

Summary
Mouse coat color mutants have led to the identification of more than 120 genes that encode proteins involved in all aspects of pigmentation, from the regulation of melanocyte development and differentiation to the transcriptional activation of pigment genes, from the enzymatic formation of pigment to the control of melanosome biogenesis and movement [Bennett and Lamoreux (2003) Pigment Cell Res. 16, 333]. One of the more perplexing of the identified mouse pigment genes is encoded at the Silver locus, first identified by Dunn and Thigpen [(1930) J. Heredity 21, 495] as responsible for a recessive coat color dilution that worsened with age on black backgrounds. The product of the Silver gene has since been discovered numerous times in different contexts, including the initial search for the tyrosinase gene, the characterization of major melanosome constituents in various species, and the identification of tumor-associated antigens from melanoma patients. Each discoverer provided a distinct name: Pmel17, gp100, gp95, gp85, ME20, RPE1, SILV and MMP115 among others. Although all its functions are unlikely to have yet been fully described, the protein clearly plays a central role in the biogenesis of the early stages of the pigment organelle, the melanosome, in birds, and mammals. As such, we will refer to the protein in this review simply as pre-melanosomal protein (Pmel). This review will summarize the structural and functional aspects of Pmel and its role in melanosome biogenesis.
Keywords: melanosome, endosome, fibril formation, ER
Identification and characterization of the Silver gene
The recessive silver (si) mutation was first described in an inbred mouse strain with graying hair that became less pigmented over time on black backgrounds (Dunn and Thigpen, 1930). Macroscopic examination of hairs from these mice showed variable loss of pigment, thought to reflect a variable reduction in the number of pigment granules (Dunn and Thigpen, 1930). The pigment loss was ascribed to a depletion of melanocytes (Quevedo et al., 1981), and immortalized melanocytes derived from B/b si/si mice were shown to be hypoproliferative in culture (Spanakis et al., 1992). Interestingly, pigment dilution was exacerbated in si/si mice that were also heterozygous at the B locus, now known to encode the melanosome component Tyrp1; a milder phenotype was observed in b/b homozygotes and in wild-type C57Bl/6 mice (Silvers, 1979) (D. Bennett, personal communication). Moreover, graying appeared to decrease with age in agouti mice (Dunn and Thigpen, 1930). These observations remain unexplained, although a possible explanation for the effect in agouti backgrounds will be discussed later.
cDNAs corresponding to the defective gene in si/si mice were isolated by expression cloning in an attempt to identify the gene for tyrosinase (Kwon et al., 1987b). The authors screened a λ-phage library containing inserts from melanocyte cDNA; the longest of the derived clones was the 17th isolated phage, and was therefore referred to as Pmel17-1 (Kwon et al., 1987a). The corresponding mRNA was expressed preferentially in both human and mouse melanocytes and melanoma cells. Cloning of the full-length human cDNA (Kwon et al., 1991) revealed a putative 668-residue polypeptide with a predicted topology of a type I integral membrane protein – an N-terminal signal sequence, a large lumenal/extracytoplasmic domain, a single membrane spanning domain, and a short cytoplasmic domain (see Figures 1 and 2 for schematic). The corresponding gene mapped near the Silver locus on chromosome 10 of the mouse (Kwon et al., 1991), and later sequencing analyses showed that Pmel cDNA from si/si mice had a nonsense mutation truncating the cytoplasmic domain (Martínez-Esparza et al., 1999; Solano et al., 2000). Moreover, an antibody to the cytoplasmic domain of the putative Pmel protein product failed to react with extracts from si/si melanocytes (Kwon et al., 1995; Solano et al., 2000), and an antibody to a melanosome matrix protein that cross-reacted with Pmel (Zhou et al., 1994) (see below) showed differential reactivity in such extracts. These studies clearly showed that Pmel was the product of the Silver locus and that the silver mutation generated an aberrant form of Pmel. The human Pmel gene, referred to by the HUGO Gene Nomenclature Committee as SILV (OMIM: 155550), was mapped to human chromosome 12q13–q14 (Kubota et al., 1995; Kwon et al., 1991), and the gene organization of 11 exons (Figure 1A) was reported by Spritz, Kwon and their colleagues (Bailin et al., 1996; Kim et al., 1996) (although only Bailin et al. identified the introns separating exons 2 through 4). There are no known diseases associated with this locus, but it is thought that some forms of albinism may be linked to Pmel deficiency by analogy to the mouse silver mutant, several mutations in chicken Pmel (Kerje et al., 2004), and a mutation in a severely hypopigmented breed of cattle (A. Oulmouden, Limoges University, Limoges, France, personal communication).
Figure 1.

Human pre-melanosomal protein (Pmel) gene and splice isoforms. (A) Organization of human SILV (Pmel) gene, according to Bailin et al. (1996). Exons are indicated by boxes, and introns by lines, drawn to scale (except where sizes of introns are indicated). The numbers at bottom indicate the codons at the start of each exon; 5′- and 3′-untranslated regions (5′UT, 3′UT) are indicated in gray. Hatched regions indicate alternatively spliced exons. (B) Schematic diagram of the products of the four known human Pmel splice isoforms. The intermediate (int.) form is the most abundant, and the short forms (short-l and short-i, including or lacking, respectively, the sequence uniquely present in the long but not the intermediate form), are the least abundant. ss, signal sequence; tm, transmembrane domain; cyt., cytoplasmic domain. Numbers indicate the amino acids at the borders of the topologic domains (ss, lumenal, tm and cyt.) or of the alternatively spliced regions with respect to Pmel-long.
Figure 2.

Domains within pre-melanosomal protein (Pmel) and their identity among vertebrates. (A) Individual domains within the human Pmel long form (identified in the text) are shown in different colors, with amino acid positions for start and end of each domain indicated below. Consensus sites for addition of N-linked oligosaccharides are indicated by branched structures; a site near the beginning of the repeat (RPT) domain is noted in gray because it is not conserved and unlikely used (Maresh et al., 1994b and our unpublished observations). Circles indicate positions of cysteine residues. TM, transmembrane domain; Cyt., cytoplasmic domain; CS, cleavage site. (B) Amino acid sequence comparison of Pmel orthologs in indicated species. The GenBank accession numbers from which the sequences were taken are indicated in the text; only the long form of human Pmel is shown. Red text indicates amino acid identities among all sequenced orthologs; orange indicates amino acid homology among most orthologs. Individual domains are indicated by bars over the sequence colored as in (A) and (C); the domain definitions are extended beyond the borders in (A) to cover the entire sequence, and were used in the analysis described in (C). (C) The amino acid sequence of each of the indicated domains of human Pmel was compared with that of the corresponding domain in each of the indicated species. The identity of the corresponding amino acid sequence to that of human Pmel is indicated graphically for each domain. Domain colors correspond to those in panel (A); also included in the comparison is the sequence of human Nmb.
Human Pmel was identified independently in screens for melanoma-associated antigens. Several groups developed monoclonal antibodies that reacted specifically with melanoma cells and not other types of tumors, including well-known antibodies NKI-beteb (Vennegoor et al., 1988), HMB-45 (Gown et al., 1986), HMB-50 (Vogel and Esclamado, 1988) and ME20 (Maresh et al., 1994a). These antibodies recognized a c. 100 kDa glycoprotein in cell lysates of melanoma cells and a c. 95 kDa glycoprotein in melanoma cell conditioned medium (Adema et al., 1994; Maresh et al., 1994a; Vennegoor et al., 1988; Vogel and Esclamado, 1988); hence it was referred to as gp100. NKI-beteb, HMB-45, and HMB-50 were subsequently shown to identify the product of the same cDNA (Adema et al., 1993). Analysis of the cDNA sequence identified a predicted product nearly identical to human Pmel17, except for a deletion of seven amino acid residues (resulting from an in-frame alternative splice removing 21 bp from the cDNA) near the predicted transmembrane domain (‘intermediate’ form in Figure 1B) and several amino acid variations that are likely due to sequence polymorphisms (Adema et al., 1994; Maresh et al., 1994a). An additional in-frame alternative splice removes a 126-bp exon, encoding 42 amino acids in the predicted lumenal domain, and can occur independently of the first alternative splice to result in four distinct products (Nichols et al., 2003) (Figure 1B). All anti-gp100 monoclonal antibodies reacted with all four products, which are co-expressed in melanocytes and melanoma cells (Adema et al., 1994; Nichols et al., 2003).1 Importantly, Pmel-derived peptides were also identified among the first characterized targets of anti-melanoma cytotoxic T cells (Bakker et al., 1994; Kawakami et al., 1994, 1995), and later helper T cells (Halder et al., 1997), from melanoma patients. Pmel expression is maintained in the majority of epidermal and uveal melanomas analyzed (Cormier et al., 1998a,b; Luyten et al., 1998; de Vries et al., 1997, 1998), making it an excellent target for anti-melanoma immunotherapy. Indeed, Pmel has been used as a target in numerous experimental immunotherapy approaches, coverage of which is beyond the scope of this review (see Yu and Restifo, 2002 for a recent review). Thus, characterization of Pmel is of significant potential importance for tumor immunotherapy.
Sequences highly homologous to human and mouse Pmel have been isolated from rat (accession no. XP_343147), horse [(Rieder et al., 2000); accession no. AAC97108], cow [called RPE1 (Kim and Wistow, 1992); accession no. XP_582778], dog (accession no. XP_538223), chicken [(Kerje et al., 2004); initially called melanosomal matrix protein 115 or MMP115 (Mochii et al., 1991); accession no. AAT58245], quail (accession no. AAD12180), the amphibians Xenopus laevis (accession no. AAH77508) and Xenopus tropicalis (accession no. AAH75473), and the teleost fish Danio rerio (zebrafish; accession no. AAT37511) and Tetraodon nigroviridis (accession no. CAG11762). Polymorphisms at the Pmel locus are linked to hypopigmentation in the chicken (Kerje et al., 2004) and cattle (A. Oulmouden, personal communication), and to the establishment of pecking order in the chicken (Keeling et al., 2004). An alignment of the predicted polypeptide sequences of the longest Pmel product in each species is shown in Figure 2B.
Characterization and expression of the Pmel gene product
The primary structure of the Pmel polypeptide provides little clue to its function (Figure 2A). Following a 24-residue N-terminal signal sequence (Maresh et al., 1994b), within which are found mutations in hypopigmented cattle (A. Oulmouden, personal communication), is a 578-residue lumenal domain (in the longest human product) that can be divided into several subdomains based on structural predictions. Immediately following the signal sequence is c. 200-residue N-terminal region (NTR) that contains three putative N-terminal glycosylation sites, all of which are used in vivo (Maresh et al., 1994b), and three cysteine residues that likely participate in disulfide bonds. The NTR lacks significant homology to other proteins within the database except for a similarly placed domain within another melanocyte-enriched protein, Nmb (see below and Figure 2C). A short region spanning a conserved cysteine residue (C60) within the NTR bears modest homology to tyrosinase (Kwon, 1993), perhaps explaining the cross-reactivity of Pmel with an anti-tyrosinase antibody (Kwon et al., 1987b). Downstream of the NTR is a polycystic kidney disease (PKD) domain with significant homology to a repeat region found within the polycystic kidney disease protein 1 (PKD1), as well as many other proteins in prokaryotes and eukaryotes (Bycroft et al., 1999). PKD domains have an immunoglobulin-like fold, and are thought to mediate protein: protein or lectin: carbohydrate interactions. Downstream and separated from the PKD domain by a conserved cysteine residue is a region rich in acidic residues, prolines, serines and threonines that consists primarily of imperfect direct repeated sequences (Kwon et al., 1991; Nichols et al., 2003; Solano et al., 2000); hence we refer to this region as the repeat (RPT) domain, and will discuss it further below. Following the RPT region in all Pmel isoforms is a highly conserved dibasic sequence that serves as a site for proteolytic cleavage by at least one member of a class of proteases called proprotein convertases (PCs) (Berson et al., 2003). As discussed in the next section, this cleavage is critical for Pmel function (Berson et al., 2003). Downstream of the cleavage site is another highly conserved region that we refer to as the Kringle-like domain (KLD). This domain is characterized by the presence of highly conserved cysteines spaced in a manner similar, but not identical to that of a Kringle domain (Magnusson et al., 1975; Sottrup-Jensen et al., 1975), an evolutionarily conserved cysteine-knot structure found most notably in blood clotting proproteases such as plasminogen and prothrombin, in which it functions in binding lysine or lysine-containing peptides. Following a highly conserved cysteine and N-glycosylation site at the C-terminus of the KLD is a poorly conserved, short proline-rich region, the length of which varies among the two major splice forms in humans that separates the KLD from the single 24-residue membrane spanning domain. The PKD domain and KLD are the most highly conserved among Pmel orthologs (Figure 2C), and the NTR is also highly conserved. These regions are also relatively well conserved in another protein of unknown function, Nmb (also called Gpnmb, osteoactivin, DC-HIL or QNR71), suggesting a related function for these two proteins. Nmb is highly expressed in pigment cells, as is Pmel, but also in other tissues such as osteoblasts, activated dendritic cells, and macrophages (Owen et al., 2003; Safadi et al., 2001; Shikano et al., 2001; Turque et al., 1996; Weterman et al., 1995). The Pmel membrane spanning domain is the site for insertions and deletions within chicken Pmel variants that result in altered plumage color (Kerje et al., 2004), suggesting that the length of this domain is critical for Pmel function. Most conserved among Pmel orthologs within the 45-residue cytoplasmic domain is a C-terminal valine residue and a sequence near the C-terminus, ExxPLL (where x refers to any amino acid), that conforms to a consensus acidic di-leucine internalization/endocytic targeting signal and that is similar to a putative melanosome targeting signal in Tyrp1 (Vijayasaradhi et al., 1995) and tyrosinase (Calvo et al., 1999; Höning et al., 1998; Simmen et al., 1999). The truncation of the cytoplasmic domain and consequent loss of the di-leucine signal by the silver mutation has been proposed to underlie the defects observed in si/si mice (Martínez-Esparza et al., 1999); our preliminary results suggest that additional determinants within the cytoplasmic domain may also contribute to Pmel transport (ACT and MSM, unpublished data).
Perhaps the most notable Pmel subdomain is the RPT region. The RPT region in humans consists of 10 imperfect direct repeats of a 13-residue sequence (see Nichols et al., 2003 for definition of the 13 amino acid repeat). Similar repeats are found in Pmel of all vertebrates, but the number and nature of the repeats differ across species and even across splice isoforms. Pmel isoforms in mammals contain 7–10 repeats, with the shortest human splice isoform having deleted three (and part of a fourth) of the 10 found in the longest splice isoform. The nature and sequence of the core repeat in Pmel from birds, amphibians and teleost fish differ from those in mammalian Pmel. Many of the serines and threonines in the repeats conform to O-glycosylation sites, consistent with biochemical analyses that suggest significant O-glycosylation of the lumenal domain (see below). Interestingly, the RPT region is the only domain lacking in Nmb, suggesting a Pmel-specific function.
Strong expression of Pmel mRNA is limited to pigmented tissues, including skin melanocytes, uveal melanocytes, and retinal and iris pigment epithelium. One report identified Pmel transcripts in all tissues but protein expression only in melanocytic cells (Brouwenstijn et al., 1997). However, scanning of homologous clones deposited in the EST database (using UniGene from the National Center for Biotechnology Information; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=unigene) reveals that significant expression is nearly exclusive to the skin and eye. Relatively rare additional clones were isolated from libraries from placenta, uterus, and embryonic stem cells, fewer still from brain, and random isolates are present in libraries from other tissues. Pmel expression is initiated in early melanoblasts (Baxter and Pavan, 2003), suggesting an important function in early melanogenesis. The only conclusive demonstration of Pmel protein expression outside of the skin and eye has been in an unusual class of clear cell tumors (Folpe et al., 2000; Lantuejoul et al., 1997) and in angiomyolipomas (Barnard and Lajoie, 2001; Yamasaki et al., 2000), likely due to dysregulation of gene expression in these transformed cell types. Pmel expression in pigment cells can be modified by melanogenic and inflammatory cytokines that affect other melanogenic proteins. For example, the Pmel gene is a target of microphthalmia-associated transcription factor (Baxter and Pavan, 2003; Du et al., 2003) and is thus activated by alpha-melanocyte stimulating hormone (Furumura et al., 1998; Kobayashi et al., 1995; Martinez-Esparza et al., 2000) and inactivated by exposure to toxins such as peroxide (Jimenez-Cervantes et al., 2001). Pmel expression is sharply downregulated in cells synthesizing pheomelanins by agouti signaling (Furumura et al., 1998; Kobayashi et al., 1995), and is antagonized by inflammatory cytokines such as transforming growth factor-β1, alpha-tumor necrosis factor, and interferon-γ (Le Poole et al., 2002; Martinez-Esparza et al., 2000). Together, the data strongly support a pigment-cell-specific function for Pmel, specifically in eumelanogenesis.
Pmel function in melanosome biogenesis and melanin polymerization
The pigment cells of mammals, birds and amphibians harbor specialized subcellular compartments, the melanosomes, in which melanins are synthesized and stored (Hearing, 2000; Marks and Seabra, 2001; Seiji et al., 1961). Mammalian and avian melanosomes, at least in skin melanocytes synthesizing predominantly eumelanins (perhaps not in those synthesizing pheomelanins; see Furumura et al., 1998; Moyer, 1966), progress through four distinct stages of maturation that have been well described at the ultrastructural level (Figure 3). The first two stages precede the deposition of melanins in later stages and appear to constitute intermediates required to generate a ‘matrix’ favorable for eumelanin polymerization. The unique morphologic features of early stage melanosomes are most apparent in fully developed stage II structures, and include an ellipsoidal shape and intralumenal ‘striations’ composed of fibrillar material. It is upon these fibrils that melanins are deposited in stages III and IV melanosomes, resulting in their blackening and thickening. The intralumenal fibrils are thus thought to provide a ‘matrix’ upon which melanin intermediates can be concentrated and perhaps stabilized or detoxified (Raposo and Marks, 2002; Seiji et al., 1963; Solano et al., 2000; Spanakis et al., 1992). Concentration of melanin intermediates could play a significant role in facilitating transfer of melanin from melanocytes to keratinocytes in the epidermis (Boissy, 2003) or in segregating melanosomes from phagolysosomes within retinal pigment epithelial cells (Schraermeyer et al., 1999).
Figure 3.
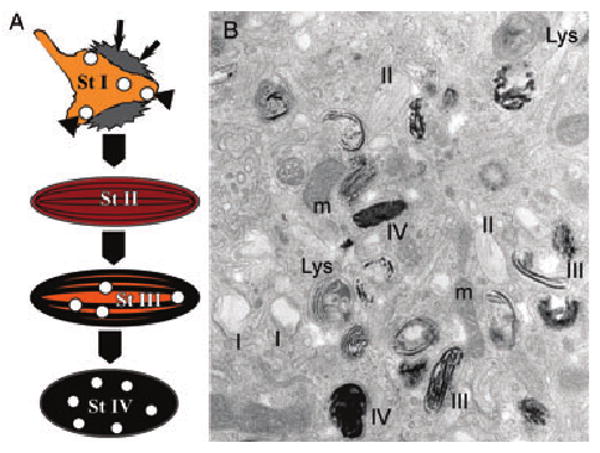
Melanosome stages of development. (A) Schematic diagram of the four melanosome stages as described in the text. (B) Representative electron micrograph of an ultrathin section of plastic-embedded MNT-1 cells showing examples of each of the melanosome stages I, II, III and IV as indicated. Lys, lysosome; m, mitochondria.
The premelanosome fibrils have been long thought to be proteinaceous as they lack the ultrastructural hallmarks of a lipid bilayer (Hearing et al., 1973; Maul, 1969; Moyer, 1966). Initial efforts to define the biochemical nature of the ‘melanosome matrix’ led to the generation of antisera that recognized a number of polypeptides within a Triton X-100-insoluble (Orlow et al., 1993) or Triton X-114-soluble (Zhou et al., 1994) fraction of melanosomes from mouse melanoma cells or melanocytes. The major polypeptide identified was a 85–94 kDa glycoprotein that was processed to a lower molecular weight form accumulating in melanosome fractions (Orlow et al., 1993). This polypeptide was absent from melan-si cells (Zhou et al., 1994), generated from si/si mice homozygous for the silver mutation (Spanakis et al., 1992), suggesting that it was the product of the Silver locus. Indeed the precursor polypeptide cross-reacted with an antiserum generated to a peptide from the predicted C-terminus of Pmel (Kobayashi et al., 1994; Zhou et al., 1994). Moreover, anti-Pmel monoclonal antibodies labeled immature melanosomes, as well as other vacuolar and vesicular profiles, by immunoelectron microscopy (Lee et al., 1996; Mochii et al., 1988; Raposo et al., 2001; Vennegoor et al., 1988). The vacuolar and vesicular profiles, containing variable amounts of internal membranes, were shown to correspond to the previously described stage I melanosomes (see below); quantitative immunogold labeling demonstrated that Pmel antibodies decorate primarily the fibrous stage II melanosomes and secondarily the multivesicular stage I melanosomes (Raposo et al., 2001). Ultimate proof that Pmel is not only a component of the fibrils but is sufficient for their formation came from transfection studies in non-melanocytic cells. Expression in HeLa cervical carcinoma cells of Pmel, as the only pigment cell-specific protein, generated fibrous structures within late endosomes that were nearly identical to those found in stage II melanosomes (Berson et al., 2001). Thus, Pmel is capable of polymerizing into fibrillar arrays that form the backbone of eumelanosomes. Interestingly, this feature is shared by fibrillogenic proteins associated with amyloid deposits in pathologies such as Parkinson's and Alzheimer's disease, making Pmel fibrillogenesis an excellent model for pathologic fibril formation (Huff et al., 2003). This property of Pmel and its lack of expression in cells synthesizing predominantly pheomelanins (Furumura et al., 1998) may explain why pheomelanosomes have a round shape and lack fibrils (Moyer, 1966), and why the silver mutation has a muted effect on pigmentation in agouti animals (Dunn and Thigpen, 1930). Indeed, ultrastructural analyses of immortalized melanocytes derived from non-agouti si/si mice support the requirement for high levels of Pmel expression in premelanosomes to generate the characteristic fibrils and elongated shape of eumelanosomes (Theos, A.C., Berson, J.F., Cromer, S.B., Harper, D.C., Herman, K.E., Sviderskaya, E.V., Bennett, D.C., Raposo, G. and Marks, M.S., unpublished data).
Immunolabeling for Pmel vanishes in more mature melanosomes (Raposo et al., 2001; Vennegoor et al., 1988), and in earlier studies was more apparent in the periphery of immature melanosomes than in the fibrous matrix (Lee et al., 1996; Mochii et al., 1988; Orlow et al., 1993; Vennegoor et al., 1988). This correlates with an increase in Pmel immunoreactivity by western blotting upon attenuation of pigmentation, interpreted as an interaction of Pmel with melanin (Donatien and Orlow, 1995). These characteristics would be expected for a protein constituent of the fibrous material upon which melanins are deposited in maturing melanosomes; deposition of melanin would ‘mask’ the Pmel epitopes on the fibrils. Consistent with this notion, two studies implicated Pmel as a stimulator of melanin polymerization – a ‘convertase’ for the melanin precursor, dihydroxyindole-2-carboxylic acid (DHICA). One study demonstrated DHICA convertase activity using affinity-purified Pmel from insect cells or human melanoma cells (Lee et al., 1996), and the other study used extracts of melanocytes from wild-type or si/si mice (Chakraborty et al., 1996). While these studies warrant further verification with purified recombinant protein and appropriate controls, they would be consistent with a model in which Pmel first forms fibrils by ordered oligomerization, and then, perhaps through a distinct domain, facilitates melanin deposition. A function for Pmel in facilitating melanin deposition and in generating fibrils, which are unique among physiologic organellar structures to melanosomes, would explain the pigment cell-specific expression of the Pmel gene product.
Pmel trafficking to melanosomes: direct from endoplasmic reticulum or from endosomes?
As a major component of the fibrillar matrix of early stage melanosomes, Pmel serves as the best-known marker to follow the intracellular trafficking steps that regulate melanosome biogenesis. Thus, significant attention has been given to understand the intracellular processing and transport of Pmel within melanocytic cells. The results of these studies have led to contradictory and controversial conclusions. Here, we will summarize the data and attempt to reconcile the contrasting views.
The ER to melanosome view
One view of premelanosome biogenesis (Figure 4A) was shaped by an early and elegant electron microscopy study. Using serial sectioning, Maul showed that structures with the characteristic morphologic features of stage II melanosomes could sometimes be observed in continuity with smooth tubular membranes near the Golgi apparatus of a human melanoma cell line (Maul, 1969). The data were interpreted as the emergence of premelanosomes from smooth endoplasmic reticulum (ER). This was an appropriate conclusion at the time; the morphology of late secretory and endocytic organelles was not yet appreciated, and smooth ER was the only smooth tubular membrane system known. The idea that Pmel, the biogenetic component of the fibrils, would traffic from ER to premelanosomes was supported by protein processing studies in mouse melanocytic cell lines (Kobayashi et al., 1994). These studies showed that mouse Pmel, detected with an antibody to the cytoplasmic domain, is modified by N-glycosylation in the ER as expected. However, further processing of the N-linked oligosaccharides in the Golgi, as occurs for late-stage melanosomal proteins such as tyrosinase, Tyrp1 and DCT was not observed. Rather, the immature protein disappeared from cell lysates with a half-life of approximately 60–90 min, as confirmed in other studies (Donatien and Orlow, 1995). Moreover, Pmel was not detected in coated vesicle fractions in which tyrosinase, Tyrp1 and DCT were enriched, suggesting a distinct sorting pathway to melanosomes (Kobayashi et al., 1994). In apparent corroboration of this notion, Hearing and coworkers identified the immature form of Pmel in a subcellular fraction of a human melanoma cell line enriched in stage II melanosomes (Kushimoto et al., 2001). Proteomic analysis of this fraction revealed the presence of traditional ER resident proteins in addition to known constituents of early and late-stage melanosomes, late endosomes/lysosomes, lipid rafts, and secretory granules (Basrur et al., 2003). Proteolytic processing of Pmel during maturation from stage II to stage III melanosomes in these cells was inferred by the loss of the full-length ER-modified precursor in fractions enriched in later stage melanosomes and the appearance in these fractions of a faster migrating band reactive with the anti-Pmel monoclonal antibody HMB-45 (Kushimoto et al., 2001), known to detect a lumenal determinant of Pmel (Berson et al., 2001, 2003; Yasumoto et al., 2004). The inability to detect Golgi processed forms of Pmel by immunoblotting in these studies (Kobayashi et al., 1994; Kushimoto et al., 2001; Yasumoto et al., 2004) has been taken as a further indication of a direct ER to premelanosome pathway for Pmel (Figure 4A).
Figure 4.
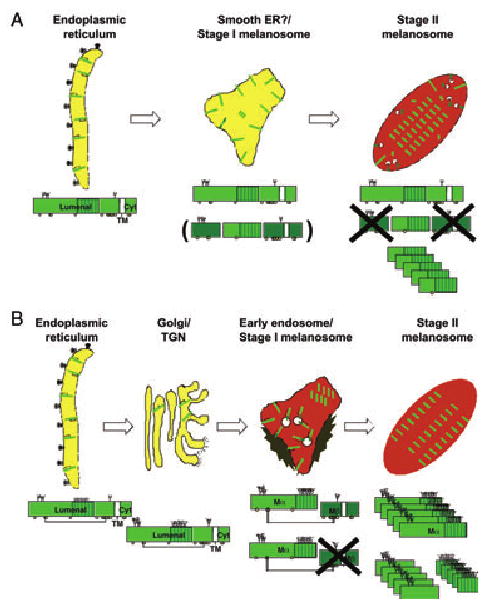
Two models for pre-melanosomal protein (Pmel) trafficking to stage I/II melanosomes. Shown are two models for Pmel trafficking through the cell, with schematic diagram of the processing intermediate for Pmel within each organelle as described in the text. (A) Based on models described in Kushimoto et al. (2001) and Yasumoto et al. (2004). (B) Based on models described in Berson et al. (2001, 2003) and Raposo et al. (2001). The Pmel lumenal, transmembrane (TM; white) and cytoplasmic (cyt) domains are labeled, and the RPT domain is indicated by vertical shading. N-glycosylation sites are indicated by branched structures, and potential O-glycosylation sites are indicated by circles; branching in panel (B) indicates processing of glycans in the Golgi. Cleavage products that are eventually lost to proteolytic degradation are indicated in dark green, and oligomerization of the final cleavage products is indicated by stacking. Note that the cleavage boundaries and responsible proteases for generating the final products (HMB-45-reactive RPT region and HMB-50/NKI-beteb-reactive N-terminal region) in stage II melanosomes have not yet been defined in either model.
The endosome to melanosome view
We have proposed an alternative model of premelanosome biogenesis based on Pmel processing and localization in human melanocytic cells (Figure 4B). This model was initially based on our electron microscopy analyses using quantitative immunogold labeling of ultrathin cryosections from pigmented human melanoma cell lines and from eumelanin-enriched primary melanocytes (Raposo et al., 2001). Immunolabeling for Pmel, using monoclonal antibodies that recognize the mature protein (see below), was enriched predominantly within stages I and II melanosomes. The stage I melanosomes are identified as early endosomal organelles based on the following criteria. First, they appear morphologically similar to tubular and vacuolar early endosomes of other cell types that are known to regulate cargo sorting to the cell surface, late endosomes, and the Golgi (Raiborg et al., 2003). In particular, they are characterized by intralumenal vesicles that form from invagination of the limiting membrane, and by a flat clathrin lattice along part of the cytoplasmic surface, known to serve as a sorting platform for cargo recruitment onto the intralumenal vesicles (Gruenberg and Stenmark, 2004). Secondly, they label for the early endosomal antigen 1, a well-characterized marker limited to early endosomes. Finally and conclusively, they are targeted by markers of fluid phase and receptor-mediated endocytosis with kinetics expected for early endosomes. The fluid phase markers proceed to classical late endosomes and lysosomes in these cells with expected kinetics. Interestingly, immunogold labeling for Pmel on stage I melanosomes was found not only on the limiting membrane, but also on the intralumenal membranes; whereas an antibody to the Pmel cytoplasmic domain also labeled these organelles (as well as earlier secretory organelles), no labeling with this antibody was observed on fully formed, striated stage II melanosomes. Passage through an endosomal intermediate, and particularly on intralumenal vesicles of endosomes, is also consistent with the localization of Pmel when expressed in non-pigment cells, such as HeLa. In these cells, Pmel accumulates primarily on the intralumenal vesicles of multivesicular late endosomes, from which it is recruited to form fibrils within those same organelles (Berson et al., 2001, 2003). Together, these data suggest that full-length Pmel traverses an early endosomal organelle en route to stage II melanosomes, and that concomitant proteolytic processing results in the loss of the cytoplasmic domain in stage II melanosomes (Figure 4B). The latter is consistent with a role for proteolytic maturation of Pmel as proposed by Hearing and coworkers (Kushimoto et al., 2001; Yasumoto et al., 2004).
If Pmel trafficks directly from the ER to stage II melanosomes, then it should not be processed by resident Golgi oligosaccharide transferases. However, analyses in human melanocytic cells similar to those performed in mouse melanocytes (Kobayashi et al., 1994) showed clear evidence for Golgi-associated glycosylation modifications as well as post-Golgi proteolytic processing (Adema et al., 1994; Berson et al., 2001; Maresh et al., 1994b; Nichols et al., 2003; Vennegoor et al., 1988; Vogel and Esclamado, 1988). By metabolic pulse/chase and immunoprecipitation analyses, the precursor form in the ER, termed P1 (Mr c. 97 000), is post-translationally modified within the Golgi apparatus to a higher Mr form, termed P2 (Mr c. 128 000). The P2 harbored mature N-linked oligosaccharides, as well as additional modifications that likely include processed O-linked oligosaccharides, was demonstrated using endoglycosidase digestions (Berson et al., 2001). However, the accumulation of P2 is limited by two factors. Once formed, P2 is rapidly proteolytically processed into two subunits, a large lumenal fragment Mα (Mr 85–95 000) and a smaller Mβ fragment (Mr 28 000) consisting of the remaining lumenal region and the transmembrane and cytoplasmic domains. That these products are formed after progression of Pmel through the Golgi complex is supported by the resistance of at least two of the N-linked oligosaccharides on Mα and the single N-linked glycan remaining on the Mβ to digestion by endoglycosidase H (Berson et al., 2001), which is acquired concomitant with terminal modification of trimmed, core oligosaccharides by N-acetylglucosaminyl transferase in the medial Golgi (Kornfeld and Kornfeld, 1985; Tarentino et al., 1974). Moreover, drugs that interfere with ER to Golgi transport, such as brefeldin A, or that block acidification of post-Golgi organelles, such as ammonium chloride and bafilomycin A1, inhibit the appearance of Mα and Mβ and stabilize P2, whereas inhibitors of lysosomal proteolysis have no effect (Berson et al., 2001). A small fraction of Mα is secreted, and two of the three N-linked glycans on purified, secreted Mα were shown by mass spectrometry to contain complex oligosaccharides resulting from Golgi modifications (Maresh et al., 1994b). Finally, the epitope for the HMB-45 antibody, which detects a Pmel fragment within stage II melanosomes (see below), is destroyed by treatment with neuraminidase, indicating that it requires terminal modification of N- or O-linked oligosaccharides with sialic acid (Chiamenti et al., 1996; Kapur et al., 1992). This modification occurs within the trans-Golgi or trans-Golgi network (TGN) (Kornfeld and Kornfeld, 1985; Van den Steen et al., 1998). Taken together, these data indicate that full-length Pmel is cleaved within an acidic, post-Golgi, and prelysosomal compartment.
A second factor that limits P2 accumulation is the disappearance of all Pmel isoforms, after a short lag period, from detergent lysates of melanocytic cells (Berson et al., 2001), concomitant with the appearance of Mα within a detergent-insoluble fraction (Berson et al., 2003). These two factors contribute to an extremely short half-life for P2 and a relatively short half-life for detergent soluble forms of its derivative Mα- and Mβ-fragments. By contrast, P1 is relatively long-lived, likely reflecting slow Pmel folding and ER exit. The long half-life of P1 and the short half-life of P2 explain the failure to observe P2 using steady state measures such as western blotting. However, the Mβ-fragment is somewhat longer lived in detergent extracts, and is quite apparent by western blotting under conditions that favor detection of small glycopeptides (Berson et al., 2001). These data strongly support the notion that Pmel indeed passes through the Golgi en route to premelanosomes, and is further processed in an acidic post-Golgi compartment. Interestingly, all processing steps are recapitulated upon ectopic expression of human Pmel in a non-pigment cell type, HeLa cells (Berson et al., 2001), suggesting that Pmel processing does not require any unconventional transport pathway specific to pigment cells.
Although Mβ can be detected by western blotting of mouse melanocytes (Berson et al., 2001), none of the processed intermediates of mouse Pmel are easily observed by metabolic pulse/chase assays (Kobayashi et al., 1994). This is likely a property of rapid kinetics of processing and insolubility inherent to the species of Pmel and not of the host cell, because human Pmel is processed with identical patterns and kinetics to that in human melanocytes when expressed in mouse melanocytes (ACT and MSM, unpublished data). Given these as-yet-unexplained and perhaps misleading kinetic differences between mouse and human Pmel, it is not surprising that the earlier studies of Pmel processing in mice led to an unconventional view of Pmel progression through the secretory pathway.
Is the post-Golgi pathway for Pmel maturation merely representative of a small fraction of improperly transported human Pmel or a requisite pathway for stage II melanosome biogenesis? Further analysis of the cleavage event suggests that the latter is indeed the case. A clue to the cleavage came from analysis of the small fraction of Mα that is secreted by melanocytic cells (c. 5–8% of the total in all cells analysed thus far; (Berson et al., 2001 and unpublished data). Peptide sequencing of this secreted form showed that the C-terminal residue was V467 (Maresh et al., 1994b), which is immediately followed in the primary sequence by two highly conserved basic residues. A dibasic recognition site is characteristic of substrates for the PC class of proteolytic processing enzymes, of which furin is the founding family member (Steiner, 1998). PCs cleave substrates within the TGN, secretory granules, and/or endocytic organelles (but not early secretory organelles) resulting in maturation of the proprotein substrate to the mature protein product. Several PC family members, including furin, are ubiquitously expressed (Steiner, 1998), and two family members that are expressed predominantly in neuroendocrine cells, PC1 and PC2, are expressed by melanocytes and cofractionate with melanosomes (Peters et al., 2000). Proprotein convertase cleavage is normally followed by a carboxypeptidase activity to remove the basic residues (Steiner, 1998); thus, production of Mα could be explained by sequential proteolytic maturation by a PC and a carboxypeptidase. This prediction was confirmed experimentally. Mutagenesis of the dibasic site in ectopically expressed Pmel, expression of Pmel in a PC-deficient cell line, or inhibition of PC activity by expression of a specific inhibitor in melanocytic cells resulted in a failure to convert P2 to Mα- and Mβ-fragments (Berson et al., 2003). Inhibition of PC cleavage had dramatic consequences on the fate of Pmel. By contrast to non-melanocytic HeLa cells expressing full-length Pmel, cells expressing an uncleavable Pmel isoform did not harbor fibrils within endosomes. Consistent with the failure to form fibrils, inhibition of PC cleavage in melanocytic cells resulted in a dramatic disruption of melanosome morphology such that fibrillar arrays no longer formed (Berson et al., 2003). Finally, western blotting of preparations of purified fibrils from a melanoma cell line showed a depletion of full-length Pmel and an enrichment of Mα-fragments (Berson et al., 2003). These data show that PC cleavage of Pmel and consequent formation of Mα is absolutely required for the generation of premelanosome fibrils. As PC cleavage occurs only in the TGN or a later, acidic post-Golgi compartment (likely an endosome such as a stage I melanosome), the data indicate that passage through the Golgi and endosomes is an essential feature of the Pmel life cycle (Figure 4B).
Interestingly, metabolic pulse/chase analyses show that Mα levels in detergent-insoluble fractions fall soon after they peak, suggesting that Mα either becomes inaccessible to antibodies or is subject to a second proteolytic cleavage. In support of the latter, the HMB-45 antibody – which detects Pmel primarily within stage II melanosomes by microscopic methods (Raposo et al., 2001) – detects primarily a series of bands ranging in Mr from 30–40 kDa and only faintly detects Mα in whole cell lysates by immunoblotting (Chiamenti et al., 1996; Kushimoto et al., 2001; Yasumoto et al., 2004). The nature of this putative second cleavage has not yet been identified, but is likely to occur within stage II melanosomes (Yasumoto et al., 2004).
Resolution of the two views
Can the data in support of the two models of Pmel maturation and transport be reconciled? While it is possible that elements of both models may be correct, we contend that the data supporting an ER to melanosome pathway can be interpreted differently. As discussed earlier, the morphologic data that initially led to the model that premelanosomes emerge from smooth ER (Maul, 1969) would be interpreted differently today by cell biologists. The membranes from which Maul observed striated structures emerging are more characteristic of the TGN or endosomes than of smooth ER (Geuze and Morre, 1991; Gruenberg, 2001; Tooze and Hollinshead, 1991). The subcellular fractionation studies, interpreted as supporting this model (Basrur et al., 2003; Kushimoto et al., 2001), did not account for contamination of stage II premelanosome fractions by ER membranes. Such contamination, which we observe in our subcellular fractionations from the same cells and are unable to resolve (MSM, unpublished observations), together with the long residence time for Pmel within the ER in these cells (Berson et al., 2001), would explain the presence of the P1 form of Pmel within stage II melanosome-enriched fractions. Indeed, reevaluation of the immunoelectron micrograph in the first of these reports, purportedly showing immunogold labeling for the Pmel cytoplasmic domain in stage II melanosomes, shows an accumulation of aggregated gold particles on only one side of the organelle (Kushimoto et al., 2001) (Figure 5B); this contrasts with evenly distributed immunogold labeling for tyrosinase on the limiting membrane (Figure 5A) and for the Pmel lumenal domain on the intralumenal fibrils (Figure 5C) of the same structures. This cytoplasmic Pmel labeling is thus likely to represent a contaminating membrane, perhaps derived from the ER, associated with the periphery of the stage II melanosome rather than an integral component of the stage II limiting membrane. By contrast, our quantitative analyses show no labeling for the Pmel cytoplasmic domain in stage II melanosomes (Raposo et al., 2001) (see Figure 5D, E) despite extensive labeling of stage I melanosomes/endosomes and earlier secretory compartments, including Golgi (Figure 5D–F). ER contamination of subcellular fractions would also explain the presence in these fractions of components of rough ER (such as calnexin, ribophorin 1 and soluble ER chaperones) and the absence of known enzymatic components of smooth ER (e.g. lipid biosynthetic enzymes and cytochrome p450) despite the undisputed derivation of stage II melanosomes from smooth membranes.
Figure 5.

Immunoelectron microscopy evidence for pre-melanosomal protein (Pmel) cytoplasmic domain in melanosome precursors. Published electron micrographs of immunogold labeled samples derived from the pigmented MNT-1 melanoma cell line. Immunogold labeling for the Pmel cytoplasmic domain is indicated by arrows, and for the lumenal domain by arrowheads. (A–C) Pre-embedding immunogold labeling of purified stage II melanosome fractions using antibodies to the tyrosinase cytoplasmic domain [(A); stars], the Pmel cytoplasmic domain [(B), αPep13h; arrows] or the Pmel lumenal domain [(C), HMB-45; arrowheads]. Note the cluster of gold between stage II melanosomes in (B), contrasting with the even distribution of gold on the internal striations in (C) and on the limiting membrane of the melanosome in (A). Data are reproduced from Figure 4 of Kushimoto et al. (2001) (D–F). Double immunogold labeling of ultrathin cryosections of MNT-1 cells using antibodies to the Pmel cytoplasmic domain (arrows) or lumenal domain (HMB-50; arrowheads). (D). Reproduced from Supplementary Figure S3 of Raposo et al., 2001. Pmel cytoplasmic domain is labeled with αPep13h and protein A-conjugated 15-nm gold (arrows), and HMB-50 labeling is developed with protein A-conjugated 10-nm gold (arrowheads). Note the labeling of the Golgi stacks by both antibodies, whereas the stage II melanosomes are labeled only in the lumen by HMB-50. (E,F). Reproduced from Figure 6 of Berson et al. (2003). Pmel cytoplasmic domain is labeled with αPep13h and protein A-conjugated 10-nm gold (arrows), and HMB-50 labeling is developed with protein A-conjugated 5-nm gold (arrowheads). In (E), note the labeling of stage II melanosomes only by HMB-50, whereas an endosomal structure shows labeling for both antibodies. (F), a typical stage I melanosome, in which cytoplasmic domain labeling is evident at the limiting membrane.
The absence of immature Pmel from stage II melanosomes is also supported by immunofluorescence microscopy experiments in formaldehyde-fixed cells. These experiments show immunolabeling for the Pmel cytoplasmic domain throughout the cytoplasm (Kushimoto et al., 2001; Yasumoto et al., 2004; our unpublished observations), coincident with labeling for the ER resident chaperone, calnexin, and characteristic of ER labeling. This contrasts with the punctate immunolabeling obtained with antibodies such as HMB-45, HMB-50 and NKI-beteb (Adema et al., 1993; Berson et al., 2001; Raposo et al., 2001; Vogel and Esclamado, 1988; Yasumoto et al., 2004) that recognize the Mα-fragment 2. The predominant labeling of the ER with antibodies to the Pmel cytoplasmic domain in these experiments is consistent with the slow ER exit of Pmel (thus high steady-state expression of the P1 form) and the relatively poor accessibility of cytoplasmic domain antibodies to the intralumenal membranes and coated regions of the stage I melanosomes by this technique. By contrast, antibody accessibility is improved upon cryosectioning of these same structures for immunoelectron microscopy analyses. Finally, the fibrils of stage II melanosomes are strongly labeled by the HMB-45 antibody (Raposo et al., 2001), which detects an epitope dependent on Golgi/TGN processing as discussed earlier. Thus, the data overwhelmingly support the second model by which Pmel traffics through the ‘normal’ biosynthetic pathway and an endosomal intermediate en route to fibrillar stage II melanosomes.
Perspectives
While much has been learned about Pmel over the last 15 years, much remains to be discovered. Our new insights into the transport of Pmel through endosomes and its fibrillogenic activity raise several important questions about how the environment of the multivesicular early endosome/stage I melanosome may facilitate fibril formation. It is of tantamount importance in understanding the spatial and temporal regulation of the deposition of large intralumenal fibers that underlies melanosome morphogenesis. The physiologic incorporation of Mα into large regular aggregates within the stage II premelanosome is thought to share similarities with the pathologic process of amyloidogenesis in certain amyloid diseases (Berson et al., 2003; Kelly and Balch, 2003). As a result Pmel may represent a novel model protein for the study of post-biosynthetic folding diseases (Dobson, 2003; Huff et al., 2003; Selkoe, 2003).
How is the fibrillogenic Mα-fragment released in stage I melanosomes from the membrane tethered Mβ-fragment to which it is disulfide bonded (Berson et al., 2001)? This release may occur either by reduction of the disulfide bond(s) or proteolytic digestion of Mβ. The latter hypothesis is more likely given the oxidative nature of the melanin intermediates, the presence of lysosomal hydrolases in melanosomes (Orlow, 1995), and evidence for further proteolytic maturation of Pmel17 in stage II premelanosome-enriched subcellular fractions, subsequent to the PC-mediated cleavage (Chiamenti et al., 1996; Kushimoto et al., 2001; Yasumoto et al., 2004), but evidence for either model is lacking. Clarification of this issue may lead to a better understanding of how the lumenal environment of the melanosome changes during maturation from the common endocytic stage I precursor to the melanosome-specific stage II precursor. It may also clarify whether the subsequent cleavage of Mα to the smaller species, detected by immunoblotting of cell lysates with HMB-45 (Chiamenti et al., 1996; Kushimoto et al., 2001; Yasumoto et al., 2004), is necessary for melanosome biogenesis.
Upon release, what is the trigger that induces Mα to form fibrils? Perhaps a conformational change induced by its PC-dependent release from Mβ and the low pH of the stage I melanosome (Raposo et al., 2001) favors the ordered oligomerization of Mα into fibrillar arrays, similar to models for pathologic amyloidogenic fibrillogenesis for gelsolin in familial amyloidoses of Finnish type (Chen et al., 2001), BRI-L in familial British dementia (Kim et al., 1999), and proislet amyloid polypeptide in type II diabetes (Badman et al., 1996). Indeed, proteolytic maturation and acidification in endosomes are common features in fibril formation by amyloidogenic peptides, including the Aβ-peptide associated with Alzheimer's disease (Ehehalt et al., 2003) and prions associated with the spongiform encephalopathies (Mayer et al., 1996; Zou and Cashman, 2002). Moreover, other proteins may regulate Pmel fibrillogenesis and perhaps contribute to fibril formation. One candidate is MART-1, a small melanocyte-specific integral membrane protein that localizes to melanosomes and endosomes (Lévy et al., 2005; de Mazière et al., 2002; Rimoldi et al., 2001), and that appears to regulate Pmel stability and fibrillogenic activity in melanoma cells (Hoashi et al., 2005). How this function is exerted is not clear, nor is it clear why MART-1 may be required for fibril formation in melanocytes but not in transfected HeLa cells (Berson et al., 2001, 2003). Additional regulators of Pmel fibrillogenesis await identification.
Thirdly, whereas it is apparent how acidification and proteolysis within the lumenal environment of the stage I melanosome might facilitate fibrillogenesis, what advantage is provided by passage through intralumenal membranes? Intralumenal membranes are characteristic of normal endosomal subdomains (Gruenberg and Stenmark, 2004), and thus participate in the maturation of both melanosomes (Berson et al., 2001; Jimbow et al., 1979; Raposo et al., 2001; Turner et al., 1975) and late endosomes in the same cell. How Pmel is incorporated into fibrils from these organelles and at the same time is segregated away from endocytic material destined for degradation is currently not understood; this process may be comparable with that which occurs in other cell types in which multivesicular endosomes serve as precursors to specialized lysosome-related organelles, including platelet α and dense granules (Heijnen et al., 1998; Youssefian and Cramer, 2000), major histocompatibility complex class II compartments (Peters et al., 1991a, 1995), Weibel–Palade bodies (Kobayashi et al., 2000) and cytotoxic granules (Peters et al., 1991b). Interestingly, multivesicular endosomes in all cell types examined can fuse with the plasma membrane, releasing their contents; the released intralumenal vesicles are then called exosomes (Février and Raposo, 2004). Exosomes are readily taken up by dendritic cells, the master antigen presenting cells of the immune system. Thus, the association of Pmel with intralumenal membranes of endosomes, and hence with exosomes, may explain its frequent recognition by the immune system in patients with melanoma. Indeed, Pmel has been identified as an exosome component released by melanoma cells (Wolfers et al., 2001). Internal membranes of multivesicular endosomes are also typically enriched in specific lipids, such as glycosphingolipids and cholesterol that also tend to be enriched in detergent-insoluble lipid rafts (Mobius et al., 2002; Wubbolts et al., 2003). Association with lipid raft-like domains is a common feature of amyloidogenic precursors and has been implicated as a factor in fibrillogenesis (Ehehalt et al., 2003; Février et al., 2004; Taraboulos et al., 1995; Zou and Cashman, 2002). Hence, the association of Pmel with intralumenal membranes of endosomes may explain its intracellular sorting route, its recognition by the immune system, and its ability to form fibrillar arrays. A test of these hypotheses awaits experimental verification.
Finally, what is the function of the Pmel fibrillar arrays within forming melanosomes? Is Pmel itself incorporated into melanin polymers, or does it serve as a scaffold for the deposition of melanins upon other substrates? To what advantage does polymerization on a scaffold provide for the melanocyte and/or pigmented epithelial cell? These questions will require careful analyses of Pmel-deficient cells using assays for melanin transfer in the epidermis or phagosome/melanosome resolution in pigment epithelia of the eye.
Certainly, while controversial and perplexing, Pmel has provided us with a great many questions to focus upon in the future, and we look forward to answer these questions in the years ahead.
Acknowledgments
We thank Danielle Tenza, Joanne Berson, Dawn Harper Maholik, and Ilse Hurbain for providing data that have contributed to our evolving view of Pmel. This work was supported by grants from the National Institutes of Health (R01 AR048155, R01 EY015625 and R03 EY014919 from the National Institutes of Arthritis and Musculoskeletal Diseases and the National Eye Institute), the Institut Curie, and the Centre National de la Recherche Scientifique. ACT and STT were supported in part by training grant T32 CA09140 from the National Cancer Institute.
Footnotes
HMB-45 reacts with melanoma and not epidermal melanocytes in paraffin-embedded human tissue (Thomson and MacKie, 1989). However, this is not necessarily true in cultured cells or in eye pigment cells, as HMB-45 reacts variably with primary cultured melanocytes and with most immortalized melanocyte cell lines. HMB-45 binds to an epitope that requires sialic acid (Chiamenti et al., 1996; Kapur et al., 1992), likely in a linkage catalyzed by enzymes that are more active in proliferating cells.
Note the claim that the HMB-50 and NKI-beteb epitopes reside within the Mβ-fragment (Yasumoto et al., 2004) is incorrect, as both antibodies immunoprecipitate secreted Mα from cell supernatants which is not associated with Mβ-fragment (Adema et al., 1994; Berson et al., 2001; Vennegoor et al., 1988; Vogel and Esclamado, 1988). Yasamoto et al. assigned these epitopes by virtue of coprecipitation of the Mβ-fragment by HMB-50 and NKI-beteb, but did not account for the covalent association of Mα and Mβ in detergent lysates of melanoma cells (Berson et al., 2001).
References
- Adema GJ, de Boer AJ, van't Hullenaar R, Denijn M, Ruiter DJ, Vogel AM, Figdor CG. Melanocyte lineage-specific antigens recognized by monoclonal antibodies NKI-beteb, HMB-50, and HMB-45 are encoded by a single cDNA. Am J Pathol. 1993;143:1579–1585. [PMC free article] [PubMed] [Google Scholar]
- Adema GJ, de Boer AJ, Vogel AM, Loenen WAM, Figdor CG. Molecular characterization of the melanocyte lineage-specific antigen gp100. J Biol Chem. 1994;269:20126–20133. [PubMed] [Google Scholar]
- Badman MK, Shennan KI, Jermany JL, Docherty K, Clark A. Processing of pro-islet amyloid polypeptide (proIAPP) by the prohormone convertase PC2. FEBS Lett. 1996;378:227–231. doi: 10.1016/0014-5793(95)01460-8. [DOI] [PubMed] [Google Scholar]
- Bailin T, Lee ST, Spritz RA. Genomic organization and sequence of D12S53E (Pmel 17), the human homologue of the mouse silver (si) locus. J Invest Dermatol. 1996;106:24–27. doi: 10.1111/1523-1747.ep12326976. [DOI] [PubMed] [Google Scholar]
- Bakker ABH, Schreurs MWJ, de Boer AJ, Kawakami Y, Rosenberg SA, Adema GJ, Figdor CG. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard M, Lajoie G. Angiomyolipoma: immunohistochemical and ultrastructural study of 14 cases. Ultrastruct Pathol. 2001;25:21–29. doi: 10.1080/019131201300004654. [DOI] [PubMed] [Google Scholar]
- Basrur V, Yang F, Kushimoto T, et al. Proteomic analysis of early melanosomes: identification of novel melanosomal proteins. J Proteome Res. 2003;2:69–79. doi: 10.1021/pr025562r. [DOI] [PubMed] [Google Scholar]
- Baxter LL, Pavan WJ. Pmel17 expression is Mitf-dependent and reveals cranial melanoblast migration during murine development. Gene Expr Patterns. 2003;3:703–707. doi: 10.1016/j.modgep.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Bennett DC, Lamoreux ML. The color loci of mice – a genetic century. Pigment Cell Res. 2003;16:333–344. doi: 10.1034/j.1600-0749.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- Berson JF, Harper D, Tenza D, Raposo G, Marks MS. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell. 2001;12:3451–3464. doi: 10.1091/mbc.12.11.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–533. doi: 10.1083/jcb.200302072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissy RE. Melanosome transfer to and translocation in the keratinocyte. Exp Dermatol. 2003;12:5–12. doi: 10.1034/j.1600-0625.12.s2.1.x. [DOI] [PubMed] [Google Scholar]
- Brouwenstijn N, Slager EH, Bakker AB, Schreurs MW, Van der Spek CW, Adema GJ, Schrier PI, Figdor CG. Transcription of the gene encoding melanoma-associated antigen gp100 in tissues and cell lines other than those of the melanocytic lineage. Br J Cancer. 1997;76:1562–1566. doi: 10.1038/bjc.1997.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft M, Bateman A, Clarke J, Hamill SJ, Sandford R, Thomas RL, Chothia C. The structure of a PKD domain from polycystin-1: implications for polycystic kidney disease. EMBO J. 1999;18:297–305. doi: 10.1093/emboj/18.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo PA, Frank DW, Bieler BM, Berson JF, Marks MS. A cytoplasmic sequence in human tyrosinase defines a second class of di-leucine-based sorting signals for late endosomal and lysosomal delivery. J Biol Chem. 1999;274:12780–12789. doi: 10.1074/jbc.274.18.12780. [DOI] [PubMed] [Google Scholar]
- Chakraborty AK, Platt JT, Kim KK, Kwon BS, Bennett DC, Pawelek JM. Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur J Biochem. 1996;236:180–188. doi: 10.1111/j.1432-1033.1996.t01-1-00180.x. [DOI] [PubMed] [Google Scholar]
- Chen CD, Huff ME, Matteson J, Page LJ, Phillips R, Kelly JW, Balch WE. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca2+ stabilization. EMBO J. 2001;20:6277–6287. doi: 10.1093/emboj/20.22.6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamenti AM, Vella F, Bonetti F, Pea M, Ferrari S, Martignoni G, Benedetti A, Suzuki H. Anti-melanoma monoclonal antibody HMB-45 on enhanced chemiluminescence-western blotting recognizes a 30–35 kDa melanosome-associated sialated glycoprotein. Melanoma Res. 1996;6:291–298. doi: 10.1097/00008390-199608000-00003. [DOI] [PubMed] [Google Scholar]
- Cormier JN, Abati A, Fetsch P, Hijazi YM, Rosenberg SA, Marincola FM, Topalian SL. Comparative analysis of the in vivo expression of tyrosinase, MART-1/Melan-A, and gp100 in metastatic melanoma lesions: implications for immunotherapy. J Immunother. 1998a;21:27–31. doi: 10.1097/00002371-199801000-00003. [DOI] [PubMed] [Google Scholar]
- Cormier JN, Hijazi YM, Abati A, Fetsch P, Bettinotti M, Steinberg SM, Rosenberg SA, Marincola FM. Heterogeneous expression of melanoma-associated antigens and HLA-A2 in metastatic melanoma in vivo. Int J Cancer. 1998b;75:517–524. doi: 10.1002/(sici)1097-0215(19980209)75:4<517::aid-ijc5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- Donatien PD, Orlow SJ. Interaction of melanosomal proteins with melanin. Eur J Biochem. 1995;232:159–164. doi: 10.1111/j.1432-1033.1995.tb20794.x. [DOI] [PubMed] [Google Scholar]
- Du J, Miller AJ, Widlund HR, Horstmann MA, Ramaswamy S, Fisher DE. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am J Pathol. 2003;163:333–343. doi: 10.1016/S0002-9440(10)63657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn LC, Thigpen LW. The silver mouse: a recessive color variation. J Heredity. 1930;21:495–498. [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer b-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Février B, Raposo G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol. 2004;16:415–421. doi: 10.1016/j.ceb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Février B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G. Cells release prions in association with exosomes. Proc Natl Acad Sci USA. 2004;101:9683–9688. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folpe AL, Goodman ZD, Ishak KG, Paulino AF, Taboada EM, Meehan SA, Weiss SW. Clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres: a novel member of the perivascular epithelioid clear cell family of tumors with a predilection for children and young adults. Am J Surg Pathol. 2000;24:1239–1246. doi: 10.1097/00000478-200009000-00007. [DOI] [PubMed] [Google Scholar]
- Furumura M, Sakai C, Potterf SB, Vieira WD, Barsh GS, Hearing VJ. Characterization of genes modulated during pheomelanogenesis using differential display. Proc Natl Acad Sci USA. 1998;95:7374–7378. doi: 10.1073/pnas.95.13.7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuze HJ, Morre DJ. Trans-Golgi reticulum. J Electron Microsc Tech. 1991;17:24–34. doi: 10.1002/jemt.1060170105. [DOI] [PubMed] [Google Scholar]
- Gown AM, Vogel AM, Hoak D, Gough F, McNutt MA. Monoclonal antibodies specific for melanocytic tumors distinguish subpopulations of melanocytes. Am J Pathol. 1986;123:195–203. [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J. The endocytic pathway: a mosaic of domains. Nat Rev Mol Cell Biol. 2001;2:721–730. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- Halder T, Pawelec G, Kirkin AF, Zeuthen J, Meyer HE, Kun L, Kalbacher H. Isolation of novel HLA-DR restricted potential tumor-associated antigens from the melanoma cell line FM3. Cancer Res. 1997;57:3238–3244. [PubMed] [Google Scholar]
- Hearing VJ. The melanosome: the perfect model for cellular responses to the environment. Pigment Cell Res. 2000;13:23–34. doi: 10.1034/j.1600-0749.13.s8.7.x. [DOI] [PubMed] [Google Scholar]
- Hearing VJ, Phillips P, Lutzner MA. The fine structure of melanogenesis in coat color mutants of the mouse. J Ultrastruct Res. 1973;43:88–106. doi: 10.1016/s0022-5320(73)90072-5. [DOI] [PubMed] [Google Scholar]
- Heijnen HF, Debili N, Vainchencker W, Breton-Gorius J, Geuze HJ, Sixma JJ. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood. 1998;91:2313–2325. [PubMed] [Google Scholar]
- Hoashi T, Watabe H, Muller J, Yamaguchi Y, Vieira WD, Hearing VJ. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J Biol Chem. 2005;280:14006–14016. doi: 10.1074/jbc.M413692200. [DOI] [PubMed] [Google Scholar]
- Höning S, Sandoval IV, von Figura K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J. 1998;17:1304–1314. doi: 10.1093/emboj/17.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff ME, Balch WE, Kelly JW. Pathological and functional amyloid formation orchestrated by the secretory pathway. Curr Opin Struct Biol. 2003;13:674–682. doi: 10.1016/j.sbi.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Jimbow K, Oikawa O, Sugiyama S, Takeuchi T. Comparison of eumelanogenesis in retinal and follicular melanocytes; role of vesiculo-globular bodies in melanosome differentiation. J Invest Dermatol. 1979;73:278–284. doi: 10.1111/1523-1747.ep12531650. [DOI] [PubMed] [Google Scholar]
- Jimenez-Cervantes C, Martinez-Esparza M, Perez C, Daum N, Solano F, Garcia-Borron JC. Inhibition of melanogenesis in response to oxidative stress: transient downregulation of melanocyte differentiation markers and possible involvement of microphthalmia transcription factor. J Cell Sci. 2001;114:2335–2344. doi: 10.1242/jcs.114.12.2335. [DOI] [PubMed] [Google Scholar]
- Kapur RP, Bigler SA, Skelly M, Gown AM. Anti-melanoma monoclonal antibody HMB45 identifies an oncofetal glycoconjugate associated with immature melanosomes. J Histochem Cytochem. 1992;40:207–212. doi: 10.1177/40.2.1552165. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, Robbins PF, Sette A, Appella E, Rosenberg SA. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3961–3968. [PubMed] [Google Scholar]
- Keeling L, Andersson L, Schutz KE, Kerje S, Fredriksson R, Carlborg O, Cornwallis CK, Pizzari T, Jensen P. Feather-pecking and victim pigmentation. Nature. 2004;431:645–646. doi: 10.1038/431645a. [DOI] [PubMed] [Google Scholar]
- Kelly JW, Balch WE. Amyloid as a natural product. J Cell Biol. 2003;161:461–462. doi: 10.1083/jcb.200304074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerje S, Sharma P, Gunnarsson U, et al. The Dominant white, Dun and Smoky color variants in chicken are associated with insertion deletion polymorphisms in the PMEL17 gene. Genetics. 2004;168:1507–1518. doi: 10.1534/genetics.104.027995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RY, Wistow GJ. The cDNA RPE1 and monoclonal antibody HMB-50 define gene products preferentially expressed in retinal pigment epithelium. Exp Eye Res. 1992;55:657–662. doi: 10.1016/0014-4835(92)90170-w. [DOI] [PubMed] [Google Scholar]
- Kim KK, Youn BS, Heng HH, Shi XM, Tsui LC, Lee ZH, Pickard RT, Kwon BS. Genomic organization and FISH mapping of human Pmel 17, the putative silver locus. Pigment Cell Res. 1996;9:42–48. doi: 10.1111/j.1600-0749.1996.tb00085.x. [DOI] [PubMed] [Google Scholar]
- Kim SH, Wang R, Gordon DJ, Bass J, Steiner DF, Lynn DG, Thinakaran G, Meredith SC, Sisodia SS. Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nat Neurosci. 1999;2:984–988. doi: 10.1038/14783. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Urabe K, Orlow SJ, Higashi K, Imokawa G, Kwon BS, Potterf B, Hearing VJ. The Pmel 17/silver locus protein. Characterization and investigation of its melanogenic function. J Biol Chem. 1994;269:29198–29205. [PubMed] [Google Scholar]
- Kobayashi T, Vieira WD, Potterf B, Sakai C, Imokawa G, Hearing VJ. Modulation of melanogenic protein expression during the switch from eu- to pheomelanogenesis. J Cell Sci. 1995;108:2301–2309. doi: 10.1242/jcs.108.6.2301. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, Parton RG, Kruithof EK, Gruenberg J. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell. 2000;11:1829–1843. doi: 10.1091/mbc.11.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Kubota R, Wang Y, Minoshima S, Kudoh J, Mashima Y, Oguchi Y, Shimizu N. Mapping of the human gene for a melanocyte protein Pmel 17 (D12S53E) to chromosome 12q13-q14. Genomics. 1995;26:430–431. doi: 10.1016/0888-7543(95)80239-i. [DOI] [PubMed] [Google Scholar]
- Kushimoto T, Basrur V, Valencia J, Matsunaga J, Vieira WD, Ferrans VJ, Muller J, Appella E, Hearing VJ. A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc Natl Acad Sci USA. 2001;98:10698–10703. doi: 10.1073/pnas.191184798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS. Pigmentation genes: the tyrosinase gene family and the pmel 17 gene family. J Invest Dermatol. 1993;100:134S–140S. doi: 10.1111/1523-1747.ep12465022. [DOI] [PubMed] [Google Scholar]
- Kwon BS, Haq AK, Pomerantz SH, Halaban R. Isolation and sequence of a cDNA clone for human tyrosinase that maps at the mouse c-albino locus. Proc Natl Acad Sci USA. 1987a;84:7473–7477. doi: 10.1073/pnas.84.21.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS, Halaban R, Kim GS, Usack L, Pomerantz S, Haq AK. A melanocyte-specific complementary DNA clone whose expression is inducible by melanotropin and isobutylmethyl xanthine. Mol Biol Med. 1987b;4:339–355. [PubMed] [Google Scholar]
- Kwon BS, Chintamaneni C, Kozak CA, Copeland NG, Gilbert DJ, Jenkins N, Barton D, Francke U, Kobayashi Y, Kim KK. A melanocyte-specific gene, Pmel 17, maps near the silver coat color locus on mouse chromosome 10 and is in a syntenic region on human chromosome 12. Proc Natl Acad Sci USA. 1991;88:9228–9232. doi: 10.1073/pnas.88.20.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS, Halaban R, Ponnazhagan S, Kim K, Chintamaneni C, Bennett D, Pickard RT. Mouse silver mutation is caused by a single base insertion in the putative cytoplasmic domain of Pmel 17. Nucleic Acids Res. 1995;23:154–158. doi: 10.1093/nar/23.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantuejoul S, Ferretti G, Negoescu A, Parent B, Brambilla E. Multifocal alveolar hyperplasia associated with lymphangioleiomyomatosis in tuberous sclerosis. Histopathology. 1997;30:570–575. doi: 10.1046/j.1365-2559.1997.4600811.x. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, Riker AI, Quevedo ME, Stennett LS, Wang E, Marincola FM, Kast WM, Robinson JK, Nickoloff BJ. Interferon-gamma reduces melanosomal antigen expression and recognition of melanoma cells by cytotoxic T cells. Am J Pathol. 2002;160:521–528. doi: 10.1016/s0002-9440(10)64871-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ZH, Hou L, Moellmann G, Kuklinska E, Antol K, Fraser M, Halaban R, Kwon BS. Characterization and subcellular localization of human Pmel 17/silver, a 100-kDa (pre)melanosomal membrane protein associated with 5,6, -dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J Invest Dermatol. 1996;106:605–610. doi: 10.1111/1523-1747.ep12345163. [DOI] [PubMed] [Google Scholar]
- Lévy F, Muehlethaler K, Salvi S, Peitrequin AL, Lindholm CK, Cerottini JC, Rimoldi D. Ubiquitylation of a melanosomal protein by HECT-E3 ligases serves as sorting signal for lysosomal degradation. Mol Biol Cell. 2005;16:1777–1787. doi: 10.1091/mbc.E04-09-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyten GP, van der Spek CW, Brand I, Sintnicolaas K, de Waard-Siebinga I, Jager MJ, de Jong PT, Schrier PI, Luider TM. Expression of MAGE, gp100 and tyrosinase genes in uveal melanoma cell lines. Melanoma Res. 1998;8:11–16. doi: 10.1097/00008390-199802000-00003. [DOI] [PubMed] [Google Scholar]
- Magnusson S, Sottrup-Jensen L, Petersen TE, Claeys H. The primary structure of prothrombin, the role of vitamin K in blood coagulation and a thrombin catalyzed “negative feedback” control mechanism for limiting the activation of prothrombin. In: Hemker HC, Veltkamp J, editors. Prothrombin and Related Coagulation Factors. Leiden, Holland: Universitaire Pers; 1975. pp. 25–46. [Google Scholar]
- Maresh GA, Marken JS, Neubauer M, Aruffo A, Hellström I, Hellström KE, Marquardt H. Cloning and expression of the gene for the melanoma-associated ME20 antigen. DNA Cell Biol. 1994a;13:87–95. doi: 10.1089/dna.1994.13.87. [DOI] [PubMed] [Google Scholar]
- Maresh GA, Wang WC, Beam KS, Malacko AR, Hellström I, Hellström KE, Marquardt H. Differential processing and secretion of the melanoma-associated ME20 antigen. Arch Biochem Biophys. 1994b;311:95–102. doi: 10.1006/abbi.1994.1213. [DOI] [PubMed] [Google Scholar]
- Marks MS, Seabra MC. The melanosome: membrane dynamics in black and white. Nat Rev Mol Cell Biol. 2001;2:738–748. doi: 10.1038/35096009. [DOI] [PubMed] [Google Scholar]
- Martínez-Esparza M, Jiménez-Cervantes C, Bennett DC, Lozano JA, Solano F, García-Borrón JC. The mouse silver locus encodes a single transcript truncated by the silver mutation. Mamm Genome. 1999;10:1168–1171. doi: 10.1007/s003359901184. [DOI] [PubMed] [Google Scholar]
- Martinez-Esparza M, Jimenez-Cervantes C, Solano F, Lozano JA, Garcia-Borron JC. Regulation of the murine silver locus product (gp87) by the hypopigmenting cytokines TGFb1 and TNF-a. Pigment Cell Res. 2000;13:120–126. doi: 10.1034/j.1600-0749.2000.130211.x. [DOI] [PubMed] [Google Scholar]
- Maul GG. Golgi-melanosome relationship in human melanoma in vitro. J Ultrastruct Res. 1969;26:163–176. doi: 10.1016/s0022-5320(69)90042-2. [DOI] [PubMed] [Google Scholar]
- Mayer RJ, Tipler C, Arnold J, Laszlo L, Al-Khedhairy A, Lowe J, Landon M. Endosome-lysosomes, ubiquitin and neurodegeneration. Adv Exp Med Biol. 1996;389:261–269. doi: 10.1007/978-1-4613-0335-0_33. [DOI] [PubMed] [Google Scholar]
- de Mazière AM, Muehlethaler K, van Donselaar E, Salvi S, Davoust J, Cerottini JC, Lèvy F, Slot JW, Rimoldi D. The melanocytic protein melan-A/MART-1 has a subcellular localization distinct from typical melanosomal proteins. Traffic. 2002;3:678–693. doi: 10.1034/j.1600-0854.2002.30909.x. [DOI] [PubMed] [Google Scholar]
- Mobius W, Ohno-Iwashita Y, van Donselaar EG, Oorschot VM, Shimada Y, Fujimoto T, Heijnen HF, Geuze HJ, Slot JW. Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J Histochem Cytochem. 2002;50:43–55. doi: 10.1177/002215540205000105. [DOI] [PubMed] [Google Scholar]
- Mochii M, Takeuchi T, Kodama R, Agata K, Eguchi G. The expression of melanosomal matrix protein in the transdifferentiation of pigmented epithelial cells into lens cells. Cell Differ. 1988;23:133–142. doi: 10.1016/0045-6039(88)90045-0. [DOI] [PubMed] [Google Scholar]
- Mochii M, Agata K, Eguchi G. Complete sequence and expression of a cDNA encoding a chicken 115-kDa melanosomal matrix protein. Pigment Cell Res. 1991;4:41–47. doi: 10.1111/j.1600-0749.1991.tb00312.x. [DOI] [PubMed] [Google Scholar]
- Moyer FH. Genetic variations in the fine structure and ontogeny of mouse melanin granules. Am Zool. 1966;6:43–66. doi: 10.1093/icb/6.1.43. [DOI] [PubMed] [Google Scholar]
- Nichols SE, Harper DC, Berson JF, Marks MS. A novel splice variant of Pmel17 expressed by human melanocytes and melanoma cells lacking some of the internal repeats. J Invest Dermatol. 2003;121:821–830. doi: 10.1046/j.1523-1747.2003.12474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlow SJ. Melanosomes are specialized members of the lysosomal lineage of organelles. J Invest Dermatol. 1995;105:3–7. doi: 10.1111/1523-1747.ep12312291. [DOI] [PubMed] [Google Scholar]
- Orlow SJ, Zhou BK, Boissy RE, Pifko-Hirst S. Identification of a mammalian melanosomal matrix glycoprotein. J Invest Dermatol. 1993;101:141–144. doi: 10.1111/1523-1747.ep12363626. [DOI] [PubMed] [Google Scholar]
- Owen TA, Smock SL, Prakash S, et al. Identification and characterization of the genes encoding human and mouse osteoactivin. Crit Rev Eukaryot Gene Expr. 2003;13:205–220. doi: 10.1615/critreveukaryotgeneexpr.v13.i24.130. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HJ. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature. 1991a;349:669–676. doi: 10.1038/349669a0. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Borst J, Oorschot V, Fukuda M, Krähenbühl O, Tschopp J, Slot JW, Geuze HJ. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med. 1991b;173:1099–1109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters PJ, Raposo G, Neefjes JJ, Oorschot V, Leijendekker RL, Geuze HJ, Ploegh HL. Major histocompatibility complex class II compartments in human B lymphoblastoid cells are distinct from early endosomes. J Exp Med. 1995;182:325–334. doi: 10.1084/jem.182.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters EMJ, Tobin DJ, Seidah NG, Schallreuter KU. Pro-opiomelanocortin-related peptides, prohormone convertases 1 and 2 and the regulatory peptide 7B2 are present in melanosomes of human melanocytes. J Invest Dermatol. 2000;114:430–437. doi: 10.1046/j.1523-1747.2000.00913.x. [DOI] [PubMed] [Google Scholar]
- Quevedo WC, Fleischmann RD, Dyckman J. Premature loss of melanocytes from hair follicles of light (Blt) and silver (si) mice. In: Seiji M, editor. Phenotypic Expression in Pigment Cells. Tokyo: Tokyo University Press; 1981. pp. 177–184. [Google Scholar]
- Raiborg C, Rusten TE, Stenmark H. Protein sorting into multivesicular endosomes. Curr Opin Cell Biol. 2003;15:446–455. doi: 10.1016/s0955-0674(03)00080-2. [DOI] [PubMed] [Google Scholar]
- Raposo G, Marks MS. The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic. 2002;3:237–248. doi: 10.1034/j.1600-0854.2002.030401.x. [DOI] [PubMed] [Google Scholar]
- Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. 2001;152:809–823. doi: 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder S, Stricker C, Joerg H, Dummer R, Stranzinger G. A comparative genetic approach for the investigation of ageing grey horse melanoma. J Anim Breed Genet. 2000;117:73–82. [Google Scholar]
- Rimoldi D, Muehlethaler K, Salvi S, Valmori D, Romero P, Cerottini JC, Lévy F. Subcellular localization of the melanoma-associated protein melan-A/MART-1 influences the processing of its HLA-A2-restricted epitope. J Biol Chem. 2001;276:43189–43196. doi: 10.1074/jbc.M103221200. [DOI] [PubMed] [Google Scholar]
- Safadi FF, Xu J, Smock SL, Rico MC, Owen TA, Popoff SN. Cloning and characterization of osteoactivin, a novel cDNA expressed in osteoblasts. J Cell Biochem. 2001;84:12–26. doi: 10.1002/jcb.1259. [DOI] [PubMed] [Google Scholar]
- Schraermeyer U, Peters S, Thumann G, Kociok N, Heimann K. Melanin granules of retinal pigment epithelium are connected with the lysosomal degradation pathway. Exp Eye Res. 1999;68:237–245. doi: 10.1006/exer.1998.0596. [DOI] [PubMed] [Google Scholar]
- Seiji M, Fitzpatrick TB, Birbeck MSC. The melanosome: a distinctive subcellular particle of mammalian melanocytes and the site of melanogenesis. J Invest Dermatol. 1961;36:243–252. doi: 10.1038/jid.1961.42. [DOI] [PubMed] [Google Scholar]
- Seiji M, Fitzpatrick TM, Simpson RT, Birbeck MSC. Chemical composition and terminology of specialized organelles (melanosomes and melanin granules) in mammalian melanocytes. Nature. 1963;197:1082–1084. doi: 10.1038/1971082a0. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Shikano S, Bonkobara M, Zukas PK, Ariizumi K. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J Biol Chem. 2001;276:8125–8134. doi: 10.1074/jbc.M008539200. [DOI] [PubMed] [Google Scholar]
- Silvers WK. A Model for Mammalian Gene Action and Interaction. New York, NY: Springer-Verlag; 1979. The Coat Colors of Mice; p. 379. [Google Scholar]
- Simmen T, Schmidt A, Hunziker W, Beermann F. The tyrosinase tail mediates sorting to the lysosomal compartment in MDCK cells via a di-leucine and a tyrosine-based signal. J Cell Sci. 1999;112:45–53. doi: 10.1242/jcs.112.1.45. [DOI] [PubMed] [Google Scholar]
- Solano F, Martinez-Esparza M, Jimenez-Cervantes C, Hill SP, Lozano JA, Garcia-Borron JC. New insights on the structure of the mouse silver locus and on the function of the silver protein. Pigment Cell Res. 2000;13:118–124. doi: 10.1111/j.0893-5785.2000.130821.x. [DOI] [PubMed] [Google Scholar]
- Sottrup-Jensen L, Zajdel M, Claeys H, Petersen TE, Magnusson S. Amino-acid sequence of activation cleavage site in plasminogen: homology with “pro” part of prothrombin. Proc Natl Acad Sci USA. 1975;72:2577–2581. doi: 10.1073/pnas.72.7.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanakis E, Lamina P, Bennett DC. Effects of the developmental colour mutations silver and recessive spotting on proliferation of diploid and immortal mouse melanocytes in culture. Development. 1992;114:675–680. doi: 10.1242/dev.114.3.675. [DOI] [PubMed] [Google Scholar]
- Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB, Avrahami D. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol. 1995;129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarentino AL, Plummer TH, Jr, Maley F. The release of intact oligosaccharides from specific glycoproteins by endo-b-N-acetylglucosaminidase H. J Biol Chem. 1974;249:818–824. [PubMed] [Google Scholar]
- Thomson W, MacKie RM. Comparison of five anti-melanoma antibodies for identification of melanocytic cells on tissue sections in routine dermatopathology. J Am Acad Dermatol. 1989;21:1280–1284. doi: 10.1016/s0190-9622(89)70344-3. [DOI] [PubMed] [Google Scholar]
- Tooze J, Hollinshead M. Tubular early endosomal networks in AtT20 and other cells. J Cell Biol. 1991;115:635–653. doi: 10.1083/jcb.115.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner WA, Jr, Taylor JD, Tchen TT. Melanosome formation in the goldfish: the role of multivesicular bodies. J Ultrastruct Res. 1975;51:16–31. doi: 10.1016/s0022-5320(75)80004-9. [DOI] [PubMed] [Google Scholar]
- Turque N, Denhez F, Martin P, Planque N, Bailly M, Begue A, Stehelin D, Saule S. Characterization of a new melanocyte-specific gene (QNR-71) expressed in v-myc-transformed quail neuroretina. EMBO J. 1996;15:3338–3350. [PMC free article] [PubMed] [Google Scholar]
- Van den Steen P, Rudd PM, Dwek RA, Opdenakker G. Concepts and principles of O-linked glycosylation. Crit Rev Biochem Mol Biol. 1998;33:151–208. doi: 10.1080/10409239891204198. [DOI] [PubMed] [Google Scholar]
- Vennegoor C, Hageman P, van Nouhuijs H, Ruiter DJ, Calafat J, Ringens PJ, Rümke P. A monoclonal antibody specific for cells of the melanocytic lineage. Am J Pathol. 1988;130:179–192. [PMC free article] [PubMed] [Google Scholar]
- Vijayasaradhi S, Xu YQ, Bouchard B, Houghton AN. Intracellular sorting and targeting of melanosomal membrane proteins: identification of signals for sorting of the human brown locus protein, gp75. J Cell Biol. 1995;130:807–820. doi: 10.1083/jcb.130.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel AM, Esclamado RM. Identification of a secreted Mr 95 000 glycoprotein in human melanocytes and melanomas by a melanocyte specific monoclonal antibody. Cancer Res. 1988;48:1286–1294. [PubMed] [Google Scholar]
- de Vries TJ, Fourkour A, Wobbes T, Verkroost G, Ruiter DJ, van Muijen GN. Heterogeneous expression of immunotherapy candidate proteins gp100, MART-1, and tyrosinase in human melanoma cell lines and in human melanocytic lesions. Cancer Res. 1997;57:3223–3229. [PubMed] [Google Scholar]
- de Vries TJ, Trancikova D, Ruiter DJ, van Muijen GN. High expression of immunotherapy candidate proteins gp100, MART-1, tyrosinase and TRP-1 in uveal melanoma. Br J Cancer. 1998;78:1156–1161. doi: 10.1038/bjc.1998.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterman MA, Ajubi N, van Dinter IM, Degen WG, van Muijen GN, Ruitter DJ, Bloemers HP. Nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int J Cancer. 1995;60:73–81. doi: 10.1002/ijc.2910600111. [DOI] [PubMed] [Google Scholar]
- Wolfers J, Lozier A, Raposo G, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7:297–303. doi: 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- Wubbolts RW, Leckie RS, Veenhuizen PT, Schwartzmann G, Möbius W, Hoernschemeyer J, Slot JW, Geuze HJ, Stoorvogel W. Proteomic and biochemical analyses of human B cell-derived exosomes: potential implications for their function and multivesicular body formation. J Biol Chem. 2003;278:10963–10972. doi: 10.1074/jbc.M207550200. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Tanaka S, Fujii H, Matsumoto T, Okuda C, Watanabe G, Suda K. Monotypic epithelioid angiomyolipoma of the liver. Histopathology. 2000;36:451–456. doi: 10.1046/j.1365-2559.2000.00848.x. [DOI] [PubMed] [Google Scholar]
- Yasumoto K, Watabe H, Valencia JC, Kushimoto T, Kobayashi T, Appella E, Hearing VJ. Epitope mapping of the melanosomal matrix protein gp100 (PMEL17): rapid processing in the endoplasmic reticulum and glycosylation in the early Golgi compartment. J Biol Chem. 2004;279:28330–28338. doi: 10.1074/jbc.M401269200. [DOI] [PubMed] [Google Scholar]
- Youssefian T, Cramer EM. Megakaryocyte dense granule components are sorted in multivesicular bodies. Blood. 2000;95:4004–4007. [PubMed] [Google Scholar]
- Yu Z, Restifo NP. Cancer vaccines: progress reveals new complexities. J Clin Invest. 2002;110:289–294. doi: 10.1172/JCI16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BK, Kobayashi T, Donatien PD, Bennett DC, Hearing VJ, Orlow SJ. Identification of a melanosomal matrix protein encoded by the murine si (silver) locus using “organelle scanning”. Proc Natl Acad Sci USA. 1994;91:7076–7080. doi: 10.1073/pnas.91.15.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou WQ, Cashman NR. Acidic pH and detergents enhance in vitro conversion of human brain PrPC to a PrPSc-like form. J Biol Chem. 2002;277:43942–43947. doi: 10.1074/jbc.M203611200. [DOI] [PubMed] [Google Scholar]