

Abstract
To date, most genome-wide association studies (GWAS) and studies of fine-scale population structure have been conducted primarily on Europeans. Han Chinese, the largest ethnic group in the world, composing 20% of the entire global human population, is largely underrepresented in such studies. A well-recognized challenge is the fact that population structure can cause spurious associations in GWAS. In this study, we examined population substructures in a diverse set of over 1700 Han Chinese samples collected from 26 regions across China, each genotyped at ∼160K single-nucleotide polymorphisms (SNPs). Our results showed that the Han Chinese population is intricately substructured, with the main observed clusters corresponding roughly to northern Han, central Han, and southern Han. However, simulated case-control studies showed that genetic differentiation among these clusters, although very small (FST = 0.0002 ∼0.0009), is sufficient to lead to an inflated rate of false-positive results even when the sample size is moderate. The top two SNPs with the greatest frequency differences between the northern Han and southern Han clusters (FST > 0.06) were found in the FADS2 gene, which associates with the fatty acid composition in phospholipids, and in the HLA complex P5 gene (HCP5), which associates with HIV infection, psoriasis, and psoriatic arthritis. Ingenuity Pathway Analysis (IPA) showed that most differentiated genes among clusters are involved in cardiac arteriopathy (p < 10−101). These signals indicating significant differences among Han Chinese subpopulations should be carefully explained in case they are also detected in association studies, especially when sample sources are diverse.
Introduction
Population stratification or ancestry differences among subpopulations poses a general concern in genome-wide association studies (GWAS) because it can cause spurious associations.1–3 Previous studies have shown that even minor stratification can have a substantial impact on population-based studies with large sample sizes.4–11 Therefore, characterization of population substructure is important in study design, statistical analysis, and interpretation of the results of association studies. However, so far most genome-wide studies of fine-scale population structure focused primarily on Europeans,3,12–17 although the majority of the global health burden is in low- and middle-income countries with populations of non-European origins. The recent release of detailed characterization of genetic diversity in non-European populations, such as Africans,18 Pacific Islanders,7 Native Americans,19 Southeast Asians,20 Indians,21 and Japanese,22 demarcated an extension of efforts on populations of other geographic origins.
Han Chinese is the largest ethnic group in the world, making up 20% of the entire global human population. However, studies on its population substructures have been largely underrepresented in similar efforts worldwide. In the next two years, China will, with a pledge of $30 million, launch a project to search at a genome-wide level for genes that contribute to common diseases such as cancer, hypertension, type 2 diabetes, and psychiatric illness in the Han Chinese, who make up 92% of the population of China. Although we have shown in previous studies, on the basis of genome-wide single-nucleotide polymorphism (SNP) data from the Human Genome Diversity Panel (HGDP),4,23 that the average of genetic differences between Han Chinese population samples (FST = 0.002) was much lower than that among European populations (FST = 0.009),24 population substructures are often expected in the Han Chinese because of the population's complex origins and long history of interaction with many surrounding ethnic groups. Furthermore, given that genetic studies are currently aiming at identifying smaller and smaller genetic effects, recognizing and controlling for population substructure, even at such a fine level within a seemingly homogeneous population, becomes imperative if one is to avoid confounding and spurious associations.25
Discerning the population structure of Han Chinese is important for the prevention of spurious associations and the identification of genetic variants whose contribution to disease risk differs across Han Chinese subpopulations. In this study, genotyping data from approximately 160,000 SNPs were collected from more than 1700 Han Chinese population samples residing in 26 of the 34 administrative regions of China. With these diverse samples encompassing both geographical populations and metropolitan populations, we conducted population-structure analyses and simulation studies to investigate whether the Han Chinese population is stratified and to what extent the stratification affects the results of the association studies. We investigated false-positive rates and statistical power in various study designs, using both empirical and simulated data. We also identified SNPs, as well as genes, that showed substantial differences among subpopulations and discussed the important issues of sample-collection strategies, optimal association-study design, and public data usage for future association studies involving Han Chinese. To our knowledge, this study is the first effort to address the issue of fine-scale population substructures and GWAS design in Han Chinese by using genome-wide SNPs.
Subjects and Methods
Populations and Samples
Overall, 1721 Han Chinese samples were studied, of which 1506 samples were collected in this study. A total of 44 samples were from the HGDP23 (34 Han and 10 Han-NChina); and 171 samples were from the International HapMap Project26 (86 CHB [Han Chinese from Beijing], 85 CHD [Han Chinese from metropolitan Denver, CO, USA]). These samples represent Han Chinese populations residing in 26 of the 34 administrative regions of China and in a region of the USA. These 26 geographical populations can be classified into northern Han Chinese (N-Han) and southern Han Chinese (S-Han), with the Yangtze River used as a geographical boundary, as proposed by previous studies27 (Figure S1A, available online). The sample size and average heterozygosity for each regional population are shown in Figures S1B and S1C, respectively. All procedures were followed in accordance with the ethical standards of the Responsible Committee on Human Experimentation (ethics committee of Fudan University) and the Helsinki. Declaration of 1975, as revised in 2000. In addition, 788 HapMap3 unrelated individuals from the other nine populations (apart from CHB and CHD) were included in the analysis, which comprised Japanese from Tokyo, Japan (JPT, n = 89); Gujarati Indians in Houston, TX, USA (GIH, n = 83);, individuals of Mexican ancestry in Los Angeles, CA, USA (MEX, n = 47); individuals of African ancestry from the southwest U.S. (ASW, n = 47); Luhya in Webuye, Kenya (LWK, n = 83); Maasai in Kinyawa, Kenya (MKK, n = 143); Yoruba in Ibadan, Nigeria (YRI, n = 108); Utah residents with northern and western European ancestry from the CEPH collection (CEU, n = 111); and Tuscans from Italy (TSI, n = 77).
Genotyping and Data Assemblage
Genotyping of 340 Han Chinese samples was performed with the Affymetrix Genome-Wide Human SNP Array 6.0 on the Affymetrix genotyping platform at Fudan University, Shanghai, in accordance with the “48 Sample Protocol” (Affymetrix, Genome-Wide Human SNP Nsp/Sty 6.0 User Guide; see Web Resources). The ∗.CEL files containing raw intensity data were analyzed with Birdsuite, version 1.5.5.28 We used the default confidence-score threshold (0.1) for Birdseed analysis; i.e., genotypes with a confidence score greater than 0.1 were considered as missing data. This default provides a good compromise between accuracy and call rate. Genotyping of 1166 Han Chinese samples was performed with the Illumina Human 610-Quad BeadChips as described elsewhere.29 Genotypic data of 44 HGDP Han Chinese samples were obtained from the HGDP database.4 The genotypic data of HapMap samples were downloaded from the International HapMap Project website (phase II+III, release no. 27). The data set was filtered for individuals with > 2% missing genotypes and SNPs with > 10% missing data, as well as Hardy-Weinberg disequilibrium (HWD) (p < 0.00001) within regional population. For the purposes of this study, only SNPs with reference sequence (rs) numbers and vendor-specified strands were used in combing data. The final data set comprised 158,015 autosomal SNPs shared by 1708 Han Chinese and 767 non-Han-Chinese samples. We investigated batch effect to ensure that it would not affect the downstream analyses by comparing samples collected from the same region but genotyped by different chips (data not shown).
Statistical Analyses, Simulation Studies, and Pathway Analysis
Unbiased estimates of FST were calculated by following Weir and Hill.30 Confidence intervals of the FST over loci were calculated by bootstrap resampling, with 1000 replications. Principal components analysis (PCA) was performed at the individual level in EIGENSOFT version 3.0.31 Great circle distance calculations were performed by following the approach of Ramachandran et al.32 Permutation tests for between-group identity by state (IBS) differences were performed in PLINK version 1.0633 with 10,000 permutations; this analysis was for identifying whether or not there are significant differences between groups of individuals.
Simulation studies of whole-genome association analyses were performed with PLINK version 1.06.33 To examine the effect of the Han Chinese population structure on a GWAS, we conducted simulations by sampling individuals from the subpopulations in different proportions between cases and controls, then evaluated possible inflation of false-positive rates with the use of the genome-wide x2 inflation factor (λ) for the genomic control (GC).34–36 The value of λ was computed as the median x2 statistic divided by 0.455, the predicted median x2 if there is no inflation. We simulated 1000 SNPs associated with disease (population odds ratio of 2.0) to evaluate statistical power in a GWAS. A uniform allele-frequency range (0∼1) was adopted. Power was calculated as the number of disease SNPs showing p < 0.05 (unadjusted or adjusted) divided by 1000.
SNPs showing substantial differences among clusters were investigated for network and functional interrelatedness with the Ingenuity Pathway Analysis (IPA) version 7.6 software tool (Ingenuity Systems). This software can help the search for information on genes and/or chemicals, their impact on diseases and cellular processes, and their role in pathways. IPA scans data generated by various large-scale technologies, including gene expression and SNP microarrays, proteomics experiments, and small-scale experiments, generating gene lists to identify networks by using information in the Ingenuity Pathways Knowledge Base, a repository of molecular interactions, regulatory events, gene-to-phenotype associations, and chemical knowledge, all pulled from the full text of the peer-reviewed life sciences literatures. With IPA, we can analyze data in the context of molecular mechanisms, identify key mechanistic differences among subpopulations, and further relate molecular events to higher-order cellular and disease processes.
Results
Cryptic Population Substructures of Han Chinese
The 1708 Han Chinese samples were studied together with 767 HapMap3 unrelated individuals from nine non-Han Chinese populations (see Subjects and Methods) via PCA with EIGENSOFT.31 The PCA was performed on the basis of 158,015 autosomal SNPs shared by all 2475 samples. The Han Chinese population shows a rather small genetic diversity when compared with worldwide populations. In the PCA plot, all Han Chinese individuals cluster closely and together with Japanese individuals, with European and African samples forming distinct clusters, at the other two angles of the triangle-like plot (Figure 1A). With 160,000 SNPs, Han Chinese and Japanese samples could be distinguished clearly as two distinct clusters when European and African samples were removed (Figure 1B). When only Han Chinese samples were analyzed, they seemed to form a homogenous cluster without observable substructure (Figure 1C). However, the difference between northern and southern populations emerged when CHB, CHD, three metropolitan populations (Beijing, Shanghai, Guangzhou), and two regional populations close to Shanghai (Anhui and Jiangsu) were excluded as the first principal component (PC1) classified all the remaining samples into two subgroups (Figure 1D), corresponding to N-Han and S-Han, with the Yangtze River serving as a geographical boundary (Figure S1).
Figure 1.
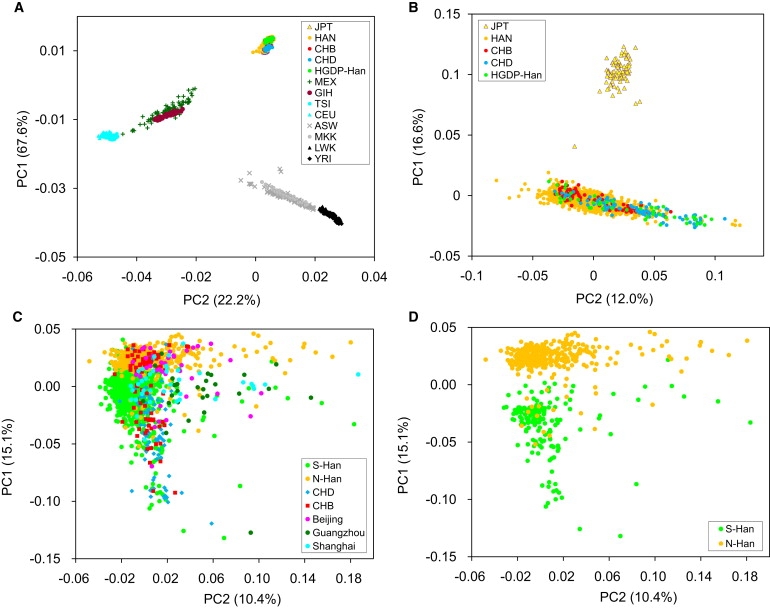
Analysis of the First Two Principal Components
The % Eigenvalue is the percentage of the total variance in the first ten PCs.
(A) 1708 Han Chinese and 767 non-Han Chinese individuals representing all samples studied.
(B) 1708 Han Chinese and 89 Japanese (JPT) individuals (excluding non-East Asian samples).
(C) Han Chinese individuals (excluding all non-Han Chinese samples).
(D) Han Chinese individuals (excluding CHB, CHD, three metropolitan populations [Beijing, Shanghai, Guangzhou], and two regional populations close to Shanghai [Anhui and Jiangsu]).
In the two-dimensional plot of the first and second PCs (Figure 2A), as well as that of the first and third PCs (Figure 2B), most of the samples from Jiangsu, Anhui, and Shanghai, which are geographically located between the N-Han and the S-Han (Figure 2C), are also genetically located between N-Han and S-Han and form the third subgroup, which is hereafter referred to as central Han Chinese (C-Han). The average within-subgroup between-region FST values were 0.00014, 0.00046, and 0.00079 for N-Han, C-Han, and S-Han, respectively, indicating higher between-region diversity within S-Han. This observation was echoed by the distribution of PC1, as shown later (Figure 2D). The pairwise FST of N-Han and S-Han (0.00116) was much larger than the FST of N-Han and C-Han (0.00039) and also larger than that of S-Han and C-Han (0.00108) (Table 1). These results indicated that the main difference occurs between N-Han and S-Han, larger than the values of the other two comparisons (p < 0.002).
Figure 2.
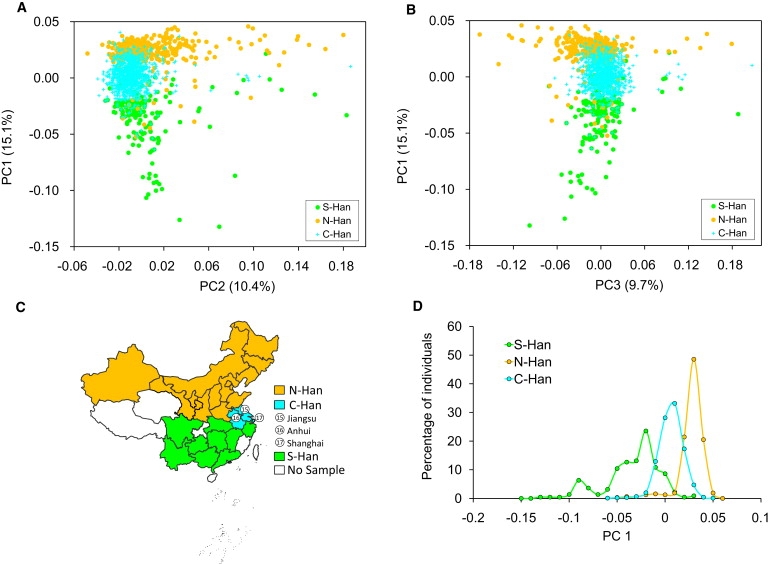
Principal Components Analysis of Han Chinese Individuals
(A) Analysis of PC1 and PC2 of Han Chinese.
(B) Analysis of PC1 and PC3 of Han Chinese.
(C) Geographical locations of three C-Han populations.
(D) Distribution of PC1 for Han Chinese individuals as classified into three subgroups.
Table 1.
Pairwise FST within and between Han Chinese Subgroups
| N-Han | C-Han | S-Han | |
|---|---|---|---|
| N-Han | 0.00014 ± 0.00027 | ||
| C-Han | 0.00039 ± 0.00037 | 0.00046 ± 0.00015 | |
| S-Han | 0.00116 ± 0.00125 | 0.00108 ± 0.00103 | 0.00079 ± 0.00091 |
Abbreviations are as follows: N-Han, northern Han Chinese; C-Han, central Han Chinese; S-Han, southern Han Chinese. Pairwise FST values within a subgroup were obtained from the average FST between populations within that subgroup. Pairwise FST values between two subgroups were obtained from the average FST between populations from the two different subgroups. The standard deviation is also shown for each pairwise FST.
Three subgroups of Han Chinese samples formed distinctive distributions of PC1, those of N-Han and S-Han almost nonoverlapping and that of C-Han lying in between and considerably overlapping with those of N-Han and S-Han, respectively (Figure 2D). We thus classified Han Chinese individuals into three clusters by K-means clustering on PC1 values (F-test, p < 10−3): the N-Han cluster (NHC), the C-Han cluster (CHC), and the S-Han cluster (SHC). As shown in Figure 3 (see Table S1 also), 92.4% of N-Han individuals were assigned to the NHC, 87.8% of S-Han individuals were assigned to the SHC, and 74.2% of the C-Han individuals were assigned to the CHC. In comparison with the FST value between Han Chinese clusters and the Japanese (0.00688), the genetic divergence among three Chinese clusters (FST [NHC−CHC] = 0.00018, FST [SHC−CHC] = 0.00032, FST [NHC−SHC] = 0.00093) is small (Table 2). However, permutation tests for between-group IBS differences showed that individuals from the same cluster were genetically much more similar than those from different clusters (p < 10−5), indicating that the N-Han, C-Han, and S-Han can be taken as separate subpopulations of Han Chinese. The smaller genetic difference between the NHC and the JPT than that between the JPT and either of the other two Han Chinese clusters (CHC and SHC) indicates that the Japanese population is more related to N-Han, and further suggests that the Japanese population either initially migrated from north of East Asia or was more affected by immigrants from northern populations of East Asia.
Figure 3.
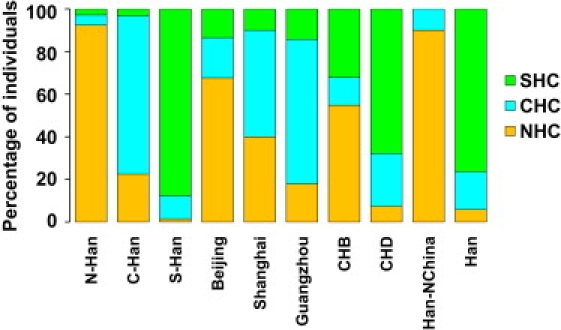
Classification of Han Chinese Individuals into the Three Clusters
For each population or group (x axis), individuals are assigned to three clusters. Percentages (y axis) are depicted by three colors representing three clusters.
Table 2.
Pairwise FST between Han Chinese Clusters and Population Samples in a Public Database
| NHC | CHC | SHC | |
|---|---|---|---|
| CHC | 0.00018 (0.00017–0.00019) | ||
| SHC | 0.00093 (0.00091–0.00095) | 0.00032 (0.00031–0.00033) | |
| Beijing | 0.00000 (0.00000–0.00005) | 0.00008 (0.00001–0.00014) | 0.00034 (0.00027–0.00041) |
| Shanghai | 0.00022 (0.00010–0.00032) | 0.00000 (0.00000–0.00000) | 0.00015 (0.00004–0.00027) |
| Guangzhou | 0.00063 (0.00055–0.00077) | 0.00010 (0.00003–0.00017) | 0.00000 (0.00000–0.00000) |
| CHB | 0.00020 (0.00017–0.00023) | 0.00004 (0.00001–0.00007) | 0.00031 (0.00027–0.00034) |
| CHD | 0.00170 (0.00164–0.00174) | 0.00084 (0.00080–0.00088) | 0.00020 (0.00016–0.00024) |
| Han-NChina | 0.00001 (0.00000–0.00012) | 0.00090 (0.00069–0.00110) | 0.00187 (0.00163–0.00209) |
| Han | 0.00182 (0.00173–0.00192) | 0.00085 (0.00079–0.00093) | 0.00000 (0.00000–0.00000) |
| JPT | 0.00688 (0.00680–0.00697) | 0.00716 (0.00708–0.00724) | 0.00777 (0.00769–0.00786) |
Abbreviations are as follows: NHC, northern Han Chinese cluster; CHC, central Han Chinese cluster; SHC, southern Han Chinese cluster. The numbers in parentheses are 95% confidence intervals for each pairwise FST.
Genetic Profiles of HapMap, HGDP, and Metropolitan Han Populations
Both HGDP and HapMap samples have been extensively used in human population-genetics studies and have facilitated the discovery of sequence variants underlying common diseases.4,6,8,9,26,32 We examined the genetic profiles of two HapMap Han Chinese population samples (CHB and CHD) and two HGDP Han Chinese population samples, referred to in HGDP as Han-NChina and Han (Figure 3, Table S1). CHB individuals are distributed widely in the NHC (54.8%), the CHC (13.1%), and the SHC (32.1%) in the PC plots (Figures S3A and S3B). CHD individuals are also distributed widely in the NHC (7.4%), the CHC (24.7%), and the SHC (67.9%) (Figures S3C and S3D). However, Han-NChina individuals were confined mostly in the NHC (90%) (Figures S4A and S4B), and Han individuals resembled the SHC clusters (76.5%) (Figures S4C and S4D).
It is expected that the residents in large cities are immigrants from different areas. We examined the genetic profiles of the populations currently residing in the three largest cities in China: Beijing, Shanghai, and Guangzhou, which are geographically located in northern, central, and southern China, respectively. Although our analyses (Table 2, Figure S5, and Table S1) indicated that metropolitan populations at Shanghai, Beijing, and Guangzhou do not, as a whole, show significant difference from the cluster of which they belong to geographically, the diverse distribution of individuals on PC plots (Fig. S5) suggested that each of them should not be taken as one single homogenous population.
Correlation of Genetic and Geographical Coordinates of Populations
Given that PC1 could classify Han Chinese individuals into northern and southern clusters, we asked whether it also applies to populations. The genetic coordinate of a population was calculated by taking the arithmetic average of the PC1 values of individuals within each population whose geographical location was unequivocal. In this analysis, HapMap samples (CHB and CHD) and HGDP samples (Han-NChina and Han) with uncertain location information were not included. As shown in Figure 4A, N-Han populations generally have PC1 values larger than 0.01, whereas S-Han populations have PC1 values smaller than −0.01, and the average PC1 values of each C-Han population were between −0.01 and 0.01. Clearly, the average PC1 values and latitudes of sample locations were highly correlated (R2 = 0.69, p = 1.27 × 10−7; Figure 4B). We did not observe significant correlation between the other PCs and geographical coordinates, although the top ten PCs were all found to be significant on the basis of the TW-test.31 We also did not observe significant correlation of FST between populations and the geographical distances between them (R2 = 0.003, p = 0.23; Figure S2).
Figure 4.
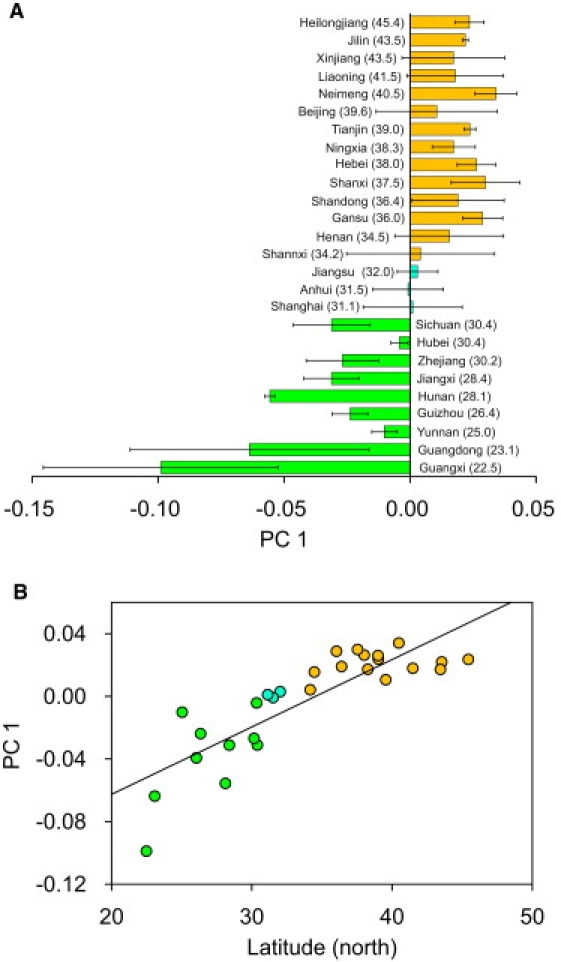
Relationship of PC1 and Latitude
(A) Average PC1 and latitude of populations. The standard deviation of PC1 is also shown for each population.
(B) Correlation of PC1 and latitude. The line in the plot shows the regression line (y = 0.0043x − 0.147). R2 for the linear regression of genetic distance on geographic distance is 0.69 (p = 1.27 × 10−7).
Highly Differentiated SNPs among Han Chinese Clusters
To identify the genomic regions that are highly differentiated, in allele frequency, among the three Han Chinese clusters, we examined the distributions of FST for all of the SNPs over the entire genome (Figure S6, Figure S7, and Figure S8). Regardless of the root cause for the difference in allele frequency, which includes genetic drift and natural selection, spurious associations are likely to occur in association studies. In spite of the low level of differentiation between the NHC and the SHC (average FST = 0.00093), a substantial proportion of SNPs were located in the tails of the distribution (empirical p < 10−3); 191 of the 158,014 SNPs have FST ≥ 0.023 (99.90 percentile). Table 3 lists the SNPs with FST ≥ 0.04 (99.99th percentile; p < 10−4). The two SNPs that showed the highest FST were rs174570 C/T (0.066) and rs174570 (0.063). The former resides within an intron on the FADS2 gene (MIM 606149) located on chromosome 11 (at Chr11:61353788), which is associated with the fatty acid composition in phospholipids37 and arachidonic acid levels, a precursor of molecules involved in inflammation and immunity processes, as well as cardiovascular disease,38 the frequencies of the C allele being 0.692 and 0.512 in the NHC and the SHC, respectively. The SNP rs2596472 (nearGene-5) is located in the HLA complex P5 (HCP5 [MIM 604676]) region on chromosome 6 (at Chr6:31536946), which is associated with HIV infection, psoriasis, and psoriatic arthritis,38–40 the frequencies of the A allele being 0.809 and 0.938 in the NHC and the SHC, respectively. In addition, a missense SNP (Val/Met) that is one of the top six highly differentiated SNPs is in the natriuretic peptide precursor A gene (NPPA [MIM 108780]), of which some common genetic variants were found to be associated with circulating natriuretic peptide concentrations that contribute to interindividual variation in blood pressure and hypertension.41–44 Genetic differentiation of the other two pairs of cluster comparisons (NHC versus CHC and CHC versus SHC) is expectedly lower; 197 of the 158,014 SNPs have FST ≥ 0.011 (99.90 percentile) between the NHC and the CHC. Next, we searched for genomic regions that showed relatively higher differentiation, using the FST value for each SNP (FST ≥ 0.033, 99.99th percentile; Table S3). Of the 158,014 SNPs, 166 have FST ≥ 0.016 (99.90th percentile) between the CHC and the SHC. The genomic regions that showed relatively higher differentiation by the FST value for each SNP (FST ≥ 0.023, 99.99th percentile) are shown in Table S4.
Table 3.
SNPs that Are Highly Differentiated between the NHC and the SHC
| SNP | FST | Raw p Value | FDR p Value | Chr. | Position | Gene | MIM Number | Category |
|---|---|---|---|---|---|---|---|---|
| rs174570 | 0.066 | 2.40 × 10−13 | 4.61 × 10−7 | 11 | 61353788 | FADS2 | 606149 | intron |
| rs2596472 | 0.063 | 7.37 × 10−13 | 7.09 × 10−7 | 6 | 31536946 | HCP5 | 604676 | nearGene-5 |
| rs2517646 | 0.055 | 2.00 × 10−11 | 1.29 × 10−5 | 6 | 30230554 | TRIM10 | 605701 | intron |
| rs174534 | 0.054 | 2.82 × 10−11 | 1.36 × 10−5 | 11 | 61306034 | C11orf9 | 608329 | intron |
| rs1265048 | 0.052 | 6.64 × 10−11 | 2.56 × 10−5 | 6 | 31189388 | PSORS1C1 | N/A | nearGene-5 |
| rs5063 | 0.050 | 1.65 × 10−10 | 5.28 × 10−5 | 1 | 11830235 | NPPA | 108780 | missense |
| rs9266629 | 0.050 | 2.11 × 10−10 | 5.80 × 10−5 | 6 | 31454801 | N/A | N/A | N/A |
| rs1419675 | 0.048 | 3.56 × 10−10 | 8.56 × 10−5 | 6 | 30200686 | N/A | N/A | N/A |
| rs198464 | 0.048 | 4.61 × 10−10 | 9.85 × 10−5 | 11 | 61278197 | C11orf9 | 608329 | nearGene-5 |
| rs9405015 | 0.045 | 1.69 × 10−9 | 3.26 × 10−4 | 6 | 31263782 | N/A | N/A | N/A |
| rs4626416 | 0.043 | 3.86 × 10−9 | 6.59 × 10−4 | 6 | 23243905 | N/A | N/A | N/A |
| rs2253907 | 0.042 | 4.11 × 10−9 | 6.59 × 10−4 | 6 | 31444849 | N/A | N/A | N/A |
| rs2076003 | 0.041 | 7.48 × 10−9 | 1.11 × 10−3 | 1 | 11806734 | CLCN6 | 602726 | intron |
| rs7537765 | 0.041 | 8.63 × 10−9 | 1.19 × 10−3 | 1 | 11809890 | CLCN6 | 602726 | intron |
| rs1264704 | 0.041 | 9.34 × 10−9 | 1.20 × 10−3 | 6 | 30173298 | N/A | N/A | N/A |
| rs17367504 | 0.040 | 1.07 × 10−8 | 1.29 × 10−3 | 1 | 11785365 | MTHFR | 607093 | intron |
| rs2236797 | 0.040 | 1.24 × 10−8 | 1.40 × 10−3 | 1 | 11815237 | CLCN6 | 602726 | intron |
| rs9295676 | 0.040 | 1.39 × 10−8 | 1.49 × 10−3 | 6 | 26036355 | SLC17A2 | 611049 | intron |
Abbreviations are as follows: FDR, p values are corrected by Benjamini and Hochberg step-up false discovery rate control; Chr., chromosome; MIM, Mendelian Inheritance in Man; N/A, not available.
With IPA, we can analyze data in the context of molecular mechanisms, identify key mechanistic differences between subpopulations or clusters, and further relate molecular events to higher-order cellular and disease processes. The results of IPA showed a considerable number of differentiated genes among clusters that are involved in cardiotoxicity, hepatotoxicity, and nephrotoxicity. The most significant association is with cardiac arteriopathy (P[NHC−CHC] = 3.76 × 10−101; P[SHC−CHC] = 5.04 × 10−128; P[NHC−SHC] = 1.39 × 10−132).
Simulation Studies of Genome-wide Association Analyses
In practical studies, it is very important to know how Han Chinese population substructures can affect the results of a GWAS, so we conducted a series of simulations to examine the effect of the population substructures on false-discovery rate and statistical power. We asked how many false-positive results there would be if Han Chinese population substructures were not considered in case-control sampling and what the statistical power would be when false-positive results due to population substructures were controlled.
We first performed a simulated GWAS by sampling individuals either as cases or as controls from different clusters; e.g., N-Han, C-Han, and S-Han. Because of the limited sample size of real data, we sampled 300 individuals as cases and 300 individuals as controls from the same cluster to evaluate the association between false-positive rates and statistical power (Table 4). The genome-wide x2 inflation factor (λ), an indicator of the inflation of false-positive rates due to the presence of population substructure,34–36 was computed as the median x2 statistic divided by 0.455, the predicted median x2 if there is no inflation. The λ values were all within an acceptable level (λ ≤ 1.1). For example, λ was 1.00 when both cases and controls were sampled from the NHC (scenario I in Table 4), λ was 1.07 when both cases and controls were sampled from the CHC (scenario II), and λ was 1.05 when both cases and controls were sampled from the SHC (scenario III).
Table 4.
Case-Control Simulation Studies
| Scenario |
Case |
Control |
λ |
False Positives (%) |
Power (%) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NHC | CHC | SHC | NHC | CHC | SHC | Unadj. | GC | BONF | FDR | Unadj. | GC | BONF | FDR | ||
| I | 300 | 300 | 1.00 | 2.90 | 2.90 | 0.00 | 0.01 | 85.9 | 85.9 | 2.6 | 25.2 | ||||
| II | 300 | 300 | 1.07 | 4.95 | 3.95 | 0.00 | 0.00 | 85.9 | 84.0 | 2.6 | 25.2 | ||||
| III | 300 | 300 | 1.05 | 4.81 | 3.80 | 0.00 | 0.00 | 85.9 | 84.3 | 2.6 | 24.8 | ||||
| IV | 300 | 300 | 1.74 | 11.61 | 3.69 | 0.03 | 0.07 | 85.9 | 71.9 | 2.6 | 38.4 | ||||
| V | 300 | 300 | 1.20 | 6.31 | 3.90 | 0.02 | 0.02 | 85.9 | 80.9 | 2.6 | 26.8 | ||||
| VI | 300 | 300 | 1.30 | 7.18 | 3.94 | 0.00 | 0.01 | 85.9 | 80.0 | 2.6 | 26.9 | ||||
| VII | 100 | 100 | 100 | 100 | 100 | 100 | 1.09 | 8.94 | 3.90 | 0.01 | 0.02 | 85.9 | 77.6 | 2.6 | 28.5 |
| VIII | 100 | 100 | 100 | 300 | 1.20 | 6.19 | 3.87 | 0.00 | 0.00 | 85.9 | 80.8 | 2.6 | 25.3 | ||
| IX | 100 | 100 | 100 | 300 | 1.09 | 5.21 | 4.05 | 0.01 | 0.01 | 85.9 | 84.0 | 2.6 | 26.2 | ||
| X | 100 | 100 | 100 | 300 | 1.26 | 6.77 | 3.90 | 0.00 | 0.01 | 85.9 | 80.2 | 2.6 | 26.0 | ||
| I | 300 (100%) | 0 (0%) | 300 | 1.00 | 2.90 | 2.90 | 0.00 | 0.01 | 85.9 | 85.9 | 2.6 | 25.2 | |||
| XI | 240 (80%) | 60 (20%) | 300 | 1.06 | 6.05 | 4.09 | 0.00 | 0.00 | 85.9 | 82.2 | 2.6 | 25.3 | |||
| XII | 210 (70%) | 90 (30%) | 300 | 1.10 | 5.21 | 3.94 | 0.00 | 0.01 | 85.9 | 83.7 | 2.6 | 25.8 | |||
| XIII | 180 (60%) | 120 (40%) | 300 | 1.15 | 6.03 | 4.13 | 0.00 | 0.00 | 85.9 | 82.5 | 2.6 | 25.0 | |||
| XIV | 150 (50%) | 150 (50%) | 300 | 1.21 | 6.40 | 3.94 | 0.00 | 0.00 | 85.9 | 80.7 | 2.6 | 25.8 | |||
| XV | 100 (33%) | 200 (67%) | 300 | 1.33 | 7.69 | 3.99 | 0.00 | 0.00 | 85.9 | 79.9 | 2.6 | 27.4 | |||
| IV | 0 (0%) | 300 (100%) | 300 | 1.74 | 11.61 | 3.69 | 0.03 | 0.07 | 85.9 | 71.9 | 2.6 | 38.4 | |||
| I | 300 (100%) | 0 (0%) | 300 | 1.00 | 2.90 | 2.90 | 0.00 | 0.01 | 85.9 | 85.9 | 2.6 | 25.2 | |||
| XVI | 240 (80%) | 60 (20%) | 300 | 1.05 | 4.59 | 3.91 | 0.00 | 0.00 | 85.9 | 84.7 | 2.6 | 25.2 | |||
| XVII | 210 (70%) | 90 (30%) | 300 | 1.06 | 4.92 | 3.99 | 0.00 | 0.00 | 85.9 | 84.2 | 2.6 | 24.9 | |||
| XVIII | 180 (60%) | 120 (40%) | 300 | 1.06 | 4.76 | 3.96 | 0.00 | 0.00 | 85.9 | 84.4 | 2.6 | 25.2 | |||
| XIX | 150 (50%) | 150 (50%) | 300 | 1.07 | 4.89 | 4.10 | 0.00 | 0.00 | 85.9 | 84.4 | 2.6 | 24.9 | |||
| XX | 100 (33%) | 200 (67%) | 300 | 1.10 | 5.15 | 3.93 | 0.00 | 0.00 | 85.9 | 83.9 | 2.6 | 24.9 | |||
| V | 0 (0%) | 300 (100%) | 300 | 1.20 | 6.31 | 3.90 | 0.02 | 0.02 | 85.9 | 80.9 | 2.6 | 26.8 | |||
Abbreviations are as follows: NHC, northern Han Chinese cluster; CHC, central Han Chinese cluster; SHC, southern Han Chinese cluster; Unadj., unadjusted; GC, p values are corrected by genomic control; BONF, p values are corrected by Bonferroni single-step method; FDR, p values are corrected by Benjamini and Hochberg step-up false discovery rate control.
Next, we investigated the scenarios of extreme situations; i.e., cases and controls were sampled from different clusters. λ was 1.74 when 300 cases were sampled from the NHC and 300 controls were sampled from the SHC (scenario IV). In this case, there were 17,843 (11.61%) SNPs showing an unadjusted p value < 0.05, there were 5667 (3.69%) SNPs showing a GC-adjusted p value < 0.05, and 874 (0.57%) SNPs showed a false-discovery rate (FDR)-adjusted p value < 0.05 with the use of the Benjamini-Hochberg method.45 λ was 1.20 when cases and controls were sampled from the NHC and the CHC, respectively (scenario V). In this case, there were 9716 (6.31%) SNPs showed an unadjusted p value < 0.05, there remained 5993 (3.90%) SNPs showing GC-adjusted p < 0.05, and there were 80 (0.05%) SNPs showing FDR-adjusted p < 0.05. λ was 1.30 when cases and controls were sampled from the CHC and the SHC, respectively (scenario VI). In this case, there were 10,906 (7.18%) SNPs showing unadjusted p < 0.05, there remained 5984 (3.94%) SNPs showing GC-adjusted p < 0.05, and 89 (0.06%) SNPs showed FDR-adjusted p < 0.05.
For the investigation of more-realistic scenarios, cases and controls were sampled from clusters in different case-control proportions. This is similar to a recent study that also investigated such scenarios in the Japanese population.22 First, we examined the condition that both cases and controls were from three clusters with a 1:1:1 ratio (scenario VII). The λ was very close to but still less than 1.1. We then examined conditions in which only the cases were from three clusters with a ratio of 1:1:1. The λ was larger than 1.2 when controls were from the NHC (scenario VIII) or the SHC (scenario X), but the λ was smaller than 1.1 when controls were from the CHC (scenario IX). We further examined to what extent differences in proportions between case individuals from two or three clusters and control individuals from the one cluster would affect λ. We conducted case-control simulations in which the control group consisted of individuals from the one cluster and the case group was a mixture of individuals from two clusters. With 300 individuals used for both cases and controls under these conditions, the λ value reached 1.1 when 30% of the cases were from the SHC (scenario XII in Table 4) or when 67% of cases were from the CHC (scenario XX in Table 4), with all of the controls coming from the NHC. If all of the controls were from the CHC, the λ value reached 1.1 when 30% of the cases were from the NHC (scenario XXII in Table S4) or when 30% of the cases were from the SHC (scenario XXVII in Table S3). If all of the controls were from the SHC, the λ value reached 1.2 even when 20% of cases were from the NHC (scenario XXXI in Table S5) or when 20% of cases were from the SHC (scenario XXXVI in Table S5). This suggests that the inflation of false-positive rates would exceed an acceptable level (λ ≤ 1.1) for a study design when the proportion of the individuals from a different cluster is larger than 30% and the sample size is 300. This indicates that including a substantial proportion of individuals from the other clusters would increase the rate of false-positive results even if the sample sizes were much smaller than 1000. Because λ is expected to increase linearly with sample size, if the sample size is larger than 300, inclusion of individuals from the other clusters could affect the results of the association study even when the proportion is small.
We also evaluated the statistical power in all of the above scenarios. We simulated 1000 SNPs associated with disease (population odds ratio of 2.0) by using a uniform allele-frequency range (0∼1). Power was calculated as the number of disease SNPs showing p < 0.05 (unadjusted or adjusted) divided by 1000. In all of our simulation studies with a sample size of 300, the unadjusted power was 85.9%. The power could be increased as the sample size increased (data not shown). As expected, a GC could result in lower power. With increasing values of λ, more power could be lost. For example, when cases and controls were sampled within the CHC cluster (λ = 1.07), the power after inclusion of a GC decreased from 85.9% to 84.0% (scenario II in Table 4), and when cases and controls were sampled from the SHC and the NHC, respectively (λ = 1.74), the power after inclusion of a GC decreased from 85.9% to 71.9% (scenario IV in Table 4).
In all of the scenarios, the statistical power of the GC was affected much more heavily than that of the FDR control when population stratification existed. However, the power decreased significantly after FDR control; i.e., most of the scenarios had less than 30% power, or only about 300 of 1000 simulated disease loci showing p < 0.05 after FDR control, although the power was still much higher than that after Bonferroni correction (2.6%; Table 4, Table S4, and Table S5).
Discussion
We have shown in the present study that the Han Chinese population, a seemingly homogeneous population, is actually complicatedly substructured, with the main observed clusters roughly corresponding to N-Han (NHC),C-Han (CHC), and S-Han (SHC). Our results showed that the greatest genetic differentiation of Han Chinese is between the NHC and the SHC. Previous studies based on analyses of archeological, anatomical, linguistic, and genetic data consistently suggested the presence of a significant boundary between the northern and southern populations in China.46 Genetic differentiation between the northern and southern populations was observed at classic markers,27,47–49 microsatellite DNA markers,50 mtDNA,51–53 and Y chromosome SNP markers.54,55 Using classic markers, Xiao et al.27 proposed a genetic boundary located approximately at the Yangtze River. Wen et al.51 found that the mtDNA haplogroup distribution showed substantial differentiation between N-Han and S-Han. It is interesting that in our data, PC1 values and latitudes of sample locations were highly correlated but we did not observe significant correlation between FST and geographical distances, suggesting that the population differentiation could have primarily resulted from isolation due to a geographical barrier such as the Yangtze River, which runs across China from west to east.
We emphasize that the intention of this classification is not to precisely define the ancestry of Han Chinese individuals. The complex origins, historical immigrations, and recent intermarriages with other ethnic groups preclude the possibility of unambiguously identifying the ancestry of Han Chinese at an individual level. In other words, the gene pool of the Han Chinese is very entangled. The genetic admixture of complex ancestries has been embedded in each individual genome, and each chromosome is fragmentally composed of mosaic ancestries. Therefore, it is almost impossible to assign a particular ancestry to a Han Chinese individual. In addition, genetic-differentiation values among these clusters (FST [NHC−CHC] = 0.0002; FST [SHC−CHC] = 0.0003; FST [NHC−SHC] = 0.0009) are much smaller than that between the Han Chinese and the Japanese population (0.007–0.008). As a matter of fact, Han Chinese (CHB) and Japanese (JPT) samples are generally combined as one sample group representing East Asia in the HapMap data set,10,11 and they formed a cluster in PC plot when worldwide population samples were jointly analyzed (Figure 1A). It may be scientifically appropriate to pool data from Han Chinese samples when the data are described as the combined analysis panel because they have very small difference in allele frequencies as compared with worldwide populations. Indeed, consistent with our previous observations,24 we found that the genetic differences among Han Chinese subpopulations were much smaller than those among European populations (FST = 0.0097, p = 3.48 × 10−10), on the basis of both HGDP and HapMap data.
However, the simulated case-control studies showed that the observed genetic differentiation could lead to an inflated rate of false-positive results even when the sample size is moderate. For example, if there were 300 individuals for both cases and controls, with all of the controls coming from the NHC, the genome-wide x2 inflation factor, λ, would reach 1.1 when 30% of the cases were from the SHC. Considering the frequent population flow of Han Chinese across the country as a result of socioeconomic and political factors, when an association study is intended to be conducted in one of the clusters, it is not uncommon for a substantial proportion of samples to be from the other clusters. Furthermore, it was expected and was observed that a linear increase of λ occurs as the sample size increases.22 The acceptable proportion of subjects from the other clusters that would prevent λ from reaching 1.1 will be even smaller when the sample size is larger than 300, because even minor stratification can have a substantial impact on population-based studies with large sample sizes36,56,57 (for example, 1000, as the current GWAS generally adopted). On the other hand, the simulated studies showed that inclusion of a higher proportion of individuals from the CHC cluster (Table 4) may not seriously affect the results of the association study when the sample size is smaller than 1000, which, as was observed in our data, may possibly explain the relatively small genetic differentiation between the NHC and CHC (Table 2). In summary, although differences in allele frequencies among Han Chinese clusters are small, our study has demonstrated the importance of accounting for population stratification in order to reduce false-positive associations.
We have shown how inclusion of different proportions of individuals from different subgroups in case and control groups would lead to spurious associations. To determine in which regions spurious associations are likely to occur, we examined the differences in allele frequencies among the three Han Chinese clusters. In our data, most genes that show great differences among clusters are involved in cardiac arteriopathy, as IPA58 revealed. However, it is not clear what kind of factors led to the differences in those genes. Given that the Han Chinese subgroups correlate with their geographical locations along the latitudes, temperature and exposure to ultraviolet light could be possible determinants, but additional studies are needed for confirmation of those hypotheses. Other factors, such as food, pathogens, and life style, could also be responsible for differentiation between Han Chinese in north and those in the south. One of the top two SNPs, rs2596472, is in the HLA complex P5 (HCP5) region (nearGene-5), which is associated with HIV infection, psoriasis, and psoriatic arthritis, suggesting that the immune systems of northern and southern populations could be subjected to different pressures from virus and other diseases. These signals showing significant differences among Han Chinese clusters should be carefully explained in case they are also detected in a single association study, especially when sample sources are diverse.
The two HGDP Han Chinese population samples resemble N-Han and S-Han, respectively. However, the small sample size and lower density of genome-wide data available limit their utilities in GWAS or other association studies, although these samples have been extensively used in population, evolutionary, and forensic genetic studies.4,6,8,9,32
The HapMap samples are the most intensively studied samples in recent years, since the International HapMap Project was established in 2002 in order to facilitate the discovery of sequence variants underlying common diseases.26 One primary usage of HapMap data in association studies is to select tag SNPs from a HapMap panel that is in ancestry close to the studied population; for example, tag SNPs were selected from CHB data when a candidate gene was to be genotyped in Han Chinese samples for association study.59,60 Another usage of HapMap data in GWAS is to impute SNPs that are not directly genotyped in studied samples but are present on a HapMap panel.61,62 In phase III of the International HapMap project, two Han Chinese population samples (CHB and CHD) were included. The CHB samples were collected from individuals residing near Beijing Normal University, which is located in northern China. Although in the PC plot CHB individuals distribute widely and across the three clusters, CHB is more similar to N-Han than to S-Han when it is taken as a representative of one single population. The other HapMap Han Chinese population samples, CHD, were collected from unrelated individuals living in metropolitan Denver, CO, USA. The CHD individuals who donated samples emigrated from many different locations of China. In the PC plot, similarly to CHB individuals, CHD individuals distribute widely and across the three clusters. However, CHD is more similar to S-Han than to N-Han when it is taken as a representative of one single population. Therefore, considering the genetic differences between and affinities of the two HapMap Han Chinese population samples, CHB and CHD should be separately chosen as N-Han and S-Han references for tagSNP selection or data imputation if an association study is performed in typical N-Han or S-Han samples.
Supplemental Data
Supplemental Data include eight figures and five tables and are available with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Affymetrix Human SNP User Guide, http://www.affymetrix.com/support/downloads/manuals/genomewidesnp6_manual.pdf
EIGENSOFT v3.0, http://genepath.med.harvard.edu/∼reich/Software.htm
The HGDP-CEPH project, http://www.cephb.fr/en/hgdp
Ingenuity Pathway Analysis (IPA 7.6) software tool, Ingenuity Systems, http://www.ingenuity.com
The International HapMap Project, http://www.hapmap.org/downloads/index.html.en
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
PLINK v1.06, http://pngu.mgh.harvard.edu/purcell/plink/
Acknowledgments
This is a report of a joint effort of three research groups with equal contribution. L.J. is the senior author for the data analysis group, and B.W. and X-J.Z. are the senior authors for two genotyping teams. The authors are grateful to many people in L.J., B.W., and X-J.Z.'s lab for their help in collecting samples. This work was supported by grants from the National Outstanding Youth Science Foundation of China (30625016), the National Science Foundation of China (30890034, 30971577), and the 863 Program (2007AA02Z312). L.J. was also supported by the Shanghai Leading Academic Discipline Project (B111) and the Center for Evolutionary Biology. S.X. was also supported by the Science and Technology Commission of Shanghai Municipality (09ZR1436400) and the Knowledge Innovation Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (2008KIP311). S.X. gratefully acknowledges the support of the SA-SIBS Scholarship Program and the K.C.Wong Education Foundation, Hong Kong.
References
- 1.Campbell C.D., Ogburn E.L., Lunetta K.L., Lyon H.N., Freedman M.L., Groop L.C., Altshuler D., Ardlie K.G., Hirschhorn J.N. Demonstrating stratification in a European American population. Nat. Genet. 2005;37:868–872. doi: 10.1038/ng1607. [DOI] [PubMed] [Google Scholar]
- 2.Helgason A., Yngvadottir B., Hrafnkelsson B., Gulcher J., Stefansson K. An Icelandic example of the impact of population structure on association studies. Nat. Genet. 2005;37:90–95. doi: 10.1038/ng1492. [DOI] [PubMed] [Google Scholar]
- 3.Price A.L., Helgason A., Palsson S., Stefansson H., St Clair D., Andreassen O.A., Reich D., Kong A., Stefansson K. The impact of divergence time on the nature of population structure: an example from Iceland. PLoS Genet. 2009;5:e1000505. doi: 10.1371/journal.pgen.1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 5.Kayser M., Lao O., Saar K., Brauer S., Wang X., Nurnberg P., Trent R.J., Stoneking M. Genome-wide analysis indicates more Asian than Melanesian ancestry of Polynesians. Am. J. Hum. Genet. 2008;82:194–198. doi: 10.1016/j.ajhg.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 7.Friedlaender J.S., Friedlaender F.R., Reed F.A., Kidd K.K., Kidd J.R., Chambers G.K., Lea R.A., Loo J.H., Koki G., Hodgson J.A. The genetic structure of Pacific Islanders. PLoS Genet. 2008;4:e19. doi: 10.1371/journal.pgen.0040019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg N.A., Mahajan S., Ramachandran S., Zhao C., Pritchard J.K., Feldman M.W. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 2005;1:e70. doi: 10.1371/journal.pgen.0010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg N.A., Pritchard J.K., Weber J.L., Cann H.M., Kidd K.K., Zhivotovsky L.A., Feldman M.W. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 10.TheInternationalHapMapConsortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelis M., Esko T., Magi R., Zimprich F., Zimprich A., Toncheva D., Karachanak S., Piskackova T., Balascak I., Peltonen L. Genetic structure of Europeans: a view from the North-East. PLoS One. 2009;4:e5472. doi: 10.1371/journal.pone.0005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian C., Plenge R.M., Ransom M., Lee A., Villoslada P., Selmi C., Klareskog L., Pulver A.E., Qi L., Gregersen P.K. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genet. 2008;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salmela E., Lappalainen T., Fransson I., Andersen P.M., Dahlman-Wright K., Fiebig A., Sistonen P., Savontaus M.L., Schreiber S., Kere J. Genome-wide analysis of single nucleotide polymorphisms uncovers population structure in Northern Europe. PLoS One. 2008;3:e3519. doi: 10.1371/journal.pone.0003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paschou P., Drineas P., Lewis J., Nievergelt C.M., Nickerson D.A., Smith J.D., Ridker P.M., Chasman D.I., Krauss R.M., Ziv E. Tracing sub-structure in the European American population with PCA-informative markers. PLoS Genet. 2008;4:e1000114. doi: 10.1371/journal.pgen.1000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novembre J., Johnson T., Bryc K., Kutalik Z., Boyko A.R., Auton A., Indap A., King K.S., Bergmann S., Nelson M.R. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seldin M.F., Shigeta R., Villoslada P., Selmi C., Tuomilehto J., Silva G., Belmont J.W., Klareskog L., Gregersen P.K. European population substructure: clustering of northern and southern populations. PLoS Genet. 2006;2:e143. doi: 10.1371/journal.pgen.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tishkoff S.A., Reed F.A., Friedlaender F.R., Ehret C., Ranciaro A., Froment A., Hirbo J.B., Awomoyi A.A., Bodo J.M., Doumbo O. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S., Lewis C.M., Jakobsson M., Ramachandran S., Ray N., Bedoya G., Rojas W., Parra M.V., Molina J.A., Gallo C. Genetic variation and population structure in native Americans. PLoS Genet. 2007;3:e185. doi: 10.1371/journal.pgen.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The-HUGO-Pan-Asian-SNP-Consortium Mapping Human Genetic Diversity in Asia. Science. 2008 doi: 10.1126/science.1177074. in press. [DOI] [PubMed] [Google Scholar]
- 21.Indian-Genome-Variation-Consortium Genetic landscape of the people of India: a canvas for disease gene exploration. J. Genet. 2008;87:3–20. doi: 10.1007/s12041-008-0002-x. [DOI] [PubMed] [Google Scholar]
- 22.Yamaguchi-Kabata Y., Nakazono K., Takahashi A., Saito S., Hosono N., Kubo M., Nakamura Y., Kamatani N. Japanese population structure, based on SNP genotypes from 7003 individuals compared to other ethnic groups: effects on population-based association studies. Am. J. Hum. Genet. 2008;83:445–456. doi: 10.1016/j.ajhg.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cann H.M., de Toma C., Cazes L., Legrand M.F., Morel V., Piouffre L., Bodmer J., Bodmer W.F., Bonne-Tamir B., Cambon-Thomsen A. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 24.Xu S., Jin L. A Genome-wide Analysis of Admixture in Uyghurs and a High-Density Admixture Map for Disease-Gene Discovery. Am. J. Hum. Genet. 2008;83:322–336. doi: 10.1016/j.ajhg.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakkula E., Rehnstrom K., Varilo T., Pietilainen O.P., Paunio T., Pedersen N.L., deFaire U., Jarvelin M.R., Saharinen J., Freimer N. The genome-wide patterns of variation expose significant substructure in a founder population. Am. J. Hum. Genet. 2008;83:787–794. doi: 10.1016/j.ajhg.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.TheInternationalHapMapConsortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 27.Xiao C.J., Cavalli-Sforza L.L., Minch E., Du R. Principal component analysis of gene frequencies of Chinese populations. Sci. China C Life Sci. 2000;43:472–481. [Google Scholar]
- 28.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X.J., Huang W., Yang S., Sun L.D., Zhang F.Y., Zhu Q.X., Zhang F.R., Zhang C., Du W.H., Pu X.M. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat. Genet. 2009;41:205–210. doi: 10.1038/ng.310. [DOI] [PubMed] [Google Scholar]
- 30.Weir B.S., Hill W.G. Estimating F-statistics. Annu. Rev. Genet. 2002;36:721–750. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- 31.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramachandran S., Deshpande O., Roseman C.C., Rosenberg N.A., Feldman M.W., Cavalli-Sforza L.L. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci. USA. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devlin B., Roeder K., Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. 2001;60:155–166. doi: 10.1006/tpbi.2001.1542. [DOI] [PubMed] [Google Scholar]
- 35.Devlin B., Roeder K., Wasserman L. Genomic control for association studies: a semiparametric test to detect excess-haplotype sharing. Biostatistics. 2000;1:369–387. doi: 10.1093/biostatistics/1.4.369. [DOI] [PubMed] [Google Scholar]
- 36.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 37.Schaeffer L., Gohlke H., Muller M., Heid I.M., Palmer L.J., Kompauer I., Demmelmair H., Illig T., Koletzko B., Heinrich J. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 2006;15:1745–1756. doi: 10.1093/hmg/ddl117. [DOI] [PubMed] [Google Scholar]
- 38.Catano G., Kulkarni H., He W., Marconi V.C., Agan B.K., Landrum M., Anderson S., Delmar J., Telles V., Song L. HIV-1 disease-influencing effects associated with ZNRD1, HCP5 and HLA-C alleles are attributable mainly to either HLA-A10 or HLA-B∗57 alleles. PLoS One. 2008;3:e3636. doi: 10.1371/journal.pone.0003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colombo S., Rauch A., Rotger M., Fellay J., Martinez R., Fux C., Thurnheer C., Gunthard H.F., Goldstein D.B., Furrer H. The HCP5 single-nucleotide polymorphism: a simple screening tool for prediction of hypersensitivity reaction to abacavir. J. Infect. Dis. 2008;198:864–867. doi: 10.1086/591184. [DOI] [PubMed] [Google Scholar]
- 40.Limou S., Le Clerc S., Coulonges C., Carpentier W., Dina C., Delaneau O., Labib T., Taing L., Sladek R., Deveau C. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02) J. Infect. Dis. 2009;199:419–426. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- 41.Conen D., Cheng S., Steiner L.L., Buring J.E., Ridker P.M., Zee R.Y. Association of 77 polymorphisms in 52 candidate genes with blood pressure progression and incident hypertension: the Women's Genome Health Study. J. Hypertens. 2009;27:476–483. doi: 10.1097/hjh.0b013e32832104c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox A.A., Collard C.D., Shernan S.K., Seidman C.E., Seidman J.G., Liu K.Y., Muehlschlegel J.D., Perry T.E., Aranki S.F., Lange C. Natriuretic peptide system gene variants are associated with ventricular dysfunction after coronary artery bypass grafting. Anesthesiology. 2009;110:738–747. doi: 10.1097/aln.0b013e31819c7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw G.M., Iovannisci D.M., Yang W., Finnell R.H., Carmichael S.L., Cheng S., Lammer E.J. Risks of human conotruncal heart defects associated with 32 single nucleotide polymorphisms of selected cardiovascular disease-related genes. Am. J. Med. Genet. A. 2005;138:21–26. doi: 10.1002/ajmg.a.30924. [DOI] [PubMed] [Google Scholar]
- 44.Newton-Cheh C., Larson M.G., Vasan R.S., Levy D., Bloch K.D., Surti A., Guiducci C., Kathiresan S., Benjamin E.J., Struck J. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat. Genet. 2009;41:348–353. doi: 10.1038/ng.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JRStatistSocB. 1995;57:289–300. [Google Scholar]
- 46.Jin L., Su B. Natives or immigrants: modern human origin in east Asia. Nat. Rev. Genet. 2000;1:126–133. doi: 10.1038/35038565. [DOI] [PubMed] [Google Scholar]
- 47.Chen R., Ye G., Geng Z., Wang Z., Kong F., Tian D., Bao P., Liu R., Liu J., Song F. Yi Chuan Xue Bao. 1993;20:389–398. [PubMed] [Google Scholar]
- 48.Du R., Xiao C., Cavalli-Sforza L.L. Genetic distances between Chinese groups calculated on gene frequencies of 38 loci. Sci. China C Life Sci. 1998;28:83–89. doi: 10.1007/BF02882691. [DOI] [PubMed] [Google Scholar]
- 49.Cavalli Sforza L.L., Menozzi P., Piazza A. Princeton University Press; Princeton, New Jersey: 1993. The history and geography of human genes. [Google Scholar]
- 50.Chu J.Y., Huang W., Kuang S.Q., Wang J.M., Xu J.J., Chu Z.T., Yang Z.Q., Lin K.Q., Li P., Wu M. Genetic relationship of populations in China. Proc. Natl. Acad. Sci. USA. 1998;95:11763–11768. doi: 10.1073/pnas.95.20.11763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wen B., Li H., Lu D., Song X., Zhang F., He Y., Li F., Gao Y., Mao X., Zhang L. Genetic evidence supports demic diffusion of Han culture. Nature. 2004;431:302–305. doi: 10.1038/nature02878. [DOI] [PubMed] [Google Scholar]
- 52.Yao Y.G., Nie L., Harpending H., Fu Y.X., Yuan Z.G., Zhang Y.P. Genetic relationship of Chinese ethnic populations revealed by mtDNA sequence diversity. Am. J. Phys. Anthropol. 2002;118:63–76. doi: 10.1002/ajpa.10052. [DOI] [PubMed] [Google Scholar]
- 53.Yao Y.G., Kong Q.P., Bandelt H.J., Kivisild T., Zhang Y.P. Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am. J. Hum. Genet. 2002;70:635–651. doi: 10.1086/338999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Su B., Xiao J., Underhill P., Deka R., Zhang W., Akey J., Huang W., Shen D., Lu D., Luo J. Y-Chromosome evidence for a northward migration of modern humans into Eastern Asia during the last Ice Age. Am. J. Hum. Genet. 1999;65:1718–1724. doi: 10.1086/302680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karafet T., Xu L., Du R., Wang W., Feng S., Wells R.S., Redd A.J., Zegura S.L., Hammer M.F. Paternal population history of East Asia: sources, patterns, and microevolutionary processes. Am. J. Hum. Genet. 2001;69:615–628. doi: 10.1086/323299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marchini J., Cardon L.R., Phillips M.S., Donnelly P. The effects of human population structure on large genetic association studies. Nat. Genet. 2004;36:512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 57.Freedman M.L., Reich D., Penney K.L., McDonald G.J., Mignault A.A., Patterson N., Gabriel S.B., Topol E.J., Smoller J.W., Pato C.N. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 58.Calvano S.E., Xiao W., Richards D.R., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 59.Zhao W., Wang L., Lu X., Yang W., Huang J., Chen S., Gu D. A coding polymorphism of the kallikrein 1 gene is associated with essential hypertension: a tagging SNP-based association study in a Chinese Han population. J. Hypertens. 2007;25:1821–1827. doi: 10.1097/HJH.0b013e328244e119. [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Barcelo M.M., Miao X., Lui V.C., So M.T., Ngan E.S., Leon T.Y., Lau D.K., Liu T.T., Lao X., Guo W. Correlation between genetic variations in Hox clusters and Hirschsprung's disease. Ann. Hum. Genet. 2007;71:526–536. doi: 10.1111/j.1469-1809.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 61.Nothnagel M., Ellinghaus D., Schreiber S., Krawczak M., Franke A. A comprehensive evaluation of SNP genotype imputation. Hum. Genet. 2009;125:163–171. doi: 10.1007/s00439-008-0606-5. [DOI] [PubMed] [Google Scholar]
- 62.Spencer C.C., Su Z., Donnelly P., Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477. doi: 10.1371/journal.pgen.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.