

Abstract
In biology, rapid oxidation and evolution of H2 is catalyzed by metalloenzymes known as hydrogenases. These enzymes have unusual active sites, consisting of iron complexed by carbonyl, cyanide, and thiolate ligands, often together with nickel, and are typically inhibited or irreversibly damaged by O2. The Knallgas bacterium Ralstonia eutropha H16 (Re) uses H2 as an energy source with O2 as a terminal electron acceptor, and its membrane-bound uptake [NiFe]-hydrogenase (MBH) is an important example of an “O2-tolerant” hydrogenase. The mechanism of O2 tolerance of Re MBH has been probed by measuring H2 oxidation activity in the presence of O2 over a range of potential, pH and temperature, and comparing with the same dependencies for individual processes involved in the attack by O2 and subsequent reactivation of the active site. Most significantly, O2 tolerance increases with increasing temperature and decreasing potentials. These trends correlate with the trends observed for reactivation kinetics but not for H2 affinity or the kinetics of O2 attack. Clearly, the rate of recovery is a crucial factor. We present a kinetic and thermodynamic model to account for O2 tolerance in Re MBH that may be more widely applied to other [NiFe]-hydrogenases.
Keywords: electrochemistry, hydrogen, hydrogenase, oxygen tolerance, Ralstonia eutropha
Hydrogenases play a crucial role in the metabolism of many microorganisms, where they catalyze the reversible oxidation and production of H2. Hydrogenases possess active sites containing either one Ni and one Fe atom (“[NiFe]-hydrogenases”), or only Fe atoms (“[FeFe]-hydrogenases”), coordinated by cysteine thiolates and the biologically unusual ligands CO and CN−. (A third class, the Hmd or [Fe]-hydrogenases (1), will not be discussed here.) Hydrogenases are highly active, with turnover frequencies for H2 oxidation (believed to occur through a heterolytic cleavage mechanism) in excess of thousands of molecules of H2 per second at 30° C (2). Hydrogenases are usually reported to be highly O2-sensitive, being inactivated or irreversibly damaged by even trace O2. It is generally considered that [FeFe]-hydrogenases react irreversibly with O2, giving rise to as yet poorly characterized inactive products, whereas [NiFe]-hydrogenases react with O2 to give products that can be reactivated upon reduction.
The well-characterized “standard” [NiFe]-hydrogenases from Desulfovibrio species, such as Desulfovibrio fructosovorans (Df), cannot oxidize H2 in the presence of O2. Exposure to O2 under electron-rich conditions produces mainly the Ready state (also known as “Ni-B”), in which an HO− ligand is bound in a bridging position between the Ni and Fe atoms (3). This state is also produced under anaerobic oxidizing conditions, and can be rapidly recovered by applying a reducing potential under H2. Exposure to O2 under electron-deficient conditions produces mainly the Unready state (also known as “Ni-A”), in which a peroxo group is believed to occupy the bridging position, although other modifications may occur, such as the oxidation of sulfur ligands (3–5). Although the Unready state can be activated by applying a reducing potential under H2, its recovery is many orders of magnitude slower than that of Ready. The term “O2 attack” encompasses its access to the [NiFe]-active site and its subsequent reaction to form an inactive state. These reactions are summarized in Fig. 1.
Fig. 1.

A simplified scheme showing the reactions of standard [NiFe]-hydrogenases. Active hydrogenase molecules catalyze H2/H+cycling, and are subject to aerobic inactivation, forming either the Ready or Unready forms, depending on the availability of electrons and protons. The Ready form can also be formed anaerobically at oxidizing potentials. (Adapted from ref. 20 with permission from the Royal Society of Chemistry.)
In contrast with Df hydrogenase, the membrane-bound [NiFe]-hydrogenase (MBH) from Ralstonia eutropha H16 (Re) can oxidize H2 [and, in vitro, also reduce protons to H2 in the reverse reaction (6)] in the presence of air (7). As a “Knallgas” bacterium, Re uses H2 as an alternative energy source with O2 as the terminal electron acceptor. An O2-tolerant hydrogenase is therefore essential for this metabolic pathway of energy conservation and, more generally, would be crucial for other microorganisms oxidizing H2 in environments where O2 may be found.
Protein film electrochemistry (PFE) has proved especially useful in probing the reactions of hydrogenases in air (2). In this technique, small amounts of enzyme are adsorbed onto an electrode such that they retain native catalytic activity. Catalytic activity is proportional to the electrical current, and is directly controlled through a precisely applied electrode potential. A key advantage of PFE over solution techniques, especially when measuring enzyme activity in air, is that soluble electron donors/acceptors (which invariably react with O2) are not required. By using PFE, we recently showed that the O2 tolerance of the MBH enzyme from Ralstonia metallidurans CH34 (Rm, closely related to Re) is so effective that the oxidation of even nM levels of H2 in air can be detected (8), consistent with the very low threshold limits for H2 uptake determined by Conrad et al. (9). A practical demonstration of O2 tolerance was provided by a membraneless fuel cell producing power from 3% H2 in air (10, 11).
Crystallographic and computational studies have suggested that hydrophobic “gas channels” provide a selective filter to restrict the access of small molecules such as O2 and CO in hydrogenases (3). Duche et al. (12) reported how enlarging the gas channel in the regulatory hydrogenase from Rhodobacter capsulatus increased its O2-sensitivity. We used a similar strategy with Re MBH, anticipating that a restricted gas channel would make the enzyme even more O2 tolerant; however, no such effect was observed. Clearly, gas channels alone cannot confer O2 tolerance. Most significantly, recent EPR and FTIR studies on Re MBH showed that reaction with O2 even under electron-deficient conditions produces only the Ready state; no Ni-A (Unready) was detected (13).
In this paper, we describe PFE studies on Re MBH that offer valuable insight into the kinetic and thermodynamic aspects of O2 tolerance and support a model that may be generally useful.
Results
Defining an O2 Tolerance Factor.
We measured the inhibitory effect of O2 on H2 oxidation activity over a wide range of pH values, O2 concentrations, electrode potentials, and temperatures. From these measurements, we obtained values for the “O2 tolerance factor,” KIO2,app, by using the method described previously (14, 15). The O2 tolerance factor is the O2 concentration required to attenuate H2 oxidation activity by 50%; therefore, a high value indicates a high level of O2 tolerance. The H2 oxidation current at a pyrolytic graphite “edge” (PGE) electrode modified with Re MBH was monitored following a succession of injections of O2 gas into the electrochemical cell headspace. After correcting for film loss over time, KIO2,app was obtained by analyzing the catalytic current that stabilizes at each O2 concentration. Details are provided in Methods and values are summarized in Table 1. Significantly, O2 tolerance increases with decreasing electrode potential (i.e., more reducing conditions) and with increasing temperature. It appears that KIO2,app may decrease at high pH, but we were unable to extend the pH range studied as the enzyme activity became too low. These observations suggest immediately that O2 tolerance does not rely on a selective filter, such as a restrictive gas channel.
Table 1.
Values of KIO2,app under a range of conditions
| Variable | KIO2,app, μM |
|---|---|
| Potential, V vs. SHE* | |
| −0.018 | 1,630 |
| +0.052 | 390 |
| +0.122 | 110 |
| +0.192 | 40 |
| +0.262 | 8 |
| pH† | |
| 4.5 | 70 |
| 5.0 | 80 |
| 5.5 | 110 |
| 6.0 | 90 |
| 6.5 | 70 |
| Temperature, ° C‡ | |
| 10 | 70 |
| 20 | 70 |
| 30 | 110 |
| 40 | 140 |
*All values were recorded at pH 5.5, 30° C.
†All values were recorded at 30° C, at a constant overpotential (driving force) of +453 mV relative to the thermodynamic H+/H2 cell potential at each pH.
‡ All values were recorded at pH 5.5, at a constant overpotential (driving force) of +453 mV relative to the thermodynamic H+/H2 cell potential at each temperature.
Michaelis Constants for H2.
The O2 tolerance of a hydrogenase in H2 oxidation is expected to depend on the enzyme's affinity for H2 because of competition for binding at the active site. We define KMH2 as the Michaelis constant for H2. We used the method described previously (14, 15) to determine KMH2 for Re MBH over a range of pH, potentials, and temperatures (Table 2). A known volume of H2-saturated buffer solution was introduced into the electrochemical cell, and the H2 was then removed by a constant flow of inert gas, typically N2. Values of KMH2 were determined by analyzing the current vs. time trace as described in refs. 14 and 15.
Table 2.
Values of KMH2 under a range of conditions
| Variable | KMH2, μM |
|---|---|
| Potential, V vs. SHE* | |
| −0.158 | 8 |
| −0.058 | 6 |
| +0.042 | 14 |
| +0.142 | ≈100 |
| +0.242 | ≈130 |
| pH† | |
| 4.5 | 6 |
| 5.0 | 8 |
| 5.5 | 6 |
| 6.0 | 15 |
| 6.5 | 10 |
| Temperature, ° C‡ | |
| 10 | 0.4 |
| 20 | 1 |
| 30 | 6 |
| 40 | 24 |
*All values were recorded at pH 5.5, 30° C.
†All values were recorded at 30° C, at a constant overpotential (driving force) of +273 mV relative to the thermodynamic H+/H2 cell potential at each pH.
‡All values were recorded at pH 5.5, at a constant overpotential (driving force) of +273 mV relative to the thermodynamic H+/H2 cell potential at each temperature.
Rates of O2 Reaction.
The rate of reaction of O2 at the active site of Re MBH was probed by analyzing the current decay following O2 introduction at a range of potentials, pH, and temperatures. A typical experiment is shown in Fig. 2A. An electrode modified with Re MBH was placed in a gas-tight electrochemical cell under an atmosphere of 50% H2 and 50% N2. The electrode was initially polarized at −0.508 V to ensure that all of the enzyme was active. After 120 s, the potential was stepped to the value at which the rate of O2 reaction was to be determined. After allowing a background current to be determined, the atmosphere was changed to 50% H2, 25% O2, and 25% N2; simultaneously, an aliquot of temperature-equilibrated buffer solution, saturated with 50% H2 and 50% O2 was injected into the cell solution. Under the experimental conditions, the mixing time is ≈0.1 s and therefore insignificant on the experimental timescale. The decrease in current that begins immediately is purely due to O2 reaction at the active site; at the potentials used, direct O2 reduction at the electrode surface is negligible, and, crucially, following the introduction of O2, neither H2 nor O2 concentrations change over the course of the measurement. After correcting data for anaerobic inactivation and film loss (Fig. 2A), the rate of O2 reaction was determined (Fig. 2B). Typically, the reaction was first order for more than two half-lives, and rate constants are presented in Table 3 (see also Table S1 of the SI Appendix; trends within sets of experiments are independent of the method used to determine the rates).
Fig. 2.

A typical experiment to determine the rate of O2 reaction with Re MBH. (A) The current vs. time trace (black) with the fit to film loss/anaerobic inactivation (gray). (B) The corrected data (black), with the fit to the initial attack by O2 (gray). Experimental conditions were pH 5.5, 10° C, +0.192 V, electrode rotation rate = 4,500 rpm.
Table 3.
Calculated rates of reaction of O2 (25%) with Re MBH under a range of conditions, electrode rotation rate = 4500 rpm
| Variable | Rate of O2 reaction, s−1 |
|---|---|
| Potential, V vs. SHE* | |
| +0.192 | 0.11 |
| +0.262 | 0.10 |
| +0.332 | 0.10 |
| +0.402 | 0.11 |
| pH† | |
| 4.5 | 0.12 |
| 5.5 | 0.11 |
| 6.5 | 0.11 |
| Temperature, ° C‡ | |
| 0 | 0.06 |
| 10 | 0.11 |
| 20 | 0.19 |
| 30 | 0.31 |
*All values were recorded at pH 5.5, 10° C.
†All values were recorded at 10° C, at a constant overpotential (driving force) of +523 mV relative to the thermodynamic H+/H2 cell potential at each pH.
‡All values were recorded at pH 5.5, at a constant overpotential (driving force) of +523 mV relative to the thermodynamic H+/H2 cell potential at each temperature.
The temperature dependence of the rate constants gave a linear fit to the Eyring equation with an activation enthalpy of ≈34 kJ mol−1. We also measured the rate of O2 reaction with the O2-sensitive [NiFe]-hydrogenase from Desulfovibrio gigas by using the same method; a rate of 0.73 s−1 was calculated at pH 6.5, 10° C, +0.192 V.
Rates of Reactivation.
Rate and potential dependencies of recovery from both anaerobic and aerobic inactivation were measured at 10° C (the rate of recovery at 30° C is so rapid that Re MBH regains full activity within the “dead time” caused by the release of electrode capacitive charge). To measure the rate of recovery after anaerobic inactivation, an electrode modified with Re MBH was placed in a gas-tight cell under a constant flow of H2. After 120 s at −0.508 V, the electrode was stepped to a high potential (+0.442 V), causing the enzyme to anaerobically inactivate. After 900 s, the electrode was stepped to the potential at which the rate of recovery was to be measured. A single exponential was fitted to the current vs. time trace (Fig. 3A; data from the first 10 s following the potential step were within the dead time and thus not included).
Fig. 3.

Reactivation of aerobically and anaerobically inactivated states of Re MBH. (A and B) Typical experiments to measure the rate of reactivation of Re MBH under 100% H2 following anaerobic (A) and aerobic (B) inactivation. In these examples, reactivation was measured at +0.192 V. (C) Rates of reactivation as a function of potential with a fit to the data overlaid. Data for the reactivation of Av MBH from the Unready state (filled squares, taken from reference 16, pH 6, 45° C) are also shown, and have been multiplied by 10 for clarity (unfilled squares). Sigmoidal fits are also shown. (D) Voltammograms (100% H2, 0.1 mV s−1) of Re MBH (bold) and Av MBH (dashed), showing recovery after aerobic inactivation at 392 mV under N2. All at pH 5.5, 10° C, electrode rotation rate = 2,500 rpm.
Experiments to measure the rate of reactivation following aerobic inactivation were more complex (Fig. 3B). Again, the electrode was held, initially, for 120 s at −0.508 V under 100% H2. The potential was then stepped to +0.092 V. (At this potential, anaerobic inactivation is negligible.) The cell headgas was then changed from 100% H2 to 80% H2 and 20% O2, causing the current to decrease as the enzyme becomes aerobically inactivated. After 900 s, the current reaches a plateau level, at which point the electrode potential was stepped to +0.442 V and the headgas changed back to 100% H2. At this potential, the enzyme does not reactivate, thus allowing a clear time window for gas exchange. After 900 s (to ensure that all O2 was removed from solution), the electrode was stepped to the potential at which the rate of recovery was to be measured, and the rate of reactivation was determined by fitting a single exponential as before.
Fig. 3C compiles the rates of reactivation following both anaerobic and aerobic inactivation, plotted as a function of potential. The rate of recovery is similar in each case, and a limiting rate of ca. 0.1 s−1 is reached at potentials below 0 V. Fitting the data to a sigmoid gives the number of electrons involved in the reactivation process, n (16). The best fit to the data yields n = 0.96 (overlaid in Fig. 3C), consistent with a single electron transfer. Also overlaid in Fig. 3C are the rates of recovery at 45° C of the Unready state of the O2-sensitive MBH from Allochromatium vinosum (Av) (data from ref. 16). The Unready state is produced in large amounts under similar conditions in Av MBH. Reactivation of Re MBH following aerobic inactivation is clearly much faster than that of Av MBH. (Note that Ready and Unready reactivate at similar potentials in Av MBH.)
Potentials of reactivation are most easily compared by using voltammetry. Fig. 3D shows reactivation of both Re MBH and Av MBH after aerobic inactivation, recorded under the same conditions. The MBH-modified electrode was initially held at −0.558 V under 100% N2. The potential was then stepped to +0.392 V and an aliquot of O2-saturated buffer was simultaneously injected into the cell solution, causing the enzyme to inactivate aerobically. After 300 s, the headgas was changed to 100% H2; then, after 900 s, the potential was slowly (0.1 mV s−1) swept back to −0.558 V, allowing the recovery to be monitored. Reactivation of Av MBH occurs at a much lower potential, requiring ≈200 mV extra driving force compared to Re MBH.
Discussion
PFE allows the detailed measurement of reactions crucial to understanding O2-tolerant H2 oxidation in [NiFe]-hydrogenases. The O2 tolerance factor, KIO2,app, depends strongly on electrode potential and temperature but is not affected by pH in the region 4.5–6.5. Greatest O2 tolerance is observed at low potentials and, significantly, at high temperatures. Standard O2-sensitive [NiFe]-hydrogenases typically have values of KIO2,app that are very low. For example, under one bar H2, Av MBH is completely inhibited by just 4 μM O2 (+0.142 V, pH 5.6, 30° C) (7), and the soluble hydrogenase from Df is completely inhibited by 5 μM O2 (+0.190 V, pH 7, 40° C) (14)—thus, even micromolar O2 concentrations are well in excess of KIO2,app for these enzymes. By contrast, for Re MBH, KIO2,app is ≈50 μM at these conditions and exceeds 1.6 mM at −0.018 V. To deconvolute the O2 tolerance factor, it is necessary to probe the potential, pH, and temperature dependencies of the various contributory reactions and incorporate these within a model. Specifically, we are inspecting whether the individual reactions that must form the basis of O2 tolerance either support, oppose, or are indifferent to the potential and temperature dependencies of KIO2,app.
We first consider H2 cycling in the absence of O2, where both the affinity for H2 and the catalytic turnover frequency (kcat) are crucial. Although it has not yet proved possible to determine kcat for Re MBH electrochemically, it is likely, by analogy with other [NiFe]-hydrogenases, that Re MBH has a turnover frequency for H2 oxidation in the order of 103 s−1 at 30° C (17).
The Michaelis constant for H2, KMH2, increases with increasing electrode potential and increasing temperature. The smaller is KMH2, the more long-lived is the Michaelis complex (Eact−H2, where Eact represents Active hydrogenase), and hence the more likely the hydrogenase should be protected against O2 binding at the active site. This is indeed the case for the potential dependence but not for the temperature dependence, showing that KMH2 cannot be the dominating factor in determining O2 tolerance. Consistent with this, KMH2 values for Re MBH at low potentials are similar to those determined for some O2-sensitive hydrogenases, such as Av MBH and the soluble enzyme from Df [which has KMH2 ≈5 μM at −0.16 V (14)]. In addition, some mutants of Re MBH exhibit a very high KMH2 but retain substantial O2 tolerance (15).
There is a natural potential dependence in KMH2, because the rate of catalysis (kcat, cf. Eq. 2, below) increases with increasing driving force (a consequence of the inherent high activity of the active site and the limit on how fast electrons can be supplied from the electrode). This dependence is evident in voltammograms for numerous hydrogenases catalyzing H2 oxidation (2, 18). (Interestingly, low H2 concentrations also increase the extent of anaerobic inactivation, potentially leading to small overestimates in KMH2.) An increase in KM with increasing temperature is a typical trend and has origins in the increase in kcat and, in this case, the increasing rate of anaerobic inactivation with increasing temperature.
The rates of reaction of Re MBH with O2 (Fig. 2) are unaffected by changing potential or pH but increase strongly with increasing temperature. If the rate of O2 attack (which includes its transport through the protein) were a controlling factor in determining KIO2,app, then at higher temperatures the hydrogenase would be less O2-tolerant, which is not the case. The lack of a pH- or potential-dependence in the rate of reaction with O2 is in excellent agreement with the findings of Léger et al. (14) in their studies on Df hydrogenase. The potential dependence of KIO2,app does not therefore arise from a dependence in the rate of O2 reaction. It is worth noting, however, that the rates of O2 reaction for Re MBH are considerably lower than for the periplasmic hydrogenase from D. gigas, whose catalytic subunit shares 67% sequence identity with the large subunit of Df hydrogenase.
We finally turn to the properties of the reactivation reaction. Our electrochemical results support the spectroscopic evidence that, in contrast with standard O2-sensitive hydrogenases, Re MBH makes only the Ready state on reaction with O2: No detectable amounts of Unready are made. All data fitted well to a single exponential, showing that only one inactive state is being recovered (recovery of Av MBH, which produces both Ready and Unready states, is biphasic). The rates of recovery of Re MBH following both anaerobic and aerobic inactivation cannot be distinguished, suggesting strongly that the same state is formed regardless of whether the active site is oxidized by O2 or anaerobically at high potential. As expected, rates increase with increasing temperature (18). The value of n (the number of electrons involved in the reductive reactivation of inactive NiIII to an active NiII state) is 0.96, implying a single electron transfer consistent with reactivation of Ready (Fig. 1). The potential of reactivation of Re MBH is more than 200 mV more positive than for the O2-sensitive [NiFe]-hydrogenases from Av and D. gigas (18), and reactivation is therefore thermodynamically more favorable. [Interestingly, in the soluble hexameric NAD+-reducing [NiFe]-hydrogenase from Re, the inactive Ready state can be reactivated by using reducing equivalents from added NAD(P)H (19).]
To bring these factors together, the catalytic cycle shown in Fig. 1B was modeled by using the kinetic cycle shown in Fig. 4(Re MBH is represented by the symbol E). The steady-state approximation was applied to all intermediate species (Eact−H2, Eact−O2, EReady, and EUnready). The following expression (Eq. 1) was obtained (see SI Appendix for derivation), in which KMH2 and KIO2 are as defined in Eqs. 2 and 3, respectively, and i is the catalytic current.
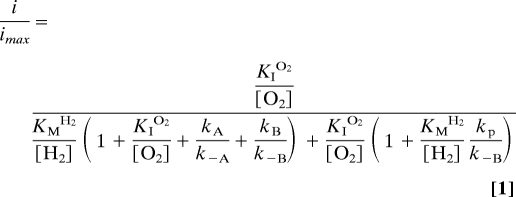 |
The term KIO2 is the inhibition constant for O2, and would vary from KIO2,app by a factor of {[H2]/KMH2 × (1+KMH2/[H2])}, if O2 were to be considered simply as a competitive inhibitor.
Fig. 4.

The kinetic scheme used to model O2 tolerance. Re MBH is abbreviated as “E” and is shown in the reduced active form (Eact), as an adduct with H2 or O2 (Eact−H2 and Eact−O2, respectively) or in the Ready (EReady) or Unready (EUnready) state.
Eq. 1 was fitted to the data to determine KIO2,app as described above at 10° C (pH 5.5, +0.122 V). Values of kcat (≈250 s−1) and k−A, the rate of reactivation of Unready (≈0.00025 s−1) were used as “best guesses”, based on analogy with standard [NiFe]-hydrogenases. The value of kP, the rate of anaerobic inactivation, is ≈0.003 s−1 at 10° C and is potential independent (see Fig. S2 of the SI Appendix). The value of k−B, the rate of reactivation of Ready (≈0.03 s−1) was determined under these conditions (Fig. 3). To simplify, the approximation was made that the rates of access of H2 and O2 (k1 and k2, respectively) depend only on their masses according to the kinetic theory of gases (vH2/vO2 = ), and hence k1 = 4 × k2, and, for the reverse reaction, k−1 = 4 × k−2. Taking typical values of KMH2 ≈4 μM and KIO2,app ≈60 μM at these conditions, the best fit to the experimental data (Fig. 5A) yielded k1 = 68 μM−1 s−1, k−1 = 22 s−1 and the rate of formation of Ready from the initial O2 adduct, kB = 0.01 s−1.
Fig. 5.

Simulations obtained by applying Eq. 1 to Re MBH. (A) Limiting H2 oxidation currents at varying O2 concentrations, determined experimentally (black) and calculated from the model (gray). (B–D) show the effect of altering the percentage O2 (B), the electrode potential (C), and kA (D) on the catalytic currents calculated from the model. All conditions are pH 5.5, 10° C. B and D are at 0.122 V, and C and D are at 10% O2.
The model allows an understanding of the roles of the different rate constants in affecting O2 tolerance. Fig. 5B shows the effect of varying the O2 concentration on the H2 oxidation activity under the conditions above. Fig. 5C shows the effect of changing the electrode potential on the H2 oxidation activity at 10% O2; the potential dependence of k−B is modeled by using Eq. 4. Fig. 5D stresses the importance, for O2 tolerance, of not producing the Unready state, i.e., of ensuring that kA is very small. It is unclear as yet why Re MBH does not produce Unready, but formation of the Ready state (kB) requires a greater immediate availability of electrons (and protons) than for producing the Unready state (kA). This requirement could be accommodated by some property of Re MBH, such as an increase in the number of redox sites able to donate an electron. Analogously, being embedded in the cytoplasmic membrane or immobilized on an electrode with a ready supply of electrons may protect the enzyme against O2 inactivation.
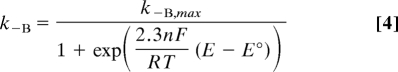 |
In essence, both the potential and temperature dependencies of KIO2,app emphasize the importance of enhancing the rate of recovery as opposed to limiting the rate of O2 attack. Reactivation from Ready is faster at low potential and at high temperature, and this matches the characteristics of the O2-tolerance factor. Interestingly, this means that Re MBH can be viewed as acting not only in H2 cycling, but also as a very slow oxidase (20), catalyzing the four-electron reduction of O2 to water with a rate constant in the order of 10−1–10−2 s−1. The positive temperature dependence could have particular significance for O2-tolerant H2 cycling in microorganisms such as Aquifex aeolicus (21), Thermococcus litoralis (22), and Pyrococcus furiosus (23) that live at high temperatures and could encounter O2.
Methods
The Re MBH enzyme was isolated and purified as described previously (15). All experiments were performed in an N2-filled anaerobic glove box (M. Braun, <2 ppm O2). A PGE rotating disk electrode (area 0.03 cm2) was used with an electrode rotator (EcoChemie Autolab Rotator), which fitted tightly above a gas-tight, glass, electrochemical cell. A three-electrode configuration was used, with a Pt wire counter electrode and a saturated calomel reference electrode (SCE) located in a reference arm containing aqueous 0.10 M NaCl and connected to the working electrode compartment via a Luggin capillary. The temperature of the working compartment was controlled by using a water jacket, whereas the reference electrode remained at ≈25° C. The potential (E) was corrected with respect to the standard hydrogen electrode (SHE) by using the following formula: ESHE = ESCE + 0.242 V at 298 K (24): All potentials are quoted relative to SHE. An Autolab PGSTAT 10 electrochemical analyzer was used with an electrochemical detection module and GPES software (EcoChemie).
All experiments were carried out in 0.05 M sodium phosphate buffer with 0.10 M NaCl as supporting electrolyte (prepared by using purified water, 18.2 MΩ cm, Millipore) and titrated to the desired pH at the experimental temperature. Gases were of Premier/Research Grade and supplied by Air Products or BOC. Mass-flow controllers (Smart-Trak, Sierra Instruments) were used to supply precise mixtures of gases to the cell headspace at a constant flow rate.
To prepare films of MBH, the PGE electrode was polished by using an aqueous slurry of 1 μm α-alumina (Buehler) on cotton wool, then sonicated for ca. 5 s and rinsed with purified water, before 1.5 μl of enzyme solution (0.1–0.2 mg/mL) was successively applied and withdrawn from the electrode surface over a period of ca. 30 s. In all experiments, the electrode was rotated at a constant rate, typically 2,500 rpm, to provide efficient supply of substrate and removal of product from the electrode surface.
To measure KIO2,app and the rate of O2 reaction, data were corrected for “film loss” over the course of the experiment by fitting anaerobic data points to a single exponential. A good fit was usually obtained (see Fig. 2) and by dividing the raw data by the exponential curve, normalized/corrected data were obtained. To obtain KIO2,app, currents were corrected for film loss, and the inverse of the limiting value of the corrected current at each O2 concentration was plotted against O2 concentration, with KI2,app given by dividing the y intercept by the gradient. Values of the Henry's constants for H2 and O2, and the temperature dependence thereof, were taken from ref. 25.
Supporting Information.
Analysis of the rates of O2 reaction, determination of the rate of anaerobic inactivation, and the derivation of Eq. 1, are available in the SI Appendix.
Supplementary Material
Acknowledgments.
The authors wish to thank Dr. M. Ludwig for preparation of the membrane-bound uptake [NiFe]-hydrogenase samples, Dr. C. F. Blanford and Mr. P. A. Cracknell for help with modeling, and Dr. K. A. Vincent for obtaining preliminary results on the reactivation rates for Ralstonia eutropha MBH. J.A.C., A.F.W., and F.A.A. are supported by the Engineering and Physical Sciences Research Council UK (Grant Supergen 5) and the Biotechnology and Biological Sciences Research Council UK (Grant BB/D52222X/1). B.F. and O.L. are supported by the Sonderforschungsbereich (Grant 498, Project C1) and the Cluster of Excellence “Unicat” from the Deutsche Forschungsgemeinschaft.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0905959106/DCSupplemental.
References
- 1.Thauer RK, Klein AR, Hartmann GC. Reactions with molecular hydrogen in microorganisms: Evidence for a purely organic hydrogenation catalyst. Chem Rev. 1996;96:3031–3042. doi: 10.1021/cr9500601. [DOI] [PubMed] [Google Scholar]
- 2.Vincent KA, Parkin A, Armstrong FA. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem Rev. 2007;107:4366–4413. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- 3.Fontecilla–Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 4.Volbeda A, et al. Structural differences between the Ready and unReady oxidized states of [NiFe] hydrogenases. J Biol Inorg Chem. 2005;10:239–249. doi: 10.1007/s00775-005-0632-x. [DOI] [PubMed] [Google Scholar]
- 5.Ogata H, et al. Activation process of [NiFe] hydrogenase elucidated by high-resolution X-Ray analyses: Conversion of the Ready to the Unready state. Structure. 2005;13:1635–1642. doi: 10.1016/j.str.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 6.Goldet G, et al. Hydrogen production under aerobic conditions by membrane-bound hydrogenases from Ralstonia species. J Am Chem Soc. 2008;130:11106–11113. doi: 10.1021/ja8027668. [DOI] [PubMed] [Google Scholar]
- 7.Vincent KA, et al. Electrocatalytic hydrogen oxidation by an enzyme at high carbon monoxide or oxygen levels. Proc Natl Acad Sci USA. 2005;102:16951–16954. doi: 10.1073/pnas.0504499102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cracknell JA, et al. Enzymatic oxidation of H2 in atmospheric O2: The electrochemistry of energy generation from trace H2 by aerobic microorganisms. J Am Chem Soc. 2008;130:424–425. doi: 10.1021/ja078299+. [DOI] [PubMed] [Google Scholar]
- 9.Conrad R. Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO) Microbiol Rev. 1996;60:609–640. doi: 10.1128/mr.60.4.609-640.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cracknell JA, Vincent KA, Armstrong FA. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem Rev. 2008;108:2439–2461. doi: 10.1021/cr0680639. [DOI] [PubMed] [Google Scholar]
- 11.Vincent KA, et al. Electricity from low-level H2 in still air—an ultimate test for an oxygen tolerant hydrogenase. Chem Commun. 2006:5033–5035. doi: 10.1039/b614272a. [DOI] [PubMed] [Google Scholar]
- 12.Duche O, Elsen S, Cournac L, Colbeau A. Enlarging the gas access channel to the active site renders the regulatory hydrogenase HupUV of Rhodobacter capsulatus O2 sensitive without affecting its transductory activity. Febs J. 2005;272:3899–3908. doi: 10.1111/j.1742-4658.2005.04806.x. [DOI] [PubMed] [Google Scholar]
- 13.Saggu M, et al. Spectroscopic insights into the oxygen-tolerant membrane-associated [NiFe]-hydrogenase of Ralstonia eutropha H16. J Biol Chem. 2009;284:16264–16276. doi: 10.1074/jbc.M805690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Léger C, et al. Inhibition and aerobic inactivation kinetics of Desulfovibrio fructosovorans NiFe hydrogenase studied by protein film voltammetry. J Am Chem Soc. 2004;126:12162–12172. doi: 10.1021/ja046548d. [DOI] [PubMed] [Google Scholar]
- 15.Ludwig M, et al. Oxygen-tolerant H2 oxidation by membrane-bound [NiFe]-hydrogenases of Ralstonia species: Coping with low-level H2 in air. J Biol Chem. 2009;284:465–477. doi: 10.1074/jbc.M803676200. [DOI] [PubMed] [Google Scholar]
- 16.Lamle SE, Albracht SP, Armstrong FA. The mechanism of activation of a [NiFe]-hydrogenase by electrons, hydrogen, and carbon monoxide. J Am Chem Soc. 2005;127:6595–6604. doi: 10.1021/ja0424934. [DOI] [PubMed] [Google Scholar]
- 17.Pershad HR, et al. Catalytic electron transport in Chromatium vinosum [NiFe]-Hydrogenase: Application of voltammetry in detecting redox- active centers and establishing that hydrogen oxidation is very fast even at potentials close to the reversible H+/H2 value. Biochem. 1999;38:8992–8999. doi: 10.1021/bi990108v. [DOI] [PubMed] [Google Scholar]
- 18.Vincent KA, et al. Electrochemical definitions of O2 sensitivity and oxidative inactivation in hydrogenases. J Am Chem Soc. 2005;127:18179–18189. doi: 10.1021/ja055160v. [DOI] [PubMed] [Google Scholar]
- 19.Burgdorf T, et al. The soluble NAD+-reducing [NiFe]-hydrogenase from Ralstonia eutropha H16 consists of six subunits and can be specifically activated by NADPH. J Bacteriol. 2005;187:3122–3132. doi: 10.1128/JB.187.9.3122-3132.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armstrong FA, et al. Dynamic electrochemical investigations of hydrogen oxidation and production by enzymes and implications for future technology. Chem Soc Rev. 2009;38:36–51. doi: 10.1039/b801144n. [DOI] [PubMed] [Google Scholar]
- 21.Guiral M, Aubert C, Giudici–Orticoni MT. Hydrogen metabolism in the hyperthermophilic bacterium Aquifex aeolicus. Biochem Soc Trans. 2005;33:22–24. doi: 10.1042/BST0330022. [DOI] [PubMed] [Google Scholar]
- 22.Rakhely G, Zhou ZH, Adams MW, Kovacs KL. Biochemical and molecular characterization of the [NiFe] hydrogenase from the hyperthermophilic archaeon, Thermococcus litoralis. Eur J Biochem. 1999;266:1158–1165. doi: 10.1046/j.1432-1327.1999.00969.x. [DOI] [PubMed] [Google Scholar]
- 23.Baker SE, et al. Hydrogen production by a hyperthermophilic membrane-bound hydrogenase in water-soluble nanolipoprotein particles. J Am Chem Soc. 2009;131:7508–7509. doi: 10.1021/ja809251f. [DOI] [PubMed] [Google Scholar]
- 24.Bard A, Faulkner L. Electrochemical Methods: Fundamentals and Applications. New York: Wiley; 2001. [Google Scholar]
- 25.Sander R. Compilation of Henry's law constants for inorganic and organic species of potential importance in environmental chemistry (Version 3) [Accessed May 29, 2009];1999 Available at www.henrys-law.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.