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by galpha13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- 9.Kourlas PJ, Strout MP, Becknell B, Veronese ML, Croce CM, Theil KS, Krahe R, Ruutu T, Knuutila S, Bloomfield CD, Caligiuri MA. Identification of a gene at 11q23 encoding a guanine nucleotide exchange factor: evidence for its fusion with MLL in acute myeloid leukemia. Proc Natl Acad Sci USA. 2000;97:2145–2150. doi: 10.1073/pnas.040569197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reuther GW, Lambert QT, Booden MA, Wennerberg K, Becknell B, Marcucci G, Sondek J, Caligiuri MA, Der CJ. Leukemia-associated Rho guanine nucleotide exchange factor, a Dbl family protein found mutated in leukemia, causes transformation by activation of RhoA. J Biol Chem. 2001;276:27145–27151. doi: 10.1074/jbc.M103565200. [DOI] [PubMed] [Google Scholar]
- 11.Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- 12.Sternweis PC, Carter AM, Chen Z, Danesh SM, Hsiung YF, Singer WD. Regulation of Rho guanine nucleotide exchange factors by G proteins. Adv Protein Chem. 2007;74:189–228. doi: 10.1016/S0065-3233(07)74006-8. [DOI] [PubMed] [Google Scholar]
- 13.Teixeira CE, Jin L, Priviero FB, Ying Z, Webb RC. Comparative pharmacological analysis of Rho-kinase inhibitors and identification of molecular components of Ca2+ sensitization in the rat lower urinary tract. Biochem Pharmacol. 2007;74:647–658. doi: 10.1016/j.bcp.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilgers RH, Todd J, Webb RC. Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. J Hypertens. 2007;25:1687–1697. doi: 10.1097/HJH.0b013e32816f778d. [DOI] [PubMed] [Google Scholar]
- 15.Gong MC, Iizuka K, Nixon G, Browne JP, Hall A, Eccleston JF, Sugai M, Kobayashi S, Somlyo AV, Somlyo AP. Role of guanine nucleotide-binding proteins--ras-family or trimeric proteins or both--in Ca2+ sensitization of smooth muscle. Proc Natl Acad Sci USA. 1996;93:1340–1345. doi: 10.1073/pnas.93.3.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kristelly R, Gao G, Tesmer JJ. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J Biol Chem. 2004;279:47352–47362. doi: 10.1074/jbc.M406056200. [DOI] [PubMed] [Google Scholar]
- 17.Derewenda U, Oleksy A, Stevenson AS, Korczynska J, Dauter Z, Somlyo AP, Otlewski J, Somlyo AV, Derewenda ZS. The crystal structure of RhoA in complex with the DH/PH fragment of PDZRhoGEF, an activator of the Ca(2+) sensitization pathway in smooth muscle. Structure. 2004;12:1955–1965. doi: 10.1016/j.str.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Worthylake DK, Rossman KL, Sondek J. Crystal structure of the DH/PH fragment of Dbs without bound GTPase. Structure. 2004;12:1078–1086. doi: 10.1016/j.str.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 19.Snyder JT, Worthylake DK, Rossman KL, Betts L, Pruitt WM, Siderovski DP, Der CJ, Sondek J. Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat Struct Biol. 2002;9:468–475. doi: 10.1038/nsb796. [DOI] [PubMed] [Google Scholar]
- 20.Rossman KL, Worthylake DK, Snyder JT, Siderovski DP, Campbell SL, Sondek J. A crystallographic view of interactions between Dbs and Cdc42: PH domain-assisted guanine nucleotide exchange. EMBO J. 2002;21:1315–1326. doi: 10.1093/emboj/21.6.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Worthylake DK, Rossman KL, Sondek J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 2000;408:682–688. doi: 10.1038/35047014. [DOI] [PubMed] [Google Scholar]
- 22.Soisson SM, Nimnual AS, Uy M, Bar-Sagi D, Kuriyan J. Crystal structure of the Dbl and pleckstrin homology domains from the human Son of sevenless protein. Cell. 1998;95:259–268. doi: 10.1016/s0092-8674(00)81756-0. [DOI] [PubMed] [Google Scholar]
- 23.Cierpicki T, Bushweller JH, Derewenda ZS. Probing the supramodular architecture of a multidomain protethe structure of syntenin in solution. Structure. 2005;13:319–327. doi: 10.1016/j.str.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 24.Karnoub AE, Worthylake DK, Rossman KL, Pruitt WM, Campbell SL, Sondek J, Der CJ. Molecular basis for Rac1 recognition by guanine nucleotide exchange factors. Nat Struct Biol. 2001;8:1037–1041. doi: 10.1038/nsb719. [DOI] [PubMed] [Google Scholar]
- 25.Berjanskii MV, Wishart DS. A simple method to predict protein flexibility using secondary chemical shifts. J Am Chem Soc. 2005;127:14970–14971. doi: 10.1021/ja054842f. [DOI] [PubMed] [Google Scholar]
- 26.Zheng M, Cierpicki T, Momotani K, Artamonov MV, Derewenda U, Bushweller JH, Somlyo AV, Derewenda ZS. On the mechanism of autoinhibition of the RhoA-specific nucleotide exchange factor PDZRhoGEF. BMC Struct Biol. 2009;9:36. doi: 10.1186/1472-6807-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003;12:1–16. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 29.Prestegard JH, Al-Hashimi HM, Tolman JR. NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q Rev Biophys. 2000;33:371–424. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- 30.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 31.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 32.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Svergun DI, Barberato C, Koch MHJ. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- 35.Takahashi H, Nakanishi T, Kami K, Arata Y, Shimada I. A novel NMR method for determining the interfaces of large protein-protein complexes. Nat Struct Biol. 2000;7:220–223. doi: 10.1038/73331. [DOI] [PubMed] [Google Scholar]
- 36.Oleksy A, Opalinski L, Derewenda U, Derewenda ZS, Otlewski J. The molecular basis of RhoA specificity in the guanine nucleotide exchange factor PDZ-RhoGEF. J Biol Chem. 2006;281:32891–32897. doi: 10.1074/jbc.M606220200. [DOI] [PubMed] [Google Scholar]
- 37.Zhou B, Zhang JL, Liu SJ, Reddy S, Wang F, Zhang ZY. Mapping ERK2-MKP3 binding interfaces by hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2006;281:38834–38844. doi: 10.1074/jbc.M608916200. [DOI] [PubMed] [Google Scholar]
- 38.Mandell JG, Falick AM, Komives EA. Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc Natl Acad Sci USA. 1998;95:14705–14710. doi: 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B, Zhang Y, Shacter E, Zheng Y. Mechanism of the guanine nucleotide exchange reaction of Ras GTPase—evidence for a GTP/GDP displacement model. Biochemistry. 2005;44:2566–2576. doi: 10.1021/bi048755w. [DOI] [PubMed] [Google Scholar]
- 40.Barbato G, Ikura M, Kay LE, Pastor RW, Bax A. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 1992;31:5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- 41.Chhatriwala MK, Betts L, Worthylake DK, Sondek J. The DH and PH domains of Trio coordinately engage Rho GTPases for their efficient activation. J Mol Biol. 2007;368:1307–1320. doi: 10.1016/j.jmb.2007.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rapley J, Tybulewicz VL, Rittinger K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Report. 2008;9:655–661. doi: 10.1038/embor.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chrencik JE, Brooun A, Zhang H, Mathews II, Hura GL, Foster SA, Perry JJ, Streiff M, Ramage P, Widmer H, Bokoch GM, Tainer JA, Weckbecker G, Kuhn P. Structural basis of guanine nucleotide exchange mediated by the T-cell essential Vav1. J Mol Biol. 2008;380:828–843. doi: 10.1016/j.jmb.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itzen A, Pylypenko O, Goody RS, Alexandrov K, Rak A. Nucleotide exchange via local protein unfolding--structure of Rab8 in complex with MSS4. EMBO J. 2006;25:1445–1455. doi: 10.1038/sj.emboj.7601044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sue SC, Cervantes C, Komives EA, Dyson HJ. Transfer of flexibility between ankyrin repeats in IkappaB* upon formation of the NF-kappaB complex. J Mol Biol. 2008;380:917–931. doi: 10.1016/j.jmb.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oleksy A, Barton H, Devedjiev Y, Purdy M, Derewenda U, Otlewski J, Derewenda ZS. Preliminary crystallographic analysis of the complex of the human GTPase RhoA with the DH/PH tandem of PDZ-RhoGEF. Acta Cryst. 2004;60:740–742. doi: 10.1107/S0907444904002318. [DOI] [PubMed] [Google Scholar]
- 47.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “Parallel” expression vectors. Protein Expr Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 48.Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–16266. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- 49.Tugarinov V, Kay LE. Quantitative NMR studies of high molecular weight proteins: application to domain orientation and ligand binding in the 723 residue enzyme malate synthase G. J Mol Biol. 2003;327:1121–1133. doi: 10.1016/s0022-2836(03)00238-9. [DOI] [PubMed] [Google Scholar]
- 50.Roessle MW, Klaering R, Ristau U, Robrahn B, Jahn D, Gehrmann T, Konarev P, Round A, Fiedler S, Hermes C, Svergun D. Upgrade of the small-angle X-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J Appl Cryst. 2007;40:s190–s194. [Google Scholar]
- 51.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 52.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1 - towards automated and web-supported small-angle scattering data analysis. J Appl Cryst. 2007;40:s223–s228. [Google Scholar]
- 53.Porod G. General theory. In: Glatter O, Kratky O, editors. Small-angle X-ray scattering. London: Academic Press; 1982. pp. 17–51. [Google Scholar]
- 54.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]
- 55.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Cryst. 2001;34:33–41. [Google Scholar]