

Abstract
Population stratification is a potential problem for genome-wide association studies (GWAS), confounding results and causing spurious associations. Hence, understanding how allele frequencies vary across geographic regions or among subpopulations is an important prelude to analyzing GWAS data. Using over 350,000 genome-wide autosomal SNPs in over 6000 Han Chinese samples from ten provinces of China, our study revealed a one-dimensional “north-south” population structure and a close correlation between geography and the genetic structure of the Han Chinese. The north-south population structure is consistent with the historical migration pattern of the Han Chinese population. Metropolitan cities in China were, however, more diffused “outliers,” probably because of the impact of modern migration of peoples. At a very local scale within the Guangdong province, we observed evidence of population structure among dialect groups, probably on account of endogamy within these dialects. Via simulation, we show that empirical levels of population structure observed across modern China can cause spurious associations in GWAS if not properly handled. In the Han Chinese, geographic matching is a good proxy for genetic matching, particularly in validation and candidate-gene studies in which population stratification cannot be directly accessed and accounted for because of the lack of genome-wide data, with the exception of the metropolitan cities, where geographical location is no longer a good indicator of ancestral origin. Our findings are important for designing GWAS in the Chinese population, an activity that is expected to intensify greatly in the near future.
Introduction
Genome-wide association studies (GWAS) have been widely employed as a tool for dissecting the genetic bases of many complex traits.1–3 Simultaneously, GWAS provide rich data with insights into important aspects of the population structure that arises through drift, selection, and migration both within and among various isolated populations or geographical regions. Dense genome-wide data also enable the detection of distant ancestry among individuals by examination of genetic distances, homozygosity, and genome-wide patterns of linkage disequilibrium.4,5 Population structure, and unusual levels of shared ancestry, can potentially cause spurious associations. Therefore, it is critical to understand the genetic structure of a population, not only for the resulting historical insights, but also for the appropriate design of association studies. Whereas the genetic geography of North America and Europe is well understood, a detailed analysis of the population structure is lacking for China, the most populated country in the world.
The Han Chinese constitute more than 90% of China's population and nearly a fifth of the human species. Although the International HapMap Project has provided a clear window into the variation present in a single representative sample of the Han Chinese in Beijing, there remains a paucity of genome-wide data from elsewhere in China or the significant overseas Chinese communities. Population genetics research in China has centered on minority populations, or has been limited to specialized molecular markers such as microsatellites,6 Y-chromosome polymorphisms,7 and mitochondrial DNA (mtDNA).8–10 Collectively, these studies support the hypothesis of an initial entry of modern humans into China from southeast Asia, followed by later expansions, chiefly those stemming from the development of agriculture in China's two main centers of domestication (rice in the south and millet in the north).11,12 Su et al., Chu et al., and the many investigations of Cavalli-Sforza et al. (summarized in 13) have observed largely distinct clusters of southern and northern populations in phylogenetic trees of the Han Chinese sampled throughout the country.6,7 Others have found no support for this “north-south” division,14 highlighting the need for denser genomic and geographic sampling of the modern Chinese population, as major initiatives for genome-wide association studies are currently being planned in China. To this end, we used data for more than 350,000 SNPs from more than 6000 Han Chinese samples across ten of China's 23 provinces. We explore Chinese population structure across these geographical locations, and we examine the potential magnitude of Chinese population stratification on GWAS via simulations.
Material and Methods
Samples
The study involved a total of 8200 samples: 6580 Han Chinese samples from ten provinces in China, 1050 Han Chinese from the Chinese metropolises of Beijing and Shanghai, and 570 overseas Chinese samples from Singapore. These were genotyped in a series of GWAS of diseases with the use of the Illumina Human 610-Quad BeadChips (Table S1, available online). The Han Chinese samples were recruited according to the location of their current residence in China, with the exception of the samples from Guangdong province. In addition to reporting origins for themselves and their parents in Guangdong, all spoke one of three major dialects: Cantonese, Hakka, or Teochew. For some analyses, data from the four reference population samples of the International HapMap Project (release 23) were used (60 Yoruba from Ibadan, Nigeria [YRI], 60 Europeans from the CEPH panel [CEU], 45 Han Chinese from Beijing [CHB], and 44 Japanese samples from Tokyo [JPT]).15 All samples underwent quality control (QC) procedures as part of their respective original studies. Samples with a call rate < 98% were removed. A detailed description of the QC procedures can be found elsewhere.2 After merging, additional QC was carried out to ensure that no duplicated or first-degree relative pairs remained. The samples for population structure and GWAS simulation analyses were selected randomly, after population outliers were removed (based on the first principal component analysis [PCA] with HapMap samples). For maximization of the number of SNPs for analysis, two data sets were derived from the combined GWAS data sets. The first data set (PS set) is for the population structure analysis, in which 1454 samples were randomly selected (from a total of 8200 samples) on the basis of locality. In the second data set (SM set), we first combined all 6580 Chinese samples from only the provinces. These include all GWAS samples (except samples from Psoriasis Singapore) and the Sichuan samples (Table S1). The 6580 samples were then grouped into northern, central, and southern samples on the basis of their geographic regions, before the selection of 1500 samples from the northern and southern regions (Table 1).
Table 1.
Summary of the Samples
| Locality | Number of Samples | Number of Samples Used in the Population Structure Analysis (PS) | Region | Number of Samples in Each Region | Number of Samples Used in the Simulation Analysis (SM) |
|---|---|---|---|---|---|
| Metropolis | |||||
| Beijing (CHB) | 45 | 45 | - | - | - |
| Shanghai | 1005 | 100 | - | - | - |
| Singapore | 570 | 200 | - | - | - |
| Province | |||||
| Hebei | 4 | 4 | North | 1213 | 1000 |
| Henan | 25 | 25 | |||
| Liaoning | 14 | 14 | |||
| Shandong | 1170 | 200 | |||
| Anhui | 3173 | 200 | Central | 3284 | 0 |
| Hubei | 48 | 48 | |||
| Jiangsu | 47 | 46 | |||
| Sichuan | 16 | 16 | |||
| Guangdong | 1977 | 450 | South | 2083 | 500 |
| Hunan | 106 | 106 | |||
| Total | 8200 | 1454 | 6580 | 1500 | |
Genome-wide SNP Data
All the samples were genotyped with the use of Illumina Human 610-Quad BeadChips, and only autosomal SNPs (chromosome 1–22) were used. All genotypes were output from the Illumina BeadStudio software, with the “top” strand option used to ensure consistent nomenclature. QC procedures prior to merging resulted in SNPs that had a minimum call rate of 90% and satisfied Hardy-Weinberg equilibrium (p value > 10−7). Merging was then performed for the common SNPs among all of the various data sets. Subsequently, filtering ensured SNP call rates > 90%. Ultimately, 420,932 SNPs were common to the 1454 samples within the PS set, used for population structure analysis. For the SM set, the combination of 6580 GWAS Chinese samples brought the common set of SNPs to 400,328 (Table S1). Then, all of the SNPs within five regions of long-range linkage disequilibrium were removed from both the PS and the SM data sets, including the HLA region on chromosome 6, inversions on chromosomes 8 and 5, and two regions on chromosome 11 (Table S2). SNPs from these regions have been shown to be highly correlated, such that they might obscure patterns of population structure. In addition, for the PS data set, correlated SNPs were removed on the basis of an r2 threshold value of 0.2, with the use of PLINK,16 for PCA. For the SM data set, SNPs that are potentially associated with the disease phenotypes (p value < 0.01 from the various GWAS analyses) were removed from the GWAS simulation analysis.
After the various QC steps, 413,477 SNPs were used for STRUCTURE and FRAPPE analysis, 107,565 SNPs for PCA, and 373,383 SNPs for GWAS simulation analysis (Table S2).
Population Structure Analysis
Population structure was examined primarily via PCA and model-based clustering algorithms implemented in FRAPPE17 and STRUCTURE,18,19 which are based on a “frequentist” maximum-likelihood model and a Bayesian Markov Chain Monte Carlo algorithm, respectively.
PCA was performed with 107,565 SNPs with the use of smartpca from the software package EIGENSTRAT (found in EIGENSOFT20). Subsequently, the principal components (PCs) were plotted in two-dimensional plots for visualization. STRUCTURE and FRAPPE algorithms were performed on 413,477 SNPs for K values 1 to 5, with STRUCTURE using 10,000 burn-ins and 20,000 repetition, under an admixture model with correlated allele frequency, and FRAPPE using 10,000 iterations. Because of their computational constraints, STRUCTURE and FRAPPE analyses were performed on several smaller subsets of the 413,477 SNPs in accordance with a methodology adapted from Jakobsson et. al.21 For this, all of the 413,477 SNPs were first ranked in order of their physical position and thinned into 26 subsets by the picking of every 26th SNP in the ordered list. This yields approximately 4% of the total 413,477 SNPs (about 15,900 SNPs) in each subset to fit the computational constraints of STRUCTURE and FRAPPE analysis. STRUCTURE and FRAPPE analysis was then performed with the use of each of the 26 subsets, and CLUMPP22 was used for identifying shared modes among the results from the 26 subsets. In CLUMPP, each subset was regarded as a single run of STRUCTURE or FRAPPE, and the resultant shared modes of the 26 runs were calculated on the basis of its GREEDY algorithm, with 500,000 repetitions. Furthermore, a PCA was also performed with the use of the same 26 subsets of SNPs (used in the STRUCTURE and FRAPPE analysis) and revealed results almost identical to those of the PCA of the full set of 413,477 SNPs. In addition, the clusters defined by STRUCTURE and FRAPPE were highly correlated. Therefore, the subset-based STRUCTURE and FRAPPE analysis did provide a true reflection of the results based on the full set of SNPs. This is an efficient method for the STRUCTURE and FRAPPE analysis, in which the full information of SNPs was analyzed at a low computational cost. DISTRUCT23 was used for producing the plots for visualization.
Wright's FST statistic24 was calculated at each of the 413,477 loci among the three major regions of China (northern, southern, and central China) and the three dialect groups in Guangdong. The average FST over all of the loci was subsequently computed. The grouping of the provinces into the three major regions of China is defined in Table 1.
Simulation of Case-Control Samples for Genome-wide Association Analysis
To evaluate the impact of population stratification within the Han Chinese on the GWAS, we performed genome-wide association analysis by using simulated case and control samples. For simulation of the case and control samples, the provinces (and their corresponding samples) were first divided into northern, central, and southern on the basis of their geographic regions (Table 1). Then 1000 samples were selected from the north and 500 from the south, randomly and without replacement. Of the 1000 northern samples (1000N), 500 individuals were randomly assigned to be the cases. The other 500 northern samples were mixed, at different ratios, with the 500 southern samples (500S) to yield five mixed controls, each with an increasing proportion of individuals from the south: (1) 500N (0%), (2) 400N+100S (20%), (3) 300N+200S (40%), (4) 200N+300S (60%), and (5) 500S (100%). In addition, the simulated genome-wide association analysis was also performed with the use of the 1000 randomly selected samples from Shanghai, one of the three metropolitan samples investigated in this study. In brief, all of the 1005 Shanghai samples were divided into two equal northern and southern clusters on the basis of the values of their first principal component (PC1) along the north-south axis of the Han Chinese population. In the association analysis, 500 samples of the northern cluster were used as cases, and 500 samples of the southern cluster were used as controls. Similarly, association analysis was also performed with the use of the 300N+200S cases and the 200N+300S controls.
Association Analysis
SNP-based association tests were performed in the simulated data with the use of the allele test (chi-square test with one degree of freedom) in PLINK.16 To correct for population stratification on the association results, we implemented PCA- (from EIGENSTRAT20) and genomic control25 (GC)-based methods. The PCA-based correction method involves an iterative three-step procedure: first, the use of PCA for identification of population stratification or ancestry, then the removal of all correlations in a candidate set of SNPs that were found to be closely related to ancestry, and, finally, the recalculation of the association statistic in these SNPs until no significant association is shown between the genotypes and ancestry. GC-based correction involves a correction of the association statistic by a uniform inflation factor across all of the SNPs. The quantile-quantile (Q-Q) plots of p values, with and without correction for population stratification, were plotted for comparison.
Results
Comparison with HapMap Reference Populations
Of over 8000 Han Chinese samples with genome-wide SNP genotypes, three provinces (Shandong, Anhui, and Guangdong) were overrepresented by a large number of samples. To balance the representation of each province in the analyses, we first randomly selected 200 individuals from the Shandong province, 200 from the Anhui province, and 150 from each of the three dialect groups of Guangdong province. To view the selected Han Chinese samples in a global context, we performed a PCA on the genome-wide SNP genotypes from the 1409 representative Chinese samples (excluding CHB) and the four main reference populations in the International HapMap 2 project (release 23).15 The two-dimensional plot of PC1 and PC2 (Figure S1A) shows a clear separation of CEU (green), YRI (yellow), and the East Asian populations (CHB, red; JPT, orange; our samples from China, blue). PC4 (Figure S1C) accounts for the small variation between the Chinese and the Japanese. The 45 HapMap CHB samples are found among the Chinese samples. With only the East Asian populations considered, the Japanese (JPT) cluster splits from the Han Chinese cluster in only PC2, which accounts for only 0.18% of the total variation (Figure S2). PC1 suggests a relationship between the Japanese and the Chinese from the northern provinces of China, as geography would suggest.
Population Structure within the Han Chinese in China
Restricting our PCA to only the Han Chinese samples, we observed a tendency for the samples from the same province to be clustered together, and various province clusters distributed along PC1 in the following order: Liaoning, Hebei, Shandong, Henan, Jiangsu, Anhui, Hubei, Sichuan, Hunan, and Guangdong (Figure 1A). In comparison to the geographic locations of the ten provinces, the subpopulation structure of the Han Chinese population along PC1 is characterized by a one-dimensional north-south trend (Figure 2), as opposed to a two-dimensional north-south and east-west stratification observed within the European population.26 The one-dimensional subpopulation structure of the Han Chinese population (along PC1) showed a close resemblance to their sampling locations on a geographic map, and there is a very high correlation of 0.93 between the mean PC1 values of samples and the median latitudes of the provinces (Table S4). There seems to be a loosely defined central region, between the two poles of the very north (red) and south (green), characterized by the mingling of the individuals from Hubei (cyan) and Hunan (dark green), as well as Jiangsu, Anhui, and Sichuan. This one-dimensional structure of the Han Chinese population is clearly characterized by a continuous genetic gradient along a north-south geographical axis, rather than a distinct clustering of northern and southern samples.
Figure 1.
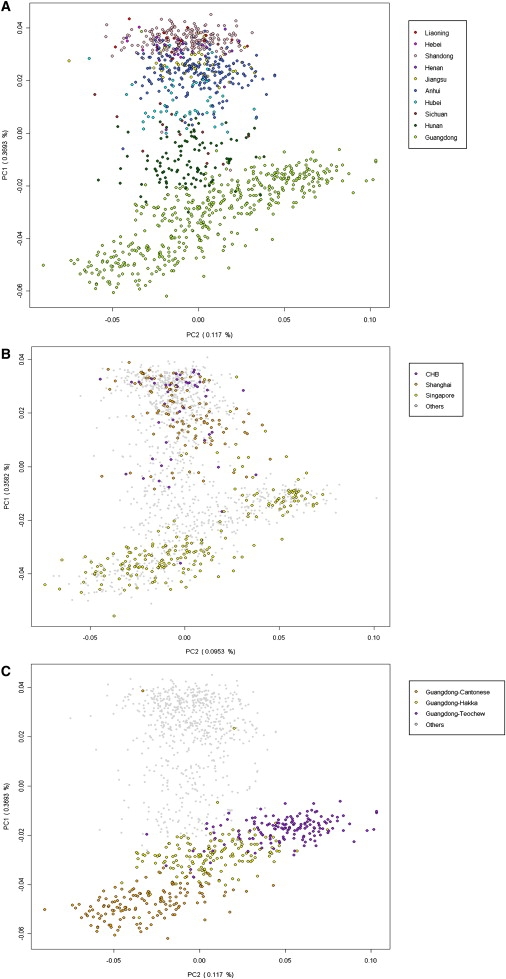
Population Stratification of the Han Chinese
The PCA plots were oriented with PC1 on the y axis and with PC2 on the x axis. These plots were obtained by using all 107,565 SNPs.
(A) The cluster and stratification of the samples from the ten provinces of China, showing an evident north-south genetic differentiation.
(B) The samples from Beijing (purple) (CHB samples from the International HapMap project), Shanghai (orange), and Singapore (yellow) were compared against the provincial samples. The majority of the Singapore samples fall in the southern (lower) sector, whereas the Beijing and Shanghai samples were largely located within the northern (upper) sector.
(C) The three dialect groups from the Guangdong province were shown against all of the Han Chinese samples from the ten provinces. There was stratification among the three dialect groups, along the same north-south trend observed for the overall Han Chinese.
Figure 2.
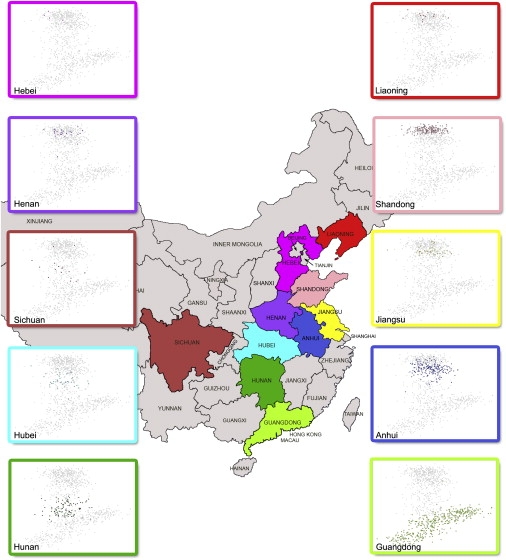
Comparison between the Geographic Map of China and the Genetic Structure of the Han Chinese
The PCA plots of the provincial samples are superimposed on the map of China to show the general north-south trend across China.
We saw little evidence for an east-west pattern in any of the PCs. Beyond PC2, little additional distinction among the individuals from the various locations is evident (Figure S3). To ensure that the east-west pattern is not masked by an oversampling from the provinces that largely represent the north-south populations, we performed PCA by using only the samples forming a west-east swathe of the central provinces of Sichuan, Hunan, Hubei, and Anhui, with an additional halving of the Anhui samples. Again, no discernible east-west pattern was observed (Figure S4).
The population structure of the Han Chinese was further examined with STRUCTURE (Figure 3) and FRAPPE (Figure S5). Both clustering methods produced similar membership coefficients across K = 2 to K = 5, as shown by the almost-perfect correlation between the clusters between both algorithms (Table S5B). Only K = 2 and K = 3 analyses showed readily interpretable results (Figure 3). With the assumption of two underlying source populations (K = 2), the clusters were anchored by JPT, segregating the more northern provinces of Liaoning, Hebei, Shandong, Henan, Jiangsu, and Anhui from the more southern provinces of Sichuan, Hubei, Hunan, and Guangdong. The very northern provinces of Liaoning, Hebei, Shandong, and Henan were denoted by the sharing of large yellow segments and small brown segments with JPT. However, it should be noted that, in terms of the membership coefficients, there was a slight difference between Sichuan and Hubei and the northern provinces or the other southern provinces of Hunan and Guangdong. In fact, when we averaged out the membership coefficients of the individuals according to their provinces, there was an incremental trend in the brown segment (conversely for the yellow segment) as we progressed from north to south (left to right) (Table S5A). At K = 3, JPT was clearly separated from the Chinese provinces, whereas the Chinese provinces showed a cluster pattern similar to that at K = 2. For the clustering both at K = 2 and K = 3, there were several aberrant individuals from Shandong and Guangdong (Figure 3), which correspond exactly with those exceptions picked out by the PCA (Figure 1). The average membership coefficients of each cluster from STRUCTURE and FRAPPE were found to be highly correlated with the median latitude of the cluster's geographic location and with each other (Tables S5A and S5B, respectively).
Figure 3.
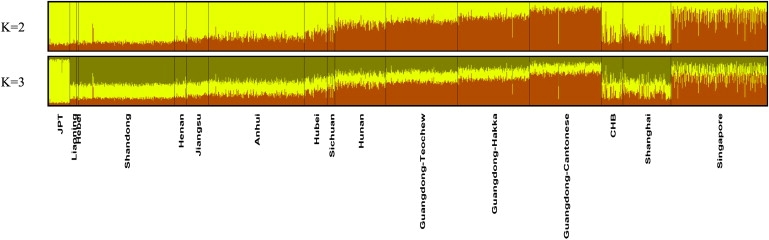
Estimated Population Structure by STRUCTURE for K = 2 and K = 3
Each individual is represented by a thin vertical line, and each province is demarcated by a thick vertical black line. The provinces are arranged from north to south, with JPT on the extreme left, representing the northernmost locality, to Liaoning, the northernmost province of China investigated in this study. The Guangdong individuals were grouped into the three dialect groups of Teochew, Hakka and Cantonese. These were then followed by the samples from the two metropolitan cities of Beijing (represented by CHB) and Shanghai, as well as the overseas Chinese community in Singapore. In K = 2, the northern provinces are clearly anchored by the JPT, with a huge membership of northern samples (represented by the yellow segment). The northern membership decreases gradually down to the southern provinces, which show a strong membership of southern samples (represented by the brown segment). At K = 3, JPT is clearly separated from the Han Chinese samples. The analysis revealed a demarcation of north-central-south similar to that shown by Figure 2. The Beijing, Shanghai and Singapore samples showed a clear mixture of southern (long brown lines) and northern (shorter brown lines) individuals, as compared to the provincial samples. The three dialect samples from the Guangdong province were also different from each other, with Teochew being more similar to individuals from the provinces of Hunan and Cantonese being the most southern representative.
We also calculated the average FST values among the three major regions: northern, central, and southern. The greatest differentiation was between the northern and southern samples, and the smallest differentiation was between the northern and central samples (Table S3).
Analysis of the Han Chinese Samples from Metropolitan Cities and Overseas Chinese Community as well as Three Dialect Groups in Guangdong
We also performed a PCA of the Han samples from the metropolitan cities of Beijing (purple) and Shanghai (orange), as well as Singapore, one of the major overseas Chinese communities (yellow), by comparing them with the rest of the Han Chinese samples. Unlike the ten provinces of China, where the samples tended to cluster together and were distributed mainly along a north-south gradient, these three metropolitan Han populations are composed of samples from throughout the north-south cline (Figure 1B). The samples of Singapore overwhelmingly belonged to the southern province clusters, such as Guangdong,27,28 but a small contribution of samples from the northern provinces was also evident (Figure 1B). The heterogeneous origin of the metropolitan Han Chinese samples was also supported by the STRUCTURE (Figure 3) and FRAPPE analyses (Figure S5), in which the metropolitan samples showed much more diverse contributions of underlying populations (K = 2 or 3) than the provincial samples. By ordering of the metropolitan samples by their membership coefficients, it is clear that the three metropolitan Han Chinese populations are mixes, to varying degrees, of the northern and southern samples (Figure S6).
The 450 samples from Guangdong province belonged to three local dialect groups (Teochew, Hakka, and Cantonese), with 150 individuals from each group. Looking at subpopulation structure at a finer scale, we examined PCA and STRUCTURE plots for the three dialect groups within Guangdong province, in the context of the overall Han Chinese population (Figure 1C and Figure 3). Even at this fine geographic scale, the three dialect groups appeared to cluster somewhat separately and to be distributed along the same north-south axis as the rest of the Han Chinese. Whereas the Cantonese group represents the southernmost cluster of the Han Chinese, the Teochew group is on the same stratum as the samples from the central provinces, such as Hunan (Figure 1C). This is also well supported by the average FST values, which show that among the three dialect groups, Cantonese is most differentiated from the northern samples and from the rest of the southern samples and that the Teochew group is the least differentiated from the central samples (Table S3). Interestingly, the PCA suggests that the samples of Singapore that were used in this study are largely Cantonese and Teochew and that few are Hakka.
Effects of Chinese Population Structure on Case-Control Association Study
Although the proportion of genetic variance responsible for the north-south stratification of the Han Chinese was small (just 0.37% of the total genetic variance of the Han Chinese is explained by PC1), it remains unclear how the north-south stratification of the Han Chinese population could affect poorly matched GWAS. Hence, we assessed the potential impact of population stratification within the Han Chinese population on GWAS via simulation studies, and we further compared the performance of GC and PCA approaches in adjusting for stratification.
We first simulated different degrees of population stratification within case-control samples by designating 500 randomly selected northern samples as the cases and various mixes of 500 samples from throughout the north and the south as the controls (0%, 20%, 40%, 60%, 80%, and 100% in terms of the percentage of the southern samples). Association analysis was then performed with the use of this synthetic case-control data set. The genome-wide χ2 inflation factor, λ, for each simulation is summarized in Table 2, and the Q-Q plot of the experimental p values from each synthetic data set is shown in Figure 4. As expected, population stratification caused inflated evidence for association, and the inflation increased exponentially with increasing degrees of stratification (Figure S7). GC- and PCA-based correction methods gave comparable results, in terms of reducing the extent of inflation, when the population stratification was moderate (20% in terms of the percentage of the southern samples in the mix controls) (Figures 4A–4C). However, PCA correction appeared to work much better than GC correction as the degree of stratification increased from 40% to 80% (with a corresponding increase of λ from 1.172 to 3.868). In the extreme case of fully stratified samples (λ > 5.5), in which the cases are from the north and the controls are from the south, a joint correction by PCA and GC methods was required for elimination of the inflation (Figure S9).
Table 2.
The Inflation Factors for Different Degrees of Stratification from the Simulated Association Analysis
| Simulation Scenario | Stratification (%) | Inflation Factor (λ) |
|---|---|---|
| 500N cases, 500N controls | 0 | 0.99 |
| 500N cases, 400N and 100S controls | 20 | 1.17 |
| 500N cases, 300N and 200S controls | 40 | 1.70 |
| 500N cases, 200N and 300S controls | 60 | 2.59 |
| 500N cases, 100N and 400S controls | 80 | 3.87 |
| 500N cases, 500S controls | 100 | 5.49 |
Figure 4.
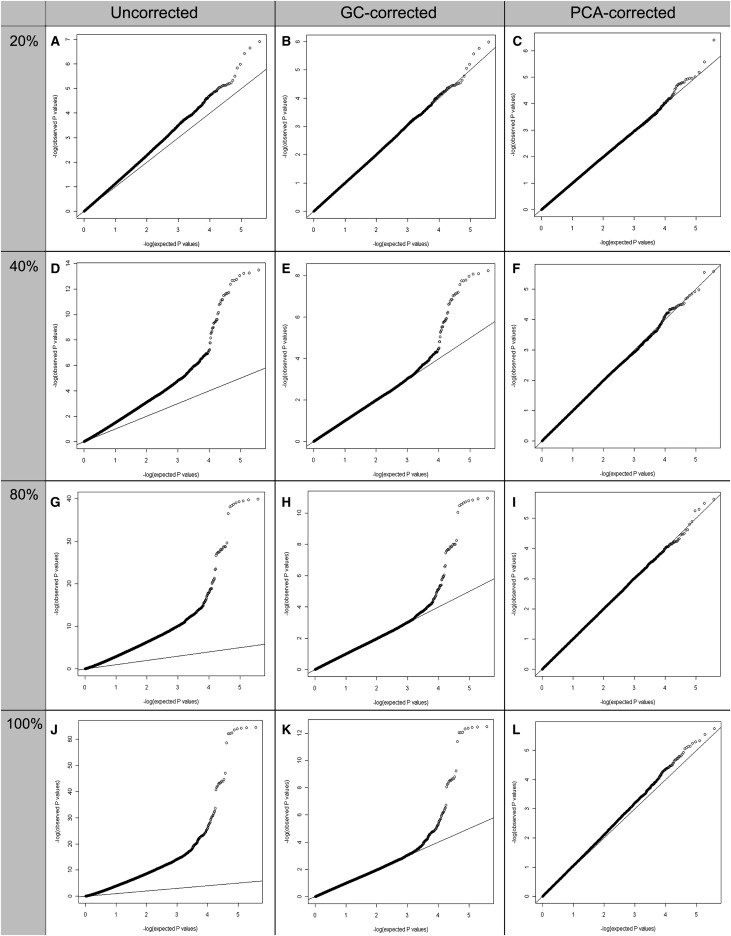
Q-Q Plots of the p Values from the Simulated Association Analyses with or without Correction for Population Stratification
The columns correspond to the Q-Q plots of the uncorrected, GC-corrected, and PCA-corrected p values. The rows correspond to 20%, 40%, 80%, and 100% stratification of the simulated case and control samples.
(A–C) 20% stratification: 500N cases, 400N and 100S controls.
(D–F) 40% stratification: 500N cases, 300N and 200S controls.
(G–I) 80% stratification: 500N cases, 100N and 400S controls.
(J–L) 100% stratification: 500N cases and 500S controls.
The impact of stratification that could exist within the metropolitan samples was also investigated, through analysis of the simulated cases and controls from the Shanghai samples. As an example of extremely stratified samples, the simulated GWAS analysis of the 500 cases from the northern cluster (500N) and the 500 controls from the southern cluster (500S) (100% stratification) yielded a high λ value of 1.66, and the Q-Q plot shows a marked inflation of association evidence (Figure S10). As expected, a moderate stratification of case and control samples (for example, using the cases of 300N+200S and the controls of 200N+300S) led to less inflation of association evidence (data not shown).
Discussion
Using genome-wide autosomal SNPs on a large number of the Han Chinese samples that were recruited according to their geographic locations of residence, we clearly demonstrated a one-dimensional north-south population structure among the Han Chinese. The degree to which the purely genetic clustering of the individuals closely corresponds to their geographical location suggests the persistence of local coancestry in the country. Large metropolises, such as Beijing and Shanghai, as well as the overseas Chinese community in Singapore are the exceptions, on the account of recent or historical large-scale migration from the countryside. We have further demonstrated, via simulations, that this north-south population structure of the Han Chinese, if left unaccounted for, can confound association studies and inflate association evidence to a substantial degree.
Although several previous studies6–10 have also detected a north-south geographical division among the Chinese, these results have been based primarily on segregation of various ethnic groups, including the Han Chinese, of which they had either chosen individuals from a few regions to represent the Han Chinese or categorized them under the generic terms of “southern Han” and “northern Han,” which can be quite subjective. The current study focused on the Han Chinese population by sampling ten provinces, two metropolitan cities, and one overseas Chinese community, allowing us to achieve denser coverage of the modern Han Chinese population. Sample recruitment by the geographic location of residence further allowed us to directly compare the genetic and geographic structure of the Han Chinese population. The existence of a north-south trend observed in previous studies was confirmed by our study. This implies the possibility of inferring genetic lineage, the lineal descent from an ancestral genetic pool, by geographic location. Although the observed trend can be explained by a myriad of population models, such as isolation by distance, it seems to concur very well with documented migratory patterns in China. The inferred north-south pattern in the genetic structure analyses suggests a primary north-south migratory pattern in China. This ties in very well with historical records indicating that the Huaxia tribes in northern China, the ancient ancestors of the Han Chinese, embarked on a long period of continuous southward expansion as a result of war and famine over the past two millenia.29 Furthermore, a broad sampling of the Han Chinese across various regions of China and the employment of genome-wide SNP-variation data allowed the current study to define the population structure at a finer scale. First, our study clearly showed that the one-dimensional north-south structure of the Han Chinese population is characterized by a continuous gradient, instead of distinct subpopulation clusters. This seems to support the hypothesis by Wen et al. for a demic diffusion of the Han Chinese.30 Second, our study was also able to reveal some fine-scale subpopulation structure within local populations of language from the same region. The three dialect groups from the Guangdong province were separated genetically along the same one-dimensional north-south axis of the overall population structure of the Han Chinese. Hakka and Teochew were found to be closer to the central provinces in terms of ancestry, whereas Cantonese, being native dwellers of Guangdong, showed a degree of genetic differentiation from the other two groups. This is consistent with historical migration records29 that show that, from the late ninth century onward, the Teochew originated from the neighboring province of Fujian, and began migrating particularly during the Song dynasty. The Hakka, who had their origins in the northern provinces, migrated to Guangdong during that time, as well as in late 17th century, particularly during the Qing dynasty. The lack of evidence for the more northerly origins of the Hakka in our study could be attributed to the early migrations of the Hakka, such that subsequent genetic exchange between the Hakka and the local population through marriage occurred long enough to make the Hakka more similar to the southern populations (instead of to their northern ancestral populations). This further exemplifies the fine-scale resolution of genome-wide autosomal SNPs and their ability to infer ancestry by geography and language.
The inclusion of samples from the Sichuan province, in western China, did not reveal evidence for an east-west stratification of the Han Chinese population. This is likely to result from an underrepresentation of samples from the more western provinces. The absence of east-west stratification could also be the result of the recent mass migration of peoples from central and southern regions, such as Hunan and Hubei, of China to Sichuan during the late Ming and early Qing dynasties.31
Our study found that large, metropolitan cities are the exceptions to this north-south pole of stratification. The widely utilized CHB samples from HapMap consisted of the Han Chinese from Beijing, which is situated in the northern part of China. It has often been used as a representative of the entire Chinese population in many genetic analyses. In our study, the CHB exhibited a clear mixture of individuals from both the northern and central provinces, but not from the southern region. This suggests caution when applying results from HapMap data to populations in southern China. Metropolitan Shanghai showed a similar pattern, with the majority of individuals coming from the northern and central provinces (Figure 1B). Although the overseas Chinese population in Singapore is often considered to be a homogenous one, our study shows a degree of heterogeneity in the ancestral origins of its Chinese population. These characteristics of the large urban centers in China and the overseas Chinese community in Singapore are expected, given the convergence of people from different regions of China resulting from modernization, as well as employment in the geographic location of residence, instead of genetic ancestry, as a basis for recruitment in the study. The inflation observed in the simulated association analysis on the Shanghai samples showed that sampling in metropolises can possibly introduce population stratification into GWAS as well.
Our results have important implications for designing GWAS in the Chinese population, an activity that is expected to intensify in the near future. Even though the genetic variance involved in the population stratification of the Han Chinese is small, the north-south stratification of the Han Chinese population, if left unaccounted for, can clearly confound association study and inflate association evidence. Although the inflation of association evidence can be effectively corrected by GC- and PCA-based methods in the genome-wide analysis, the stratification will probably have some adverse impact on validation studies, as well as candidate-gene studies, in which population stratification cannot be directly assessed and corrected because of the lack of genome-wide data. At this juncture, it is noted that the results of the simulation analysis might be largely dominated by the samples from Shandong and Guangdong in the northern and southern cohorts, respectively, as a result of sample overrepresentations (Table 1), implying that geographic matching by province can effectively eliminate the adverse impact of stratification. However, the high degree of resemblance between the genetic and geographic structure of the Han Chinese, as revealed by this study, can further imply that geographic matching by region might be a good proxy for genetic matching as well. Geographic matching, however, will not as work well in the large metropolitan cities of China, where geographical location is no longer a good indicator of ancestral origin. Our study also suggested that for the regions of China that are populated by various dialect groups, additional matching by language might be necessary.
Supplemental Data
Supplemental Data include ten figures and six tables and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for the data presented herein are as follows:
CLUMPP v1.1.2, DISTRUCT v1.1, and STRUCTURE v2.2, http://pritch.bsd.uchicago.edu/structure.html
EIGENSOFT, http://genepath.med.harvard.edu/∼reich/Software.htm
FRAPPE, http://smstaging.stanford.edu/tanglab/software/frappe.html
HapMap, http://www.hapmap.org
PLINK v1.06 (by Shaun Purcell), http://pngu.mgh.harvard.edu/purcell/plink/
Acknowledgments
This is a report of a joint effort of three research groups with equal contribution. L.J. is the senior author of the data analysis team, and X.Z. and Y-X.Z. are the senior authors of the two genotyping teams. We want to thank C. Zhang, J.W. Han, C. Quan. X.B. Zuo, H. Cheng, Z. Zhang, and F.S. Zhou from the Institute of Dermatology and the Department of Dermatology at the No.1 Hospital, Anhui Medical University and X.Y. Chen, H.B. Toh, K.K. Heng, and Y.W. Meah from the Genome Institute of Singapore for their supports in genotyping analysis. We also want to thank Erwin Tantoso from the Genome institute of Singapore for his assistance in data analysis. This work was funded by the General Program of National Natural Science Foundation of China (30530300, 30671895, and 30771943/C030116), the Key Project of Natural Science Foundation of China (30530670 and u0732005), the High-Tech Research and Development Program of China (863 Program) (2007AA02Z161), the National Basic Research Program of China (973 Program) (2004CB518604, 2006AA02A404 and 2007CB512301), the Shandong Provincial Research Fund of Science and Technology (2006GG2302029), and the Agency for Science & Technology and Research of Singapore (A∗STAR).
References
- 1.The Wellcome Trust Case-Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang X.J., Huang W., Yang S., Sun L.D., Zhang F.Y., Zhu Q.X., Zhang F.R., Zhang C., Zheng H.F., Liu J.J. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat. Genet. 2009;41:205–210. doi: 10.1038/ng.310. [DOI] [PubMed] [Google Scholar]
- 3.Liu X.G., Tan L.J., Lei S.F., Liu Y.J., Shen H., Wang L., Yan H., Guo Y.F., Xiong D.H., Deng H.W. Genome-wide association and replication studies identified TRHR as an important gene for lean body mass. Am. J. Hum. Genet. 2009;84:418–423. doi: 10.1016/j.ajhg.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakkula E., Rehnström K., Varilo T., Pietiläinen O.P., Paunio T., Pedersen N.L., deFaire U., Järvelin M.R., Saharinen, Peltonen L. The genome-wide patterns of variation expose significant substructure in a founder population. Am. J. Hum. Genet. 2008;83:787–794. doi: 10.1016/j.ajhg.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian C., Plenge R.M., Ransom M., Lee A., Villoslada P., Selmi C., Klareskog L., Pulver A.E., Qi L., Gregersen P.K., Seldin M.F. Analysis and application of European genetic substructure using 300K SNP information. PLoS Genet. 2008;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu J.Y., Huang W., Kuang S.Q., Wang J.M., Xu J.J., Chu Z.T., Yang Z.Q., Lin K.Q., Li P., Wu M. Genetic relationship of populations in China. Proc. Natl. Acad. Sci. USA. 1998;95:11763–11768. doi: 10.1073/pnas.95.20.11763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su B., Xiao J., Underhill P., Deka R., Zhang W., Akey J., Huang W., Shen D., Lu D., Jin L. Y-chromosome Evidence for a Northward Migration of Modern Humans into Eastern Asia during the Last Ice Age. Am. J. Hum. Genet. 1999;65:1718–1724. doi: 10.1086/302680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao Y.G., Kong Q.P., Bandelt H.J., Kivisild T., Zhang Y.P. Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am. J. Hum. Genet. 2002;70:635–651. doi: 10.1086/338999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao Y.G., Kong Q.P., Man X.Y., Bandelt H.J., Zhang Y.P. Reconstructing the evolutionary history of China: a caveat about inferences drawn from ancient DNA. Mol. Biol. Evol. 2003;20:214–219. doi: 10.1093/molbev/msg026. [DOI] [PubMed] [Google Scholar]
- 10.Yao Y.G., Kong Q.P., Wang C.Y., Zhu C.L., Zhang Y.P. Different matrilineal contributions to genetic structure of ethnic groups in the silk road region in china. Mol. Biol. Evol. 2004;21:2265–2280. doi: 10.1093/molbev/msh238. [DOI] [PubMed] [Google Scholar]
- 11.Piazza A. Towards a genetic history of China. Nature. 1998;395:636–637. doi: 10.1038/27071. 639. [DOI] [PubMed] [Google Scholar]
- 12.Jin L., Su B. Natives or immigrants: modern human origin in east Asia. Nat. Rev. Genet. 2000;1:126–133. doi: 10.1038/35038565. [DOI] [PubMed] [Google Scholar]
- 13.Cavalli-Sforza L.L., Menozzi P., Piazza A. Princeton Univ. Press; Princeton, New Jersey: 1994. The History and Geography of Human Genes. [Google Scholar]
- 14.Ding Y.C., Wooding S., Harpending H.C., Chi H.C., Li H.P., Fu Y.X., Pang J.F., Yao Y.G., Xiang Yu J.G., Moyzis R., Zhang Y.P. Population structure and history in East Asia. Proc. Natl. Acad. Sci. USA. 2000;97:14003–14006. doi: 10.1073/pnas.240441297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang H., Peng J., Wang P., Risch N.J. Estimation of Individual Admixture. Analytical and Study Design Considerations. Genet. Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- 18.Pritchard J.K., Stephens M., Donnelly P. Inference of Population Structure Using Multilocus Genotype Data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falush D., Stephens M., Pritchard J.K. Inference of Population Structure Using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 21.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Singleton A.B. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 22.Jakobsson M., Rosenberg N.A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg N.A. Dros. Inf. Serv.TRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes. 2004;4:137–138. [Google Scholar]
- 24.Wright S. The genetical structure of populations. Ann. Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 25.Devlin B., Roeder K. Genomic Control for Association Studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 26.Novembre J., Johnson T., Bryc K., Kutalik Z., Bokyo A.R., Auton A., Indap A., King K.S., Bergmann S., Nelson M.R. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saw S.H. Second Edition. Institute of Southeast Asian Studies; Singapore: 2007. The Population of Singapore. [Google Scholar]
- 28.Tong C.K. Chinese in Singapore. In: Ember C.R., Ember M., Skoggard I.A., editors. Encyclopedia of Diasporas: Immigrant and Refugee Cultures around the World. Volume 1. Springer, in conjunction with the Human Relations Area Files, Yale University; New York, USA: 2004. pp. 723–741. [Google Scholar]
- 29.Ge J.X., Wu S.D., Cao S.J. Fujian People's Publishing House; Fuzhou, China: 1997. Zhongguo Yimin Shi (The migration history of China) ([In Chinese.]) [Google Scholar]
- 30.Wen B., Li H., Lu D., Song X., Zhang F., He Y., Li F., Gao Y., Mao X., Zhang L., Jin L. Genetic evidence supports demic diffusion of Han culture. Nature. 2004;431:302–305. doi: 10.1038/nature02878. [DOI] [PubMed] [Google Scholar]
- 31.Lee K.C. World Scientific; 2005. Pioneers of Modern China: Understanding the Inscrutable Chinese. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.