

Abstract

The longstanding challenge posed by the complex diterpene vinigrol has been answered for the first time. The notorious difficulty in synthesizing vinigrol stems from its unprecedented decahydro-1,5-butanonaphthalene ring system, eight contiguous stereocenters, and highly congested functionality. This communication delineates a stereocontrolled 23-step route to vinigrol that is scalable (>5 grams prepared of a late stage intermediate), minimally reliant on protecting group chemistry, and facilitated by a number of unique and chemoselective transformations.
The total synthesis of the biologically significant diterpene vinigrol (1)1 has stood for over two decades as a major unsolved challenge for organic synthesis.2 The extreme difficulty in preparing this molecule stems from its unprecedented and highly congested decahydro-1,5-butanonaphthalene ring system containing eight contiguous stereocenters (shown in four different views in Figure 1A). In this Communication we report a solution to this longstanding problem in complex terpene synthesis.3
Figure 1.
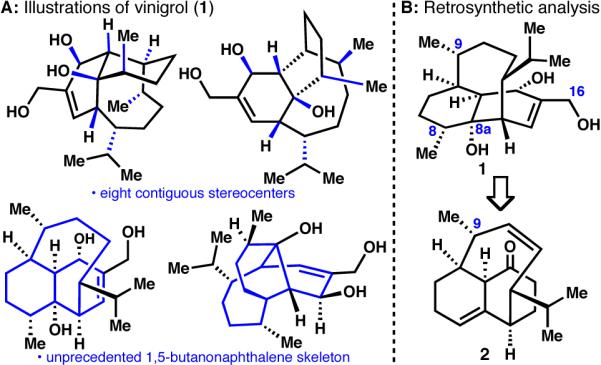
A: Illustrations of vinigrol (1) and B: Retrosynthetic analysis.
Last year our laboratory reported a short route to the core skeleton of 1, featuring a simple sequence of classic transforms such as the Diels-Alder and Grob fragmentation, that proved to be capable of delivering structures similar to 2 (Figure 1B) albeit lacking the C-9 methyl group.2w In accord with the well-documented reluctance of late-stage intermediates to be converted to 1,2 many seemingly logical routes to 1 from 2 and related intermediates failed in our hands. The final route presented herein was formed from a detailed series of experiments.
The route to 1 commences from 3 (Scheme 1), an intermediate available in decagram quantities in seven steps from commercially available materials. The C-9 methyl group was installed by alkylation (LDA, MeI) and, following silyl group removal (TBAF), the adjacent alcohol stereochemistry was established using Evans' Me4NBH(OAc)3–mediated,4 hydroxyl-directed reduction to deliver 4 as a single diastereomer in 72% yield over the three step sequence. Non-directed reductions (i.e. DIBAL) afforded mainly the undesired alcohol diastereomer at C-11 due to the shielding effect of the C-9 methyl group. The correct stereochemistry is critical for the ensuing Grob fragmentation that furnished 2 (see Figure 1B for structure) after mesylation and treatment with KHMDS (85% yield over 2 steps).
Scheme 1. Total synthesis of vinigrol (1).

Reagents and conditions: (a) LDA (1.3 equiv.), MeI (1.6 equiv.), THF, −78 to 0 °C, 3.3 h; (b) TBAF (3.2 equiv.), THF, 50 °C, 3 h; (c) Me4NBH(OAc)3 (4.0 equiv.), AcOH:MeCN:THF = 1:1:1, 23 °C, 1.5 h, 72% over 3 steps; (d) MsCl (1.2 equiv.), Pyridine, 0 °C, 2.5 h; (e) KHMDS (1.1 equiv.), THF, 0 to 23 °C, 35 min, 85% over 2 steps; (f) KHCO3 (3.0 equiv.), Br2 C=NOH (1.5 equiv.), EtOAc, 23 °C, 45 min, 88%; (g) DIBAL (1.2 equiv.), DCM, −78 °C, 1 h, 95%; (h) Crabtree's catalyst (0.2 equiv.), B(O-iPr)3 (1.0 equiv.), H2 (1 atm), DCE, 80 °C, 8 h, 87%; (i) NaH (10 equiv.), CS2 (20 equiv.), MeI (40 equiv.), THF, 0 to 23 °C, 15 h, 88%; (j) o-DCB, 180 °C, 3 h, 96%; (k) LiAlH4 (20 equiv.), THF, 0 to 23 °C, 12 h; HCOOH (2.0 equiv.), CDMT (2.1 equiv.), NMM (2.2 equiv.), DMAP (0.1 equiv.), DCM, 23 °C, 1 h, 81%; (l) COCl2 (1.0 equiv.), Et3N (15 equiv.), DCM, −20 °C, 20 min, 76%; (m) AIBN (3.0 equiv.), Bu3SnH (9.8 equiv.), Toluene, 100 °C, 2.5 h, 91%; (n) OsO4 (0.1 equiv.), NMO (1.3 equiv.), Acetone:H2O = 3:1, 23 °C, 12 h, 95%; (o) NaOCl (1.5 equiv.), TEMPO (0.1 equiv.), KBr (0.1 equiv.), a.q 5% NaHCO3:DCM = 2:5, 0 °C, 1.5 h, 85%; (p) TrisNHNH2 (2.0 equiv.), DCM, 23 °C, 5 h; n-BuLi (4.0 equiv.), (CH2O)n (30 equiv.), TMEDA:THF = 2:1, −78 to 23 °C, 3 h, 51% overall.
Installation of the C-8 methyl and C-8a hydroxyl group proved to be a challenge due to their cis orientation. The methyl group cannot arise from the simple hydrogenation of an exocyclic olefin because reagents approach from the less hindered (and wrong) diastereoface.2 In essence, a hypothetical transform to achieve the cis-addition of the −CH3 and −OH groups of methanol across an olefin was required. After extensive exploration, the formal equivalent of such a reaction was developed. Thus, exposure of 2 to bromonitrile oxide5 (generated in situ from dibromoformaldoxime and KHCO3) resulted in a dipolar cycloaddition leading to the formation of 5 as a single isomer in 88% yield on a gram-scale. This cycloaddition proceeds with complete control over regio- and positional selectivity to produce a single diastereomer of 5 (verified by X-ray crystallography). Ketone reduction with DIBAL followed by directed olefin hydrogenation (20% Crabtree's catalyst, H2, B(O-iPr)3)6 furnished 6 in 83% yield. It should be noted that olefin hydrogenation was confounded by the C-9 methyl and C-12 isopropyl groups flanking the disubstituted olefin on the face in which most hydrogenations would be expected to occur from. In our hands this was the only intermediate and the only set of conditions that succeeded; dozens of hydrogenation conditions on several different intermediates failed.
Xanthate formation (NaH, CS2, MeI) and subsequent Chugaev elimination (180 °C) furnished olefin 7 in 85% overall yield. The bromoisoxazole was unveiled to the desired tertiary alcohol 9 by the Saegusa deamination sequence:7 1. Reduction with LiAlH4 and immediate formylation of the crude amine, 2. Dehydration to a primary isonitrile, and 3. Treatment with Bu3SnH in the presence of AIBN in 56% overall yield. The robustness of this overall route is evident from the fact that >5 grams of 9 have been easily prepared and all the steps leading to this key intermediate have been conducted on a gram-scale.
Access to large quantities of key intermediates such as 9 was critical since, as alluded to above, we encountered a maze of unpredictable failures en route to 1, a small sampling of which are shown in Figure 2. Thus, allylic oxidation of 9 led to 13 which could not be productively functionalized further. Although olefin 9 reacted with bromonitrile oxide to furnish 14, it's downstream product 15 and and related structures could not be converted to 1. Finally, 2,3-dihydrovinigrol (16) and derivatives thereof could not be dehydrogenated to 1.
Figure 2.
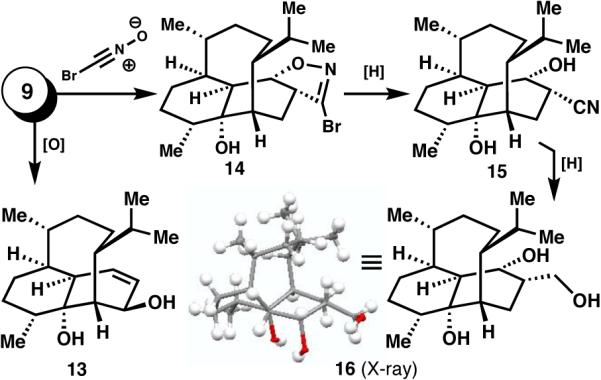
A small sampling of “dead-end” intermediates.
Ultimately, a simple route to 1 from 9 was developed. Thus, dihydroxylation of 9 with OsO4 and chemoselective oxidation of the resulting diol (NaOCl, TEMPO)8 led to α-hydroxy ketone 10 in 81% overall yield. A Shapiro reaction9 via trisylhydrazone 11 took place presumably via the trianionic species 12 to deliver (±)−1 (spectroscopically identical in all respects to a natural sample of 1, with the exception of optical rotation).
The venerable challenge posed by vinigrol (1) has been addressed by a 23 step route in 3% overall yield from commercially available materials. Aside from a minimal use of protecting group chemistry, nearly complete stereocontrol over all eight stereocenters, and the scalability of the route, notable aspects include: (1) simple formation of the decahydro-1,5-butanonaphthalene ring system by way of inter- and intramolecular Diels-Alder reactions followed by Grob fragmentation, (2) highly selective functionalization of 2 by way of an unusual dipolar cycloaddition, and (3) a Shapiro reaction that takes place via trianion 12. Obvious areas for refinement to the current route to 1 include a minimization of non-strategic redox fluctuations and an enantioselective variant of the first step.
Supplementary Material
Acknowledgement
We thank Dr. D.-H. Huang and Dr. L. Pasternack for NMR spectroscopic assistance, and Dr. G. Siuzdak for assistance with mass spectroscopy. Dr. Arnold Rheingold and Dr. Curtis Moore are acknowledged for X-ray crystallographic assistance. We thank Astellas Pharma Inc. (Japan) for a natural sample of 1. The JSPS provided support for a postdoctoral fellowship to S. A. and the ACS (to T. J. M.) and Bristol-Myers Squibb (to T.J.M. and J. S.) funded graduate student fellowships. Financial support for this work was provided by Amgen, Bristol-Myers Squibb, the NIH (CA134785), and the Skaggs Institute.
Footnotes
Supporting Information Available: Detailed experimental procedures, copies of all spectral data and full characterization. This material is available via the Internet at http://pubs.acs.org.
References
- (1).For isolation, see: Uchida I, Ando T, Fukami N, Yoshida K, Hashimoto M, Tada T, Koda S, Morimoto Y. J. Org. Chem. 1987;52:5292–5293.; For biological activity studies, see: Ando T, Tsurumi Y, Ohata N, Uchida I, Yoshida K, Okuhara M. J. Antibiotics. 1988;41:25–30. doi: 10.7164/antibiotics.41.25.; Ando T, Yoshida K, Okuhara M. J. Antibiotics. 1988;41:31–35. doi: 10.7164/antibiotics.41.31.; Norris DB, Depledge P, Jackson AP. PCT Int. Appl. WO 9107 953. 1991; Nakajima H, Yamamoto N, Kaizu T, Kino T. Jpn. Kokai Tokkyo Koho JP 07-206668. 1995; Keane JT. PCT Int. Appl. WO 2001000229. 2001; Onodera H, Ichimura M, Sakurada K, Kawabata A, Ota T. PCT Int. Appl. WO 2006077954. 2006
- (2).Studies towards the total synthesis of 1: Devaux JF, Hanna I, Lallemand JY. J. Org. Chem. 1993;58:2349–2350.; Guevel R. Ph.D. Thesis. Ohio State University; 1994. ; Devaux JF, Hanna I, Lallemand JY, Prange T. J. Chem. Res. Syn. 1996:32–33.; Mehta G, Reddy KS. Synlett. 1996:625–627.; Kito M, Sakai T, Haruta N, Shirahama H, Matsuda F. Synlett. 1996:1057–1060.; Kito M, Sakai T, Shirahama H, Miyashita M, Matsuda F. Synlett. 1997:219–220.; Devaux JF, Hanna I, Lallemand JY. J. Org. Chem. 1997;62:5062–5068.; Matsuda F, Sakai T, Okada N, Miyashita M. Tetrahedron Lett. 1998;39:863–864.; Matsuda F, Kito M, Sakai T, Okada N, Miyashita M, Shirahama H. Tetrahedron. 1999;55:14369–14380.; Goodman SN. Ph.D. Thesis. Harvard University; 2000. ; Efremov IV. Ph.D. Thesis. Ohio State University; 2001. ; Paquette LA, Guevel R, Sakamoto S, Kim IH, Crawford J. J. Org. Chem. 2003;68:6096–6107. doi: 10.1021/jo0301301.; Gentric L, Hanna I, Ricard L. Org. Lett. 2003;5:1139–1142. doi: 10.1021/ol034217k.; Morency L, Barriault L. Tetrahedron Lett. 2004;45:6105–6107.; Paquette LA, Efremov I, Liu ZS. J. Org. Chem. 2005;70:505–509. doi: 10.1021/jo048458x.; Paquette LA, Efremov I. J. Org. Chem. 2005;70:510–513. doi: 10.1021/jo0484575.; Paquette LA, Liu ZS, Efremov I. J. Org. Chem. 2005;70:514–518. doi: 10.1021/jo048456c.; Morency L, Barriault L. J. Org. Chem. 2005;70:8841–8853. doi: 10.1021/jo051318i.; Morency L. Ph.D. Thesis. University of Ottawa; 2006. ; Grise CM, Tessier G, Barriault L. Org. Lett. 2007;9:1545–1548. doi: 10.1021/ol0702977.; Tessier G, Barriault L. Org. Prep. Proc. Int. 2007;39:311–353.; Souweha MS, Enright GD, Fallis AG. Org. Lett. 2007;9:5163–5166. doi: 10.1021/ol702202b.; Maimone TJ, Voica A-F, Baran PS. Angew. Chem. Int. Ed. 2008;47:3054–3056. doi: 10.1002/anie.200800167.; Brekan JA. Ph.D. Thesis. State University of New York; 2008. ; Morton JGM, Kwon LD, Freeman JD, Njardarson JT. Tetrahedron Lett. 2009;50:1684–1686.; Morton JGM, Kwon LD, Freeman JD, Njardarson JT. Synlett. 2009:23–27.; Morton JGM, Draghici C, Kwon L, Njardarson JT. Org. Lett. 2009;11:4492–4495. doi: 10.1021/ol901519k.; Maimone TJ. Ph.D. Thesis. The Scripps Research Institute; 2009. .
- (3).For a review of modern approaches to terpene synthesis, see: Maimone TJ, Baran PS. Nat. Chem. Biol. 2007;7:396–407. doi: 10.1038/nchembio.2007.1.
- (4).Evans DA, Chapman KT. Tetrahedron Lett. 1986;49:5939–5942. [Google Scholar]
- (5).Vyas DM, Chiang Y, Doyle TW. Tetrahedron Lett. 1984;25:487–490. [Google Scholar]
- (6).Crabtree RH, Davis MW. J. Org. Chem. 1986:2655–2661. [Google Scholar]
- (7).Saegusa T, Kobayashi S, Ito Y, Yasuda N. J. Am. Chem. Soc. 1968;90:4182. [Google Scholar]
- (8).Burns NZ, Baran PS. Angew. Chem. Int. Ed. 2007;46:205–208. doi: 10.1002/anie.200704576. [DOI] [PubMed] [Google Scholar]
- (9).Shapiro RH, Heath MJ. J. Am. Chem. Soc. 1967;89:5734–5735. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.