

Abstract
DNA sustains a wide variety of damage, such as the formation of abasic sites, pyrimidine dimers, alkylation adducts, or oxidative lesions, upon exposure to UV radiation, alkylating agents, or oxidative conditions. Since such damage may be acutely toxic or mutagenic and potentially carcinogenic, it is of interest to gain insight into how their structures impact biochemical processing of DNA, such as synthesis, transcription, and repair. Lesion-specific molecular probes have been used to study polymerase-mediated translesion DNA synthesis of abasic sites and TT dimers, while other probes have been developed to specifically investigate the alkylation adduct O6-Bn-G and the oxidative lesion 8-oxo-G. In this review recent examples of lesion-specific molecular probes are surveyed; their specificities of incorporation opposite target lesions compared to unmodified nucleotides are discussed, and limitations of their applications under physiologically relevant conditions are assessed.
Within years of the discovery that DNA1 was responsible for transmitting hereditary information (1), and before publication of the double helical structure of DNA (2), researchers began to probe its susceptibility to various types of damage (3). Exposure of DNA to UV radiation, alkylating agents, and reactive oxidative species leads to the formation of abasic sites, pyrimidine dimers, alkylation adducts, and oxidative damage products. Each of these types of lesions can be mutagenic, sometimes carcinogenic, and this chemical process, therefore, is highly relevant to cellular biology and disease. For example, UV radiation damage-induced skin cancer was estimated to result in over one million new cases of the disease in the United States in 2008 (4). Further, DNA alkylation by tobacco-derived carcinogens contributes to lung cancer, which is estimated will result in about 160,000 deaths in the United States in 2009 (5, 6). These examples of the biological consequences of DNA damage underscore the continued need for understanding the underlying molecular mechanisms.
Cellular factors such as base-excision repair (7, 8) mitigate some damage to DNA; however, certain lesions evade repair and, through a variety of biochemical pathways, these DNA adducts may wreak havoc on normal cellular replication and its regulation. For example, misincorporation of nucleotides across from a lesion during polymerase-mediated DNA synthesis, in which the damaged DNA strand may act as a template, could lead to frame-shift or substitution mutations (9). The ability of various polymerases to perform translesion synthesis has been studied in great detail over the last decade, yielding important mechanistic information regarding origins of mutagenicity (10, 11). Another mode of disruptive lesion formation involves DNA alkylation followed by hydrolytic base excision, i.e. depurination or depyrimidination. This reactivity mode has been demonstrated for various genotoxins such as the natural product leinamycin (12, 13), the natural product derivatives acylfulvene (14, 15) and azinomycin epoxide (16), the tobacco-derived nitrosamine NNK (17), and endogenous estrogen metabolites (18). In many cases it is unclear whether cytotoxicity arises from initially formed DNA adducts or subsequently generated abasic sites. Finally, DNA lesions such as inter- and intra-strand crosslinks formed by chemotherapeutic alkylating agents (i.e. nitrogen mustards and cisplatin) are biochemically disruptive by stalling DNA replication and triggering apoptosis (19).
To elucidate biochemical mechanisms underlying the important role DNA lesions play in mutagenesis and carcinogenesis, their biological prevalence and chemical structure need to be determined. It is therefore of interest to: 1) identify and quantify DNA lesions in model experimental systems and in vivo, 2) determine influences of lesions on physical properties of DNA, such as helical structure and thermal stability, and 3) characterize the impact of the lesion on DNA function, i.e., enzyme-mediated processes such as replication. With regards to identification and quantification, several approaches for detecting DNA lesions exist and have been reviewed; examples include 32P-post-labeling, electrochemical or fluorescence detection, immunoassay, mass spectrometry, accelerator mass spectrometry, and quantitative PCR (20–23). Described herein are emerging chemical and biochemical approaches for probing how specific adduct types affect molecular structure and function. Lesion-specific nucleoside probes have been reported for abasic sites, TT photodimers, an alkylation adduct (O6-Bn-G), and an oxidative lesion (8-oxo-G). These have been used to probe abasic sites in DNA, as well as those formed in RNA by ribosome-inactivating proteins, to elucidate mechanistic aspects of polymerase-mediated translesion synthesis, including the impact of stereoelectronic properties of the incoming nucleotide on polymerase activity and exo/endonuclease activity opposite TT photodimers. Finally, lesion-specific probes under development for the study of the DNA adducts 8-oxo-G and O6-Bn-G have been reported, and will be discussed here.
ABASIC SITES
DNA abasic site formation resulting from nucleotide depurination or depyrimidination (i.e. AP site formation) is estimated to occur at a background physiological rate of about 10,000 times per human genome per day (24). The depurination rate at physiological pH, ionic strength, and temperature has been estimated to be 3 × 10−11 s−1, or one purine per E. coli genome per generation (25, 26), and depyrimidination is about twenty times slower than depurination (27). Abasic sites impact the local and global structure of DNA, and, depending on the context, single base loss destabilizes the duplex on the order of 3–11 kcal/mol (28). Base loss can occur spontaneously or be promoted by DNA alkylation or oxidation. Furthermore, abasic sites are intermediates during excision-repair of damaged nucleotides(29), and healthy cells typically have a robust capacity for repair (30). Deficiencies in this process can alter normal biochemical processing of DNA, leading to mutation or acute toxicities (27). Because of the extensive role of AP sites in normal and disease-related processes, it is important to be able to detect their presence and better understand how they influence biochemical processing of DNA.
Enzymes involved in the repair of abasic sites exist in prokaryotes and eukaryotes; these utilize polymerases that recognize abasic sites as intermediates in base excision repair (BER) (8, 31–33). Briefly, AP endonucleases cleave at the 5′-position of the damaged DNA strand, forming a 5′-phosphodeoxyribose (dRP) and liberating a 3′-hydroxyl group. The deoxyribose phosphate group is excised by 2-deoxyribose-5-phosphate lyase, resulting in a single-nucleotide gap. Then, a DNA polymerase, such as pol β, inserts the missing nucleotide, which is finally ligated into the DNA strand by a DNA ligase (32).
Abasic sites are manifested in several chemical structures, beyond the hemiacetal moiety directly resulting from nucleobase hydrolysis (AP, Figure 1). For example, one-electron nucleotide oxidation results in the formation of 2′-deoxyribonolactone (L, Figure 1) (34, 35). Despite the structural similarity between L and AP, it was found that T is primarily incorporated opposite the L lesion by E. coli (36, 37), while A is typically inserted opposite AP sites (a phenomenon often referred to as the “A-rule”) (36–41). Another abasic site variant, C4-AP (Figure 1), is formed after treatment of DNA with antitumor antibiotics such as bleomycin; similar to AP, it was found to block primer extension by Klenow fragment polymerases (42). Due to lack of stability and difficulty in synthesis for each of these types of abasic sites, the lesions are often modeled by the stabilized tetrahydrofuran analog (THF, Figure 1) to investigate their biochemical impacts. Such studies have been carried out using lesion-specific molecular probes, including chemically reactive probes, non-natural nucleotide probes, and other non-nucleotide molecular probes.
FIGURE 1.

Structures of different forms of abasic DNA lesions.
Chemically reactive abasic site probes
The classic approach for detecting abasic sites involves aldehyde-reactive probes (ARPs). AP sites exist as an equilibrium mixture between the closed-ring hemiacetal and the corresponding open-chain aldehyde. ARPs consisting of a nucleophilic O-substituted hydroxylamine linked to a reporter substrate react with the aldehyde to covalently bind the abasic site (43, 44). Methoxyamine was originally utilized as an AP site probe in BER studies. It was discovered that when methoxyamine binds the aldehyde, it blocks AP endonuclease and inhibits DNA cleavage 5′ of the AP site (43). Studies with colon cancer xenografts showed that when methoxyamine was administered together with a DNA methylating drug such as temozolomide, the alkylation-induced AP sites reacted with methoxyamine, blocking BER and thus potentiating the therapeutic potential of the drug (45).
Various ARPs have been designed for quantitative analysis of abasic sites. For example, following incubation with DNA, the biotin-tagged reagent 1 (Figure 2) was treated with an avidin-biotinylated horseradish peroxidase complex, and any unbound DNA was removed by washing. In this ELISA-like assay, conjugated peroxidase activity colorimetrically quantified correlates with bound open-chain abasic sites in DNA (46). ARP 1, now commercially available, was recently used to measure abasic sites formed from depurination of dibenzo[a,l]pyrene adducts in calf-thymus DNA (47). In other studies, fluorescence intensities of the solutions of DNA incubated with luminescent ARPs, such as those containing dansyl or lissamine-rhodamine fluorophores (2–3, Figure 2) were spectroscopically measured to determine the level of ARP adduct formation (48, 49). The accuracy of the fluorescent ARP assay may be improved by utilizing capillary electrophoresis to separate any unreacted probe from the DNA sample, increasing the signal-to-noise ratio of the fluorescence detection (50).
FIGURE 2.

Chemically-reactive probes for abasic lesions.
Chemically-reactive probes have been designed for detecting oxidized abasic site products as well. The biotin-tagged cysteine probe 4 (Figure 2) selectively reacts with the butenolide formed from treating 2′-deoxyribonolactone L with N,N′-dimethylethylenediamine, creating an adduct that subsequently could be quantified by ELISA-like assays similar to those discussed above (51). Similarly, probe 5 (Figure 2) was designed to react with C4-AP and DOB abasic sites and be detected by an ELISA-like assay (52). These chemically reactive probes are useful for quantifying abasic sites in bulk DNA.
Abasic site nucleotide probes
In seminal studies involving synthetic DNA, Matray and Kool introduced the pyrene nucleotide (dPTP, Figure 3) as a base surrogate that pairs opposite AP sites and the model THF (53). Across from an abasic deoxyribose in a template DNA strand, the pyrene nucleotide was preferentially incorporated over the four natural nucleotides by Klenow fragment (Kf) of Escherichia coli DNA polymerase I (exo mutant). This phenomenon underscored the role of shape complementarity in molecular recognition in duplex DNA, and also demonstrated that hydrogen bonding is not essential for stable pairing. Additionally, the pyrene probe could be used like a dideoxynucleotide terminator for sequencing abasic DNA because elongation stalled after dPTP incorporation. Synthetic 43-mer oligonucleotides containing one or multiple THFs were sequenced using a combination of canonical dideoxynucleotides and dPTP. Incorporation of dPTP was observed for DNA samples diluted to contain as low as 1% damaged strands among undamaged oligonucleotides. Further studies with the pyrene nucleotide have probed the abasic-like site generated when adenine-specific DNA methyltransferase M•Taq1 binds to DNA (54), affirmed the significance of nucleobase shape complementarity in closed polymerase-DNA complexes (55), demonstrated the importance of pi-stacking during template-independent nucleotide additions onto blunt-end DNA (56) and in DNA duplexes in aqueous solution (57), and revealed details regarding the base-flipping mechanisms of uracil DNA glycosylase (58) and UV-induced TT-dimer translesion DNA synthesis (vide supra).
FIGURE 3.

Nucleoside probes for abasic lesions and TT dimers. Ph: phenyl, Nap: naphthyl, Cy: cyclohexyl, Ey: ethylene, Me: methyl, Et: ethyl, CE: cyclohexenyl.
Emissive nucleosides have recently emerged as an attractive strategy for spectroscopic detection and evaluation of abasic sites in DNA and RNA. For example, Greco and Tor reported a fluorescent thymidine analog (6a, Figure 3), which contained an appended conjugated furan ring at the 5-position of the nucleotide. Synthetic 12-mers in which analog 6a was paired with either A or THF opposite the probe exhibited identical thermal stabilities, but the duplex containing 6a opposite the THF exhibited a seven-fold increase in emission intensity compared to that with 6a opposite A (59). As a further example, thiophene-based analogue (7, Figure 3) was used to spectroscopically detect RNA abasic sites produced by ribosome-inactivating proteins (RIPs). Toxic RIPs catalyze N-deglocosylation of the conserved α-sarcin/ricin stem-loop domain region of ribosomal RNA (rRNA), and treating this RNA sequence with the toxin saporin generates RNA abasic sites. When synthetic oligonucleotide hybridization probes containing 7 were paired opposite an abasic site, up to 6.5-fold enhancements in emission intensities were observed compared to 7:A. These results demonstrate the effectiveness of these fluorescent nucleotides in signaling the occurrence of abasic sites in RNA, and probes such as 7 are expected to be useful in detecting ribosome-inactivating proteins that act on other specific RNA substrates, or for the discovery of RIP inhibitors or antidotes (60).
Insight regarding detailed molecular mechanisms of polymerase-mediated synthesis template by damaged DNA has been gained from various recent studies (10), including experiments involving nucleotide probes. Before discussing some significant examples in this area, a very brief overview of polymerase-mediated DNA synthesis is warranted (10, 61, 62). As shown in Scheme 1, the polymerase binds DNA (step 1), followed by reversible binding of an incoming dNTP (step 2). Following a proposed conformational change from the open to the catalytically active closed form of the polymerase (step 3), phosphodiester bond formation between the incoming dNTP and 3′ end of the oligonucleotide primer occurs (step 4). The polymerase relaxes to the open form (step 5) and releases the inorganic pyrophosphate byproduct of phosphodiester bond formation (step 6). Finally, the polymerase-oligonucleotide complex may dissociate (step 7) or continue directly to another round of elongation (step 8) in a processive fashion. Some polymerase-lesion combinations allow non-natural nucleotide incorporation via this DNA synthesis pathway, thus facilitating a means of testing molecular aspects polymerase-mediated translesion DNA synthesis.
SCHEME 1.

Polymerase-mediated DNA synthesis.
Exonuclease-deficient T4 DNA polymerase (gp43 exo−)-mediated synthesis past abasic sites has been examined in studies carried out by Berdis and coworkers using a series of hydrophobic base analogs as polymerase substrates (63). Nucleoside triphosphates with varying pi-surface areas, shapes, dipole moments, and energies of solvation, were used as substrates in T4-mediated DNA synthesis templated by THF-containing DNA. The non-natural substituted indole nucleotides included 5-NITP (64), 5-PhITP (65), 5-NapITP (66), 5-CyITP, 5-EyITP, 5-MeITP and 5-EITP (Figure 3) (63). Analogs with higher pi-electon surface areas, such as 5-NITP, 5-PhITP, and 5-NapITP, possessed high binding affinities and were quickly incorportated opposite an abasic site, but were poorly incorporated opposite templating nucleobases (64–66). Selectivity diminished for non-natural nucleotides with pi-electron surface areas over 200 Å2, which was attributed to the larger nucleobases non-selectively intercalating into the DNA. However, non-natural nucleotides with pi-electron surface areas below 180 Å2, such as 5-CyITP, 5-EyITP, 5-EtITP and 5-MeITP, also demonstrated high catalytic efficiency opposite abasic sites. For these nucleotides, overall catalytic efficiencies for incorporation opposite templating nucleobases was 100-fold lower than opposite an abasic template (63).
Kinetic data obtained from the study of 5-CyITP, 5-EyITP, 5-EtITP and 5-MeITP supported Berdis’ proposed model for gp43 exo− polymerase-mediated synthesis, which posited that a templating base is oriented extrahelically within the polymerase, creating an abasic-like void in the DNA. Following association of an incoming nucleotide, the polymerase undergoes a conformational change that positions the templating base intrahelically for proper alignment during phosphoryl transfer; the rate constant for polymerization, kpol, reflects the rate constant for this conformational change (64–68). It was observed that the presence of a templating nucleobase modestly influenced the binding affinities of incoming dNTPs of the smaller non-natural nucleotides; Kd values measured opposite T were about three-fold higher compared to those measured opposite an abasic site. However, a templating thymine opposite incoming 5-CyITP, 5-EyITP, 5-MeITP, and 5-EtITP led to 30–60-fold lower kpol values compared to those opposite an abasic site. These data are consistent with discrimination between incoming nucleotides being largely controlled during the phosphoryl transfer step, instead of during the initial ground state binding (63). Thus, although increased pi-electron surface area of an incoming nucleotide leads to favorable stacking interactions within the abasic site that reduce Kd, the more important contributor to efficiency and selectivity of polymerase-mediated synthesis opposite templating or abasic sites is the overall size of the incoming nucleotide, which sterically interacts with templating bases, controlling kpol.
Nucleobase-derived abasic probes
With the goal of potentiating the activity of anti-cancer methylating agents, nucleobase-derived probes with the capacity to hinder BER of abasic sites induced by the drugs have been investigated. Demeunynck and co-workers designed a series of heterodimeric compounds consisting of purine nucleobases linked to an acridine moiety through polyamine or guanidine-containing tethers. NMR studies, in which the probes were added to duplex DNA containing abasic sites modeled by THF, showed that the purine nucleobases insert within the abasic site pocket and hydrogen-bond to a partner T. The tethered acridine intercalates into the DNA duplex adjacent to the abasic site (69). Cytotoxicity and the affinity of the probe for binding duplex DNA containing abasic sites were modulated by varying the length and chemical identity of the tether (70). In cancer cell lines and in a murine P388 leukemia mouse model, co-administration of the heterodimeric probes with the anti-cancer DNA-alkylating drug bis-chloroethyl nitrosourea potentiated the anti-cancer activity of the drug (69–71).
Bulky triphosphate probes of abasic DNA
With the aim of determining the minimal structural requirements of DNA polymerase substrates, Maga and co-workers investigated the suitability of nucleoside-free triphosphates for incorporation in DNA. A series of probes (8a–c, Figure 4) consisting of bulky hydrophobic groups linked to a triphosphate, lacking any bases or sugar moieties, were investigated as DNA synthesis substrates for human polymerases α, β, and λ, Saccharomyces cerevisiae polymerase IV, Escherichia coli polymerase I, and HIV-1 reverse transcriptase (72). These were screened for their ability to incorporate the analogues opposite either templating bases or THF. Generally, the non-nucleoside triphosphates competitively inhibited natural nucleotide binding and blocked template extension by human polymerases α, β, and λ, and Saccharomyces cerevisiae polymerase IV, while Escherichia coli polymerase I and HIV-1 reverse transcriptase were not affected. Polymerase λ, which was known to perform translesion synthesis opposite abasic sites, was able to incorporate 8a across from THF but not opposite templating nucleobases. Additionally, the mutant polymerase λ Y505A, which possesses a larger nucleotide binding site than the wild-type enzyme, incorporated both 8a and 8b opposite the abasic sites but not opposite normal substrates. It was rationalized that although the non-nucleoside triphosphates generally accessed polymerase binding pockets, causing polymerase inhibition, the polymerases did not undergo transition from the catalytically inactive open position to the active closed position, and thus were unable to align the non-nucleoside analog with the primer strand and form a phosphodiester bond. However, polymerase λ and λ Y505A, which adopt a closed structure with or without nucleotide substrates, were able to catalyze phosphodiester bond formation between the probes and primers. Since the probes contain no sugar moiety and thus no 3′OH group, no further extension is possible; therefore, these non-nucleoside analogues may have potential biotechnological applications as chain-terminators for specifically detecting abasic sites.
FIGURE 4.
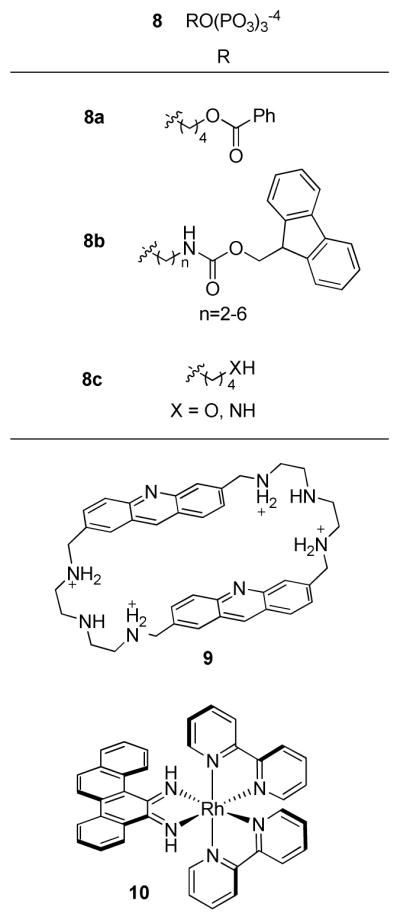
Non-nucleoside probes for abasic lesions.
Other non-nucleotide probes
Various non-nucleotide probes that do not fit into the categories described above have been reported; some notable recent examples are illustrated in Figure 4. Cyclobisacridine (CBA, 9, Figure 4) selectively intercalates into duplex DNA at an abasic site and, upon irradiation, photocleaves both strands (73). Photo-induced cleavage at an abasic site was also demonstrated with [Δ-Rh(bpy)2(chrysi)]3+ (10, Figure 4), a sterically bulky rhodium complex that inserts (not intercalates) at abasic sites or single base bulges in DNA and ejects the mismatched base pair into the major groove before cleavage (74). Finally, several recent non-nucleosbase abasic site probes include fluorescent intercalators (75), a hybridization probe detected using electrochemical methods (76), a naphthyridine-based hydrogen-bonding fluorescent probe (77), and a ferrocenyl aminonapthyridine-based hydrogen-bonding probe that can be detected both spectroscopically and electrochemically (78).
UV-INDUCED DNA DAMAGE
An important class of DNA lesions consists of those induced by UV radiation, a critical contributor to skin cancer etiology. This correlation was initially made in studies of xeroderma pigmentosa (XP) patients, who exhibit high incidences of skin cancer, and whose somatic cells, unable to repair pyrimidine dimers, were hypersensitive to killing by UV radiation (79). Further, the action spectra (a plot correlating radiation frequency to relative biological response) for killing, mutating, and transforming cells were similar to the action spectra for pyrimidine dimerization, which falls within the DNA absorbance spectrum (80, 81). Interestingly, it was also shown that reversal of pyrimidine dimers by photolyases reduced tumors in experimental animals (82, 83), which suggested that pyrimidine dimers were at least partially responsible for UV-induced biological effects (84).
The location and identity of UV-induced DNA lesions can be determined by HPLC analysis with UV spectroscopic detection (85), immunofluorescence (86), 32P-postlabeling (87), gas-chromatography-mass spectrometry (GC-MS) (88), and HPLC-electrospray ionization MS/MS (89). Cyclobutane pyrimidine dimers, which may occur at TT, CC, CT, or TC dipyrimine sites within the same DNA strand, are the most common UV-induced photolesions (70–80% occurance) (90). 6–4 photoproducts are less frequent (20–30% occurance) (90), and it was discovered that sunlight causes them to photoisomerize to Dewar valence isomers (Scheme 2) (91). While the cis-syn thymine dimer is most prevalent and was implicated in early studies linking UV-induced DNA damage to skin cancer, it has been suggested that the less common 6–4 photoproducts play a more significant role in mutagenesis and lethality 92–95). DNA repair mechanisms, such as nucleotide excision repair or photoreactivation, undoubtedly also have a large impact on UV-induced mutagenesis; the biological effects of various lesions depend on which genes are damaged and what repair mechanisms are active in affected cells (84, 94, 96).
SCHEME 2.

Pathways to thymine dimers.
Photodimer translesion DNA synthesis
Mechanistic aspects of TT and CC cis-syn dimer-induced mutagenesis have been deduced through studies of translesion DNA synthesis by various polymerases, as reviewed (97). It has been observed that E. coli DNA polymerase I (pol I) bypasses the TT cis-syn dimer, and that in this process, the only nucleotide incorporated across from each T in the dimer was its natural pair, A (97). A putative mechanism accounting for this observation is that the specificity of incorporation of AA opposite the TT dimer results not from correct coding, but from the inherent tendency for polymerases to incorporate A across from non-instructional (i.e., non-hydrogen bonding) DNA sites, as per the A-rule. Contradicting the non-coding hypothesis was NMR data for a DNA octamer containing a central TT dimer that appeared, in fact, to hydrogen bond to the complementary AA in the opposite strand (98). Further evidence that UV-induced lesions were templating, rather than non-instructional, was that E. coli under SOS conditions also incorporated AA opposite a TT dimer at a much higher frequency than A was incorporated opposite a non-instructional abasic site (99–101). Additionally, GA was found to be selectively incorporated opposite 6–4 TT photoproducts (101). In DNA synthesis reactions mediated by E. coli polymerase V (pol V), similar selectivities were observed (102). However, other polymerases, including T7 exo− (103), E. coli pol III (102), and those found in yeast (104) and human cell extracts (105), incorporated AA opposite both cis-syn TT dimers and 6–4 TT photoproducts. Collectively, these data suggested that while lesion bypass mechanisms vary among different polymerases, generally lesion bypass involves enzyme-based molecular interactions that dictate the specificity of translesion nucleotide incorporation.
Probing TT dimer translesion synthesis
Detailed information regarding the unique mechanisms for TT dimer bypass by specific enzymes has been gained through studies utilizing nucleotide probes. A crystal structure of the T7 exo− DNA polymerase bound to a DNA template in the presence of ddATP showed that the 5′ end of the template strand was bent out of the active site (106). Based on this structure, and in light of the fact that T7 exo− incorporates AA across a cis-syn TT dimer, Lloyd and coworkers proposed a mechanism for the bypass of the dimer in which the 3′-T, bent out of the active site due to its covalent bond to the downstream 5′-T, acts as an abasic-like non-instructive lesion, while the 5′-T, once moved back into the active site, acts as a templating base. Each insertion would be expected to template an A, as per the A rule for the first insertion, and through normal nucleobase specificity for the second insertion (103). To test this hypothesis, Lloyd and co-workers took advantage of the pyrene nucleotide (dPTP in Figure 1), which was previously shown by Kool and Matray to be incorporated opposite abasic sites and analogues by T7 DNA polymerase, in preference to all four natural nucleotides (53). Assuming that the 3′-T of a TT dimer was indeed bent out of the active site of the polymerase, thus presenting an abasic site on the template strand, it was expected that dPTP would be selectively incorporated rather than adenine. Primer extension analyses confirmed that dPTP was in fact inserted in over 12:1 preference to dATP across the 3′-T of a template strand cis-syn TT dimer. However, dPTP was very inefficiently incorporated opposite the 5′-T of the dimer while dATP was preferentially inserted, suggesting that the 5′-T was instructional during polymerase bypass of the lesion. Similar results with 6–4 TT photoproducts, their Dewar isomers, and trans-syn II TT dimmers corroborated the transient abasic site model for T7 DNA polymerase UV-lesion bypass (103).
To evaluate whether the transient abasic site model proposed for T7 DNA polymerase applies to other high-fidelity polymerases, Berdis and co-workers carried out primer-extension assays with the bacteriophage T4 DNA polymerase (gp43) (67). In these studies, the rates of gp43-mediated incorporation of various natural and non-natural nucleotide probes across either a cis-syn TT dimer or a THF abasic site analogue in an oligonucleotide template were compared. It was expected that if gp43 employed the transient abasic site mechanism, then it would incorporate analogues such as 5-NITP, 5-PhITP, and 5-NapITP, which were previously demonstrated to have high binding affinities and incorporation rates across from abasic sites (64–66), opposite both the TT dimer and the abasic site with nearly equal kinetic parameters.
Different rates of incorporation of nucleotide probes across from TT dimers and abasic sites were observed in experiments involving exonuclease-deficient gp43 exo−. Particularly, nucleotides which were readily inserted opposite abasic sites, such as IndTP, 5-FITP, 5-AITP (Figure 3), 5-NITP, and dATP, were poorly inserted opposite the TT dimer. Additionally, although dATP was the only of the natural nucleotides to be incorporated across the TT dimer, it was done so about 21-fold slower than its rate of incorportation opposite the abasic site, even at high 1 mM concentrations (107). These results implied that gp43 exo− does not entirely follow the A-rule or the transient abasic site model for TT dimer bypass. The significant differences between incorporation rates for each nucleotide probe opposite the TT dimers indicated that π-electron density and the overall size of the analogue both play a large role in the incorporation efficiency. On the basis of these studies, Berdis and co-workers concluded that the TT dimer was templating the incorporation of A via molecular interactions with T, not as an abasic-like site.
To test the templating TT dimer hypothesis, the rates of incorporation of the nucleotide probes across from an unmodified T were also evaluated. Comparison of the kinetic parameters showed that analogues with large π-electron surface area (5-CEITP, Figure 3), 5-PhITP, and 5-NapITP) were incorporated opposite the TT dimer more efficiently than across T, while those with smaller π-electron surface area (5-NITP, 5-FITP, and 5-CyITP) were more effectively incorporated opposite T. These observations led to the proposal that the TT dimer is replicated by gp43 exo− via a hybrid mechanism. For unmodified DNA, the templating nucleobases are oriented in an extrahelical position, creating an abasic-like void that can readily accept a bulky nucleotide probe like 5-PhITP. However, the bulk of the probe would hinder the repositioning of the extrahelical templating nucleobase back into its intrahelical position, diminishing the incorporation rate of the probe. In contrast, the kpol of incorporation of the probes opposite abasic sites are much faster since there is no extrahelical base to reposition. The kinetics of incorporation of nucleotide probes opposite TT dimers support the model that the 3′-T of the dimer is partially extrahelical, creating an abasic-like site into which the probe is incorporated at a lower kpol than at an actual abasic site.
Probing TT dimer-induced exo/endonuclease activity
In addition to comparing the rates of gp43 exo−-mediated incorporation of nucleotides opposite a TT dimer and abasic site, Berdis and co-workers also examined the ability of gp43 exo+ to excise nucleotides opposite these lesions or T (67). It had been previously shown that gp43 removed A opposite an abasic site about 30-fold faster than A opposite T (108). On this basis, it was hypothesized that altered hydrogen-bonding and stacking interactions at the abasic site distort the primer-template geometry such that exonuclease activity increases. However, while gp43 exo+ removed 5-PhITP opposite an abasic site, it did not remove the probe opposite a TT dimer, even though the exonuclease easily excised A opposite either lesion. It was hypothesized that the diminished reactivity was due to aberrant stacking interactions caused by the proximity of the phenyl moiety of 5-PhITP to the 5′-T of the TT dimer, which may have hindered the gp43 proofreading ability.
The mechanism of base excision for T4 endonuclease V (T4 endo V) has also been evaluated by a similar approach using the fluorescent nucleotide probe 2-aminopurine (109). T4 endo V is a DNA glycosylase that selectively binds to a cis-syn TT dimer and cleaves the glycosidic bond of the damaged base. The crystal structure of E23Q (a catalytically inactive T4 endo V mutant), co-crystallized with a substrate DNA, showed that the adenine opposite the 5′-T of a TT dimer was flipped extrahelically into an enzyme pocket, and that the DNA substrate had a 60° bend at the dimer (110). To study the role of base-flipping in T4 endo V recognition of thymine dimer damage, Lloyd and co-workers utilized 2-Ap for monitoring the binding of the enzyme and the relative position of the 2-Ap in the T4 endo V-substrate DNA complex. The probe was selected because it had been previously shown to form a Watson-Crick base-pair with thymine (111) and its fluorescence was significantly quenched in duplex DNA compared to singled-stranded DNA (112).
Lloyd and co-workers showed that placing 2-AP in either the 3′- or 5′-position opposite a TT dimer in oligonucleotides does not alter binding or catalytic activity of wild-type T4 endo V or E23Q (109). The specificity of base-displacement observed in the co-crystal structure was corroborated by titration assays, which showed no fluorescence enhancement (which would indicate extrahelical displacement of the probe) upon addition of E23Q to a duplex containing 2-Ap opposite the 3′-T of the TT dimer, while the converse was true for a duplex with 2-Ap opposite the 5′-T of the dimer. The specificity of E23Q for inducing base-flipping solely in damaged DNA was established by a control experiment in which no fluorescence enhancements were observed for a duplex containing 2-Ap opposite an undamaged T (109). Thus, data regarding the TT dimer-induced endo- and exonuclease activity of T4 endo V and gp43 exo+ obtained from experiments involving 2-AP and 5-PhITP, respectively, provided insight into enzyme-lesion interactions involved in these processes.
DNA AKYLATION ADDUCTS
Despite the typically low abundance and cellular repair of bulky DNA alkylation adducts, they can severely impact DNA structure and function, resulting in dramatic consequences such as mutation and apoptosis. Representative alkylation pathways are shown in Scheme 3. Nitrosamines are important DNA alkylators due to human exposures from diet, drinking water, and occupational hazards, and their link to carcinogenesis. Among the earliest characterizations of DNA lesion formation is methylation by N-nitrosodimethylamine at the 3- and 1- positions of A, and the 3-, 7-, and O6- positions of G (113, 114). Other nitrosamine alkylating agents include metabolites of nicotine, such as α-hyroxylated NNK, which breaks down to generate the methanediazonium ion that subsequently methylates the 7- and O6- positions of G and O4- position of T (115). NNK may also be metabolized to produce pyridyloxobutyl (POB) adducts with the 7-, N2-, and O6- positions of G, and the O2- positions of T and C (116). Benzylation of purine bases, at the 3-position of adenine and the 7-, N2-, N6-, and O6- positions of G, by carcinogens such as hydroxylated N-nitrosobenzylmethylamine (NBzMA) (117) or N-nitroso-N-benzylurea (BzNU), also occurs (118). Of these, O6-alkyl-G adducts have been paid particular attention due to their high mutagenicity (119–121).
SCHEME 3.

Representative nitrosamine alkylation pathways.
Guengerich and co-workers have extensively evaluated O6-alkyl-G lesion bypass by both replicative polymerases and Y-family polymerases, which are known to perform translesion bypass. They showed that human replicative polymerase δ, human Y-family polymerases η and κ, and model replicative polymerase HIV-RT all have similar efficiencies of incorporation of C and T opposite both O6-Me- and O6-Bn-G, while human Y-family polymerase ι and model replicative E. coli polymerase T7− preferentially incorporate T opposite O6-Me- and O6-Bn-G (122, 123). The model Y-family polymerase, Sulfolobus solfataricus DNA polymerase Dpo4, was shown to preferentially incorporate C opposite O6-Me-G and O6-Bn-G, at rates 14- and 62-fold slower, respectively, than incorporation of C opposite unmodified G (124, 125). Crystal structures of ternary Dpo4 complexes revealed O6-Me- and O6-Bn-G in a “wobble pairs” opposite incoming dCTP, leading to the suggestion that the open, flexible active site of Dpo4 accomodates the wobble pair, favoring incorporation of C, while more rigid replicative polymerases enclose O6-alkyl-G:T mispairs and force a greater fraction of T incorporation (124, 125). While it is difficult to know which polymerases were responsible for translesion bypass in cells, it has been shown that in both human cells and E. coli, thymine is preferentially inserted opposite O6-alkyl-G adducts during translesion DNA synthesis, leading to G to A transitions (126).
The mutagencity of O6-alkyl-G adducts in cells also depends on their ability to be repaired by O6-alkyl-G-DNA alkyltransferases (AGTs), as reviewed by Pegg et al. (Pegg, Dolan et al. 1995). The timing of repair by AGTs, which function by directly transferring the alkyl group to a reactive cysteine within the protein (127), may be crucial to whether the adduct persists, leading to mutation. In their excellent review of DNA alkylation damage, He et al. noted that while human AGT (hAGT) can repair O6-alkyl-G paired opposite thymine, it was unclear whether E. coli AGT (C-Ada) could perform the same function, though it was known to remove alkyl lesions when the G adduct was paired across from C (128, 129); (130).
O6-alkyl-G adduct probe
Towards understanding the occurrence, properties, and biological impacts of O6-alkyl-G adducts in DNA, and to investigate the molecular requirements of a nucleotide partner for these important adducts, Gong and Sturla have carried out studies using O6-Bn-G as a representative adduct (131). A diaminonaphthyl-derived adduct-specific nucleotide probe (dNap, Figure 5) was designed to form a stable pair with O6-Bn-G, and the specificity of this probe:adduct pair was evaluated by comparing the melting temperatures of 15-mer DNA duplex containing O6-Bn-G or dNap paired opposite of each other or natural bases. A 15-mer duplex with the O6-Bn-G:dNap pair in the place of X:Y in the sequences 5′-TTGTCGGTATAXCGG-3′ and 5′-CCGYTATACCGACAA-3′ was more thermally stable than any pairing of O6-Bn-G opposite a natural base by 5.6 °C on average, as well as more thermally stable than any dNap:natural nucleoside pair by an average of 5.5 °C.
FIGURE 5.

Anti-dNap: O6-Bn-G and syn-dNap: O6-Bn-G base-pairing. R: deoxyribose or the DNA backbone.
It was hypothesized that the O6-Bn-G:dNap pair is stabilized by a combination of pi-pi stacking and hydrophobic interactions with the benzyl portion of the adduct, while the urea moiety provides points of contact for hydrogen-bonding with the guanosine 1- and N2-hydrogens. Although NOESY and X-ray analysis showed that the free nucleoside possesses a syn glycosidic torsion angle (in which the urea hydrogen-bonding moieties are oriented over the ribose subunit), as opposed to the anti conformation that would allow for the hypothesized hydrogen-bonding and pi-pi stacking interactions, energy barriers between nucleoside conformations are usually low (132). Further, conformation depends on the chemical environment, be it free solution or solid state, in a mixture with other nucleosides, or in duplex DNA (133–135).
While O6-Bn-G:dNap is the first example of a stable adduct:probe pair, limitations in the stability and specificity of the pair underscore the need for further modifications to this probe. For example, a dNap:dNap self-pair also had similar thermal stability to that of the G:C duplex, which is sometimes the case for hydrophobic nucleobase surrogates (136, 137). Generally, the realization of synthetic nucleotides that pair with bulky adducts is at an early stage compared with studies involving the detection and evaluation of abasic sites and TT dimers. Thus, fundamental information regarding the basis of physical interactions in the DNA duplex or the compatibility of biochemically mediated processes such as synthesis and repair, are not understood. This situation also holds true for probes under investigation for the key DNA oxidative-lesion 8-oxo-G, discussed in the next section.
OXIDATIVE DAMAGE
Cellular reactive oxygen species, such as hydroxyl and superoxide radicals, react with DNA and induce single strand breaks, DNA-protein crosslinks, abasic sites, and base-oxidized lesions. In the case of oxidation-induced lesions, there are about fifty known adducts (138) and 8-oxoguanosine (8-oxo-G, Figure 6) is considered the most abundant (139). The oxidation potential of G in DNA is 1.3 V; upon oxidation to 8-oxo-G the oxidation potential is lowered to 0.7 V, making this species more reactive towards oxidation compared to other bases and prone to oxidation under physiological conditions (140, 141). Thus, further oxidation and other chemical transformations of 8-oxo-G yield secondary DNA adducts. For example, peroxynitrite, an endogeneous product of the reaction of nitric oxide and superoxide, oxidizes 8-oxo-dG to yield several mutagenic compounds such as cyanuric acid, oxaluric acid, and oxazolone (Figure 6) (142–146). Furthermore, 5-guanidinohydantoin and 2-imino-5,5′-spirohydantoin (Figure 6), generated from one electron oxidation of 8-oxo-G or direct oxidation or phototoxidation of G, are potentially mutagenic, leading to G→T and G→C transversion mutations in E. Coli based mutagenesis assays (147–152). If unrepaired before replication, 8-oxo-G itself can lead to G→T transversion mutations, a common somatic mutation in human carcinomas (153, 154). Thus 8-oxo-G is considered to be important in human carcinogenesis, and has emerged also as a biomarker for cellular oxidative damage (138, 155, 156).
FIGURE 6.

8-Oxo-G and other oxidation products. dR: deoxyribose.
8-Oxo-G probes
There is extensive literature concerning methods for detecting 8-oxo-G and related oxidation adducts, including LC-MS, accelerator mass spectrometry, 32P-postlabelling, and immunochemical assays (157–163). These approaches can be extremely sensitive and specific; however, because they are generally sample-destructive and high background oxidation can be quite problematic during sample preparation, there is a continued need for in-situ detection strategies involving 8-oxo-G-specific molecular probes.
Toward the goal of developing an 8-oxo-G specific nucleotide probe, Sasaki and co-workers have reported cytosine analogs with the potential to preferentially bind 8-oxo-G over G (164). The structure of cbz-8-oxo-G-clamp 11 (Figure 7) was optimized through testing analogues with various terminal substituents, and its selectivity for binding 8-oxo-G was shown by 1H NMR to result from multiple hydrogen bonds between the adduct and probe. The tricyclic probe 11 has an emission maximum at 450 nm upon excitation at 365 nm, which permits the binding characteristics of the probe to be detected by flourescence changes. Binding studies were carried out by titrating silyl-protected nucleosides into buffered chloroform solutions of the probe. The emission intensity of 11 was drastically decreased when 8-oxo-G was added, while G caused no significant quenching. Under more physiologically relevant aqueous conditions, adding 8-oxo-G to a solution of 11 in water (solubilized by the detergent Triton X-100) resulted in fluorescence quenching patterns similar to those observed in chloroform.
Figure 7.

Proposed 11:8-oxo-G complex. dR: deoxyribose.
Despite the favorable binding properties of free 11 with 8-oxo-G, mixed results were obtained when the system was evaluated in an oligonucleotide context (165). Thermal denaturation data indicated that the stability of a duplex containing G-clamp 11 opposite 8-oxo-G was, on average, 2.5 °C lower than a duplex with 11 opposite G. This result led to the conclusion that the steric hindrance from the benzyloxycarbonyl group of the probe precluded it from forming the desired hydrogen bonding contacts in the context of duplex DNA. However, the fluorescence quenching of the probe in a duplex opposite 8-oxo-G was greater than that of the probe across from unaltered nucleotides, although some quenching was observed opposite G. Again, steric bulk of the probe was postulated to be the cause of the diminished ability of 8-oxo-G to quench the fluorescence of the probe relative to quenching by G. Therefore, derivatives of 11 with alternative terminal groups are being investigated to improve selectivity and efficacy while minimizing detrimental steric interactions in the oligonucleotide system.
Recently, Tor and co-workers reported the application of 5-furanyl deoxycytosine 6b (Figure 3), the deoxyribonucleotide version of their previously reported abasic site probe 6a, as an emissive probe that distinguishes between G, 8-oxo-G, and T in duplex DNA (166). Minimal disruptions to thermal stability (<1 °C) occured when the probe was placed opposite G, 8-oxo-G, or T in 13-mer oligonucleotide duplexes, relative to similar duplexes containing C instead of the probe. However, notable differences in the fluorescence intensities of duplexes containing the probe were observed around 440 nm. The emission intensity of the duplex with the probe:8-oxo-G pair was quenched 2-fold relative to the duplex with the probe:G pair, while the emission intensity of the duplex containing the probe:T pair was 4-fold higher than that containing the probe:G pair. The quenching of the probe opposite 8-oxo-G relative to G was ascribed to 8-oxo-G’s lower redox potential, which gives the probe higher excited-state quenching ability. The drastic increase of the emission intensity of the probe:T pair was attributed to the probe’s likely extrahelical placement within the duplex, where it would be exposed to a more polar environment (thermal denaturation and emission studies were performed in aqueous buffer). It was speculated that the 5-furanyl deoxycytosine probe could be used in future non-destructive real-time fluorescence-based methods for monitoring 8-oxo-G formation in vitro.
CONCLUSION
DNA lesions, such as abasic sites, TT dimers, bulky alkylation adducts, and 8-oxo-G, play important biological roles due to disruptions in critical cellular processes. An emerging experimental strategy for elucidating mechanistic details regarding how these lesions physically interact with and affect biochemical processing of nucleic acids involves the use of chemical probes. Lesion-specific probes are reviewed here, organized on the basis of target lesions, and emphasizing chemical concepts behind the development of the probes as well as their application and pitfalls in biochemical studies. Much attention has been paid to non-natural nucleotide probes, especially for abasic sites, due to information that can be gained as these are incorporated opposite lesions in a polymerase-mediated manner. The mechanism of translesion DNA synthesis opposite TT dimers was discussed; in light of studies using non-natural nucleotide probes, it appears that TT dimers either act as transient abasic sites or as templates, depending on the polymerase. Finally, recent reports involve the design and evaluation of molecular probes for the oxidative lesion 8-oxo-G and the representative bulky alkylation adduct O6-Bn-G; these examples represent new classes of molecular probes for nucleobase adducts, but require further structure optimization and a better understanding of the physical basis of interactions with DNA and proteins before they can be effectively utilized for elucidating biochemical mechanisms or for novel damage detection strategies.
Footnotes
Abbreviations: A, adenine; 2-Ap, 2-aminopurine; AP, apurinic; ARP, aldehyde-reactive probe; BER, base-excision repair; Bn, benzyl; BzNU, N-nitroso-N-benzylurea; C, cytosine; ddATP, dideoxyadenosine triphosphate; DNA, deoxyribonucleic acid; dNTP, deoxyribonucleoside triphosphate; dPTP, deoxyribopyrene triphosphate; ELISA, enzyme-linked immunosorbent assay; exo−, exonuclease-deficient; exo+, exonuclease-proficient; G, guanine; GC, gas-chromatography; Gh, 5-guanidinohydantoin; gp43, T4 DNA polymerase; HPLC, high-performance liquid-chromatography; Kf, Klenow fragment; MS, mass-spectrometry; NBzMA, N-nitrosobenzylmethylamine; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; 8-oxo-G, 8-oxoguanosine; PCR, polymerase chain reaction; POB, pyridyloxobutyl; RIP, ribosome-inactivating protein; RNA, ribonucleic acid; Sp, 2-imino-5,5′-spirohydantoin; T, thymine; T4 endo V, T4 endonuclease V; THF, tetrahydrofuran; UV, ultraviolet; XP, xeroderma pigmentosa.
This work was supported by the National Cancer Institute (CA-108604, CA-123007).
References
- 1.Avery OT, MacLeod CM, McCarty M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: inductmion of transformation by a desoxyribonucleic acid fraction isolated from pnuemococcus type III. J Exp Med. 1944;79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watson JD, Crick FHC. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 3.Zamenhof S, Alexander HE, Leidy G. Studies on the chemistry of the transforming activity: 1. Resistance to physical and chemical agents. J Exp Med. 1953;98:373–397. doi: 10.1084/jem.98.4.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Chemical Society. Cancer Facts & Figures 2008. American Cancer Society; Atlanta: 2008. [Google Scholar]
- 5.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 6.Horner MJ, Ries LAG, Krapcho M, Neyman N, Aminou R, Howlader N, Altekruse SF, Feuer EJ, Huang L, Mariotto A, Miller BA, Lewis DR, Eisner MP, Stinchcomb DG, Edwards BK. SEER Cancer Statistics Review 1975–2006. National Cancer Institute; Bethesda, MD: 2009. [Google Scholar]
- 7.Gillet LCJ, Schärer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 8.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loechler EL. The role of adduct site-specific mutagenesis in understanding how carcinogen-DNA adducts cause mutations: perspective, prospects and problems. Carcinogenesis. 1996;17:895–902. doi: 10.1093/carcin/17.5.895. [DOI] [PubMed] [Google Scholar]
- 10.Guengerich FP. Interactions of carcinogen-bound DNA with individual DNA polymerases. Chem Rev. 2006;106:420–452. doi: 10.1021/cr0404693. [DOI] [PubMed] [Google Scholar]
- 11.Yang W, Woodgate R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc Natl Acad Sci. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nooner T, Dutta S, Gates KS. Chemical properties of the leinamycin:guanine adduct in DNA. Chem Res Toxicol. 2004;17:942–949. doi: 10.1021/tx049964k. [DOI] [PubMed] [Google Scholar]
- 13.Gates KS, Nooner T, Dutta S. Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem Res Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- 14.Gong J, Vaidyanathan VG, Yu X, Kensler TW, Peterson LA, Sturla SJ. Depurinating acylfulvene:DNA adducts: characterizing cellular chemical reactions of a selective antitumor agent. J Am Chem Soc. 2007;129:2101–2111. doi: 10.1021/ja0665951. [DOI] [PubMed] [Google Scholar]
- 15.Neels JF, Gong J, Yu X, Sturla SJ. Quantitative correlation of drug bioactivation and deoxyadenosine alkylation by acylfulvene. Chem Res Toxicol. 2007;20:1513–1519. doi: 10.1021/tx7001756. [DOI] [PubMed] [Google Scholar]
- 16.David-Cordonnier MH, Casely-Hayford M, Kouach M, Briand G, Patterson LH, Bailly C, Searcey M. Stereoselectivity, sequence specificity and mechanism of action of the azinomycin epoxide. Chem Bio Chem. 2006;7:1658–1661. doi: 10.1002/cbic.200600244. [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Cheng G, Sturla SJ, Yongli S, McIntee EJ, Villalta PW, Upadhyaya P, Hecht SS. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem Res Toxicol. 2003;16:616–626. doi: 10.1021/tx034003b. [DOI] [PubMed] [Google Scholar]
- 18.Cavalieri ELSDE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden K, Ramanathan R, Cerny RL, Rogan EG. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Nat Acad Sci USA 94. 1997:10937–10942. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Turteltaub KW, Felton JS, Gledhill BL, Vogel JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC. Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc Nat Acad Sci USA. 1990;87:5288–5292. doi: 10.1073/pnas.87.14.5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy MV. Methods for testing compounds for DNA adduct formation. Regul Toxicol Pharmacol. 2000;32:256–263. doi: 10.1006/rtph.2000.1430. [DOI] [PubMed] [Google Scholar]
- 22.Singh R, Farmer PB. Liquid chromatography-electrospray ionization-mass spectrometry: the future of DNA adduct detection. Carcinogenesis. 2006;27:178–196. doi: 10.1093/carcin/bgi260. [DOI] [PubMed] [Google Scholar]
- 23.Phillips DH. Detection of DNA modifications by the 32P-postlabelling assay. Mut Res. 1997;378:1–12. doi: 10.1016/s0027-5107(97)00092-4. [DOI] [PubMed] [Google Scholar]
- 24.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 25.Lindahl TNB. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 26.Lindahl T. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog Nucleic Acid Res Mol Biol. 1979;22:135–192. doi: 10.1016/s0079-6603(08)60800-4. [DOI] [PubMed] [Google Scholar]
- 27.Kunkel TA. Mutational specificity of depurination. Proc Nat Acad Sci USA. 1984;81:1494–1498. doi: 10.1073/pnas.81.5.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelfand CA, Plum GE, Grollman AP, Johnson F, Breslauer KJ. Thermodynamic consequences of an abasic lesion in duplex DNA are strongly dependent on base sequence. Biochemistry. 1998;37:7321–7327. doi: 10.1021/bi9803372. [DOI] [PubMed] [Google Scholar]
- 29.McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu Rev Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 30.Dianov GL, Sleeth KM, Dianova II, Allinson SL. Repair of abasic sites in DNA. Mut Res. 2003;531:157–163. doi: 10.1016/j.mrfmmm.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Parikh SSMCD, Tainer JA. Base excision repair enzyme family portrait: integrating the structure and chemistry of an entire DNA repair pathway. Structure. 1997;5:1543–1550. doi: 10.1016/s0969-2126(97)00303-1. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava DK, Vande Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 33.Wilson SH. Mammalian base excision repair and DNA polymerase beta. Mut Res. 1998;407:203–215. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- 34.Kappen LS, Goldberg IH. Identification of 2-deoxyribonolactone at the site of neocarzinostatin-induced cytosine release in the sequence d(AGC) Biochemistry. 1989;28:1027–1032. doi: 10.1021/bi00429a016. [DOI] [PubMed] [Google Scholar]
- 35.Bales BC, Pitie M, Meunier B, Greenberg MM. A minor groove binding copper-phenanthroline conjugate produces direct strand breaks via β-elimination of 2-deoxyribonolactone. J Am Chem Soc. 2002;124:9062–9063. doi: 10.1021/ja026970z. [DOI] [PubMed] [Google Scholar]
- 36.Faure V, Constant JF, Dumy P, Saparbaev M. 2′-Deoxyribonolactone lesion produces G→A transitions in Escherichia coli. Nucl Acids Res. 2004;32:2937–2946. doi: 10.1093/nar/gkh622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kroeger KM, Jiang YL, Kow YW, Goodman MF, Greenberg MM. Mutagenic effects of 2-deoxyribonolactone in Escherichia coli. An abasic lesion that disobeys the A-rule. Biochemistry. 2004;43:6723–6733. doi: 10.1021/bi049813g. [DOI] [PubMed] [Google Scholar]
- 38.Strauss B, Rabkin S, Sagher D, Moore P. The role of DNA polymerase in base substitution mutagenesis on non-instructional templates. Biochimie. 1982;64:829–838. doi: 10.1016/s0300-9084(82)80138-7. [DOI] [PubMed] [Google Scholar]
- 39.Schaaper R, Kunkel T, Loeb L. Infidelity of DNA synthesis associated with bypass of apurinic sites. Proc Nat Acad Sci USA. 1983;80:487–491. doi: 10.1073/pnas.80.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boiteux S, Laval J. Coding properties of poly(deoxycytidylic acid) templates containing uracil or apyrimidinic sites: in vitro modulation of mutagenesis by deoxyribonucleic acid repair enzymes. Biochemistry. 1982;21:6746–6751. doi: 10.1021/bi00269a020. [DOI] [PubMed] [Google Scholar]
- 41.Strauss B. The “A” rule revisited: polymerases as determinants of mutational specificity. DNA Repair. 2002;1:125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- 42.Greenberg MM, Weledji YN, Kroeger KM, Kim J, Goodman MF. In vitro effects of a C4′-oxidized abasic site on DNA polymerases. Biochemistry. 2004;43:2656–2663. doi: 10.1021/bi036028f. [DOI] [PubMed] [Google Scholar]
- 43.Talpaert-Borl M, Liuzzi M. Reaction of apurinic/apyrimidinic sites with [14C]methoxyamine: A method for the quantitative assay of AP sites in DNA. Biochim Biophys Acta. 1983;740:410–416. doi: 10.1016/0167-4781(83)90089-1. [DOI] [PubMed] [Google Scholar]
- 44.Weinfeld M, Liuzzi M, Paterson MC. Response of phage T4 polynucleotide kinase toward dinucleotides containing apurinic sites: design of a phosphorus-32-postlabeling assay for apurinic sites in DNA. Biochemistry. 1990;29:1737–1743. doi: 10.1021/bi00459a011. [DOI] [PubMed] [Google Scholar]
- 45.Liu L, Gerson S. Therapeutic impact of methoxyamine: Blocking repair of abasic sites in the base excision repair pathway. Curr Opin Invest Dr. 2004;5:623–627. [PubMed] [Google Scholar]
- 46.Ide H, Akamatsu K, Kimura Y, Michiue K, Makino K, Asaeda A, Takamori Y, Kubo K. Synthesis and damage specificity of a novel probe for the detection of abasic sites in DNA. Biochemistry. 1993;32:8276–8283. doi: 10.1021/bi00083a031. [DOI] [PubMed] [Google Scholar]
- 47.Chakravarti D, Badawi AF, Venugopal D, Meza JL, Crandall LZ, Rogan EG, Cavalieri EL. Improved measurement of dibenzo[a,l]pyrene-induced abasic sites by the aldehyde-reactive probe assay. Mut Res. 2005;588:158–165. doi: 10.1016/j.mrgentox.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Boturyn D, Boudali A, Constant J-F, Defrancq E, Lhomme J. Synthesis of fluorescent probes for the detection of abasic sites in DNA. Tetrahedron. 1997;53:5485–5492. [Google Scholar]
- 49.Boturyn D, Constant JF, Defrancq E, Lhomme J, Barbin A, Wild CP. A simple and sensitive method for in vitro quantitation of abasic sites in DNA. Chem Res Toxicol. 1999;12:476–482. doi: 10.1021/tx980275g. [DOI] [PubMed] [Google Scholar]
- 50.Fundador E, Rusling J. Detection of labeled abasic sites in damaged DNA by capillary electrophoresis with laser-induced fluorescence. Anal Bioanal Chem. 2007:1883–1890. doi: 10.1007/s00216-006-1041-x. [DOI] [PubMed] [Google Scholar]
- 51.Sato K, Greenberg MM. Selective detection of 2-deoxyribonolactone in DNA. J Am Chem Soc. 2005;127:2806–2807. doi: 10.1021/ja0426185. [DOI] [PubMed] [Google Scholar]
- 52.Dhar S, Kodama T, Greenberg MM. Selective detection and quantification of oxidized abasic lesions in DNA. J Am Chem Soc. 2007;129:8702–8703. doi: 10.1021/ja073014e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matray TJ, Kool ET. A specific partner for abasic damage in DNA. Nature. 1999;399:704–708. doi: 10.1038/21453. [DOI] [PubMed] [Google Scholar]
- 54.Singh I, Beuck C, Bhattacharya A, Hecker W, Parmar VS, Weinhold E, Seitz O. Abasic site stabilization by aromatic DNA base surrogates: High-affinity binding to a base-flipping DNA-methyltransferase. Pure Appl Chem. 2004;76:1563–1570. doi: 10.1002/anie.200219972. [DOI] [PubMed] [Google Scholar]
- 55.Dzantiev L, Alekseyev YO, Morales JC, Kool ET, Romano LJ. Significance of nucleobase shape complementarity and hydrogen bonding in the formation and stability of the closed polymerase-DNA complex. Biochemistry. 2001;40:3215–3221. doi: 10.1021/bi002569i. [DOI] [PubMed] [Google Scholar]
- 56.Fiala KA, Brown JA, Ling H, Kshetry AK, Zhang J, Taylor JS, Yang W, Suo Z. Mechanism of template-independent nucleotide incorporation catalyzed by a template-dependent DNA polymerase. J Mol Bio. 2007;365:590–602. doi: 10.1016/j.jmb.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guckian KM, Schweitzer BA, Ren RXF, Sheils CJ, Tahmassebi DC, Kool ET. Factors contributing to aromatic stacking in water: evaluation in the context of DNA. J Am Chem Soc. 2000;122:2213–2222. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang YL, Stivers JT, Song F. Base-flipping mutations of uracil DNA glycosylase: substrate rescue using a pyrene nucleotide wedge. Biochemistry. 2002;41:11248–11254. doi: 10.1021/bi026227j. [DOI] [PubMed] [Google Scholar]
- 59.Greco NJ, Tor Y. Simple fluorescent pyrimidine analogues detect the presence of DNA abasic sites. J Am Chem Soc. 2005;127:10784–10785. doi: 10.1021/ja052000a. [DOI] [PubMed] [Google Scholar]
- 60.Srivatsan SG, Greco NJ, Tor Y. A highly emmisive fluorescent nucleoside that signals the activity of toxic ribosome-inactivating proteins. Angew Chem Int Ed. 2008;47:6661–6665. doi: 10.1002/anie.200802199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 62.Keller DJ, Brozik JA. Framework model for DNA polymerases. Biochemistry. 2005;44:6877–6888. doi: 10.1021/bi0477079. [DOI] [PubMed] [Google Scholar]
- 63.Vineyard D, Zhang X, Donnelly A, Lee I, Berdis AJ. Optimization of non-natural nucleotides for selective incorporation opposite damaged DNA. Org Biomol Chem. 2007;5:3623 – 3630. doi: 10.1039/b712480e. [DOI] [PubMed] [Google Scholar]
- 64.Reineks EZ, Berdis AJ. Evaluating the contribution of base stacking during translesion DNA replication. Biochemistry. 2004;43:393–404. doi: 10.1021/bi034948s. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, Lee I, Berdis AJ. The use of nonnatural nucleotides to probe the contributions of shape complementarity and π-electron surface area during DNA polymerization. Biochemistry. 2005;44:13101–13110. doi: 10.1021/bi050585f. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Donnelly A, Lee I, Berdis AJ. Rational attempts to optimize non-natural nucleotides for selective incorporation opposite an abasic site. Biochemistry. 2006;45:13293–13303. doi: 10.1021/bi060418v. [DOI] [PubMed] [Google Scholar]
- 67.Devadoss B, Lee I, Berdis AJ. Is a thymine dimer replicated via a transient abasic site intermediate? A comparative study using non-natural nucleotides. Biochemistry. 2007;46:4486–4498. doi: 10.1021/bi602438t. [DOI] [PubMed] [Google Scholar]
- 68.Zhang X, Lee I, Berdis AJ. Evaluating the contributions of desolvation and base-stacking during translesion DNA synthesis. Org Biomol Chem. 2004;2:1703 – 1711. doi: 10.1039/b401732c. [DOI] [PubMed] [Google Scholar]
- 69.Belmont P, Jourdan M, Demeunynck M, Constant JF, Garcia J, Lhomme J, Carez D, Croisy A. Abasic site recognition in DNA as a new strategy to potentiate the action of anticancer alkylating drugs? J Med Chem. 1999;42:5153–5159. doi: 10.1021/jm9901428. [DOI] [PubMed] [Google Scholar]
- 70.Alarcon K, Demeunynck M, Lhomme J, Carrez D, Croisy A. Potentiation of BCNU cytotoxicity by molecules targeting abasic lesions in DNA. Bioorg Med Chem. 2001;9:1901–1910. doi: 10.1016/s0968-0896(01)00097-9. [DOI] [PubMed] [Google Scholar]
- 71.Alarcon K, Demeunynck M, Lhomme J, Carrez D, Croisy A. Diaminopurine-acridine heterodimers for specific recognition of abasic site containing DNA. Influence on the biological activity of the position of the linker on the purine ring. Bioorg Med Chem Lett. 2001;11:1855–1858. doi: 10.1016/s0960-894x(01)00310-9. [DOI] [PubMed] [Google Scholar]
- 72.Crespan E, Zanoli S, Khandazhinskaya A, Shevelev I, Jasko M, Alexandrova L, Kukhanova M, Blanca G, Villani G, Hubscher U, Spadari S, Maga G. Incorporation of non-nucleoside triphosphate analogues opposite to an abasic site by human DNA polymerases β and λ. Nucl Acids Res. 2005;33:4117–4127. doi: 10.1093/nar/gki723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berthet N, Michon J, Lhomme J, Teulade-Fichou MP, Vigneron JP, Lehn JM. Recognition of abasic sites in DNA by a cyclobisacridine molecule. Chem Eur J. 1999;5:3625–3630. [Google Scholar]
- 74.Zeglis BM, Boland JA, Barton JK. Targeting abasic sites and single base bulges in DNA with metalloinsertors. J Am Chem Soc. 2008;130:7530–7531. doi: 10.1021/ja801479y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benner K, Granzhan A, Ihmels H, Viola G. Targeting abasic sites in DNA by aminoalkyl-substituted carboxamidoacridizinium derivatives and acridizinium-adenine conjugates. Eur J Org Chem. 2007;2007:4721–4730. [Google Scholar]
- 76.Buzzeo MC, Barton JK. Redmond Red as a redox probe for the DNA-mediated detection of abasic sites. Bioconjugate Chem. 2008;19:2110–2112. doi: 10.1021/bc800339y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoshimoto K, Atsumi H, Saito S, Okuma M, Maeda M, Nagasaki Y. Fluorescence-based affinity labeling of nucleobase by hydrogenbond forming metal complex. Nucleic Acids Symp Ser (Oxf) 2007;51:303–304. doi: 10.1093/nass/nrm152. [DOI] [PubMed] [Google Scholar]
- 78.Morita K, Sato Y, Seino T, Nishizawa S, Teramae N. Fluorescence and electrochemical detection of pyrimidine/purine transversion by a ferrocenyl aminonaphthyridine derivative. Org Biomol Chem. 2008;6:266–268. doi: 10.1039/b716682f. [DOI] [PubMed] [Google Scholar]
- 79.Cleaver JE, Bootsma D. Xeroderma pigmentosum: biochemical and genetic characteristics. Annu Rev Genet. 1975;9:19–38. doi: 10.1146/annurev.ge.09.120175.000315. [DOI] [PubMed] [Google Scholar]
- 80.Doniger J, Jacobson ED, Krell K, DiPaolo JA. Ultraviolet light action spectra for neoplastic transformation and lethality of Syrian hamster embryo cells correlate with spectrum for pyrimidine dimer formation in cellular DNA. Proc Nat Acad Sci USA. 1981;78:2378–2382. doi: 10.1073/pnas.78.4.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sutherland BM, Delihas NC, Oliver RP, Sutherland JC. Action spectra for ultraviolet light-induced transformation of human cells to anchorage-independent growth. Cancer Res. 1981;41:2211–2214. [PubMed] [Google Scholar]
- 82.Hart RW, Setlow RB, Woodhead AD. Evidence that pyrimidine dimers in DNA can give rise to tumors. Proc Nat Acad Sci USA. 1977;74:5574–5578. doi: 10.1073/pnas.74.12.5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ley RD. Photoreactivation of UV-induced pyrimidine dimers and erythema in the marsupial Monodelphis domestica. Proc Nat Acad Sci USA. 1985;82:2409–2411. doi: 10.1073/pnas.82.8.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ananthaswamy HN, Pierceall WE. Molecular mechanisms of ultraviolet radiation carcinogenesis. Photochem Photobiol. 1990;52:1119–1136. doi: 10.1111/j.1751-1097.1990.tb08452.x. [DOI] [PubMed] [Google Scholar]
- 85.Cadet J, Gentner NE, Rözga B, Paterson MC. Rapid quantitation of ultraviolet-induced thymine-containing dimers in human cell DNA by reversed-phase high-performance liquid chromatography. J Chromatogr A. 1983;280:99–108. doi: 10.1016/s0021-9673(00)91543-7. [DOI] [PubMed] [Google Scholar]
- 86.Ley RD. Immunological detection of two types of cyclobutane pyrimidine dimers in DNA. Cancer Res. 1983;43:41–45. [PubMed] [Google Scholar]
- 87.Bykov VJ, Kumar R, Forsti A, Hemminki K. Analysis of UV-induced DNA photoproducts by 32P-postlabelling. Carcinogenesis. 1995;16:113–118. doi: 10.1093/carcin/16.1.113. [DOI] [PubMed] [Google Scholar]
- 88.Podmore ID, Cooke MS, Herbert KE, Lunec J. Quantitative determination of cyclobutane thymine dimers in DNA by stable isotope-dilution mass spectrometry. Photochem Photobiol. 1996;64:310–315. doi: 10.1111/j.1751-1097.1996.tb02463.x. [DOI] [PubMed] [Google Scholar]
- 89.Douki T, Court M, Sauvaigo S, Odin F, Cadet J. Formation of the main UV-induced thymine dimeric lesions within isolated and cellular DNA as measured by high performance liquid chromatography-tandem mass spectrometry. J Biol Chem. 2000;275:11678–11685. doi: 10.1074/jbc.275.16.11678. [DOI] [PubMed] [Google Scholar]
- 90.Clingen PH, Arlett CF, Cole J, Waugh APW, Lowe JE, Harcourt SA, Hermanova N, Roza L, Mori T, Nikaido O, Green MHL. Corrlelation of UVC and UVB cytotoxicity with the induction of specific photoproducts in T-lymphocytes and fibroblasts from normal human donors. Photochem Photobiol. 1995;61:163–170. doi: 10.1111/j.1751-1097.1995.tb03955.x. [DOI] [PubMed] [Google Scholar]
- 91.Taylor JS, Cohrs MP. DNA, light, and Dewar pyrimidinones: the structure and biological significance to TpT3. J Am Chem Soc. 1987;109:2834–2835. [Google Scholar]
- 92.Kelly GE, Meilke WD, Moore DE. Enhancement of UV-induced skin carcinogenesis by azathioprine: role of photochemical sensitisation. Photochem Photobiol. 1989;49:59–65. doi: 10.1111/j.1751-1097.1989.tb04078.x. [DOI] [PubMed] [Google Scholar]
- 93.Mitchell DL, Nairn RS. The biology of the (6–4) photoproduct. Photochem Photobiol. 1989;49:805–819. doi: 10.1111/j.1751-1097.1989.tb05578.x. [DOI] [PubMed] [Google Scholar]
- 94.Mitchell DL. The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem Photobiol. 1988;48:51–57. doi: 10.1111/j.1751-1097.1988.tb02785.x. [DOI] [PubMed] [Google Scholar]
- 95.Cleaver JECF, Karentz D, Lutze LH, Morgan WF, Player AN, Vuksanovic L, Mitchell DL. The relative biological importance of cyclobutane and (6-4) pyrimidine-pyrimidone dimer photoproducts in human cells: evidence from a xeroderma pigmentosum revertant. Photochem Photobiol. 1988;48:41–49. doi: 10.1111/j.1751-1097.1988.tb02784.x. [DOI] [PubMed] [Google Scholar]
- 96.Carell T. Sunlight-damaged DNA repaired with sunlight. Angew Chem Int Ed. 1995;34:2491–2494. [Google Scholar]
- 97.Taylor JS. Unraveling the Molecular Pathway from Sunlight to Skin Cancer. Acc Chem Res. 1994;27:76–82. [Google Scholar]
- 98.Kemmink J, Boelens R, Koning T, van der Marel GA, van Boom JH, Kaptein R. 1H NMR study of the exchangeable protons of the duplex d(GCGTTGCG). d(CGCAACGC) containing a thymine photodimer containing a thymine photodimer. Nucl Acids Res. 1987;15:4645–4653. doi: 10.1093/nar/15.11.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lawrence CW, Banerjee SK, Borden A, LeClerc JE. T-T Cyclobutane dimers are misinstructive, rather than non-instructive, mutagenic lesions. Mol Gen Genet. 1990;222:166–168. doi: 10.1007/BF00283040. [DOI] [PubMed] [Google Scholar]
- 100.Banerjee SK, Borden A, Christensen RB, LeClerc JE, Lawrence CW. SOS-dependent replication past a single trans-syn T-T cyclobutane dimer gives a different mutation spectrum and increased error rate compared with replication past this lesion in uninduced cells. J Bacteriol. 1990;172:2105–2112. doi: 10.1128/jb.172.4.2105-2112.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smith CA, Wang M, Jiang N, Che L, Zhao X, Taylor J-S. Mutation spectra of M13 vectors containing site-specific cis-syn, trans-syn-I, (6-4), and Dewar pyrimidone photoproducts of thymidyl-(3′→5′)-thymidine in Escherichia coli under SOS conditions. Biochemistry. 1996;35:4146–4154. doi: 10.1021/bi951975c. [DOI] [PubMed] [Google Scholar]
- 102.Tang M, Pham P, Shen X, Taylor J-S, O’Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 103.Smith CA, Baeten J, Taylor JS. The ability of a variety of polymerases to synthesize past site-specific cis-syn, trans-syn-II, (6-4), and dewar photoproducts of thymidylyl-(3′→5′)-thymidine. J Biol Chem. 1998;273:21933–21940. doi: 10.1074/jbc.273.34.21933. [DOI] [PubMed] [Google Scholar]
- 104.Gibbs PEM, Borden A, Lawrence CW. The T-T pyrimidine (6-4) pyrimidinone UV photoproduct is much less mutagenic in yeast than in Escherichia coli. Nucl Acids Res. 1995;23:1919–1922. doi: 10.1093/nar/23.11.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carty MP, Lawrence CW, Dixon K. Complete replication of plasmid DNA containing a single UV-induced lesion in human cell extracts. J Biol Chem. 1996;271:9637–9647. doi: 10.1074/jbc.271.16.9637. [DOI] [PubMed] [Google Scholar]
- 106.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 107.Berdis AJ. Dynamics of translesion DNA synthesis catalyzed by the bacteriophage T4 exonuclease-deficient DNA polymerase. Biochemistry. 2001;40:7180–7191. doi: 10.1021/bi0101594. [DOI] [PubMed] [Google Scholar]
- 108.Zhang X, Lee I, Berdis AJ. A potential chemotherapeutic strategy for the selective inhibition of promutagenic DNA synthesis by nonnatural nucleotides. Biochemistry. 2005;44:13111–13121. doi: 10.1021/bi050584n. [DOI] [PubMed] [Google Scholar]
- 109.McCullough AK, Dodson ML, Schärer OD, Lloyd RS. The role of base-flipping in damage recognition and catalysis by T4 endonuclease V. J Biol Chem. 1997;272:27210–27217. doi: 10.1074/jbc.272.43.27210. [DOI] [PubMed] [Google Scholar]
- 110.Vassylyev DG, Kashiwagi T, Mikami Y, Ariyoshi M, Iwai S, Ohtsuka E, Morikawa K. Atomic model of a pyrimidine dimer excision repair enzyme complexed with a DNA substrate: Structural basis for damaged DNA recognition. Cell. 1995;83:773–782. doi: 10.1016/0092-8674(95)90190-6. [DOI] [PubMed] [Google Scholar]
- 111.McCullough AK, Schärer O, Verdine GL, Lloyd RS. Structural determinants for specific recognition by T4 endonuclease V. J Biol Chem. 1996;271:32147–32152. doi: 10.1074/jbc.271.50.32147. [DOI] [PubMed] [Google Scholar]
- 112.Raney KD, Sowers LC, Millar DP, Benkovic SJ. A fluorescence-based assay for monitoring helicase activity. Proc Nat Acad Sci USA. 1994;91:6644–6648. doi: 10.1073/pnas.91.14.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lawley PD, Brookes P, Magee PN, Craddock VM, Swann PF. Methylated bases in liver nucleic acids from rats treated with dimethylnitrosamine. Biochim Biophys Acta. 1968;157:646–648. doi: 10.1016/0005-2787(68)90167-6. [DOI] [PubMed] [Google Scholar]
- 114.Craddock VM. The pattern of methylated purines formed in DNA of intact and regenerating liver of rats treated with the carcinogen dimethylnitrosamine. Biochim Biophys Acta. 1973;312:202–210. doi: 10.1016/0005-2787(73)90365-1. [DOI] [PubMed] [Google Scholar]
- 115.Hecht SS. DNA adduct formation from tobacco-specific N-nitrosamines. Mut Res. 1999;424:127–142. doi: 10.1016/s0027-5107(99)00014-7. [DOI] [PubMed] [Google Scholar]
- 116.Lao Y, Villalta PW, Sturla SJ, Wang M, Hecht SS. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatograpy-electrospray ionization-tandem mass spectrometry. Chem Res Toxicol. 2006;19:674–682. doi: 10.1021/tx050351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Peterson LA. N-Nitrosobenzylmethylamine is activated to a DNA benzylating agent in rats. Chem Res Toxicol. 1997;10:19–26. doi: 10.1021/tx9601014. [DOI] [PubMed] [Google Scholar]
- 118.Moschel RC, Hudgins WR, Dipple A. Aralkylation of guanosine by the carcinogen N-nitroso-N-benzylurea. J Org Chem. 1980;45:533–535. [Google Scholar]
- 119.Pegg AE, Dolan ME, Moschel RC, Waldo EC, Kivie M. Progress in Nucleic Acid Research and Molecular Biology. Academic Press; 1995. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase; pp. 167–223. [DOI] [PubMed] [Google Scholar]
- 120.Margison GP, Santibanez Koref MF, Povey AC. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis. 2002;17:483–487. doi: 10.1093/mutage/17.6.483. [DOI] [PubMed] [Google Scholar]
- 121.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Choi JY, Chowdhury G, Zang H, Angel KC, Vu CC, Peterson LA, Guengerich FP. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J Biol Chem. 2006;281:38244–38256. doi: 10.1074/jbc.M608369200. [DOI] [PubMed] [Google Scholar]
- 123.Woodside AM, Guengerich FP. Effect of the O6 substituent on misincorporation kinetics catalyzed by DNA polymerases at O6-methylguanine and O6-benzylguanine. Biochemistry. 2002;41:1027–1038. doi: 10.1021/bi011495n. [DOI] [PubMed] [Google Scholar]
- 124.Eoff RL, Angel KC, Egli M, Guengerich FP. Molecular basis of selectivity of nucleoside triphosphate incorporation opposite O6-benzylguanine by Sulfolobus solfataricus DNA polymerase Dpo4: steady-state and pre-steady state kinetics and X-ray crystallography of correct and incorrect pairing. J Biol Chem. 2007;282:13573–13584. doi: 10.1074/jbc.M700656200. [DOI] [PubMed] [Google Scholar]
- 125.Eoff RL, Irimia A, Egli M, Guengerich FP. Sulfolobus solfataricus DNA polymerase Dpo4 is partially inhibited by “wobble” pairing between O6-methylguanine and cytosine, but accurate bypass is preferred. J Biol Chem. 2007;282:1456–1467. doi: 10.1074/jbc.M609661200. [DOI] [PubMed] [Google Scholar]
- 126.Pauly GT, Moschel RC. Mutagenesis by O6-methyl-, O6-ethyl-, and O6-benzylguanine and O4-methylthymine in human cells: effects of O6-alkylguanine-DNA alkyltransferase and mismatch Repair. Chem Res Toxicol. 2001;14:894–900. doi: 10.1021/tx010032f. [DOI] [PubMed] [Google Scholar]
- 127.Lindahl T, Sedgwick B, Sekiguchi M, Nakabeppu Y. Regulation and expression the adaptive response to alkylating agents. Annu Rev Biochem. 1988;57:133–157. doi: 10.1146/annurev.bi.57.070188.001025. [DOI] [PubMed] [Google Scholar]
- 128.Mishina Y, Duguid EM, He C. Direct reversal of DNA alkylation damage. Chem Rev. 2006;106:215–232. doi: 10.1021/cr0404702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Graves RJ, Li BFL, Swann PF. Repair of O6-methylguanine, O6-ethylguanine, O6-isopropylguanine, and O4-methylthymine in synthetic oligodeoxynucleotides by Escherichia coli ada gene O6-alkylguanine-DNA-alkyltransferase. Carcinogenesis. 1989;10:661–666. doi: 10.1093/carcin/10.4.661. [DOI] [PubMed] [Google Scholar]
- 130.Lips J, Kaina B. Repair of O6-methylguanine is not affected by thymine base pairing and the presence of MMR proteins. Mut Res. 2001;487:59–66. doi: 10.1016/s0921-8777(01)00105-7. [DOI] [PubMed] [Google Scholar]
- 131.Gong J, Sturla SJ. A synthetic nucleoside probe that discerns a DNA adduct from unmodified DNA. J Am Chem Soc. 2007;129:4882–4883. doi: 10.1021/ja070688g. [DOI] [PubMed] [Google Scholar]
- 132.Rosemeyer H, Toth G, Golankiewicz B, Kazimierczuk Z, Bourgeois W, Kretschmer U, Muth HP, Seela F. Syn-anti conformational analysis of regular and modified nucleosides by 1D 1H NOE difference spectroscopy: a simple graphical method based on conformationally rigid molecules. J Org Chem. 1990;55:5784–5790. [Google Scholar]
- 133.Haschemeyer AE, Sobell HM. The crystal structure of a hydrogen bonded complex of deoxyguanosine and 5-bromodeoxycytidine. Acta Crystallogr. 1965;19:125–130. doi: 10.1107/s0365110x65002864. [DOI] [PubMed] [Google Scholar]
- 134.Guckian KM, Krugh TR, Kool ET. Solution structure of a nonpolar, non-hydrogen-bonded base pair surrogate in DNA. J Am Chem Soc. 2000;122:6841–6847. doi: 10.1021/ja994164v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guckian KM, Morales JC, Kool ET. Structure and base pairing properties of a replicable nonpolar isostere for deoxyadenosine. J Org Chem. 1998;63:9652–9656. doi: 10.1021/jo9805100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McMinn DL, Ogawa AK, Wu Y, Liu J, Schultz PG, Romesberg FE. Efforts toward expansion of the genetic alphabet: DNA polymerase recognition of a highly stable, self-pairing hydrophobic base. J Am Chem Soc. 1999;121:11585–11586. [Google Scholar]
- 137.Henry AA, Yu C, Romesberg FE. Determinants of unnatural nucleobase stability and polymerase recognition. J Am Chem Soc. 2003;125:9638–9646. doi: 10.1021/ja035398o. [DOI] [PubMed] [Google Scholar]
- 138.Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, Sauvaigo S. Hydroxyl radicals and DNA base damage. Mut Res. 1999;424:9–21. doi: 10.1016/s0027-5107(99)00004-4. [DOI] [PubMed] [Google Scholar]
- 139.Kasai H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mut Res. 1997;387:147–163. doi: 10.1016/s1383-5742(97)00035-5. [DOI] [PubMed] [Google Scholar]
- 140.Steenken S, Jovanovic SV, Bietti M, Bernhard K. The trap depth (in DNA) of 8-oxo-7,8-dihydro-2′-deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J Am Chem Soc. 2000;122:2373–2374. [Google Scholar]
- 141.Yanagawa H, Ogawa Y, Ueno M. Redox ribonucleosides. Isolation and characterization of 5- hydroxyuridine, 8-hydroxyguanosine, and 8-hydroxyadenosine from Torula yeast RNA. J Biol Chem. 1992;267:13320–13326. [PubMed] [Google Scholar]
- 142.Tretyakova NY, Niles JC, Burney S, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced reactions of synthetic oligonucleotides containing 8-oxoguanine. Chem Res Toxicol. 1999;12:459–466. doi: 10.1021/tx980235c. [DOI] [PubMed] [Google Scholar]
- 143.Gasparutto D, Da Cruz S, Bourdat AG, Jaquinod M, Cadet J. Synthesis and biochemical properties of cyanuric acid nucleoside-containing DNA oligomers. Chem Res Toxicol. 1999;12:630–638. doi: 10.1021/tx980255e. [DOI] [PubMed] [Google Scholar]
- 144.Duarte V, Gasparutto D, Jaquinod M, Cadet J. In vitro DNA synthesis opposite oxazolone and repair of this DNA damage using modified oligonucleotides. Nucl Acids Res. 2000;28:1555–1563. doi: 10.1093/nar/28.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Duarte V, Gasparutto D, Jaquinod M, Ravanat JL, Cadet J. Repair and mutagenic potential of oxaluric acid, a major product of singlet oxygen-mediated oxidation of 8-oxo-7,8-dihydroguanine. Chem Res Toxicol. 2001;14:46–53. doi: 10.1021/tx0001629. [DOI] [PubMed] [Google Scholar]
- 146.Henderson PT, Delaney JC, Gu F, Tannenbaum SR, Essigmann JM. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry. 2002;41:914–921. doi: 10.1021/bi0156355. [DOI] [PubMed] [Google Scholar]
- 147.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 148.Delaney S, Neeley WL, Delaney JC, Essigmann JM. The substrate specificity of MutY for hyperoxidized guanine lesions in vivo. Biochemistry. 2007;46:1448–1455. doi: 10.1021/bi061174h. [DOI] [PubMed] [Google Scholar]
- 149.Luo W, Muller JG, Burrows CJ. The pH-dependent role of superoxide in riboflavin-catalyzed photooxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 150.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 151.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 152.Adam W, Arnold MA, Grune M, Nau WM, Pischel U, Saha-Moller CR. Spiroiminodihydantoin Is a major product in the photooxidation of 2′-deoxyguanosine by the triplet states and oxyl radicals generated from hydroxyacetophenone photolysis and dioxetane thermolysis. Org Lett. 2002;4:537–540. doi: 10.1021/ol017138m. [DOI] [PubMed] [Google Scholar]
- 153.Wood ML, Esteve A, Morningstar ML, Kuziemko GM, Essigmann JM. Genetic effects of oxidative DNA damage: comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in Escherichia coli. Nucl Acids Res. 1992;20:6023–6032. doi: 10.1093/nar/20.22.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Grollman AP, Moriya M. Mutagenesis by 8-oxoguanine: an enemy within. Trends in Genetics. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 155.Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mut Res. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- 156.Helbock HJ, Beckman KB, Ames BN, Lester P. Methods Enzymol. Academic Press; 1999. 8-Hydroxydeoxyguanosine and 8-hydroxyguanine as biomarkers of oxidative DNA damage; pp. 156–166. [DOI] [PubMed] [Google Scholar]
- 157.Nakae Y, Stoward P, Bespalov I, Melamede R, Wallace S. A new technique for the quantitative assessment of 8-oxoguanine in nuclear DNA as a marker of oxidative stress. Application to dystrophin-deficient DMD skeletal muscles. Histochem Cell Biol. 2005;124:335–345. doi: 10.1007/s00418-005-0037-5. [DOI] [PubMed] [Google Scholar]
- 158.Soultanakis RP, Melamede RJ, Bespalov IA, Wallace SS, Beckman KB, Ames BN, Taatjes DJ, Janssen-Heininger YMW. Fluorescence detection of 8-oxoguanine in nuclear and mitochondrial DNA of cultured cells using a recombinant Fab and confocal scanning laser microscopy. Free Radical Biol Med. 2000;28:987–998. doi: 10.1016/s0891-5849(00)00185-4. [DOI] [PubMed] [Google Scholar]
- 159.Siomek A, Rytarowska A, Szaflarska-Poplawska A, Gackowski D, Rozalski R, Dziaman T, Czerwionka-Szaflarska M, Olinski R. Helicobacter pylori infection is associated with oxidatively damaged DNA in human leukocytes and decreased level of urinary 8-oxo-7,8-dihydroguanine. Carcinogenesis. 2006;27:405–408. doi: 10.1093/carcin/bgi238. [DOI] [PubMed] [Google Scholar]
- 160.Gielazyn ML, Ringwood AH, Piegorsch WW, Stancyk SE. Detection of oxidative DNA damage in isolated marine bivalve hemocytes using the comet assay and formamidopyrimidine glycosylase (Fpg) Mut Res. 2003;542:15–22. doi: 10.1016/j.mrgentox.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 161.Mugweru A, Wang B, Rusling J. Voltammetric sensor for oxidized DNA using ultrathin films of osmium and ruthenium metallopolymers. Anal Chem. 2004;76:5557–5563. doi: 10.1021/ac049375j. [DOI] [PubMed] [Google Scholar]
- 162.Dennany L, Forster RJ, White B, Smyth M, Rusling JF. Direct electrochemiluminescence detection of oxidized DNA in ultrathin films containing [Os(bpy)2(PVP)10]2+ J Am Chem Soc. 2004;126:8835–8841. doi: 10.1021/ja048615+. [DOI] [PubMed] [Google Scholar]
- 163.Hah SS, Mundt JM, Kim HM, Sumbad RA, Turteltaub KW, Henderson PT. Measurement of 7,8-dihydro-8-oxo-2′-deoxyguanosine metabolism in MCF-7 cells at low concentrations using accelerator mass spectrometry. Proc Nat Acad Sci. 2007;104:11203–11208. doi: 10.1073/pnas.0701733104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nakagawa O, Ono S, Li Z, Tsujimoto A, Sasaki S. Specific fluorescent probe for 8-oxoguanosine. Angew Chem Int Ed. 2008;47:8983. doi: 10.1002/anie.200700671. [DOI] [PubMed] [Google Scholar]
- 165.Nasr T, Li Z, Nakagawa O, Taniguchi Y, Ono S, Sasaki S. Selective fluorescence quenching of the 8-oxoG-clamp by 8-oxodeoxyguanosine in ODN. Bioorg Med Chem Lett. 2009;19:727–730. doi: 10.1016/j.bmcl.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 166.Greco NJ, Sinkeldam RW, Tor Y. An emissive C analog distinguishes between G, 8-oxoG, and T. Org Lett. 2009;11:1115–1118. doi: 10.1021/ol802656n. [DOI] [PMC free article] [PubMed] [Google Scholar]