

1. Introduction
For almost two decades, there has been interest in using viruses to deliver genes into cells. One particular approach consists of oncolytic viruses (OVs), which can selectively enter and replicate in neoplastic cells leading to their lytic destruction with minimal damage to surrounding normal tissue. OVs include a wide range of viruses that have been selected or genetically engineered such that viral replication is limited to permissive cancer cells with specific mutated cellular pathways. OVs have been designed to replicate only in tumors that have either activation of specific oncogenes or inactivation of specific tumor suppressor pathways.1–7 Table 1 presents an overview of specific OVs along with their salient properties that are being studied for the treatment of malignant gliomas.
Table 1. Features of Oncolytic Viruses Being Used for Glioma Therapy.
| OV | genome | Structure | salient properties |
|---|---|---|---|
| herpes simplex virus | dsDNA, 120–200 kb | enveloped, 150-200 nm |
|
| adenovirus | dsDNA, 36–38 kb | nonenveloped, icosahedral, 70–90 nm |
|
| Newcastle disease virus | ssRNA, 16–20 kb | enveloped, helical, 150–300 nm |
|
| reovirus | dsRNA, 22–27 kb | nonenveloped, icosahedral, 60–80 nm |
|
Some OVs demonstrate selective tropism for entry into tumor cells.8–12 Second generation viruses that are “armed” by incorporation of prodrug activating genes,13–20 imaging genes,21,22 immunostimulatory genes,23–27 and antiangiogenesis genes28,29 are currently being investigated for safety and efficacy.
The appropriate route of delivery of OVs remains to be defined in terms of advantages and disadvantages. For example, intratumoral viral delivery has the advantage of circumventing rapid viral clearance within the bloodstream due to antibody and complement neutralization of the virus, clearance by the liver, viral binding to nontumor cells that contain receptors for the virus, and barriers to migration across the vascular endothelium. However, intravenous administration is the route of choice for the treatment of both primary tumors that are not locally confined and metastatic disease. Methods of avoiding these limitations to viral administration will be discussed later in this review and include the development of various stealth agents and carrier cells to achieve nonimmunogenic viral delivery.
With China's recent approval of the first oncolytic virus, adenovirus H101,30 a number of clinical trials are underway in the United States and Europe. Table 2 presents a summary of the glioma clinical trials that have been performed to date.31–38 Through the process of testing OVs in the clinic, however, a number of questions must be addressed. For instance, the pharmacokinetics of viral infection, replication, and spread should be ascertained noninvasively. Two novel oncolytic measles viruses are attempting to answer these questions. First, a measles virus encoding the soluble extracellular human carcinoembryonic antigen (CEA) allows for noninvasive analysis of viral propagation by measuring CEA levels.39,40 Second, by incorporating the thyroidal sodium iodide symporter in the measles vector, clinicians are able to use radioactive iodine tracers in order to monitor the status of viral infection using single-photon-emission computed tomography or positron-emission tomography.21,41–43 Beyond questions related to pharmacokinetics, clinical implementation of OVs is hampered by technical challenges in producing large amounts of high-titer virus. Lastly, performance of phase III clinical trials to assess clinical utility and guide the future directions of basic research in the field of OV therapy is needed.
Table 2. Clinical Trials Using Oncolytic Viruses for the Treatment of Malignant Gliomasa.
| OV | virus | genetic alteration | patients | route of delivery | highest dose | median survival months) | ref |
|---|---|---|---|---|---|---|---|
| G207 | HSV-1 (strain F) | deletion of both γ34.5 copies and disruption of ICP6/RR | 21 | i.t. | 1 × 109 pfu | 6 | 31 |
| 6 | 2 doses: i.t. and I.A.B. post-tumor resection | 1.15 × 109 pfu | 6.6 | 32 | |||
| 1716 | HSV-1 (Glasgow strain 17) | deletion of both γ34.5 copies | 9 | i.t. | 1 × 105 pfu | NI | 33 |
| 12 | i.t. 4–9 days prior to resection | 1 × 105 pfu | NI | 34 | |||
| 12 | IAB post-tumor resection | 1 × 105 pfu | NI | 35 | |||
| ONYX-015 | adenovirus | deleted E1B gene | 24 | IAB post-tumor resection | 1 × 1010 pfu | 6 | 36 |
| NDV-HuJ | Newcastle disease virus | none | 14 | iv | 55 BIU | 8 | 37 |
| reolysin | reovirus | none | 12 | i.t | 1 × 109 pfu | 5 | 38 |
Legend: BIU, billion infectious units; IAB., injected into adjacent brain; i.t., intratumoral; iv, intravenous; NI, not included; pfu, plaque-forming units.
For instance, preclinical data suggests that the ability of OVs to amplify within cancer cells should lead to increased intratumoral titers independent of the initial inoculum.1,27,44–48 While these findings have been corroborated by numerous in vitro findings, clinical efficacy has been limited due to significantly attenuated in vivo viral replication.49–56 In fact, a recent clinical trial shows replication of inoculated virus in tumor, albeit at levels that appear to be fairly reduced.32,57 Attenuated in vivo viral replication may be due to inefficient intratumoral viral dispersal, to barriers imposed by the tumor microenvironment, or to rapid viral clearance by host immune responses. Future clinical trials will need to take these host factors into account in order to achieve maximal OV-mediated tumoricidal activity while simultaneously avoiding systemic toxicity to the host. Elucidation of a variety of tumor- and host-based factors that limit viral infection, replication, and propagation could lead to the design of combinatorial molecular approaches combining oncolysis with pharmacologic agents designed to circumvent such host barriers to OV lysis of tumors. Additionally, certain classes of pharmacological agents can alter cellular homeostasis and activate cellular cascades that provide an environment conducive for viral replication. In this review, we will briefly describe both the current state of knowledge of host responses that limit OV therapy and the cellular pathways that can be targeted to enhance OV efficacy, followed by a review of potential pharmacologic and chemical approaches that could be employed to circumvent these obstacles.
2. The Immediate Host Response Following OV Infection
A major assumption in the area of OV therapy of tumors has been that even a small initial dose of a replication-competent OV will amplify through successive rounds of viral replication, resulting in eventual infection and eradication of the entire tumor. In reality, however, viral distribution appears stunted, and viral yields within tumors actually decrease as a function of time.1,57 In order to fully explain this, one needs to evaluate how oncolytic viruses function within the context of the tumor microenvironment; in fact, it is critical to understand what is required for effective viral-mediated tumor killing and what could limit viral replication.
Limitations imposed within the tumor microenvironment render tumor clearance by replicating OV difficult as a single modality.58 Sequential steps predicted to occur during OV killing include (a) infection of individual cells, replication within them, and subsequent cell death, (b) induction of an adaptive antitumor immune response triggered by viral infection, and (c) stimulation of localized inflammation. A consequence of the immunosuppressive nature of certain tumor types, particularly gliomas with their low MHC I/II expression,59 has required investigators to create novel methods for modulating host immunity in order to achieve potent antitumor immune responses. For instance, the lack of immunostimulatory signaling present on intracranial glioma cells was circumvented using an oncolytic herpes simplex virus (HSV) vector expressing IL-4. This virus was able to mediate antitumor efficacy not only through viral oncolysis but also by induction of a CD4 and CD8 antitumor response.58 An additional barrier toward achieving significant antitumor immunity in the context of OV is the ability of certain viral vectors, such as HSV-1 through its ICP47 protein, to circumvent the host response by blocking antigen presentation on MHC I upon viral infection.60–62 In order to address this limitation, a novel HSV was produced that lacked the gene encoding ICP47, and when it was injected into an intracranial glioma model, significant T cell stimulation was achieved.63 As a result, through a combination of viral tumor clearance and immune modulation, a “perfect storm” of tumor killing could potentially occur: replication of the virus in a dying tumor, secretion of pro-inflammatory cytokines, and recruitment of inflammatory cells into the tumor microenvironment leading to tumor cell destruction.58,63–65
However, while viral replication can work synergistically with the immune system to elicit tumor killing, the immune system also functions as a “double-edged sword”. Upon viral infection, a series of antiviral mechanisms are activated via the initial innate immune response to the virus. This response has been demonstrated to limit successful viral propagation and tumor clearance.66–70 Consequently, these barriers must be circumvented to achieve viral replication and the subsequent activation of an adaptive antitumor immune response.2 These barriers consist of intracellular signaling and antiviral defenses,6,68,71,72 extracellular tumor environmental barriers,65,72,73 and the active host response to ongoing oncolytic virotherapy1,12,51,54,65,69,74–80 (Figure 1). In this section, we will overview the effect of each of these barriers on oncolysis.
Figure 1.
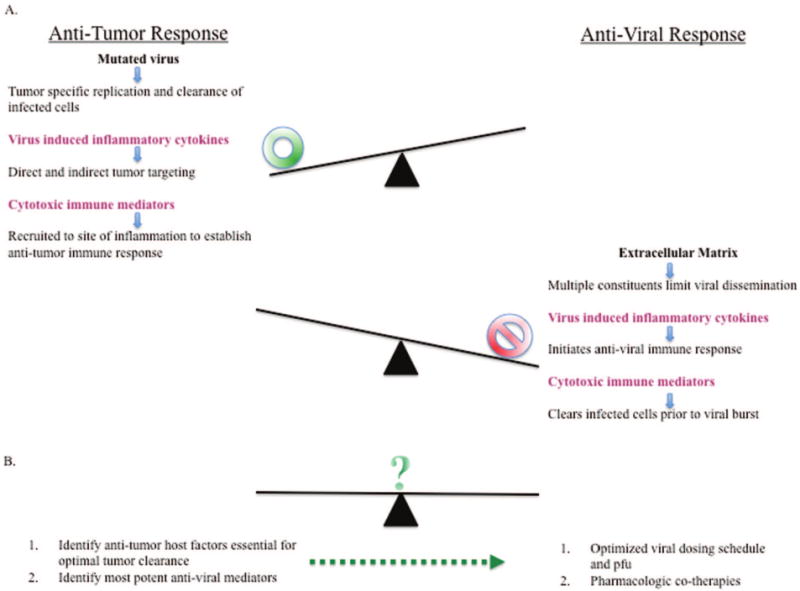
(a) The host response to OV therapy represents a unique interplay between host factors that have the capacity to both limit viral efficacy and elicit enhanced tumor killing. For instance, the inflammatory cytokine milieu following viral infection can consist of tumor necrosis factor (TNF)-α, which has the capacity to culminate in tumor regression, while inducible nitric oxide synthase (iNOS) and interferon (IFN)-γ are potent antiviral mediators. Similarly, CD8 cytotoxic T cells have the ability to selectively recognize and lyse tumor cells via a CD8-dependent mechanism. However, natural killer (NK) cells are among the rapid responders to viral infection that attempt to limit viral spread. (b) Due to the dichotomous nature of virus elicited host responses, continuing efforts are needed to clarify the contribution of components of the tumor microenvironment that both limit and enhance viral replication and spread. As the most critical factors are elucidated, they must be translated into pharmacological targets that can be paired with OVs to result in additive or synergistic tumor cell killing. In order to meet this objective, extensive studies will need to determine appropriate quantities of virus and drug along with proper dosing schedules that result in tumor clearance and limited host toxicity.
2.1. Tumor Cell Antiviral Response
Pattern recognition receptors have evolved to detect invading pathogens, and they fall into two broad categories: toll-like receptors (TLRs) and RIG-I-like helicases (Figure 2). TLRs are abundantly expressed on plasmacytoid dendritic cells (pDCs), are found on either cell surfaces or endosomes where they detect a variety of pathogen-associated molecular patterns (PAMP),81 and transmit their downstream signals through their cytoplasmic Toll/interleukin (IL)-1 (TIR) domain. The response of ligand binding to a TLR depends on the TIR adapter protein that is associated with each TLR. With the exception of TLR-3, myeloid differentiation primary response protein 88 (MyD88) associates with the TIR domain of each TLR and ultimately leads to the downstream activation of nuclear factor kappa B (NF-κB) and the production of various inflammatory cytokines, including tumor necrosis factor α (TNF-α), IL-6, and IL-1β. TLR-3, however, recognizes double-stranded RNA (dsRNA) and rather than signaling through MyD88, it associates with Toll/IL-1 receptor domain containing adaptor protein inducing interferon β (TRIF) to activate both NF-κB and interferon regulatory factor 3 (IRF-3). IRF-3 activation leads to its translocation into the nucleus and the induction of IFN-β expression.82,83
Figure 2.
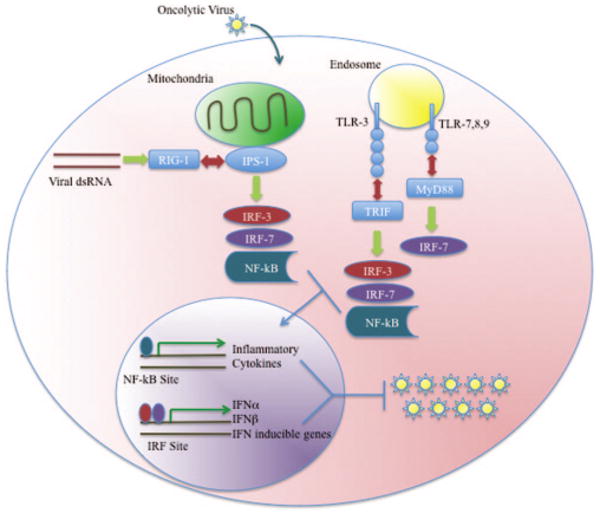
Viral infection elicits a variety of antiviral cellular responses. Following viral infection, viral pathogen-associated molecular patterns are detected through both TLR and RIG pathways as described in the text. Following the activation of each pathway, signals are relayed to interferon regulatory factors and NF-κB, leading to their translocation from the cytoplasm into the nucleus. Upon arrival in the nucleus, they activate the transcription of a variety of antiviral mediators that limit viral replication and spread.
pDCs function as the host's professional interferon (IFN) producing cells due to their TLR expression, their ability to actively produce type I interferons (IFN-I), and their critical role in limiting viral infection.84 RIG-I, however, is a critical mediator of IFN production and viral clearance in the majority of cell types, including fibroblasts, epithelial cells, and conventional dendritic cells.84 This pattern recognition receptor is ubiquitously expressed, located in the cytosol where it detects dsRNA that is unique to virally infected cells, and signals through the mitochondrial membrane-associated interferon promoter stimulator 1 (IPS-1) adaptor protein. Once dsRNA binds to RIG-I, downstream signaling events reach IPS-1 and branch out into either NF-κB, IRF-3, or IRF-7 activation. IRF-3 is a constitutively expressed protein that shuttles between the cytoplasm and nucleus. Once IRF-3 is phosphorylated at its C-terminus, it remains localized in the nucleus where it serves as a transcription factor for IFN-β, IFN-α1, and RANTES (regulated on activation normal T cell expressed and secreted).85,86 While IRF-7 also requires a C-terminal phoshorylation event to become activated,87,88 it differs from IRF-3 in several ways: it is only constitutively expressed in B cells and DCs, while its expression elsewhere only occurs following viral infection or IFN induction; it has a half-life of 30 min;87,89,90 and it actively transcribes IFN-α4,7,14.86
Because the host has a variety of methods for detecting invading pathogens and ultimately producing IFN-1, this underscores the importance of IFN-I production as an antiviral mediator. For instance, experiments that have depleted the IFN ligand demonstrated the necessity of IFN-I in abrogating initial viral replication.91 While IFN-I is not intrinsically antiviral, its production is able to induce a variety of changes through binding to nearby IFN receptors leading to the downstream activation of IRF92–94 and interferon-stimulated genes (ISG) that are responsible for creating an antiviral state.93,95–97
In addition to the PAMPs previously described, the double-stranded RNA-activated protein kinase (PKR) is activated following IFN-I production.5 PKR recognizes foreign and abnormal nucleic acid structures that accompany viral infection.98 Binding of PKR to a dsRNA leads to an activated form of PKR that has the ability to phosphorylate eIF2α and halt cellular protein synthesis. Through PKR-induced abrogation of protein translation, the host cell is no longer able carry out viral protein production.
Most viruses have evolved mechanisms to counter intracellular defense responses. Interestingly, antiviral defenses are often disrupted in tumor cells. This can provide researchers with stratagems for engineering attenuated OVs that can selectively replicate only in cells that lack antiviral defense response. For example, vesicular stomatitis virus (VSV),6 reovirus,8 and myxoma virus9 are naturally sensitive to IFN so their replication is selective for tumor cells where this pathway has been reported to be defective. Similarly, reovirus, VSV, and oncolytic HSV-1 have been reported to selectively replicate in tumor cells with an activated Ras/MEK pathway, which can counter the activation of antiviral PKR in cells.99–102 However, evidence exists that despite altered IFN signaling pathways in certain tumors, oncolytic viruses are still subject to control by the innate immune defenses of human tumor cells.103 As a result, specific approaches aimed at circumventing these antiviral defenses may lead to enhanced viral replication and spread.
2.2. Extracellular Tumor Microenvironment Barriers
The extracellular tumor microenvironment (ECM) or “the cancer field” consists of secreted proteins, proteases, growth factors, stromal and immune cells, and tumor vasculature. The significance of the ECM in governing tumor growth and also its response to therapy is increasingly being appreciated. Recent studies investigating the complex interactions between OV therapy and tumor ECM have uncovered the highly significant impact of tumor microenvironment on oncolysis. After viral replication and lysis of the infected cell, the progeny OVs resulting from the “virus burst” have to spread from one infected cell to the next. The extracellular tumor microenvironment consists of secreted proteins and proteoglycans, which form an inhibitory scaffold limiting the spread of OV particles within the solid tumor.104 Apart from the physical inhibition, the acidic tumor microenvironment and high interstitial tumor pressure present additional obstacles for viral propagation and spread in the tissue105–108 (Figure 3).
Figure 3.
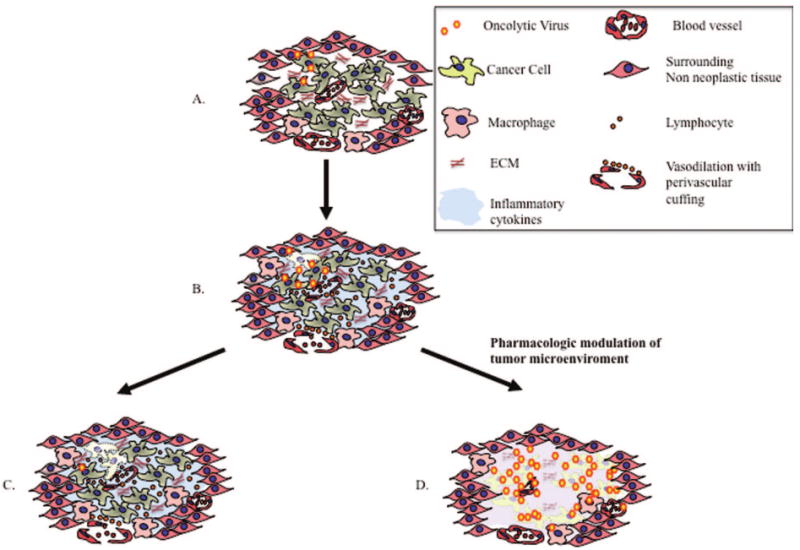
(A) Viral inoculation results in the infection of cancer cells and surrounding non-neoplastic tissue; however, only cancerous cells will support active viral replication. Just hours following viral inoculation, the tumor microenvironment undergoes a series of dynamic changes (B) that create a barrier for efficient viral replication and spread: (i) an angiogenic response with vasodilation and leakage of inflammatory cellular responders; (ii) elaboration of inflammatory cytokines that create an environment that is limiting for viral replication; (iii) the recruitment and activation of cells from the innate immune system; (iv) components of the ECM create an environment with high interstitial pressure that limits viral dissemination between individual cancer cells. If these responses to viral infection are not addressed, viral clearance will be seen within days of viral administration with limited tumor killing (C); however, each component of the host responses also provides a drug target that can be used to enhance OV efficacy using cotherapy. By tailoring OV therapy with pharmacologic agents, viral replication and spread can be enhanced with increased tumor killing (D).
Following wild-type viral infection, a classical physiologic response is vasodilation and hyperpermeability.109,110 This places blood vessels as an integral intermediary in the inflammatory host response during infection.111 During inflammation, peripheral leukocytes and monocytes extravasate into tissue by initially adhering to endothelial cells that line the vascular walls, ultimately leading to endothelial activation. This activation leads to subsequent hyperpermeability in the vascular walls and increased tissue edema, ultimately enhancing perivascular inflammatory cell infiltration.111,112 This vascular leakage can become detrimental to OV replication and spread within the tumor,80 thereby accentuating an additional factor that must be addressed in order to achieve successful OV therapy. Various approaches have been examined to enhance virotherapy by stabilizing tumor vasculature, reducing the neovascular response, and reducing the inflammatory cellular infiltrate.74,80
2.3. Immune Responses to Oncolytic Viruses and Its Cellular Mediators
Likely the most significant limitation to virotherapy is the active innate immune response to the virus that can occur fairly rapidly after OV infection. The innate immune system provides an initial potent line of defense that limits initial viral infection, replication, and spread; signals for the maturation of antigen-presenting cells; and activates the cellular components of the adaptive immune system. The importance of this concept has been elucidated in several models, including VSV, wherein initial intratumoral viral replication is followed by a dramatic decline in viral titers over the following days.113 Since antiviral antibodies were not produced until 5 days post-OV infection, the innate immune system response (including granulocytes, natural killer (NK) cells, NKT cells, and macrophages) that is recruited to the site of infection is considered a major player in limiting viral propagation.114 Depletion of mononuclear cells1 or antiviral cytokine mediators such as IFN-γ78 has been shown to cause a significant increase in intratumoral viral titers and anticancer effects.
While neutrophils are the first antiviral responders that are recruited to a site of infection, efficient viral clearance at the cellular level requires both NK cells and monocyte-derived cells. Activated NK cells14 mediate direct lysis of infected target cells by releasing cytotoxic granules containing lytic enzymes or by binding to apoptosis-inducing receptors on target cells.115 NK cell-mediated preferential lysis of HSV or vaccinia virus-infected cells has been shown to prevent viral dissemination to neighboring cells.116 While recruitment of NK cells to infected tumor tissue is limiting to viral spread and OV efficacy, IFN-γ production by NK cells has also been shown to set the stage for subsequent adaptive immune response.117,118
Apart from NK cells, macrophages also play a critical role in OV clearance. Upon viral infection, resident or recruited macrophages initially secrete IL-12 to activate NK cells, while NK cells complete the feedback loop by secreting IFN-γ, the prototypic macrophage activator, without which macrophages cannot clear microbes.117 In fact, recruitment of infiltrating monocytic cells has been shown to coincide with clearance of over 80% of HSV-derived oncolytic viral particles.54,78,119 Increased intratumoral presence of macrophage/microglia cells has also been reported in human patients treated with the adenovirus120,121 or HSV1-derived OV122 indicating the global significance of macrophages in OV therapy.
It is important to note that while the OV-mediated induction of an antiviral inflammatory state is thought to be detrimental toward oncolysis, a recent study indicated that it could also contribute to tumor killing. During inflammatory reactions, activated neutrophils adopt a “rigid” phenotype, which can result in clogging of small capillaries.13 Systemic delivery of VSV and vaccinia virus has been shown to initiate very robust recruitment of neutrophils from the vascular system into the tumor. So robust was this immune cell infiltration that it resulted in a choking of the blood vessels. The consequential increase in tumor hypoxia induced tumor cell apoptosis and contributed to tumor cell killing.65
While neutrophil-mediated choking of tumors may be beneficial, antibody-mediated neutrophil depletion facilitated extensive viral replication and spreading throughout the tumor.65 Additional studies with a variety of tumor models and oncolytic viruses will need to be performed in order to determine whether this mechanism of tumor cell death and inhibition of viral spread is dependent on tumor type, virus type, or route of OV administration.
Despite their antiviral properties, neutrophils and NK cells have pleiotropic effects that may also be critical in tumor killing. For instance, neutrophils, in addition to CD8 T cells, have been shown to contribute to HSV,49 VSV,50,65 and measles virus47,123 related virotherapy efficiency. Similarly, NK cells have been shown to augment the tumoricidal effects of oncolytic HSV. In a melanoma model, NK cells have been defined as an essential cellular component for VSV efficacy.50 In this model, NK cells functioned synergistically with the adaptive immune antitumor response, launched in response to viral antigens expressed by tumor cells. Therefore, it appears that NK cells can serve a dual function, both as potential inhibitors of viral replication and as critical mediators to establish an effective antitumor immunity following viral antigen presentation within the tumor cells. These findings further confirm the need for a refined approach to manipulate individual cell populations in order to maximize therapeutic regimens.
2.4. Intracellular Pathways Affecting Virotherapy
The metabolic/replication potential of a cell has a tremendous impact on OV replication. Intracellular changes in cell signaling cascades can transform a cell into a host that encourages or discourages OV propagation. For instance, the cellular stress response, induced by pharmacological treatment, results in a variety of changes that have significant impact on viral infection and dissemination. These can range from alterations in protein expression to changes in cell cycle status.
The acquisition and conservation of cellular genes by wild-type viruses for specific tasks has been historically documented.124,125 A defining feature of OV therapy adopts a similar approach whereby genetically engineered viruses frequently lack a specific gene whose function must be provided by the host cell in order to achieve successful viral propagation. An example of such a gene encoded by HSV-1 is γ34.5. The protein product of this gene, ICP34.5, precludes the shutoff of host protein synthesis and premature cell death.126 Notably, however, the carboxyl terminus of ICP34.5 has significant homology to the carboxyl terminus of mammalian growth arrest and DNA damaging inducible protein (GADD34),126 a cellular stress protein that circumvents apoptosis by suppressing cell division during DNA repair.127–129 GADD34 recruits protein phosphatase-1 and dephosphorylates the inactivated mRNA translation initiation factor eIF2α allowing for viral protein synthesis to occur. In the context of HSV OV therapy where the viral γ34.5 gene is frequently deleted to limit unintended virulence, the function of ICP34.5 appears to be provided in trans by GADD34.129 Taken together, identifying ways of enhancing GADD34 induction in the presence of HSV lacking γ34.5 may provide a useful strategy for enhancing virulence within the tumor targets. A similar type of engineering is provided by the finding that an activated MEK pathway in cells can substitute for the lack of γ34.5 function and allow robust replication of the γ34.5 mutant HSV-1 in vitro and in vivo. 130,131
The use of drugs to induce DNA damage also results in the stimulation of numerous cellular pathways. While different classes of these chemotherapeutic agents induce various DNA repair mechanisms, cisplatin will serve as a representative example. The DNA adducts formed from cisplatin treatment leads to activation of cell cycle checkpoints and a temporary induction of S-phase arrest followed by an extended G2/M arrest.132,133 When mild to moderate DNA damage is induced, cytotoxicity is not fully achieved since nucleotide excision repair is activated to remove DNA adducts and promote cellular survival. However, if extensive DNA damage is achieved, DNA repair fails to keep pace with DNA damage, repetitive futile rounds of mismatch repair create single-strand DNA breaks, and the serine/threonine kinase ATM and Rad3-related (ATR) is activated during S phase.134 ATR targets a variety of substrates, including cell-cycle checkpoint kinases and DNA repair proteins.135 If ATR fails to arrest the cell cycle, single-strand breaks are converted into double-strand breaks during subsequent cell cycles resulting in the activation of serine/threonine kinase ataxia-telangiectasia mutated (ATM), cell-cycle arrest, and apoptosis.136
Interestingly, the DNA repair pathway has a drastically different impact on adenovirus and HSV. For instance, while DNA damage machinery is an obstacle to adenovirus replication,137,138 DNA damaging agents have been demonstrated to reactivate HSV-1 from latency.135 Wild-type HSV infection with subsequent viral gene expression is dependent upon the activation of ATM, the recruitment of downstream DNA repair complexes such as Mre11, and the formation of stable replication structures.135 Adenovirus can be combined with chemotherapeutic agents that induce a G2 arrest rather than DNA repair. Various groups have demonstrated that infection with wild-type or E1 adenovirus mutants cause a dose dependent G2 arrest that is favorable for DNA replication139–142 due to the ample supply of nucleotides that are present in this phase of the cell cycle. In total, these findings demonstrate that certain chemotherapeutic agents have the potential for multimodal therapy with OVs; however, strategies must be developed to select appropriate viruses that will synergize with specific drug-induced cellular effects.
In an effort to pair particular viruses with appropriate cellular responses, a variety of intracellular signaling cascades that are prototypically dysregulated in cancer can be used for OV targeting. For instance, a hyperactive Ras pathway has been demonstrated as a tumor-selective target for oncolytic HSV lacking ICP34.5. An additional hallmark of tumor cells is the presence of angiogenesis with accompanying vascular endothelial growth factor (VEGF) production. Signaling cascades within the tumor and accompanying VEGF secretion into the tumor microenvironments creates an angiogenic milieu that can be further exacerbated by OV administration; thereby, creating an environment that limits OV potency. Lastly, the Akt pathway is canonically upregulated in many tumors, and the activation status of this cellular kinase is a critical factor in determining permissiveness to myxoma virus infection. The myxoma viral protein M-T5 physical interacts with Akt, further enhances the Akt activation status, and facilitates completion of the myxoma virus replication cycle.143 Collectively, these are just a few examples of cell signaling pathways that have been associated with OV efficacy. By understanding these pathways more fully, it will be possible to design combinatorial approaches that alter specific cellular cascades in the presence of administered virus.
3. Pharmacological Modulation of Host Factors To Enhance OV Therapy
The growing body of literature on the limitations induced by the various intra- and extracellular host defense responses to OV therapy has led to the development of several strategies to combat these undesirable changes to enhance tumor oncolysis. While “armed viruses” expressing genes that facilitate evasion of immune responses or destruction of tumor stroma have been constructed and shown to be efficacious in several preclinical studies,104,144–146 we will not discuss those in this review. On the other hand exploiting pharmacological agents to manipulate cancer cells and their microenvironment to enhance OV therapy is another promising approach. Results from the preclinical testing of several pharmacological drugs in combination with OV therapy have revealed the potential of this strategy to synergize with OV therapy. In the following sections, we will discuss various pharmacologic approaches that have been shown to augment virotherapy (Table 3).
Table 3. Oncolytic Viral Cotherapiesa.
| antitumor activity | ||||||||
|---|---|---|---|---|---|---|---|---|
| class | drug | DNA alkylation | Immune response | cell signaling | anti-angiogenic | FDA approved | OV enhancement | cotherapy |
| immune modulators | CVF | no | no | no | no | no | complement (C3) depletion | herpes |
| CPA | yes | yes | no | no | yes | IgM reduction; Treg modulation | herpes, adenovirus, measles, reovirus | |
| clodronate | no | no | no | no | no | macrophage depletion | herpes | |
| VPA | no | yes | yes | no | yes | attenuation of IFN-responsive genes | herpes | |
| HDACi | TSA | no | no | yes | yes | no | inhibition of cyclin D1 and VEGF | herpes, VSV |
| cilengitide | no | no | no | yes | no | integrin antagonist stabilizing tumor vasculature | herpes | |
| antiangiogenic agents | TSP-1 | no | no | no | yes | no | limit neovascularization | herpes |
| bevacizumab | no | no | yes | yes | yes | abrogating VEGF-receptor signaling | adenovirus | |
| cisplatin | yes | no | no | no | yes | GADD34 upregulation complements γ34.5 deficiency | herpes | |
| DNA alkylators | TMZ | yes | no | no | no | yes | activation of DNA repair machinery (herpes); G2 arrest (adenovirus) | herpes, adenovirus |
| rapamycin | no | no | yes | no | yes | mTOR inhibition; immunosuppressant | myxoma, adenovirus, VSV | |
| cellular kinase inhibitors | erlotinib | no | no | yes | yes | yes | blocks EGFR signaling | herpes |
Legend: CPA, cyclophosphamide; CVF, cobra venom factor; HDACi, histone deacetylase inhibitors; TMZ, temozolomide; TSA, trichostatin A; TSP-1, thrombospondin-1; VPA, valproic acid.
3.1. Immune Modulators
Recent studies investigating the impact of combating the antiviral host immune responses with pharmacologic agents has led to the identification of several drugs that synergize with OV therapy. The effects of cobra venom factor (CVF)-mediated depletion of serum complement proteins, cyclophosphamide (CPA)-mediated depletion of peripheral blood mononuclear cells (PBMC), and clodronate liposome (CL)-mediated exhaustion of phagocytic cells have been shown to increase OV persistence. In this section, we will discuss these drugs and their mechanism of OV enhancement.
The complement system consists of a series of serum proteases that result in the destruction of virions/infected cells through a number of routes, including the formation of membrane attack complexes on the surface of infected cells and enveloped viruses; production of anaphylatoxins that recruit additional immune mediators to the site of infection; phagocytosis of opsonized virions and infected cells; and the direct neutralization of virus following complement binding to the virion surface.147 Therefore, drugs that can temporarily inhibit complement could provide a therapeutic advantage to OV therapy. CVF is the prototypical complement inhibitor that depletes the C3 component of the complement system. In fact, in vivo depletion of complement by systemic administration of CVF has been shown to facilitate OV infection.76 However the benefits of CVF are short-lived, since there was no evidence of increased OV persistence in tumors after infection.12
Upon antigen recognition, the Fc region of antibody binds complement C1 and activates the complement cascade. Treatment of animals with CPA has been shown to reduce the serum neutralization of virus, partly due to reduction in IgM and anti-HSV antibody levels in treated animals.148 In contrast to CVF, treatment of animals with CPA prior to OV therapy also reduced viral clearance and increased viral propagation in vivo.12,75 This translated into increased cancer cell killing in vivo even at very low doses.52 CPA is also a DNA alkylating agent leading to DNA damage and tumor cell apoptosis.12 However in vitro treatment of glioma cells with 4-hydroperoxy-CPA (the activated form of CPA) did not increase OV replication, indicating that the observed augmentation in OV efficacy was not a direct effect of CPA on viral replication. The increase in therapeutic efficacy has been attributed to CPA-mediated reduction in PBMC counts that can limit the antiviral cytokine response, ultimately contributing to the enhanced anticancer efficacy.12 This is corroborated by recent findings showing diminished intratumoral infiltration of macrophages/microglia and NK cells and lower levels of IFN-γ in gliomas treated with OV and CPA.78 Collectively, these findings indicate that CPA-mediated improvement in OV efficacy is a product of the immunosuppressive action of CPA rather than synergistic cell killing between CPA-mediated cellular apoptosis and OV-mediated lytic destruction of cancer cells.
Among the pleiotropic immunomodulatory effects associated with CPA, lower doses of CPA have also been shown to enhance the immune response against tumors149–151 by transiently depleting regulatory T cells (Tregs) that suppress antitumor CD8 T cells.149–156 In order to achieve this biphasic response, it will be critical to establish a dosing schedule for modulating the different phases of the immune response. This will allow for an initial enhancement of viral oncolysis followed by the production of a delayed immuno-enhancing effect by suppressing Tregs, a step that is critical for the later adaptive immune response and vaccine-like effect against the tumor.2 As a result, CPA is impacting the tumor microenvironment by limiting both the influx of antiviral cellular mediators and the antiviral cytokine milieu, hence setting the stage for reduced viral clearance and maximizing oncolysis.
Apart from HSV-1 derived OVs, CPA has also been shown to increase the oncolytic capacity of other OVs derived from HSV-2,53 adenovirus,54 and reovirus.157,158 Based on the very promising preclinical results seen with CPA and OVs, the combination of CPA with measles virus is currently being evaluated for safety and efficacy in human patients.159
More recently CLs have been used to investigate the importance of macrophages in OV clearance in vivo. Clodronate encapsulated in liposomes is engulfed by phagocytic cells resulting in intracellular accumulation of apoptosis inducing clodronate.55 CL-mediated depletion of peripheral phagocytic cells resulted in a 5-fold increase in OV titers in intracranial glioma. While these findings partly recapitulated the effect of CPA on OV replication, they were unable to achieve the enhanced survival demonstrated with CPA.1 A potential reason for these findings may relate to the inability of clodronate to cross the blood–brain barrier, thereby limiting its ability to deplete phagocytic microglial cells in addition to peripheral macrophages.
3.2. Histone Deacetylase Inhibitors
Histone acetylation/deacetylation is a major factor in regulating chromatin structural dynamics during transcription. Histone deacetylase inhibitors (HDACi) have been shown induce cellular apoptosis, exert antiangiogenic activities, and also interfere with transcriptional activation of antiviral genes after IFN stimulation or viral infection.56,160–162 They are currently being pursued as potential anticancer agents163–168 alone and in conjunction with chemotherapy. 169–171 HDAC activity is critical for IRF-3 gene expression in virus-infected cells,160 and its inhibition can prevent the transcriptional activation of ISG in response to viral infections.160–162,160,162,172–178 Given the strong antiviral and antitumorigenic effects of HDACi, they are currently being investigated as potential agents to modulate OV efficacy. We will discuss the use of valproic acid (VPA) and trichostatin A (TSA) in conjunction with OV therapy.
VPA is an inhibitor of HDAC and is clinically used as an anticonvulsant and mood-stabilizing drug. VPA has also been shown to have anticancer effects in animal models and is currently being evaluated as an antineoplastic agent for several human malignancies. Apart from its direct anticancer effects, treatment of glioma cells with VPA has been shown to enhance the oncolytic efficacy of oncolytic HSV-1.179 This has been attributed to VPA-mediated inhibition of IFN-β and IFN-mediated proteins signal transducer and activator of transcription 1 (STAT1), PKR, and promyelocytic leukemia (PML) in infected cells.179 The significance of this finding is heightened since STAT1 is a key transcription factor that mediates IFN signaling and its activation is responsible for establishing an intracellular antiviral state.180
TSA is a promising HDACi that functions as a potent inhibitor of cyclin D1 and arrests cell-cycle progression.181–183 Similar to VPA, treatment of cancer cells with TSA, in combination with OV therapy, has also been shown to increase oncolysis. Combination of HDACi with VSV in a variety of cancer cells enhanced antitumor efficacy primarily by TSA-mediated increase in mitochondrial depolymerization and cleavage of caspases 3 and 9.56 Enhanced antitumoral and antiangiogenic effects of TSA in conjunction with oncolytic HSV have also been reported.184 However unlike VPA, TSA treatment did not affect the IFN response and the observed synergistic killing has been attributed to enhanced degradation of cyclin D1 and VEGF inhibition.184 Reduction in VEGF expression by TSA may also contribute to enhanced OV efficacy. TSA treatment of cancer cells has been shown to upregulate expression of cell surface receptors that are critical mediators of adenoviral cell entry: coxsackie–adenovirus receptor (CAR) and αv integrins. Consistent with this, TSA has been shown to enhance antitumor efficacy of conditionally replication competent adenovirus in glioblastoma cells.185–187
Considering the diversity of cellular pathways that are targeted by HDACi, it is not surprising that studies evaluating the effect of these drugs in conjunction with OVs have uncovered a variety of cellular effects contributing to oncolysis with different OVs. Future studies will elucidate critical cellular pathways that should be targeted for further study in order to enhance OV therapy.
3.3. Antiangiogenic Agents
Increased angiogenesis is one of the hallmarks of solid tumor growth and has been shown to be an essential prerequisite for cancer growth. Changes in the tumor “secretome” (secreted proteins) after OV therapy have been shown to disrupt the homeostasis maintained between angiogenic and angiostatic factors resulting in increased growth of blood vessels after OV therapy.74,80,188 Antiangiogenic agents are therapeutic drugs that destroy tumor vasculature resulting in increased hypoxia. While hypoxia-mediated “choking” of cancer cells has antitumor efficacy, hypoxia has also been shown to induce intracellular changes that support viral replication.189 Apart from direct effects of hypoxia, increased vascularity is associated with an enhanced inflammatory response suggesting that antiangiogenic agents can be used to reduce antiviral inflammation in tumors. Thus, antiangiogenic agents have been investigated as a potential avenue for reducing the antiviral state in the tumor microenvironment and improving both OV infection and replication. We will discuss the use of antiangiogenic agents used in conjunction with OV therapy: cilengitide (cRGD), thrombospondin-1 (TSP-1) peptides, and bevacizumab.
cRGD is a cyclic RGD peptide that was originally identified as an antagonist for the integrins αvβ3 and αvβ5.190 These integrins are overexpressed in proliferating cancer cells and tumor endothelium,191 and their interaction with the extracellular matrix mediates various intracellular signals involved in adhesion, migration, and proliferation. cRGD has been shown to function as an antiangiogenic factor that induces endothelial cell death and disrupts the enzymatic activity of matrix metalloproteases (MMPs).192 In preclinical studies, cRGD has been found to have significant antitumor efficacy in the treatment of glioblastoma in animal models,193 and is currently being evaluated in clinical trials for efficacy in human patients. cRGD has also been shown to limit leukocyte recruitment to synovial sites of chronic inflammation,194 reduce myeloid cell adhesion, and reduce transendothelial cell migration.195,196
Its promising activity as an antiangiogenic and antineoplastic agent combined with its role as an anti-inflammatory agent suggested that it would enhance OV efficacy. Kurozumi et al. have tested this hypothesis in a syngeneic rat glioma model.197 Consistent with its known function, treatment of animals with cRGD led to a significant reduction in the number of blood vessels and reduced OV-induced vascular permeability in vivo.80 Notably, cRGD pretreatment also participated in limiting OV-induced pro-inflammatory cytokine profile, including IFN-γ and INF-γ-induced proteins, such as CXCL9 and CXCL11, in vivo. This reduction in OV-induced inflammatory cytokine expression was accompanied by a decrease in infiltrating CD45 leukocytes80 and increased OV propagation in vivo. More significantly, cRGD administered to animals prior to OV therapy was able to significantly enhance therapeutic efficacy of OVs in animals with intracranial tumors.80 Future studies will elucidate the impact cRGD on the interplay between its antiangiogenic and anti-inflammatory responses and whether other antiangiogenic drugs can recapitulate the findings of cRGD when administered with OVs.
While blood vessels serve as entry points for circulating “soldiers” of the immune system, viral infection is also often accompanied by the secretion of several pro-angiogenic factors74,198,199 that can induce angiogenesis and encourage growth of residual tumor after viral clearance. Corneal infection of wild-type HSV-1 has also been linked to increased expression of angiogenic factors such as VEGF, MMP9, and Cox-2 and reduced expression of antiangiogenic factors such as TSP-1 and TSP-2.74,188,198,200–202 Consistent with these studies, we and others have recently reported a significant increase in cysteine-rich 61 and reduction of antiangiogenic TSP-1 after oncolytic HSV-1 treatment.188,203 Reduction of TSP-1 (antiangiogenic ligand for CD36 on endothelial cells) levels have been implicated in tumoral angiogenesis of residual tumor that regrows after OV-mediated tumor destruction and viral clearance.204,205 TSP-1 and TSP-2 are critical targets of HSV-induced keratitis due to their post-transcriptional downregulation in keratinocytes and subsequent neovascularization following ocular HSV infection.198
In the context of oncolytic HSV G207 and subcutaneous glioblastoma model, viral infection significantly reduced levels of TSP-1 and TSP-2 while concomitantly resulting in increased microvessel density.74 To mitigate this response, G207 treatment was combined with a recombinant peptide composed of the three type-1 repeats (3TSR) of TSP-1. In this two-armed treatment approach, the HSV-induced angiogenic response was limited, while the resumption of tumor growth was also delayed.74 Moreover, since several TSP-1 and TSP-2 derived angiogenesis inhibitors have already undergone phase I clinical trials,206 this data suggests that the combined treatment of tumors with G207 and a TSP-1 derived angiogenesis inhibitor has the potential of increasing tumor cell death by viral replication while negating the unwanted angiogenic response that accompanies HSV infection.
Bevacizumab is a humanized anti-VEGF monoclonal antibody that interferes with VEGF signaling.207 It is the first antiangiogenic drug that has been approved by the FDA for treatment of tumors. The aberrant angiogenic signaling in tumors results in a vasculature that is leaky and tortuous resulting in high interstitial pressure and poor circulation, both of which present obstacles for efficient delivery of therapeutics. Treatment of tumors with bevacizumab has been shown to result in a transient normalization of the abnormal tumor vasculature, reduced interstitial pressure, and improved drug delivery into tumor tissue.208 Since a previously noted obstacle to OV therapy is an aberrant angiogenic vasculature, cotreatment of bevacizumab and OVs was hypothesized to improve viral distribution within an anaplastic thyroid carcinoma model. Bevacizumab was not able to individually induce a reduction in tumor growth, confirming the lack of antitumor activity against this thyroid tumor model.209 However, when combined with oncolytic adenovirus, the cotherapy significantly reduced tumor growth compared with single treatments.210 Bevacizumab-induced reduction in interstitial fluid pressure is thought to have aided in improved viral distribution.210
Therefore, it seems that a variety of angiogenesis modulators may help stimulate the ability of oncolytic viruses to destroy tumors effectively.
3.4. Cisplatin and Temozolomide as Examples of DNA Alkylating Agents
DNA alkylating agents destroy the genome of dividing cancer cells resulting in cancer cell apoptosis. However increased production of DNA repair enzymes combats the antitumor efficacy of these agents and often results in chemoresistance. While this resistant population is refractory to further chemotherapy, these cells are often excellent vehicles for viral replication and are sensitized to OV therapy. In this section, we will discuss the use of DNA damaging drugs such as cisplatin and temozolomide (TMZ) in conjunction with OV therapy.
Cisplatin mediates apoptosis and cell-cycle arrest through the formation of platinum–DNA adducts in replicating cancer cells. However, this apoptotic effect is accompanied by a triad of toxic side effects—nephrotoxicity, ototoxicity, and neurotoxicity—which limit its maximal dosing.211 The inevitable build up of drug-induced chemoresistance further limits its efficacy. Oncolytic HSV-1 NV1066, with a deleted γ34.5 locus, has been shown to synergize with cisplatin for cancer cell killing in vitro and hence permit dose reductions of both agents.212 Cisplatin-induced GADD34213 has been implicated as the reason for this synergy. Consistent with this, inhibition of GADD34 with small interfering RNA (siRNA) eliminated the synergism between it and NV1066.212 This finding suggests that the mechanism of enhanced efficacy was due to GADD34 substituting in part for the γ34.5 deletion in NV1066.
While this has significant implications for future clinical trials, it is important to note that only low doses of cisplatin synergized with OV therapy and high doses of cisplatin antagonized OV therapy by limiting viral replication.214 Cellular stress response initiated by low-dose cisplatin activates antiapoptotic prosurvival pathways that create a cellular environment that facilitates viral replication, while high dose had the opposite effect. The evolution of resistant cancer cells thus sets up the stage for effective oncolysis.212
TMZ is a DNA alkylating agent and is FDA approved for the treatment of malignant glioma.215 TMZ spontaneously converts to its activated metabolite 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide, which then methylates guanine nucleotides at the O6 and N7 positions136 in DNA. During DNA replication, methylated G eventually results in a G →A transition causing genetic instability and ultimately cell death.216 This effect is countered by the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT), which demethylates alkylated guanine. Thus MGMT repairs TMZ-induced DNA damage, and its expression negatively correlates with response to therapy.217,218
While DNA damage done by TMZ treatment is cytotoxic, it also results in the induction of DNA repair genes such as GADD34 and ribonucleotide reductase (RR). Induction of these cellular DNA repair enzymes would be predicted to support enhanced viral replication of an oncolytic HSV-1 deficient in both γ34.5 and RR. Consistent with this, Aghi et al. found strong anticancer synergy of γ34.5- and RR-negative HSV (G207) in MGMT negative cells, which was reduced upon MGMT reconstitution.216 Interestingly treating cells with O6-benzylguanine, an inhibitor of MGMT, rendered resistant cells sensitive to G207.219,220 Since both GADD34 and RR lead to build up of TMZ resistance, increased OV efficacy would be predicted in patients who have failed prior TMZ treatment.216
Interestingly oncolytic adenovirus has also been shown to synergize with TMZ in a melanoma model. This enhancement has been attributed to TMZ-induced cell-cycle arrest in G2 phase, which favors adenoviral replication.221,222 This interesting discovery points to the differing roles of TMZ-induced synergy with viral oncolysis. Whereas DNA repair pathways are beneficial for HSV replication,135 they inhibit adenoviral replication.138 As a result, TMZ likely enhances adenoviral replication in a DNA repair independent pathway.
3.5. Cellular Kinase Inhibitors
Cellular kinases play a key role in the regulation of signaling events that govern multiple pathways affecting growth, proliferation, migration and angiogenesis. In cancer, these pathways are usurped to support unchecked cellular replication. Advances in understanding of the various check points in these signaling cascades has led to the identification of several small-molecule therapeutics that target specific kinases to disrupt the protumorigenic signaling in cancer cells. In this section, we will discuss the effects of two small-molecule kinase inhibitors, rapamycin and erlotinib, on oncolysis.
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase with pleiotropic cellular effects encompassing activation of protein kinase C signaling, transcription and translation regulation, actin reorganization, and membrane trafficking.223 Rapamycin (Sirolimus) is an inhibitor of mTOR and has been shown to have efficacy as (a) an antineoplastic agent,224 (b) an antiangiogenic agent, and (c) a licensed immunosuppressant based on its cytostatic effect on T cells and its ability to decrease the production of neutralizing antibodies.225 In this latter capacity, rapamycin has been used as an alternative to cyclosporine in the treatment of transplant patients.226
Conditionally replicating adenovirus causes nonapoptotic programmed cell death in tumor cells by inducing autophagy.227 Autophagy is a protein degradation system observed in cells experiencing environmental stress induced by amino acid starvation or viral or bacterial infections.228–230 Interestingly, rapamycin has also been shown to induce autophagy,231 suggesting that adenovirus-based OV therapy would be augmented by rapamycin cotreatment. Oncolytic adenovirus delta-24-RGD led to an upregulation of Atg5, a critical component of the autophagy pathway,232 and in combination with RAD001 (an analog of rapamycin) led to a synergistic antitumor effect along with induced autophagy in vitro.232 Beyond inducing autophagy, RAD001 in uninfected cells result in a slower rate of tumor progression thereby allowing the virus to have more time to initiate its anticancer effect. Interestingly myxoma virus has been shown to synergize with rapamycin albeit the mechanism of synergy is thought to be different.233 Pretreatment with rapamycin has been demonstrated to increase the levels of activated Akt, which creates an environment more conducive for myxoma virus tropism and virus spread even in a variety of human tumor cell lines that are normally not permissive for myxoma infection.233 Future studies will delineate whether the immunosuppressant and antiangiogenic effects of mTOR inhibition also contribute to the increased oncolysis seen in vivo.
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that controls cell-signaling molecules involved in diverse cellular functions, including cell proliferation, differentiation, motility, and survival. EGFR overexpression or activating mutations have been implicated in multiple malignancies.234 Erlotinib is a small-molecule inhibitor that blocks the activation of EGFR tyrosine kinase and has been demonstrated to have antitumor efficacy in several human malignancies.235–238 Antitumor activity of erlotinib in combination with oncolytic HSV, was recently tested in malignant peripheral nerve sheath tumors (MP-NSTs).239 Notably, MPNST has aberrant EGFR signaling,240,241 making this a suitable model for this cotherapy. Despite the evidence of additive efficacy in vitro, oncolytic HSV and erlotinib cotreatment demonstrated only a trend toward increased antitumor efficacy in vivo.239 Together these findings underscore the need to test the dosing and scheduling of different therapeutic regimens within in vivo animal models in order to identify cotherapies that will augment each other without increasing toxicity.
3.6. ECM Modulating Agents
One of the major barriers for effective drug delivery within the tumor parenchyma is the ubiquitous ECM secreted by glioma cells. This matrix forms a complex scaffold that modulates tumor cell proliferation, cell adhesion, and motility. Increased expression and extracellular accumulation of ECM increases the fractional volume and tortuosity of the extracellular space resulting in reduced interstitial space and increased internal pressure in the tumor.242,243 All of these together present a formidable barrier toward passive molecular diffusion and spread of macromolecular therapeutics such as OVs.244,245 Selective targeting of these components of the ECM can be exploited to enhance virotherapy. In this section, we will discuss the approaches used to modulate tumor ECM and enhance OV dissemination in tumor.
The limiting nature of tumor ECM on virotherapy was first observed in studies wherin treatment of tumors with trypsin or a mixture of collagenase and dispase was found to increase the spread and therapeutic efficacy of a nonreplicative viral vector.246 However, the nonspecific nature of these enzymes precludes any conclusions about the mechanism of this enhancement.104 Fibrillar collagen is thought to be a major barrier to macromolecular transport in the tumor interstium.247–249 Hence, direct degradation of the fibrillar collagen was tested as a possible mechanism of improving viral distribution.104 McKee et al. tested the co-injection of an oncolytic herpes virus (MGH2) with collagenase in a melanoma model. Collagenase treatment resulted in broad, uniform distribution of viral particles through the tumor along with substantial tumor regression and enhanced efficacy.104 Hyaluronic acid is also a major component of the ECM that is enriched in multiple tumor types.250 Coadministration of hyaluronidase (rHuPH20) with OVs has been shown to increase viral transduction and improved antitumor immunity.246,251–253
The widespread ability of MMPs to degrade multiple different components of the ECM represents another ideal agent for enhancing extracellular viral spread.254 Using a soft tissue sarcoma model treated with oncolytic HSV, Mok et al. were able to demonstrate that MMP-1 and -8 expression lead to a selective reduction in tumor sulfated GA content.146 MMP-8 may prove particularly useful since it is able to both improve viral spread through ECM modification and also decrease the dissemination of metastases.255
It is important to note that despite promising findings of increased OV spread and efficacy in preclinical models, increased intratumoral hemorrhages in tumors treated with collagenase has also been noted.104 This strategy has to be carefully evaluated for its impact on tumor microenvironment prior to successful application in human patients.
3.7. Stealth Agents
Systemic delivery of OVs is a prerequisite for successful targeting of disseminated cancer. However, pharmacokinetic studies have revealed that OV present in the circulating plasma is rapidly neutralized and cleared by the liver.256 Apart from rapid clearance of virus particles in serum, nonspecific cellular entry also poses a significant challenge for this approach of OV delivery. To overcome this problem both cellular and polymer-based stealth agents have been exploited to enhance systemic delivery of OVs to the target tissue. In this section, we will discuss cellular and polymer-based methods exploited to secretly deliver “OV cargo” hidden from the inhibitory effects of the circulatory system.
Soluble polymers based on N-(2-hydroxypropyl)methacrylamide (HPMA) have been used as a drug carrier in several preclinical and clinical investigations.257 Conjugation of such polymers to small molecules has been shown to increase their antitumor activity compared with the free drug.258 Polymer coating of conditionally replicating adenovirus has been shown to provide the virus with steric protection from subsequent serum neutralization, antibody binding, and clearance by the innate immune system.256 This resulted in increased bioavailability of the OV, along with reduction in toxicity compared with the uncoated OV in vivo.256
While this coating is promising, it also ablates viral binding to its endogenous receptors. Such tropism-ablated pHPMA viruses can be linked to a targeting ligand or biological effector molecules to enhance tissue tropism and penetration, respectively.259 Such an approach would maximize systemic viral propagation and minimize non-target-cell uptake of the therapeutic virus. Examples of potential targeting ligands that have been used include basic fibroblast growth factor, VEGF, and oligopeptides.260–262
Tumor-specific retargeting of OV by this technology has been tested for safety and efficacy using amino-reactive copolymers of HPMA covalently linked to epidermal growth factor (EGF). EGF is a ligand for EGFR, a receptor overexpressed in a variety of cancers.263–265 Amino-reactive copolymers of HPMA covalently linked to EGFR revealed preclinical efficacy in ovarian cancer models in vivo.266 This approach revealed a significant antitumor efficacy accompanied by a significant reduction of all toxicities, including peritoneal adhesion formation and bowel obstruction.267 Polymer coating of the virus did not inhibit viral unpackaging and permitted virotherapy after cell entry. Notably, this polymer coating is not inherited by progeny virus upon replication, allowing subsequent OV particles to infect using normal cell surface receptors. This avoids the possibility of creating new pathology, and maximizes the likelihood of neutralization of virus that escapes from the tumor into the bloodstream or ascitic fluid.266 This strategy has shown promising preclinical results, and future trials will uncover the safety and efficacy of this approach.
An additional approach for avoiding systemic antiviral immunity is the use of a carrier cell.268 In this method, cells with tumor homing abilities are exploited as vehicles to “smuggle” OV to the tumor site. This approach has demonstrated promising results for herpes virus,269 parvovirus,270 and VSV.73 Cytokine-induced killer (CIK) cells have been shown to be effective carriers of oncolytic vaccinia virus in vivo.271,272 Infected CIK cells could efficiently deliver OVs to tumor sites and enhanced antitumor efficacy in several animal models. Similarly, mesenchymal progenitor and circulating endothelial cells have also been used as vehicles to carry OV to tumor sites.273 The ability of transformed cells to support viral replication has led to some very innovative studies investigating the use of immortalized human cells as possible delivery agents for OV.268 While these studies have established the feasibility of this approach, there are obvious concerns about the tumorigenic potential of these cells, and future studies will delineate the safety and efficacy of this approach.
A particular application of this “Trojan Horse approach” exploits adoptive T cell therapy. Qiao et al. have identified a way to use autologous T cells as a platform for carrying viral vectors to lymph nodes. Since cancer cell trafficking mimics T cell trafficking to lymph nodes, T cells could be exploited as carriers to target OV particles to metastatic cancer cells in the lymph nodes.274 Autologous T cells loaded with oncolytic VSV could effectively purge lymph nodes and the spleen of metastatic cells. The antitumor efficacy of this approach was also dependent on an intact immune system indicating that apart from direct oncolysis this approach was also able to activate protective antitumor immunity.275
Collectively these findings demonstrate the significant strides that are being made in the realm of systemic OV delivery to enhance oncolysis. This will open the doors for OV therapy to be used against localized and disseminated metastatic disease.
4. Summary and Future Directions
Although a significant body of evidence exists that delineates the multivariate host response to OVs, there is clearly a need for additional studies in this field. Just as the contributions of specific populations of immune cells to OVs must be increasingly examined, there is also a need to define a particular set of antiviral effectors responsible for limiting OV survival. These factors include cytokines, neutralizing antibodies, intracellular signaling cascades, cell-cycle checkpoints, and angiogenesis. As these mechanisms are increasingly defined, the next step will be assessing a variety of multimodal treatments that are able to complement the individual mechanisms of action for drug and virus to synergize for enhanced tumor clearance while limiting unwanted toxicity. Additionally, as these factors are assessed, they must be placed in the context of both different tumor types and OV candidates. As data is collected, this will call for a further requirement to design each OV regimen for the particular context of a specific tumor.
As the key sets of antiviral responses are defined, however, it is also critically important to understand the clinical implications that accompany the use of these drugs. First, while many of the drugs that have been listed are promising in preclinical studies, approaches, for example, that use trypsin to modulate the extracellular matrix are not clinically feasible due to its relative nonspecific activity. Similarly, macrophage attenuation in the clinical setting will need to be accomplished using a pharmacological approach other than clodronate liposomes. Second, inhibition of these antiviral defense mechanisms raises safety concerns. A delicate balance must be achieved in identifying specifically targeted drugs that limit the essential antiviral pathways while also leaving the host uncompromised to defend against disseminated viral infection and replication. Taken together, the challenge will be to determine which viruses work best for specific cancers while defining the proper dose, schedule, route of administration, and appropriate cotreatments that ultimately lead to enhanced efficacy in the clinic.
Biographies

Christopher Alvarez-Breckenridge is an M.D./Ph.D. student at The Ohio State University where he is working in the laboratory of Dr. E. Antonio Chiocca. His research interests focus on identifying properties of the host immune response to oncolytic viral therapy for glioblastoma. He graduated from The Ohio State University in 2005 with a B.A. in classics and a B.S. in biology and was a member of the early admissions pathway program in The Ohio State University Medical School. His undergraduate research was with Dr. Charis Eng where he focused on lipid signaling in the context of breast cancer.

Dr. Balveen Kaur majored in physics at Delhi University and proceeded to obtain a M.S. in biotechnology at Banaras Hindu University. She subsequently carried out her Ph.D. at Emory University followed by a postdoctoral fellowship in the laboratory of Dr. Erwin Van Meir at Emory University where she studied the role of angiogenesis in the context of glioma progression. Much of her work focused on the antiangiogenic and antitumorigenic properties of vasculostatin. She joined the faculty of The Ohio State University in 2005 as an Assistant Professor where her laboratory is currently studying the role of the tumor microenvironment and angiogenesis as limiting factors for glioma virotherapy.

Dr. E. Antonio Chiocca is the Dardinger Family Professor of Neurosurgical Oncology and Department Chair of Neurological Surgery at The Ohio State University. Dr. Chiocca carried out his M.D. and Ph.D. training at the University of Texas prior to embarking on a neurological surgical residency at Massachusetts General Hospital. During residency, Dr. Chiocca began to study herpes simplex (HSV) as a potential modality for glioma therapy. During that time, he has made seminal discoveries in the field of oncolytic viral research that ranged from the creation of novel oncolytic viral mutants to the discovery of cyclophosphamide's potential as a cotherapy for enhancing oncolytic viral efficacy. During his time at The Ohio State University, he has been able to successfully combine his research interests with his clinical practice by securing multiple grants that fund the preclinical development of oncolytic HSV and its use in phase I clinical trials.
References
- 1.Fulci G, Dmitrieva N, Gianni D, Fontana EJ, Pan X, Lu Y, Kaufman CS, Kaur B, Lawler SE, Lee RJ, Marsh CB, Brat DJ, van Rooijen N, Stemmer-Rachamimov AO, Hochberg FH, Weissleder R, Martuza RL, Chiocca EA. Cancer Res. 2007;67:9398. doi: 10.1158/0008-5472.CAN-07-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiocca EA. Curr Opin Mol Ther. 2008;10:38. [PubMed] [Google Scholar]
- 3.Hiscott J. J Biol Chem. 2007;282:15325. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 4.Uematsu S, Akira S. J Biol Chem. 2007;282:15319. doi: 10.1074/jbc.R700009200. [DOI] [PubMed] [Google Scholar]
- 5.Mohr I. Int ReV Immunol. 2004;23:199. doi: 10.1080/08830180490265600. [DOI] [PubMed] [Google Scholar]
- 6.Barber GN. Oncogene. 2005;24:7710. doi: 10.1038/sj.onc.1209042. [DOI] [PubMed] [Google Scholar]
- 7.Hardcastle J, Kurozumi K, Chiocca EA, Kaur B. Curr Cancer Drug Targets. 2007;7:181. doi: 10.2174/156800907780058880. [DOI] [PubMed] [Google Scholar]
- 8.Kim M, Chung YH, Johnston RN. J Microbiol. 2007;45:187. [PubMed] [Google Scholar]
- 9.Johnston JB, Nazarian SH, Natale R, McFadden G. Virology. 2005;332:235. doi: 10.1016/j.virol.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 10.Kodukula P, Liu T, Rooijen NV, Jager MJ, Hendricks RL. J Immunol. 1999;162:2895. [PubMed] [Google Scholar]
- 11.Fujioka N, Akazawa R, Ohashi K, Fujii M, Ikeda M, Kurimoto M. J Virol. 1999;73:2401. doi: 10.1128/jvi.73.3.2401-2409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakimoto H, Fulci G, Tyminski E, Chiocca EA. Gene Ther. 2004;11:214. doi: 10.1038/sj.gt.3302143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns AR, Smith CW, Walker DC. Physiol Rev. 2003;83:309. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 14.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Annu Rev Immunol. 1999;17:189. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 15.Pawlik TM, Nakamura H, Yoon SS, Mullen JT, Chandrasekhar S, Chiocca EA, Tanabe KK. Cancer Res. 2000;60:2790. [PubMed] [Google Scholar]
- 16.Pawlik TM, Nakamura H, Mullen JT, Kasuya H, Yoon SS, Chandrasekhar S, Chiocca EA, Tanabe KK. Cancer. 2002;95:1171. doi: 10.1002/cncr.10776. [DOI] [PubMed] [Google Scholar]
- 17.Boviatsis EJ, Park JS, Sena-Esteves M, Kramm CM, Chase M, Efird JT, Wei MX, Breakefield XO, Chiocca EA. Cancer Res. 1994;54:5745. [PubMed] [Google Scholar]
- 18.Chase M, Chung RY, Chiocca EA. Nat Biotechnol. 1998;16:444. doi: 10.1038/nbt0598-444. [DOI] [PubMed] [Google Scholar]
- 19.Aghi M, Chou TC, Suling K, Breakefield XO, Chiocca EA. Cancer Res. 1999;59:3861. [PubMed] [Google Scholar]
- 20.Tyminski E, Leroy S, Terada K, Finkelstein DM, Hyatt JL, Danks MK, Potter PM, Saeki Y, Chiocca EA. Cancer Res. 2005;65:6850. doi: 10.1158/0008-5472.CAN-05-0154. [DOI] [PubMed] [Google Scholar]
- 21.Dingli D, Peng KW, Harvey ME, Greipp PR, O'Connor MK, Cattaneo R, Morris JC, Russell SJ. Blood. 2004;103:1641. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 22.Hasegawa K, Pham L, O'Connor MK, Federspiel MJ, Russell SJ, Peng KW. Clin Cancer Res. 2006;12:1868. doi: 10.1158/1078-0432.CCR-05-1803. [DOI] [PubMed] [Google Scholar]
- 23.Hellums EK, Markert JM, Parker JN, He B, Perbal B, Roizman B, Whitley RJ, Langford CP, Bharara S, Gillespie GY. Neuro-Oncol. 2005;7:213. doi: 10.1215/S1152851705000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Proc Natl Acad Sci U.S.A. 2000;97:2208. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ino Y, Saeki Y, Fukuhara H, Todo T. Clin Cancer Res. 2006;12:643. doi: 10.1158/1078-0432.CCR-05-1494. [DOI] [PubMed] [Google Scholar]
- 26.Todo T, Martuza RL, Dallman MJ, Rabkin SD. Cancer Res. 2001;61:153. [PubMed] [Google Scholar]
- 27.Todo T, Rabkin SD, Sundaresan P, Wu A, Meehan KR, Herscowitz HB, Martuza RL. Hum Gene Ther. 1999;10:2741. doi: 10.1089/10430349950016483. [DOI] [PubMed] [Google Scholar]
- 28.Liu TC, Zhang T, Fukuhara H, Kuroda T, Todo T, Canron X, Bikfalvi A, Martuza RL, Kurtz A, Rabkin SD. Clin Cancer Res. 2006;12:6791. doi: 10.1158/1078-0432.CCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 29.Liu TC, Zhang T, Fukuhara H, Kuroda T, Todo T, Martuza RL, Rabkin SD, Kurtz A. Mol Ther. 2006;14:789. doi: 10.1016/j.ymthe.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 30.Garber K. J Natl Cancer Inst. 2006;98:298. doi: 10.1093/jnci/djj111. [DOI] [PubMed] [Google Scholar]
- 31.Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore C, Tufaro F, Martuza RL. Gene Ther. 2000;7:867. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 32.Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA, Parker JN, Whitley RJ, Gillespie GY. Mol Ther. 2009;17:199. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, Petty R, MacLean A, Harland J, McKie E, Mabbs R, Brown M. Gene Ther. 2000;7:859. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 34.Papanastassiou V, Rampling R, Fraser M, Petty R, Hadley D, Nicoll J, Harland J, Mabbs R, Brown M. Gene Ther. 2002;9:398. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 35.Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, Hadley D, Patterson J, Brown SM, Rampling R. Gene Ther. 2004;11:1648. doi: 10.1038/sj.gt.3302289. [DOI] [PubMed] [Google Scholar]
- 36.Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, Kracher J, Grossman SA, Fisher JD, Carson K, Rosenblum M, Mikkelsen T, Olson J, Markert J, Rosenfeld S, Nabors LB, Brem S, Phuphanich S, Freeman S, Kaplan R, Zwiebel J. Mol Ther. 2004;10:958. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 37.Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E, Rozenman-Yair S, Panet A, Libson E, Irving CS, Galun E, Siegal T. Mol Ther. 2006;13:221. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 38.Forsyth P, Roldan G, George D, Wallace C, Palmer CA, Morris D, Cairncross G, Matthews MV, Markert J, Gillespie Y, Coffey M, Thompson B, Hamilton M. Mol Ther. 2008;16:627. doi: 10.1038/sj.mt.6300403. [DOI] [PubMed] [Google Scholar]
- 39.Peng KW, Facteau S, Wegman T, O'Kane D, Russell SJ. Nat Med. 2002;8:527. doi: 10.1038/nm0502-527. [DOI] [PubMed] [Google Scholar]
- 40.Peng KW, Hadac EM, Anderson BD, Myers R, Harvey M, Greiner SM, Soeffker D, Federspiel MJ, Russell SJ. Cancer Gene Ther. 2006;13:732. doi: 10.1038/sj.cgt.7700948. [DOI] [PubMed] [Google Scholar]
- 41.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Cancer Res. 2002;62:4656. [PubMed] [Google Scholar]
- 42.Phuong LK, Allen C, Peng KW, Giannini C, Greiner S, TenEyck CJ, Mishra PK, Macura SI, Russells SJ, Galanis EC. Cancer Res. 2003;63:2462. [PubMed] [Google Scholar]
- 43.Peng KW, Ahmann GJ, Pham L, Greipp PR, Cattaneo R, Russell SJ. Blood. 2001;98:2002. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 44.Toda M, Rabkin SD, Kojima H, Martuza RL. Hum Gene Ther. 1999;10:385. doi: 10.1089/10430349950018832. [DOI] [PubMed] [Google Scholar]
- 45.Nakano K, Todo T, Chijiiwa K, Tanaka M. Mol Ther. 2001;3:431. doi: 10.1006/mthe.2001.0303. [DOI] [PubMed] [Google Scholar]
- 46.Steinman RM, Turley S, Mellman I, Inaba K. J Exp Med. 2000;191:411. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauvrit A, Brandler S, Sapede-Peroz C, Boisgerault N, Tangy F, Gregoire M. Cancer Res. 2008;68:4882. doi: 10.1158/0008-5472.CAN-07-6265. [DOI] [PubMed] [Google Scholar]
- 48.Meng Y, Carpentier AF, Chen L, Boisserie G, Simon JM, Mazeron JJ, Delattre JY. Int J Cancer. 2005;116:992. doi: 10.1002/ijc.21131. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Dutuor A, Fu X, Zhang X. J Gene Med. 2007;9:161. doi: 10.1002/jgm.1005. [DOI] [PubMed] [Google Scholar]
- 50.Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, Valdes M, Barber G, Vile RG. Cancer Res. 2007;67:2840. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 51.Wakimoto H, Johnson PR, Knipe DM, Chiocca EA. Gene Ther. 2003;10:983. doi: 10.1038/sj.gt.3302038. [DOI] [PubMed] [Google Scholar]
- 52.Kambara H, Saeki Y, Chiocca EA. Cancer Res. 2005;65:11255. doi: 10.1158/0008-5472.CAN-05-2278. [DOI] [PubMed] [Google Scholar]
- 53.Li H, Zeng Z, Fu X, Zhang X. Cancer Res. 2007;67:7850. doi: 10.1158/0008-5472.CAN-07-1087. [DOI] [PubMed] [Google Scholar]
- 54.Lamfers ML, Fulci G, Gianni D, Tang Y, Kurozumi K, Kaur B, Moeniralm S, Saeki Y, Carette JE, Weissleder R, Vandertop WP, van Beusechem VW, Dirven CM, Chiocca EA. Mol Ther. 2006;14:779. doi: 10.1016/j.ymthe.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Rooijen N, Sanders A. J Immunol Methods. 1994;174:83. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS, Yasmeen A, Bismar TA, Kirn D, Falls T, Snoulten VE, Vanderhyden BC, Werier J, Atkins H, Vaha-Koskela MJ, Stojdl DF, Bell JC, Hiscott J. Proc Natl Acad Sci U S A. 2008;105:14981. doi: 10.1073/pnas.0803988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aghi MK, Chiocca EA. Mol Ther. 2009;17:8. doi: 10.1038/mt.2008.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andreansky S, He B, van Cott J, McGhee J, Markert JM, Gillespie GY, Roizman B, Whitley RJ. Gene Ther. 1998;5:121. doi: 10.1038/sj.gt.3300550. [DOI] [PubMed] [Google Scholar]
- 59.Tada M, Sawamura Y, Sakuma S, Suzuki K, Ohta H, Aida T, Abe H. Cancer Immunol Immunother. 1993;36:251. doi: 10.1007/BF01740907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC. Cell. 1994;77:525. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 61.Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Nature. 1995;375:411. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 62.Fruh K, Ahn K, Djaballah H, Sempe P, van Endert PM, Tampe R, Peterson PA, Yang Y. Nature. 1995;375:415. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 63.Todo T, Martuza RL, Rabkin SD, Johnson PA. Proc Natl Acad Sci U S A. 2001;98:6396. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bennett JJ, Malhotra S, Wong RJ, Delman K, Zager J, St Louis M, Johnson P, Fong Y. Ann Surg. 2001;233:819. doi: 10.1097/00000658-200106000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA, Stojdl DF, Daneshmand M, Speth K, Kirn D, McCart JA, Atkins H, Bell JC. Mol Ther. 2007;15:1686. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- 66.Abordo-Adesida E, Follenzi A, Barcia C, Sciascia S, Castro MG, Naldini L, Lowenstein PR. Hum Gene Ther. 2005;16:741. doi: 10.1089/hum.2005.16.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balachandran S, Thomas E, Barber GN. Nature. 2004;432:401. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- 68.Balachandran S, Barber GN. Cancer Cell. 2004;5:51. doi: 10.1016/s1535-6108(03)00330-1. [DOI] [PubMed] [Google Scholar]
- 69.Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Cancer Res. 2006;66:2314. doi: 10.1158/0008-5472.CAN-05-2661. [DOI] [PubMed] [Google Scholar]
- 70.Lowenstein PR. Mol Ther. 2005;12:185. doi: 10.1016/j.ymthe.2005.06.439. [DOI] [PubMed] [Google Scholar]
- 71.Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. Cancer Cell. 2003;4:263. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 72.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. Nat Med. 2000;6:821. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 73.Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, Stojdl DF, Forsyth PA, Atkins H, Bell JC. Mol Ther. 2007;15:123. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 74.Aghi M, Rabkin SD, Martuza RL. Cancer Res. 2007;67:440. doi: 10.1158/0008-5472.CAN-06-3145. [DOI] [PubMed] [Google Scholar]
- 75.Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, Harsh GRt, Louis DN, Bartus RT, Hochberg FH, Chiocca EA. Nat Med. 1999;5:881. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 76.Ikeda K, Wakimoto H, Ichikawa T, Jhung S, Hochberg FH, Louis DN, Chiocca EA. J Virol. 2000;74:4765. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wakimoto H, Ikeda K, Abe T, Ichikawa T, Hochberg FH, Ezekowitz RA, Pasternack MS, Chiocca EA. Mol Ther. 2002;5:275. doi: 10.1006/mthe.2002.0547. [DOI] [PubMed] [Google Scholar]
- 78.Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, Kaur B, Louis DN, Weissleder R, Caligiuri MA, Chiocca EA. Proc Natl Acad Sci U S A. 2006;103:12873. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Herrlinger U, Kramm CM, Aboody-Guterman KS, Silver JS, Ikeda K, Johnston KM, Pechan PA, Barth RF, Finkelstein D, Chiocca EA, Louis DN, Breakefield XO. Gene Ther. 1998;5:809. doi: 10.1038/sj.gt.3300643. [DOI] [PubMed] [Google Scholar]
- 80.Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, Hochberg FH, Weissleder R, Carson W, Chiocca EA, Kaur B. J Natl Cancer Inst. 2007;99:1768. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- 81.Kawai T, Akira S. Nat Immunol. 2006;7:131. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 82.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. Nature. 2000;408:740. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 83.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. J Exp Med. 2004;199:1651. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Immunity. 2005;23:19. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 85.Kumar KP, McBride KM, Weaver BK, Dingwall C, Reich NC. Mol Cell Biol. 2000;20:4159. doi: 10.1128/mcb.20.11.4159-4168.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin R, Genin P, Mamane Y, Hiscott J. Mol Cell Biol. 2000;20:6342. doi: 10.1128/mcb.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. FEBS Lett. 1998;441:106. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 88.Lin R, Mamane Y, Hiscott J. J Biol Chem. 2000;275:34320. doi: 10.1074/jbc.M002814200. [DOI] [PubMed] [Google Scholar]
- 89.Au WC, Moore PA, LaFleur DW, Tombal B, Pitha PM. J Biol Chem. 1998;273:29210. doi: 10.1074/jbc.273.44.29210. [DOI] [PubMed] [Google Scholar]
- 90.Marie I, Durbin JE, Levy DE. EMBO J. 1998;17:6660. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. J Exp Med. 1999;189:663. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mamane Y, Heylbroeck C, Genin P, Algarte M, Servant MJ, LePage C, DeLuca C, Kwon H, Lin R, Hiscott J. Gene. 1999;237:1. doi: 10.1016/s0378-1119(99)00262-0. [DOI] [PubMed] [Google Scholar]
- 93.Sato M, Taniguchi T, Tanaka N. Cytokine Growth Factor Rev. 2001;12:133. doi: 10.1016/s1359-6101(00)00032-0. [DOI] [PubMed] [Google Scholar]
- 94.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. Annu Rev Immunol. 2001;19:623. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 95.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. J Leukocyte Biol. 2001;69:912. [PubMed] [Google Scholar]
- 96.Le Page C, Genin P, Baines MG, Hiscott J. Rev Immunogenet. 2000;2:374. [PubMed] [Google Scholar]
- 97.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. Annu Rev Biochem. 1998;67:227. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 98.Medzhitov R, Janeway CA., Jr Science. 2002;296:298. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 99.Norman KL, Lee PW. Drug Discovery Today. 2005;10:847. doi: 10.1016/S1359-6446(05)03483-5. [DOI] [PubMed] [Google Scholar]
- 100.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. EMBO J. 1998;17:3351. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Strong JE, Lee PW. J Virol. 1996;70:612. doi: 10.1128/jvi.70.1.612-616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coffey MC, Strong JE, Forsyth PA, Lee PW. Science. 1998;282:1332. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 103.Haralambieva I, Iankov I, Hasegawa K, Harvey M, Russell SJ, Peng KW. Mol Ther. 2007;15:588. doi: 10.1038/sj.mt.6300076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, Bawendi MG, Boucher Y, Breakefield XO, Jain RK. Cancer Res. 2006;66:2509. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- 105.Shen BH, Bauzon M, Hermiston TW. Gene Ther. 2006;13:986. doi: 10.1038/sj.gt.3302736. [DOI] [PubMed] [Google Scholar]
- 106.Parkins CS, Stratford MR, Dennis MF, Stubbs M, Chaplin DJ. Br J Cancer. 1997;75:319. doi: 10.1038/bjc.1997.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hay JG. Curr Opin Mol Ther. 2005;7:353. [PubMed] [Google Scholar]
- 108.Pipiya T, Sauthoff H, Huang YQ, Chang B, Cheng J, Heitner S, Chen S, Rom WN, Hay JG. Gene Ther. 2005;12:911. doi: 10.1038/sj.gt.3302459. [DOI] [PubMed] [Google Scholar]
- 109.Millan J, Ridley AJ. Biochem J. 2005;385:329. doi: 10.1042/BJ20041584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Davis JJ, Fang B. J Gene Med. 2005;7:1380. doi: 10.1002/jgm.800. [DOI] [PubMed] [Google Scholar]
- 111.Frantz S, Vincent KA, Feron O, Kelly RA. Circ Res. 2005;96:15. doi: 10.1161/01.RES.0000153188.68898.ac. [DOI] [PubMed] [Google Scholar]
- 112.Jackson JR, Seed MP, Kircher CH, Willoughby DA, Winkler JD. FASEB J. 1997;11:457. [PubMed] [Google Scholar]
- 113.Shinozaki K, Ebert O, Kournioti C, Tai YS, Woo SL. Mol Ther. 2004;9:368. doi: 10.1016/j.ymthe.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 114.Guidotti LG, Chisari FV. Annu Rev Immunol. 2001;19:65. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 115.Anderson BD, Nakamura T, Russell SJ, Peng KW. Cancer Res. 2004;64:4919. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 116.Baraz L, Khazanov E, Condiotti R, Kotler M, Nagler A. Bone Marrow Transplant. 1999;24:179. doi: 10.1038/sj.bmt.1701825. [DOI] [PubMed] [Google Scholar]
- 117.Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, Carson WE, Caligiuri MA. Blood. 2001;97:3146. doi: 10.1182/blood.v97.10.3146. [DOI] [PubMed] [Google Scholar]
- 118.Hickey WF. Semin Immunol. 1999;11:125. doi: 10.1006/smim.1999.0168. [DOI] [PubMed] [Google Scholar]
- 119.Thomas DL, Fraser NW. Mol Ther. 2003;8:543. doi: 10.1016/s1525-0016(03)00236-3. [DOI] [PubMed] [Google Scholar]
- 120.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Science. 1991;252:854. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- 121.Fulci G, Chiocca EA. Front Biosci. 2003;8:e346. doi: 10.2741/976. [DOI] [PubMed] [Google Scholar]
- 122.Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA, Parker JN, Whitley RJ, Gillespie GY. Mol Ther. 2009;17:199. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grote D, Cattaneo R, Fielding AK. Cancer Res. 2003;63:6463. [PubMed] [Google Scholar]
- 124.Larder BA, Kemp SD, Darby G. EMBO J. 1987;6:169. doi: 10.1002/j.1460-2075.1987.tb04735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Science. 1990;248:1230. doi: 10.1126/science.2161559. [DOI] [PubMed] [Google Scholar]
- 126.Chou J, Roizman B. Proc Natl Acad Sci U S A. 1994;91:5247. doi: 10.1073/pnas.91.12.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Selvakumaran M, Reed JC, Liebermann D, Hoffman B. Blood. 1994;84:1036. [PubMed] [Google Scholar]
- 128.Zhan Q, Lord KA, Alamo I, Jr, Hollander MC, Carrier F, Ron D, Kohn KW, Hoffman B, Liebermann DA, Fornace AJ., Jr Mol Cell Biol. 1994;14:2361. doi: 10.1128/mcb.14.4.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.He B, Chou J, Liebermann DA, Hoffman B, Roizman B. J Virol. 1996;70:84. doi: 10.1128/jvi.70.1.84-90.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smith KD, Mezhir JJ, Bickenbach K, Veerapong J, Charron J, Posner MC, Roizman B, Weichselbaum RR. J Virol. 2006;80:1110. doi: 10.1128/JVI.80.3.1110-1120.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Veerapong J, Bickenbach KA, Shao MY, Smith KD, Posner MC, Roizman B, Weichselbaum RR. Cancer Res. 2007;67:8301. doi: 10.1158/0008-5472.CAN-07-1499. [DOI] [PubMed] [Google Scholar]
- 132.Shi L, Nishioka WK, Th'ng J, Bradbury EM, Litchfield DW, Greenberg AH. Science. 1994;263:1143. doi: 10.1126/science.8108732. [DOI] [PubMed] [Google Scholar]
- 133.He Q, Liang CH, Lippard SJ. Proc Natl Acad Sci U S A. 2000;97:5768. doi: 10.1073/pnas.100108697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Caporali S, Falcinelli S, Starace G, Russo MT, Bonmassar E, Jiricny J, D'Atri S. Mol Pharmacol. 2004;66:478. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 135.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. Proc Natl Acad Sci U S A. 2005;102:5844. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA. Biochemistry. 1994;33:9045. doi: 10.1021/bi00197a003. [DOI] [PubMed] [Google Scholar]
- 137.Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. EMBO J. 2003;22:6610. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stracker TH, Carson CT, Weitzman MD. Nature. 2002;418:348. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 139.Wersto RP, Rosenthal ER, Seth PK, Eissa NT, Donahue RE. J Virol. 1998;72:9491. doi: 10.1128/jvi.72.12.9491-9502.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Steinwaerder DS, Carlson CA, Lieber A. Hum Gene Ther. 2000;11:1933. doi: 10.1089/10430340050129549. [DOI] [PubMed] [Google Scholar]
- 141.Hodge LD, Scharff MD. Virology. 1969;37:554. doi: 10.1016/0042-6822(69)90273-6. [DOI] [PubMed] [Google Scholar]
- 142.Goodrum FD, Ornelles DA. J Virol. 1997;71:548. doi: 10.1128/jvi.71.1.548-561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang G, Barrett JW, Stanford M, Werden SJ, Johnston JB, Gao X, Sun M, Cheng JQ, McFadden G. Proc Natl Acad Sci U S A. 2006;103:4640. doi: 10.1073/pnas.0509341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Altomonte J, Wu L, Chen L, Meseck M, Ebert O, Garcia-Sastre A, Fallon J, Woo SL. Mol Ther. 2008;16:146. doi: 10.1038/sj.mt.6300343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Altomonte J, Wu L, Meseck M, Chen L, Ebert O, Garcia-Sastre A, Fallon J, Mandeli J, Woo SL. Cancer Gene Ther. 2008;16:266. doi: 10.1038/cgt.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mok W, Boucher Y, Jain RK. Cancer Res. 2007;67:10664. doi: 10.1158/0008-5472.CAN-07-3107. [DOI] [PubMed] [Google Scholar]
- 147.Favoreel HW, Van de Walle GR, Nauwynck HJ, Pensaert MB. J Gen Virol. 2003;84:1. doi: 10.1099/vir.0.18709-0. [DOI] [PubMed] [Google Scholar]
- 148.Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, Harsh GR, Louis DN, Bartus RT, Hochberg FH, Chiocca EA. Nat Med. 1999;5:881. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 149.Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. Eur J Immunol. 2004;34:336. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- 150.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Cancer Immunol Immunother. 2007;56:641. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Di Paolo NC, Tuve S, Ni S, Hellstrom KE, Hellstrom I, Lieber A. Cancer Res. 2006;66:960. doi: 10.1158/0008-5472.CAN-05-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Glaser M. Cell Immunol. 1979;48:339. doi: 10.1016/0008-8749(79)90128-x. [DOI] [PubMed] [Google Scholar]
- 153.Rollinghoff M, Starzinski-Powitz A, Pfizenmaier K, Wagner H. J Exp Med. 1977;145:455. doi: 10.1084/jem.145.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, Knolle PA, Thomas RK, von Bergwelt-Baildon M, Debey S, Hallek M, Schultze JL. Blood. 2005;106:2018. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- 155.Ikezawa Y, Nakazawa M, Tamura C, Takahashi K, Minami M, Ikezawa Z. J Dermatol Sci. 2005;39:105. doi: 10.1016/j.jdermsci.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 156.Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Blood. 2005;105:2862. doi: 10.1182/blood-2004-06-2410. [DOI] [PubMed] [Google Scholar]
- 157.Kottke T, Thompson J, Diaz RM, Pulido J, Willmon C, Coffey M, Selby P, Melcher A, Harrington K, Vile RG. Clin Cancer Res. 2009;15:561. doi: 10.1158/1078-0432.CCR-08-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Qiao J, Wang H, Kottke T, White C, Twigger K, Diaz RM, Thompson J, Selby P, de Bono J, Melcher A, Pandha H, Coffey M, Vile R, Harrington K. Clin Cancer Res. 2008;14:259. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, Reid JM, Federspiel M, Ames MM, Dingli D, Schweikart K, Welch A, Dispenzieri A, Peng KW, Russell SJ. Clin Pharmacol Ther. 2007;82:700. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marie I, Levy DE. Proc Natl Acad Sci U S A. 2004;101:9578. doi: 10.1073/pnas.0400567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nusinzon I, Horvath CM. Proc Natl Acad Sci U S A. 2003;100:14742. doi: 10.1073/pnas.2433987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Vlasakova J, Novakova Z, Rossmeislova L, Kahle M, Hozak P, Hodny Z. Blood. 2007;109:1373. doi: 10.1182/blood-2006-02-003418. [DOI] [PubMed] [Google Scholar]
- 163.Marks PA, Richon VM, Rifkind RA. J Natl Cancer Inst. 2000;92:1210. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 164.He LZ, Tolentino T, Grayson P, Zhong S, Warrell RP, Jr, Rifkind RA, Marks PA, Richon VM, Pandolfi PP. J Clin Invest. 2001;108:1321. doi: 10.1172/JCI11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Johnstone RW. Nat Rev Drug Discovery. 2002;1:287. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 166.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Nat Med. 2001;7:437. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 167.Fouladi M. Cancer Invest. 2006;24:521. doi: 10.1080/07357900600814979. [DOI] [PubMed] [Google Scholar]
- 168.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Nat Rev Cancer. 2001;1:194. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 169.Dowdy SC, Jiang S, Zhou XC, Hou X, Jin F, Podratz KC, Jiang SW. Mol Cancer Ther. 2006;5:2767. doi: 10.1158/1535-7163.MCT-06-0209. [DOI] [PubMed] [Google Scholar]
- 170.Qi H, Ratnam M. Cancer Res. 2006;66:5875. doi: 10.1158/0008-5472.CAN-05-4048. [DOI] [PubMed] [Google Scholar]
- 171.Piacentini P, Donadelli M, Costanzo C, Moore PS, Palmieri M, Scarpa A. Virchows Arch. 2006;448:797. doi: 10.1007/s00428-006-0173-x. [DOI] [PubMed] [Google Scholar]
- 172.Genin P, Morin P, Civas A. J Virol. 2003;77:7113. doi: 10.1128/JVI.77.12.7113-7119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Joseph J, Mudduluru G, Antony S, Vashistha S, Ajitkumar P, Somasundaram K. Oncogene. 2004;23:6304. doi: 10.1038/sj.onc.1207852. [DOI] [PubMed] [Google Scholar]
- 174.Kelly WK, Marks PA. Nat Clin Pract Oncol. 2005;2:150. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- 175.Mehnert JM, Kelly WK. Cancer J. 2007;13:23. doi: 10.1097/PPO.0b013e31803c72ba. [DOI] [PubMed] [Google Scholar]
- 176.Minucci S, Pelicci PG. Nat Rev Cancer. 2006;6:38. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 177.Nusinzon I, Horvath CM. Mol Cell Biol. 2006;26:3106. doi: 10.1128/MCB.26.8.3106-3113.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Taplin ME. Nat Clin Pract Oncol. 2007;4:236. doi: 10.1038/ncponc0765. [DOI] [PubMed] [Google Scholar]
- 179.Otsuki A, Patel A, Kasai K, Suzuki M, Kurozumi K, Chiocca EA, Saeki Y. Mol Ther. 2008;16:1546. doi: 10.1038/mt.2008.155. [DOI] [PubMed] [Google Scholar]
- 180.Katze MG, He Y, Gale M., Jr Nat Rev Immunol. 2002;2:675. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 181.Knudsen KE, Diehl JA, Haiman CA, Knudsen ES. Oncogene. 2006;25:1620. doi: 10.1038/sj.onc.1209371. [DOI] [PubMed] [Google Scholar]
- 182.Alao JP. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hu J, Colburn NH. Mol Cancer Res. 2005;3:100. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 184.Liu TC, Castelo-Branco P, Rabkin SD, Martuza RL. Mol Ther. 2008;16:1041. doi: 10.1038/mt.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kitazono M, Goldsmith ME, Aikou T, Bates S, Fojo T. Cancer Res. 2001;61:6328. [PubMed] [Google Scholar]
- 186.Okada T, Uchibori R, Iwata-Okada M, Takahashi M, Nomoto T, Nonaka-Sarukawa M, Ito T, Liu Y, Mizukami H, Kume A, Kobayashi E, Ozawa K. Mol Ther. 2006;13:738. doi: 10.1016/j.ymthe.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 187.Bieler A, Mantwill K, Dravits T, Bernshausen A, Glockzin G, Kohler-Vargas N, Lage H, Gansbacher B, Holm PS. Hum Gene Ther. 2006;17:55. doi: 10.1089/hum.2006.17.55. [DOI] [PubMed] [Google Scholar]
- 188.Kurozumi K, Hardcastle J, Thakur R, Shroll J, Nowicki M, Otsuki A, Chiocca EA, Kaur B. Mol Ther. 2008;16:1382. doi: 10.1038/mt.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Aghi MK, Liu TC, Rabkin S, Martuza RL. Mol Ther. 2009;17:51. doi: 10.1038/mt.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Arap W, Pasqualini R, Ruoslahti E. Science. 1998;279:377. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 191.Brooks PC, Clark RA, Cheresh DA. Science. 1994;264:569. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 192.Rhim JH, Tosato G. J Natl Cancer Inst. 2007;99:1739. doi: 10.1093/jnci/djm234. [DOI] [PubMed] [Google Scholar]
- 193.Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE. Neurosurgery. 2006;59:1304. doi: 10.1227/01.NEU.0000245622.70344.BE. discussion, p 1312. [DOI] [PubMed] [Google Scholar]
- 194.Wahl SM, Allen JB, Hines KL, Imamichi T, Wahl AM, Furcht LT, McCarthy JB. J Clin Invest. 1994;94:655. doi: 10.1172/JCI117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Elitok S, Brodsky SV, Patschan D, Orlova T, Lerea KM, Chander P, Goligorsky MS. Am J Physiol Renal Physiol. 2006;290:F159. doi: 10.1152/ajprenal.00227.2005. [DOI] [PubMed] [Google Scholar]
- 196.Zhang X, Chen A, De Leon D, Li H, Noiri E, Moy VT, Goligorsky MS. Am J Physiol Heart Circ Physiol. 2004;286:H359. doi: 10.1152/ajpheart.00491.2003. [DOI] [PubMed] [Google Scholar]
- 197.Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, Hochberg FH, Weissleder R, Carson W, Chiocca EA, Kaur B. J Natl Cancer Inst. 2007;99:1768. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- 198.Choudhary A, Hiscott P, Hart CA, Kaye SB, Batterbury M, Grierson I. Mol Vision. 2005;11:163. [PubMed] [Google Scholar]
- 199.Biswas PS, Rouse BT. Microbes Infect. 2005;7:799. doi: 10.1016/j.micinf.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 200.Kefalides NA, Ziaie Z. Lab Invest. 1986;55:328. [PubMed] [Google Scholar]
- 201.London FS, Brinker JM, Ziaie Z, Kefalides NA. Lab Invest. 1990;62:189. [PubMed] [Google Scholar]
- 202.Ziaie Z, Friedman HM, Kefalides NA. Collagen Relat Res. 1986;6:333. doi: 10.1016/s0174-173x(86)80004-8. [DOI] [PubMed] [Google Scholar]
- 203.Aghi MK, Liu TC, Rabkin S, Martuza RL. Mol Ther. 2008;17:51. doi: 10.1038/mt.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.de Fraipont F, El Atifi M, Gicquel C, Bertagna X, Chambaz EM, Feige JJ. J Clin Endocrinol Metab. 2000;85:4734. doi: 10.1210/jcem.85.12.7012. [DOI] [PubMed] [Google Scholar]
- 205.Armstrong LC, Bornstein P. Matrix Biol. 2003;22:63. doi: 10.1016/s0945-053x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 206.Hoekstra R, de Vos FY, Eskens FA, Gietema JA, van der Gaast A, Groen HJ, Knight RA, Carr RA, Humerickhouse RA, Verweij J, de Vries EG. J Clin Oncol. 2005;23:5188. doi: 10.1200/JCO.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 207.Ferrara N, Mass RD, Campa C, Kim R. Annu Rev Med. 2007;58:491. doi: 10.1146/annurev.med.58.061705.145635. [DOI] [PubMed] [Google Scholar]
- 208.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Cancer Res. 2004;64:3731. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 209.Salnikov AV, Heldin NE, Stuhr LB, Wiig H, Gerber H, Reed RK, Rubin K. Int J Cancer. 2006;119:2795. doi: 10.1002/ijc.22217. [DOI] [PubMed] [Google Scholar]
- 210.Libertini S, Iacuzzo I, Perruolo G, Scala S, Ierano C, Franco R, Hallden G, Portella G. Clin Cancer Res. 2008;14:6505. doi: 10.1158/1078-0432.CCR-08-0200. [DOI] [PubMed] [Google Scholar]
- 211.Boulikas T, Vougiouka M. Oncol Rep. 2003;10:1663. [PubMed] [Google Scholar]
- 212.Adusumilli PS, Chan MK, Chun YS, Hezel M, Chou TC, Rusch VW, Fong Y. Cancer Biol Ther. 2006;5:48. doi: 10.4161/cbt.5.1.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Siddik ZH. Oncogene. 2003;22:7265. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 214.Stanziale SF, Petrowsky H, Adusumilli PS, Ben-Porat L, Gonen M, Fong Y. Clin Cancer Res. 2004;10:3225. doi: 10.1158/1078-0432.ccr-1083-3. [DOI] [PubMed] [Google Scholar]
- 215.Stupp R, van den Bent MJ, Hegi ME. Curr Neurol Neurosci Rep. 2005;5:198. doi: 10.1007/s11910-005-0047-7. [DOI] [PubMed] [Google Scholar]
- 216.Aghi M, Rabkin S, Martuza RL. J Natl Cancer Inst. 2006;98:38. doi: 10.1093/jnci/djj003. [DOI] [PubMed] [Google Scholar]
- 217.Belanich M, Pastor M, Randall T, Guerra D, Kibitel J, Alas L, Li B, Citron M, Wasserman P, White A, Eyre H, Jaeckle K, Schulman S, Rector D, Prados M, Coons S, Shapiro W, Yarosh D. Cancer Res. 1996;56:783. [PubMed] [Google Scholar]
- 218.Friedman HS, McLendon RE, Kerby T, Dugan M, Bigner SH, Henry AJ, Ashley DM, Krischer J, Lovell S, Rasheed K, Marchev F, Seman AJ, Cokgor I, Rich J, Stewart E, Colvin OM, Provenzale JM, Bigner DD, Haglund MM, Friedman AH, Modrich PL. J Clin Oncol. 1998;16:3851. doi: 10.1200/JCO.1998.16.12.3851. [DOI] [PubMed] [Google Scholar]
- 219.Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM. J Neurosurg. 2003;99:1047. doi: 10.3171/jns.2003.99.6.1047. [DOI] [PubMed] [Google Scholar]
- 220.Gerson SL. Nat Rev Cancer. 2004;4:296–307. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- 221.Quirin C, Mainka A, Hesse A, Nettelbeck DM. Int J Cancer. 2007;121:2801. doi: 10.1002/ijc.23052. [DOI] [PubMed] [Google Scholar]
- 222.Bernt KM, Steinwaerder DS, Ni S, Li ZY, Roffler SR, Lieber A. Cancer Res. 2002;62:6089. [PubMed] [Google Scholar]
- 223.Schmelzle T, Hall MN. Cell. 2000;103:253. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 224.Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK. Nat Med. 2002;8:128. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- 225.Homicsko K, Lukashev A, Iggo RD. Cancer Res. 2005;65:6882. doi: 10.1158/0008-5472.CAN-05-0309. [DOI] [PubMed] [Google Scholar]
- 226.Morris PJ. Curr Opin Immunol. 1991;3:748. doi: 10.1016/0952-7915(91)90107-c. [DOI] [PubMed] [Google Scholar]
- 227.Ito H, Aoki H, Kuhnel F, Kondo Y, Kubicka S, Wirth T, Iwado E, Iwamaru A, Fujiwara K, Hess KR, Lang FF, Sawaya R, Kondo S. J Natl Cancer Inst. 2006;98:625. doi: 10.1093/jnci/djj161. [DOI] [PubMed] [Google Scholar]
- 228.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Proc Natl Acad Sci U S A. 2002;99:190. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamagu-chi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Science. 2004;306:1037. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 230.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Cell. 2004;119:753. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 231.Yao KC, Komata T, Kondo Y, Kanzawa T, Kondo S, Germano IM. J Neurosurg. 2003;98:378. doi: 10.3171/jns.2003.98.2.0378. [DOI] [PubMed] [Google Scholar]
- 232.Alonso MM, Jiang H, Yokoyama T, Xu J, Bekele NB, Lang FF, Kondo S, Gomez-Manzano C, Fueyo J. Mol Ther. 2008;16:487. doi: 10.1038/sj.mt.6300400. [DOI] [PubMed] [Google Scholar]
- 233.Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G. J Virol. 2007;81:1251. doi: 10.1128/JVI.01408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tortora G, Ciardiello F, Gasparini G. Nat Clin Pract Oncol. 2008;5:521. doi: 10.1038/ncponc1161. [DOI] [PubMed] [Google Scholar]
- 235.Philip PA, Mahoney MR, Allmer C, Thomas J, Pitot HC, Kim G, Donehower RC, Fitch T, Picus J, Erlichman C. J Clin Oncol. 2005;23:6657. doi: 10.1200/JCO.2005.14.696. [DOI] [PubMed] [Google Scholar]
- 236.Hainsworth JD, Sosman JA, Spigel DR, Edwards DL, Baughman C, Greco A. J Clin Oncol. 2005;23:7889. doi: 10.1200/JCO.2005.01.8234. [DOI] [PubMed] [Google Scholar]
- 237.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L. N Engl J Med. 2005;353:123. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 238.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. J Clin Oncol. 2004;22:77. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 239.Mahller YY, Vaikunth SS, Currier MA, Miller SJ, Ripberger MC, Hsu YH, Mehrian-Shai R, Collins MH, Crombleholme TM, Ratner N, Cripe TP. Mol Ther. 2007;15:279. doi: 10.1038/sj.mt.6300038. [DOI] [PubMed] [Google Scholar]
- 240.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Nature. 1992;356:713. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 241.DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, Vass WC, Viskochil D, Ratner N. J Clin Invest. 2000;105:1233. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Vargova L, Homola A, Zamecnik J, Tichy M, Benes V, Sykova E. Glia. 2003;42:77. doi: 10.1002/glia.10204. [DOI] [PubMed] [Google Scholar]
- 243.Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. Int J Biochem Cell Biol. 2004;36:1046. doi: 10.1016/j.biocel.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 244.Sugimura T, Kato F, Mimatsu K, Takenaka O, Iwata H. Spine. 1996;21:161. doi: 10.1097/00007632-199601150-00001. [DOI] [PubMed] [Google Scholar]
- 245.Moon LD, Asher RA, Fawcett JW. J Neurosci Res. 2003;71:23. doi: 10.1002/jnr.10449. [DOI] [PubMed] [Google Scholar]
- 246.Kuriyama N, Kuriyama H, Julin CM, Lamborn KR, Israel MA. Cancer Res. 2001;61:1805. [PubMed] [Google Scholar]
- 247.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Cancer Res. 2000;60:2497. [PubMed] [Google Scholar]
- 248.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, Brown EB, Izumi Y, Campbell RB, Berk DA, Jain RK. Proc Natl Acad Sci U S A. 2001;98:4628. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Brown E, McKee T, diTomaso E, Pluen A, Seed B, Boucher Y, Jain RK. Nat Med. 2003;9:796. doi: 10.1038/nm879. [DOI] [PubMed] [Google Scholar]
- 250.Bourguignon LY. Semin Cancer Biol. 2008;18:251. doi: 10.1016/j.semcancer.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Brekken C, Bruland OS, de Lange Davies C. Anticancer Res. 2000;20:3503. [PubMed] [Google Scholar]
- 252.Shuster S, Frost GI, Csoka AB, Formby B, Stern R. Int J Cancer. 2002;102:192. doi: 10.1002/ijc.10668. [DOI] [PubMed] [Google Scholar]
- 253.Victor R, Chauzy C, Girard N, Gioanni J, d'Anjou J, Stora De Novion H, Delpech B. Int J Cancer. 1999;82:77. doi: 10.1002/(sici)1097-0215(19990702)82:1<77::aid-ijc14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 254.Brinckerhoff CE, Matrisian LM. Nat Rev Mol Cell Biol. 2002;3:207. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 255.Montel V, Kleeman J, Agarwal D, Spinella D, Kawai K, Tarin D. Cancer Res. 2004;64:1687. doi: 10.1158/0008-5472.can-03-2047. [DOI] [PubMed] [Google Scholar]
- 256.Green NK, Herbert CW, Hale SJ, Hale AB, Mautner V, Harkins R, Hermiston T, Ulbrich K, Fisher KD, Seymour LW. Gene Ther. 2004;11:1256. doi: 10.1038/sj.gt.3302295. [DOI] [PubMed] [Google Scholar]
- 257.Thomson AH, Vasey PA, Murray LS, Cassidy J, Fraier D, Frigerio E, Twelves C. Br J Cancer. 1999;81:99. doi: 10.1038/sj.bjc.6690657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Duncan R. Anticancer Drugs. 1992;3:175. doi: 10.1097/00001813-199206000-00001. [DOI] [PubMed] [Google Scholar]
- 259.Romanczuk H, Galer CE, Zabner J, Barsomian G, Wadsworth SC, O'Riordan CR. Hum Gene Ther. 1999;10:2615. doi: 10.1089/10430349950016654. [DOI] [PubMed] [Google Scholar]
- 260.Fisher KD, Stallwood Y, Green NK, Ulbrich K, Mautner V, Seymour LW. Gene Ther. 2001;8:341. doi: 10.1038/sj.gt.3301389. [DOI] [PubMed] [Google Scholar]
- 261.Parker AL, Fisher KD, Oupicky D, Read ML, Nicklin SA, Baker AH, Seymour LW. J Drug Target. 2005;13:39. doi: 10.1080/10611860400020449. [DOI] [PubMed] [Google Scholar]
- 262.Stevenson M, Hale AB, Hale SJ, Green NK, Black G, Fisher KD, Ulbrich K, Fabra A, Seymour LW. Cancer Gene Ther. 2007;14:335. doi: 10.1038/sj.cgt.7701022. [DOI] [PubMed] [Google Scholar]
- 263.Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS, Cavenee WK. J Biol Chem. 1997;272:2927. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 264.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. N Engl J Med. 2004;350:2129. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 265.Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. J Clin Invest. 2007;117:2051. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Morrison J, Briggs SS, Green N, Fisher K, Subr V, Ulbrich K, Kehoe S, Seymour LW. Mol Ther. 2008;16:244. doi: 10.1038/sj.mt.6300363. [DOI] [PubMed] [Google Scholar]
- 267.Buller RE, Runnebaum IB, Karlan BY, Horowitz JA, Shahin M, Buekers T, Petrauskas S, Kreienberg R, Slamon D, Pegram M. Cancer Gene Ther. 2002;9:553. doi: 10.1038/sj.cgt.7700472. [DOI] [PubMed] [Google Scholar]
- 268.Power AT, Bell JC. Mol Ther. 2007;15:660. doi: 10.1038/sj.mt.6300098. [DOI] [PubMed] [Google Scholar]
- 269.Coukos G, Makrigiannakis A, Kang EH, Caparelli D, Benjamin I, Kaiser LR, Rubin SC, Albelda SM, Molnar-Kimber KL. Clin Cancer Res. 1999;5:1523. [PubMed] [Google Scholar]
- 270.Raykov Z, Balboni G, Aprahamian M, Rommelaere J. Int J Cancer. 2004;109:742. doi: 10.1002/ijc.20013. [DOI] [PubMed] [Google Scholar]
- 271.Thorne SH, Contag CH. Gene Ther. 2008;15:753. doi: 10.1038/gt.2008.42. [DOI] [PubMed] [Google Scholar]
- 272.Thorne SH, Negrin RS, Contag CH. Science. 2006;311:1780. doi: 10.1126/science.1121411. [DOI] [PubMed] [Google Scholar]
- 273.Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mol Cancer Ther. 2006;5:755. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- 274.Qiao J, Wang H, Kottke T, Diaz RM, Willmon C, Hudacek A, Thompson J, Parato K, Bell J, Naik J, Chester J, Selby P, Harrington KJ, Melcher A, Vile RG. Gene Ther. 2008;15:604. doi: 10.1038/sj.gt.3303098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Qiao J, Kottke T, Willmon C, Galivo F, Wongthida P, Diaz RM, Thompson J, Ryno P, Barber GN, Chester J, Selby P, Harrington K, Melcher A, Vile RG. Nat Med. 2008;14:37. doi: 10.1038/nm1681. [DOI] [PubMed] [Google Scholar]