

Abstract
Pancratistatin is a phenanthridone-type natural product isolated from several plants of the Amaryllidaceae family. Its potent antiproliferative, antivascular, antiviral and antiparasitic properties have attracted the attention of synthetic, biological and medicinal chemists. Pancratistatin's low natural availability and complex structure have steered many of these research projects toward the preparation of its simplified synthetic analogs with useful levels of activity. In this work we have developed synthetic chemistry aimed at the preparation of pancratistatin analogs with a truncated cyclitol portion of the molecule. The described synthetic pathways are based on a highly anti-diastereoselective arylcuprate conjugate addition to γ-alkoxy-α,β-enoates and syn-selective azidation at the α-position of ester enolates. Analogs with the formally cleaved C3-C4 bond, and thus containing an open ring C, as well as a compound containing a truncated lactol moiety in lieu of the cyclitol, were prepared. Several of the analogs exhibited weak antiproliferative activity, with the highest potency observed in the case of the lactol analog. From these results implications for the design of future pancratistatin analogs are discussed. Furthermore, the synthetic pathways can be used to construct pancratistatin-mimetic libraries, in which the cyclitol moiety is replaced by other cyclic motifs.
Introduction
The medicinal value of Amaryllidaceae plant extracts has been recognized for a long time. It dates back to at least the fourth century BC, when Hippocrates of Cos used oil from the daffodil Narciclasus poeticus L. for the treatment of cancer.1 In more recent times, more than 100 structurally diverse alkaloids, possessing a wide spectrum of biological activities have been isolated from Amaryllidaceae species.2 Lycorine (Figure 1), shown to possess antitumor and antiviral activity,3 was the first member of this family isolated in 1877.4 Later, the phenanthridone narciclasine attracted considerable interest due to its antineoplastic properties,5 and most recently due to its unique mode of action in glioblastoma cells and high potential in treating various forms of brain cancer.6
Figure 1.

Structures of selected Amaryllidaceae constituents with promising anticancer activities.
Structurally similar phenanthridones (+)-pancratistatin, isolated by Pettit and coworkers,7 and (+)-7-deoxypancratistatin (Figure 1), isolated by Ghosal and coworkers8 have also received considerable attention from both synthetic9 and medicinal chemistry communities.10 Pancratistatin has been found to exhibit strong in vitro antiproliferative effect against the US National Cancer Institute (NCI) panel of cancer cell lines as well as a number of in vivo experimental cancer systems.11 A number of recent reports demonstrate that pancratistatin is specifically toxic to cancer cells as opposed to normal ones, whereas the currently used anticancer drugs, such as taxol and etoposide, are equally toxic to both cell types.12 Powerful antiviral13 and antiparasitic14 activities of pancratistatin constitute a related area of promise. Importantly, it has been noted that pancratistatin and 7-deoxypancratistatin are the only known agents (other than an interferon inducer) to show a significant chemotherapeutic efficacy in a Japanese encephalitis virus-infected mouse model.13 Although less potent, (+)-7-deoxypancratistatin exhibits a better therapeutic index in in vitro antiviral (RNA) assays due to reduced toxicity.13 Unfortunately, studies directed at elucidation of pancratistatin's mode of action have been hampered by its low natural abundance (ca. 0.039% of dry Pancratium littorale bulbs) and, therefore the extremely small quantity of material available from isolation.7 This lack of supply, as well as poor water solubility (< 53 μg/mL), have also plagued the clinical development of pancratistatin either as an anticancer or an antiviral agent. Although the water solubility problem has been addressed by developing phosphate prodrugs, this was accomplished utilizing syntheses from the already poorly available natural product.10o,p
In view of the limited supply and the promising activities associated with these natural products, extensive synthetic studies have been undertaken and several total syntheses of both pancratistatin and its 7-deoxy congener have been reported. An overview of this body of work suggests that these molecules represent formidable synthetic challenges and that they are difficult to construct in a short practical way.9 Therefore, research programs directed at preparation of synthetic analogs with simplified structures have been pursued by a number of laboratories. So far such efforts have not resulted in identification of truncated pancratistatin analogs with comparable levels of biological activities. Thus, Hudlicky and coworkers reported that the reduction in oxygenation of ring A or its bioisosteric substitution with an indole moiety, lead to a significant drop in potency.10c-e Ring B opening, either by cleaving the C10a-C10b bond or removing the amide functionality, also resulted in inactive analogs as reported by Chapleur's and our group, respectively.10i,j Seco-B,C-pancratistatin analogs, synthesized by McNulty and coworkers and containing opened B and C rings, were shown to be inactive.10b In addition, systematic removal of hydroxyl groups in ring C leads to compounds with significantly diminished activity as well. Thus, analogs containing two hydroxyl groups in the cyclitol portion of the molecule were prepared and biologically evaluated by McNulty and Hudlicky's groups, respectively (Figure 2).10c,k,l
Figure 2.

Selected dehydroxylated pancratistatin analogs.
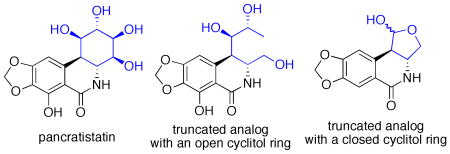
Compound 1 was found to be inactive, 2 is on average 2-3 orders of magnitude less potent than pancratistatin in the NCI 60 cell line screen, while 3 only showed low micromolar growth-inhibitory potency against the murine P388 cell line but was completely inactive against a panel of human cancer cell lines. To date, 7-deoxy-trans-dihydronarciclasine (4), possessing three hydroxyl groups in ring C, represents the only ring C dehydroxylated analog with significant levels of activity.11b,15 Therefore, we wondered whether structural simplification of pancratistatin could be achieved not by removing hydroxyl functionality, as was attempted with the above-mentioned efforts, but by modification of the carbon skeleton of the ring C while still preserving at least three hydroxyl groups. We set out to develop a synthetic pathway to ring C-opened analogs of pancratistatin and 7-deoxypancratistatin shown in Figure 3 (5 and 6). In addition, we considered a possibility of cyclizing either compound 5 or 6 into a lactol (e.g. 7), which would have a considerably truncated, but closed, ring C moiety. In this article, we describe the results of this endeavor and the biological evaluation of the first pancratistatin analogs with the truncated cyclitol portion of the molecule.
Figure 3.

Proposed pancratistatin analogs with a truncated cyclitol ring.
Results and Discussion
Because of the successful application of the Banwell variant of the Bischler-Napierlaski cyclization in this area of research,9,16 we considered the preparation of compounds 5 and 6 from the required carbamates A, which in turn could be obtained from azides B by way of an appropriate functional group interconversion (FGI, Figure 4). Significant literature precedent for syn-selective introduction of nitrogen functionality (including azidation)17 at the α-position of esters boded well for the conversion of esters C to azido esters B with the required stereochemistry. Previously, we have studied arylcuprate addition with γ-alkoxy-α,β-unsaturated esters in detail and derived a reductive-elimination based model to predict the stereochemical outcome in such reactions.18 Therefore, we had a high degree of optimism that we would be able to accomplish a highly anti-selective transformation D → C, utilizing the required arylcuprates. Finally, asymmetric dihydroxylation of commercially available inexpensive sorbate esters has been previously reported by Sharpless and coworkers.19
Figure 4.

Retrosynthetic analysis of pancratistatin analogs with an open cyclitol ring.
Indeed, dihydroxyenoate 8 was obtained as a single R,R-enantiomer when ethyl sorbate was reacted with AD-mix-β as was established by comparing its optical rotation with the literature value.19
Selective protection of the distant hydroxyl as a TBDPS ether, followed by the treatment of the resulting alcohol 9 with MOMCl, gave enoate 10 in good yield. The arylcuprate addition of a Grignard-derived organocopper reagent in the presence of TMSCl gave ester 11 as the exclusive anti-diastereomer. This highly favorable stereochemical outcome was expected on the basis of the previous systematic studies of this process in our laboratory. Syn-selective azidation of 11 was achieved by treatment with KHMDS and trisyl azide, following the precedent of Hanessian and coworkers.17 However, the yields and selectivities were not reproducible and depended on the source of the KHMDS reagent. Therefore, we employed freshly prepared LDA (BuLi + i-Pr2NH) as a base. This gave us somewhat lower syn-selectivity (6:1), but allowed for a facile scale-up and was highly reproducible. Simultaneous reduction of the ester and azide functionalities in 12 with LiAlH4, followed by the treatment of the crude product with methyl chloroformate resulted in carbamate 13. In preparation for the Bischler-Napieralski cyclization to form the lactam ring B, we attempted to replace the acid sensitive protecting groups in carbamate 13 with acetates. Unexpectedly, all efforts to remove the bulky TBDPS group resulted in mixtures of products that appeared to contain no methoxycarbamate as was established by NMR analysis. While this rather curious setback did not seem insurmountable, we needed to prepare more material by scaling-up the synthesis from diol 8. In view of the difficulty of the deprotection of the silyl ether in carbamate 13 we chose not to repeat the same reaction sequence, but rather to modify our protecting group strategy. We thus masked the two hydroxyl groups in diol 8 with an isopropylidene functionality (Scheme 2) to obtain compound 14.
Scheme 2.

Synthesis of Bischler-Napieralski precursors 19a and 19b.
Despite significant conformational constraints, enoate 14 underwent the organocopper addition reactions with cuprates derived from a and b with exclusive anti selectivity. The syn selectivity in the azidation of the enolates derived from 15a and 15b was, however, somewhat lower than in the corresponding transformation 11 → 12. Reduction of the azide and ester functionalities in the epimeric mixtures 16a and 16b, was followed by their conversion to epimeric mixtures of carbamates 17a and 17b as described before. In each of these mixtures the desired major epimer was purified from the minor one after isopropylidene deprotection, to give pure triols 18a and 18b. These were then converted into their corresponding triacetates 19a and 19b in preparation for the Bischler-Napieralski cyclization. While the unambiguous stereochemical assignment of the reaction intermediates would be obtained later (X-ray structure of 23, vide infra), at this stage the identities of the major and minor epimers in 16a were confirmed by conversion to the corresponding lactones 20 and 21 (Figure 5) and NMR analysis of these conformationally constrained cyclic systems. The large (J = 12 Hz) coupling constant between Hα and Hβ in the major lactone 20 and the smaller one (J = 8 Hz) in the minor one indicated the corresponding trans and cis positioning of Hα and Hβ in 20 and 21, in accordance with the assigned stereochemistry.
Figure 5.

Confirmation of azidation stereochemistry.
The commonly utilized Banwell conditions for the Bischler-Napieralski condensation9,16 provided only modest yields of the cyclized products when either 19a or 19b were subjected to the treatment with Tf2O and DMAP in CH2Cl2 (Scheme 3). After many unsuccessful optimization attempts we opted to test whether the recently described use of 2-chloropyridine as a base, in a related cyclization of amides to dihydroisoquinolines,20 would work on our carbamate substrates. Gratifyingly, the reaction yields were raised to respectable levels without a change in regioselectivity. Imidate 22 was the only isolated regioisomer in the cyclization of 19a, while the preponderance of the regioisomer resulting from the attack para to the methylenedioxy functionality was obtained in the cyclization of 19b. The regiochemistries of 22, 24 and 25 were established by 1H NMR analysis of the aromatic portions of the molecules and both of these regiochemical outcomes are consistent with the literature precedent on related systems.9c The imidates were smoothly converted to the corresponding amides with either acidic hydrolysis or treatment with Me3SiI. Notably, the phenolic methyl ether was concomitantly removed to give directly triacetate 27 when Me3SiI was utilized. Alternatively, 27 was also obtained under these conditions from the product of hydrolysis, lactam 26. The acetates were subsequently removed in both lactams 23 and 27 to give the desired pancratistatin truncated analogs 5 and 6 with open cyclitol rings. In addition, the vicinal diol functionality in 6 was smoothly cleaved with NaIO4 to give the corresponding aldehyde that closed to the single anomeric lactol form 7 in DMSO-d6, as was established by 1H NMR analysis.
Scheme 3.

Completion of the synthesis of truncated pancratistatin analogs 5, 6 and 7.
Analogs 5 and 6, their triacetates 23 and 27, as well as lactol 7, were evaluated for antiproliferative activity utilizing HeLa and MCF-7/AZ (models for cervical and breast adenocarcinomas) human cancer cell lines. Due to the unavailability of pancratistatin, the biological evaluation was performed with the use of phenathridone narciclasine as the positive control (see experimental part for its isolation from a plant source). This choice is justified by the similar activity profiles and potencies of narciclasine and pancratistatin.11 In contrast to narciclasine, which exhibited potent cell killing properties, the synthetic compounds manifested useful activity only at high micromolar concentrations. At a concentration of 300 μM compound 5, containing the phenolic hydroxyl reduced the viability of HeLa and MCF7/AZ cells to 70% and 82% respectively (Table 1). Its triacetate 27 exhibited slightly enhanced activity (60 and 64%). It is likely that masking the three hydroxyl groups in 5 improves cell penetration, while the acetates undergo rapid hydrolytic removal within cells. Indeed, under the same testing conditions narciclasine tetraacetate displayed antiproliferative activity which was similar in magnitude to that of narciclasine, as we reported previously.21 Trihydroxy analog 6 or its triacetate were virtually inactive. However, transformation of the ring C-opened analog 6 into lactol 7 gave the highest potency (59 and 48% cell viability) among the synthesized compounds.
Table 1.
Antiproliferative Activity of 5, 6, 7, 23 and 27.
| compound | % cell viabilitya | compound | % cell viabilitya | ||
|---|---|---|---|---|---|
| HeLa | MCF-7/AZ | HeLa | MCF-7/AZ | ||
| 6 | 78 ± 3 | 83 ± 5 | 23 | 89 ± 4 | 92 ± 3 |
| 5 | 70 ± 9 | 82 ± 11 | 27 | 60 ± 3 | 64 ± 6 |
| 7 | 59 ± 8 | 48 ± 3 | narciclasine | 6 ± 1 | 8 ± 2 |
% cell viability after 48 h of treatment with indicated compounds at 300 μM relative to DMSO control ± SD from two independent experiments, each performed in 8 replicates, determined by MTT assay.
While the antiproliferative potencies are rather low, we believe that these results are instructive. The lactam ring B in pancratistatin is strained because of the sp2-character of the four atoms at positions N5, C6, C6a and C10a, which are forced out of planarity by the trans B-C ring fusion. This strain is relieved upon ring C opening, leading to a significant change in the overall geometry. The X-ray structure of triacetate 23 (Figure 6a) illustrates this point. The planarity requirement in ring B forces the C1-C11 and C2-C18 bonds to adopt a quasidiaxial orientation with a torsion angle of 170° for the bond C11-C1-C2-C18. Using the crystallographic structure of 23 and the published X-ray structure of pancratistatin22 for initial geometries, we performed DFT conformational optimizations of ring C-open analog 6, pancratistatin and ring C-closed analog 7 (Figure 6b, 6c and 6d). It can be readily seen that the planar molecular framework of pancratistatin (c) is drastically distorted in analog 6 (b), but not in 7 (d). Thus, the crystallographic and computational data provide a likely explanation for a virtually complete loss of activity upon ring C opening in pancratistatin and retention of a small fraction of antiproliferative effect in a significantly truncated lactol ring C analog.
Figure 6.
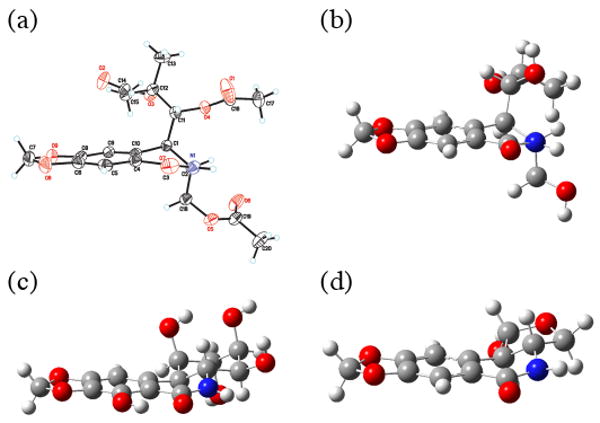
(a) X-ray structure of analog 23 (thermal ellipsoids are shown at 25% probability). DFT optimizations of molecular structures of (b) analog 6, (c) pancratistatin and (d) analog 7.
The results of the current investigation represent a contribution to the previously reported structure-activity relationship data associated with the C-ring of pancratistatin.10b,c,k,l Thus, it appears that not only the number and positioning of the hydroxyl groups are important for the potent anticancer properties of pancratistatin analogues, but also the cyclic nature of the cyclitol portion of the molecule is a key component of the natural product's cytotoxic pharmacophore. The opening of the cyclitol ring increases conformational flexibility and, perhaps more importantly, results in a drastic conformational change that distorts the overall planarity of the carbon framework. It is becoming increasingly apparent that the only structural changes of the ring C that lead to potent compounds are modifications at C1 and C10b positions (Figure 1). This includes the C1-C10b double bond, found in narciclasine and 7-deoxynarciclasine (known as lycoricidine),5b as well as the removal of the C1 hydroxyl, as found in trans-dihydronarciclasine and 7-deoxy-trans-dihydronarciclasine (4, Figure 2). All these compounds possess nanomolar antiproliferative activities toward human cancer cells.5b,11b,15 Furthermore, Pettit and coworkers showed that the antiproliferative potency of C1-benzoate ester of pancratistatin exceeds that of pancratistatin by 10-100 times depending on the cell type.23 However, it is not clear whether the benzoate moiety is an important part of the cytotoxic pharmacophore or it enhances anticancer activity by facilitating the intracellular delivery followed by hydrolysis within cells.
Because of a significant conformational change brought about by the C-ring opening, it is possible that the observed low levels of activity found with the C-ring truncated compounds synthesized in this work arise from an independent mode of action. However, a change in mechanism is a possibility for all truncated pancratistatin analogs, whose structures are significantly different from the natural product. The solution to this potential obstacle will be achieved after the biological mechanism(s) responsible for the anticancer effect of pancratistatin are unravelled, making it possible to evaluate its analogs in a direct mechanism-based assay.
Conclusions
Due to the significant medicinal potential and low natural abundance of the Amaryllidaceae constituent pancratistatin, there is a great demand for synthetic analogs with drastically simplified structures that can be rapidly accessed from commercially available inexpensive starting materials. Synthetic chemistry described in this work leads to a facile preparation of pancratistatin analogs with the truncated cyclitol portion of the molecule. The results of the biological evaluation of these compounds, complemented with DFT computations, suggest that the polyhydroxylated portion of the molecule must remain cyclic for antiproliferative activity to be observed. This also preserves the overall planarity of the carbon framework, which is evidently critical. Since the cellular macromolecular target for pancratistatin is yet to be identified, this study provides further guidance in the quest for structurally simplified potent pancratistatin analogs. Compounds 5 and 6 could thus be used as starting points for the construction of pancratistatin-mimetic libraries, incorporating diverse cyclic moieties in lieu of the six-membered cyclitol ring. The fact that the replacement of the stereochemically complex cyclitol part of the molecule with a simple five-membered lactol results in retention of small levels of activity bodes well for these future endeavors.
Experimental Section
Isolation of Narciclasine
Fresh bulbs (7.5 kg) of Narcissus pseudonarcissus (var. King Alfred) were dried at 40 °C for 3-4 days. The dried material (3 kg) was milled and extracted with 0.1N NaOH according the previously reported procedure.24 The combined basic extract were acidified to pH 2 with 96% H2SO4 and extracted with EtOAc (3 × 15 L). The combined organic extracts were dried (Na2SO4) and evaporated under reduced pressure. The residue (61 g) was separated in aliquots of 15 g and fractionated by silica gel column (CHCl3-i-PrOH 7:3 v/v). The column fractions, which by TLC analysis contained narciclasine, were combined in two groups. These were further purified by preparative TLC, using CHCl3-i-PrOH 7:3 or CHCl3-MeOH 9:1 v/v affording pure narciclasine as amorphous solid (513 mg, 142.5 mg/kg).
(4R,5R,E)-Ethyl 5-(tert-Butyldiphenylsilyloxy)-4-hydroxyhex-2-enoate (9)
Diol 819 (1 g, 5.7 mmol) was dissolved in DMF (7 mL) and cooled to 0 °C. To the mixture were added DMAP (7 mg, 0.57 mmol), imidazole (0.43 g, 6.31 mmol) and tert-butyl(chloro)diphenylsilane (1.7 g, 6.31 mmol). The resulting mixture was stirred overnight at room temperature. The reaction mixture was quenched with water (10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with brine, dried (MgSO4) and evaporated under reduced pressure. The residual oil was presorbed on silica gel and purified by column chromatography (5% - 15% EtOAc/hexanes) to afford compound 9 in 69% yield as a colorless oil; Rf 0.58 (20% EtOAc/hexanes); 1H NMR (CDCl3) δ 7.64 – 7.71 (m, 4 H), 7.37 – 7.44 (m, 6H), 6.90 (dd, J = 15.7, 4.4 Hz, 1H), 6.12 (dd, J = 15.7, 1.7 Hz, 1H), 4.20 (q, J = 7.0 Hz, 2H), 4.12 (m, 1H), 3.82 (m, 1H), 2.59 (d, OH), 1.28 (t, J = 7.0 Hz, 3H), 1.05 (s, 12H); 13C NMR (CDCl3) δ 166.3, 147.1, 135.9, 130.0, 129.7, 127.7, 127.7, 122.2, 75.4, 72.4, 60.4, 53.5, 27.0, 19.8, 19.4, 14.3; HRMS m/z (ESI) calcd for C24H32O4SiNa (M+Na)+ 435.1964, found 435.1968.
(4R,5R,E)-Ethyl 5-(tert-Butyldiphenylsilyloxy)-4-(methoxymethoxy)hex-2-enoate (10)
To a solution of 9 (1.3 g, 3.15 mmol) in CH2Cl2 (60 mL) at 0 °C was added i-Pr2NEt (4 g, 31.5 mmol) followed by the dropwise addition of chloro(methoxy)methane (1.27 g, 15.8 mmol). The resulting solution was stirred for 20 h while it was allowed to warm to room temperature. The reaction mixture was quenched with sat. NH4Cl (30 mL). The aqueous phase was extracted with ether (3 × 50 mL). The combined organic layers were washed with brine, dried (MgSO4) and evaporated to afford dark yellow oil. The crude material was presorbed on silica gel and purified by column chromatography (5% - 15% EtOAc/hexanes) to afford enoate 10 in 76 % yield as a colorless oil; Rf 0.65 (20% EtOAc/hexanes); 1H NMR (CDCl3) δ 7.34 – 7.67 (m, 10H), 7.06 (dd, J = 15.7, 5.0 Hz, 1H), 6.06 (dd, J = 15.7, 1.7 Hz, 1H), 4.45 (d, J = 10.6 Hz, 1H), 4.39 (d, J = 10.6 Hz, 1H), 4.21 (q, J = 7.2 Hz, 2H), 4.16 (m, 1H), 3.96 (m, 1H), 3.18 (s, 1H), 3.30 (t, J = 7.2 Hz, 3H), 1.07 (s, 1H), 1.01 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 145.2, 136.3, 131.0, 128.1, 127.6, 123.1, 95.4, 78.6, 70.5, 60.5, 55.7, 27.1, 18.1, 14.3; HRMS m/z (ESI) calcd for C26H36O5SiNa (M+Na)+ 479.2230, found 479.2230.
General Procedure for Arylcuprate Addition
A few drops of the corresponding aryl bromide saturated solution in THF were added to crushed Mg turnings (7.03 mmol) in THF (10 mL) under nitrogen atmosphere. Once the reaction started the solution warmed up and slightly darkened. The rest of the aryl bromide (7.03 mmol total) was added dropwise to allow a gentle reaction. The reaction mixture was allowed to cool to room temperature and then cannulated to a slurry of CuI (3.52 mmol) in THF (10 mL) at −78 °C. The mixture was stirred at −78 °C for 40 min. Me3SiCl (7.03 mmol) and the enoate (0.703 mmol in 10 mL of THF) were added sequentially dropwise at −78 °C. The yellow-brown suspension was stirred overnight while slowly warming up to room temperature. The reaction mixture was quenched with a mixture of conc. NH4OH and sat. NH4Cl (1 : 9, 30 mL) and extracted with ether (3 × 30 mL). The combined organic layers were washed with brine, dried with MgSO4 and concentrated under reduced pressure. The residue was absorbed on silica gel and purified by column chromatography (5%–30% EtOAc/hexanes) to yield corresponding addition product as an oil.
(3S,4R,5R)-Ethyl-3-(benzo[d][1,3]dioxol-5-yl)-5-(tert-butyldiphenylsilyloxy)-4-(methoxymethoxy)hexanoate (11)
74%, Rf 0.48 (20% EtOAc/hexanes); 1H NMR (CDCl3) δ 7.58 (d, J = 6.9 Hz, 2H), 7.38 (d, J = 6.9 Hz, 2H), 7.34 (m, 6H), 6.66 (m, 3H), 5.87 (d, J = 10.5 Hz, 2H), 4.59 (d, J = 6.9 Hz, 1H), 4.52 (d, J = 6.9 Hz, 1H), 4.00 (q, J = 7.2 Hz, 2H), 3.89 (m, 1H), 2.88 (dd, J = 14.9, 3.85 Hz, 1H), 2.54 (dd, J = 14.9, 9.0 Hz, 1H), 1.12 (t, J = 7.2 Hz, 3H), 1.01 (d, J = 6.9 Hz, 3 H), 0.98 (s,9H); 13C NMR (CDCl3) δ 172.7, 147.6, 146.1, 136.6, 136.0, 135.9, 134.6, 133.8, 129.6, 127.6, 127.4, 121.7, 108.7, 108.1, 100.8, 98.2, 85.5, 70.4, 60.2, 56.1, 42.3, 38.6, 27.1, 19.2, 14.2.; HRMS m/z (ESI) calcd for C33H42O7SiNa (M+Na)+ 601.2595, found 601.2598.
Ethyl (3S)-3-(1,3-Benzodioxol-5-yl)-3-[(4R,5R)-2,2,5-trimethyl-1,3-dioxolan-4-yl]propanoate (15a)
84%, Rf 0.61 (20% EtOAc/hexanes); [α]D22 +40.2 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 6.63 – 6.75 (m, 3H), 5.92 (s, 2H), 4.05 (q, J = 7.2 Hz, 2H)), 3.78 (m, 1H), 3.56 (t, J = 8.3 Hz, 1H), 3.13 (m, 1H), 2.98 (dd, J = 15.7, 5.0 Hz, 1H), 2.58 (dd, J = 15.4, 10.2 Hz, 1H), 1.30 (s, 6H), 1.12 (t, J = 7.2 Hz, 3H), 0.76 (d, J = 6.1 Hz, 3H); 13C NMR (CDCl3) δ 172.2, 147.8, 146.7, 133.7, 121.5, 108.42, 108.4, 101.1, 85.4, 60.3, 46.0, 38.6, 27.5, 27.1, 18.8, 14.2; HRMS m/z (ESI) calcd for C18H24O6Na (M+Na)+ 359.1471, found 359.1471.
Ethyl (3S)-3-(7-Methoxy-1,3-benzodioxol-5-yl)-3-[(4R,5R)-2,2,5-trimethyl-1,3-dio-xolan-4-yl]propanoate (15b)
74%; Rf 0.21 (20% EtOAc/hexanes); [α]D22 +34.8 (c 0.09, CHCl3); 1H NMR (CDCl3) δ 6.40 (d, J = 1.4 Hz, 1H), 6.36 (d, J = 1.1 Hz, 1H), 5.92 (s, 2H), 4.11 – 3.75 (m, 3H), 3.87 (s, 3H), 3.57 (t, J = 8.3 Hz, 1H), 3.09 (m, 1H), 2.96 (dd, J = 15.7, 4.9 Hz, 1H), 2.56 (q, J = 9.9 Hz, 1H), 1.37 (s, 6H), 1.13 (t, J = 7.43 Hz, 3H), 0.78 (d, J = 6.0 Hz, 3H); 13C NMR (CDCl3) δ 172.2, 149.0, 143.5, 134.5, 108.0, 102.0, 101.5, 85.3, 60.4, 56.7, 46.3, 38.6, 27.4, 27.0, 18.7, 14.1; HRMS m/z (ESI) calcd for C19H26O7Na (M + Na)+ 389.1576, found 389.1583.
General Procedure for Azidation with LDA
To a solution of freshly distilled diisopropylamine (8.19 mmol) in 10 mL dry THF at -78 °C was added 7.93 mmol of n-BuLi dropwise under N2 atmosphere. After stirring the solution at this temperature for 45 min, 1.3 mmol of the respective arylesters in THF (7 mL) was added slowly and the resulting solution was warmed up to -30 °C and stirred for 2 h. The solution was cooled back to -78 °C and HMPA (20.8 mmol) was added in one portion followed by the dropwise addition of precooled trisylazide (5.2 mmol). The resulting mixture was stirred at -78 °C for 2 h and then glacial acetic acid (19 mmol) was added. The reaction mixture was allowed to warm up to room temperature and allowed to stir at room temperature for 12 h. 50 mL of sat. NaHCO3 was added and extracted with (3 × 50 mL) CH2Cl2. The combined organic layers were washed with brine (1×30 mL) and water (2×30 mL) dried (MgSO4) and evaporated under reduced pressure. The residual oil was presorbed on silica gel and purified by column chromatography (2% - 5% EtOAc/hexanes) to yield a corresponding azide addition product.
(2R,3R,4R,5R)-Ethyl-2-azido-3-(benzo[d][1,3]dioxol-5-yl)-5-(tert-butyldiphenylsilyloxy)-4-(methoxymethoxy)hexanoate (12)
46%, Rf 0.49 (20% EtOAc/hexanes); 1H NMR (CDCl3) δ 7.23 – 7.58 (m, 10H), 6.62 (m, 3H), 5.86 (d, J = 10.5 Hz, 2H), 4.80 (d, J = 6.4 Hz, 1H), 4.74 (m, 2H), 4.13 (q, J = 7.2 Hz, 2H), 3.72 (m, 3H), 3.43 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H), 0.96 (s, 12H); 13C NMR (CDCl3) δ 169.9, 147.4, 146.9, 136.0, 135.8, 135.8, 133.9, 129.8, 129.7, 129.6, 127.5, 127.3, 123.5, 110.2, 107.9, 101.0, 98.9, 83.4, 69.6, 64.0, 61.7, 56.4, 47.8, 27.1, 20.0, 19.3, 14.3; HRMS m/z (ESI) calcd for C33H41N3O7SiNa (M+Na)+ 642.2610, found 642.2611.
Ethyl (2R + S,3R)-2-azido-3-(1,3-benzodioxol-5-yl)-3-[(4R,5R)-2,2,5-trimethyl-1,3-dioxolan-4-yl]propanoate (16a)
77% (anti:syn = 2.5:1); Rf 0.61 (20% EtOAc/hexanes); [α]22D +27.86 (c 0.05, CHCl3); 1H NMR (CDCl3) δ 6.67 – 6.89 (m, 3H), 5.94 (m, 2H), 4.69 (d, J = 4.9 Hz, 1H), 4.07 – 4.29 (m, 3H), 3.91 (m, 1H), 3.77 (m, 1H), 3.31 (dd, J = 9.6, 3.6 Hz, 1H), 3.17 (dd, J = 9.9, 4.6 Hz, 1H), 1.42 (m, 6H), 1.29 (t, J = 7.2 Hz, 3H), 1.18 (t, J = 7.2 Hz, 3H), 0.77 (d, J = 6.1 Hz, 3H), 0.72 (d, J = 6.1 Hz, 3H); 13C NMR (CDCl3) δ 169.4, 169.3, 148.1, 147.7, 147.5, 131.1, 128.5, 123.0, 122.4, 109.6, 109.1, 108.5, 108.3, 108.2, 101.3, 101.2, 81.6, 65.6, 64.3, 61.9, 61.8, 51.3, 27.5, 27.2, 26.8, 19.1, 19.0, 14.2; HRMS m/z (ESI) calcd for C18H23N3O6Na (M + Na)+ 400.1486, found 400.1485.
Ethyl (2R + S,3R)-2-azido-3-(7-methoxy-1,3-benzodioxol-5-yl)-3-[(4R,5R)-2,2,5-tri-methyl-1,3-dioxolan-4-yl]propanoate (16b)
74% (anti:syn = 1:1); Rf 0.28 (20% EtOAc/hexanes); selected 1H NMR (CDCl3) data for the anti-isomer δ 6.52 (d, J = 1.6 Hz, 1H), 6.40 (d, J = 0.8 Hz, 1H), 5.92 (s, 2H), 4.63 (d, J = 5.2 Hz, 1H), 4.11 – 3.72 (m, 4H), 3.84 (s, 3H), 3.10 (dd, J = 10.5, 5.2 Hz, 1H), 1.39 (s, 3H), 1.38 (s, 3H), 1.15 (t, J = 6.9 Hz, 3H), 0.72 (d, J = 5.8 Hz, 3H); selected 1H NMR (CDCl3) data for the syn-isomer δ 6.46 (m, 2H), 5.90 (m, 2H), 4.28 – 3.91 (m, 5H), 3.87 (s, 3H), 3.26 (dd, J = 9.9, 3.6 Hz, 1H), 1.36 (s, 3H), 1.32 (s, 3H), 1.26 (t, J = 7.7 Hz, 3H), 0.76 (d, J = 6.0 Hz, 3H); selected 13C NMR (CDCl3) δ 169.4, 149.1, 143.6, 135.1, 131.5, 129.2, 109.3, 108.4, 103.5, 102.9, 81.6, 65.6, 64.3, 61.9, 56.8, 51.5, 27.4, 19.3, 14.3; HRMS m/z (ESI) calcd for C19H25N3O7Na (M + Na)+ 430.1590, found 430.1588.
General Procedure for Carbamate Formation
To a suspension of LiAlH4 (2.5 mmol) in anhydrous ether (20 mL) at 0 °C was added a solution of the azide (1 mmol in 5 mL) dropwise. The reaction mixture was stirred for 2.5 h at room temperature. After the completion of the reaction, monitored by TLC, 20 mL of ether was added to the reaction mixture and recooled to 0 °C. Sat. NaSO4 (15 mL) was added slowly and the reaction was stirred for 30 min at room temperature. 15 mL of water was added and extracted with EtOAc (3 × 25 mL). Combined organic layers were washed with brine, dried (MgSO4) and evaporated under reduced pressure to yield the respective aminoalcohol, which was used without further purification. To the amine (1.03 mmol) in dioxane (12 mL) and water (12 mL) were added K2CO3 (2.06 mmol) and methylchloroformate (1.24 mmol) at room temperature. The solution was stirred for 2 h and dioxane was evaporated under vacuum and the aqueous layer was extracted with EtOAc (3 × 10mL). The combined organic layers were washed with brine, dried and evaporated under reduced pressure. The residual oil was presorbed on silica gel and purified by column chromatography (15% - 30% EtOAc/hexanes) to yield corresponding carbamate.
Methyl-(2R,3R,4R,5R)-3-(benzo[d][1,3]dioxol-5-yl)-5-(tert-butyldiphenylsilyloxy)-1-hydroxy-4-(methoxymethoxy)hexan-2-ylcarbamate (13)
65%, Rf 0.52 (60% EtOAc/hexanes); 1H NMR (CDCl3) δ 7.26 – 7.50 (m, 10H), 6.68 (d, J = 8.0 Hz, 1H), 6.49 (m, 2H), 5.90 (d, J = 19.0 Hz, 2H), 4.75 (m, 3H), 4.31 (m, 1H), 3.86 (m, 1H), 3.71(d, J = 10.0 Hz, 1H), 3.64 (s, 3H), 3.43 (m, 1H), 3.39 (s, 3H), 3.35 (m, 1H), 1.00 (m, 3H), 0.96 (s, 9H); 13C NMR (CDCl3) δ 147.8, 146.7, 135.9, 135.8, 134.8, 134.1, 129.5, 127.5, 127.3, 122.9, 109.9, 108.4, 101.1, 98.9, 83.1, 70.1, 64.8, 56.4, 52.3, 46.1, 27.1, 19.9, 19.2; HRMS m/z (ESI) calcd for C33H43NO8SiNa (M+Na)+ 632.2650, found 632.2656.
Methyl N-(1R + S,2R)-2-(1,3-benzodioxol-5-yl)-1-(hydroxymethyl)-2-[(4R,5R)-2,2, 5-trimethyl-1,3-dioxolan-4-yl]ethylcarbamate (17a)
86%; Rf 0.50 (60% EtOAc/hexanes); [α]22D -5.24 (c 0.56, CHCl3); 1H NMR (CDCl3) δ 6.54 – 6.82 (m, 3H), 5.94 (s, 2H), 4.12 (m, 1H), 3.78 (m, 2H), 3.64 (m, 3H). 3.54 (s, 3H), 2.82 (m, 1H), 1.42 (s, 6H), 0.76 (d, J = 5.8 Hz, 3H); 13C NMR (CDCl3) δ 156.7, 148.0, 147.1, 122.3, 122.2, 109.2, 108.6, 108.0, 101.3, 82.7, 64.1, 56.1, 52.3, 50.9, 27.4, 27.1, 18.9; HRMS m/z (ESI) calcd for C18H25NO7Na (M + Na)+ 390.1528, found 390.1529.
Methyl N-(1R + S,2R)-1-(hydroxymethyl)-2-(7-methoxy-1,3-benzodioxol-5-yl)-2-[(4R,5R)-2,2,5-trimethyl-1,3-dioxolan-4-yl]ethylcarbamate (17b)
71%; Rf 0.47 (10% MeOH/CHCl3); 1H NMR (CDCl3) δ 6.37 (m, 2H), 5.93 (s, 2H), 4.07 – 3.41 (m, 13H), 2.84 (m, 1H), 1.38 (m, 6H), 0.73 (d, J = 6.0 Hz, 3H); 13C NMR (CDCl3) δ 156.9, 149.3, 143.7, 134.8, 132.4, 108.4, 108.0, 103.1, 101.6, 83.3, 82.7, 64.0, 56.9, 52.3, 50.9, 27.4, 27.1, 18.7; HRMS m/z (ESI) calcd for C19H27NO8Na (M + Na)+ 420.1634, found 420.1644.
General Procedure for Isopropylidene Deprotection
Dowex-50 acid resin (450 mg) was added to a solution of the corresponding carbamate (1.06 mmol, 420 mg) in MeOH (40 mL). The reaction mixture was refluxed for 10 h. The resin was removed from the mixture and washed with methanol (3 × 30 mL). The combined organic fractions were evaporated under reduced pressure to yield corresponding pure triol as a semisolid.
Methyl N-[(1R,2R,3R,4R)-2-(1,3-benzodioxol-5-yl)-3,4-dihydroxy-1-(hydroxy-methyl)pentyl] carbamate (18a)
56%; Rf 0.26 (60% EtOAc/hexanes); [α]D22 +9.41 (c 0.26, CHCl3); 1H NMR (CDCl3) δ 6.59 – 6.74 (m, 3H), 5.92 (s, 2H), 5.16 (m, 1H), 4.28 (m, 1H), 3.69 (s, 3H), 3.42 (m, 3H), 3.07 (m, 1H). 1.11 (d, J = 5.5 Hz, 3H); 13C NMR (CDCl3) δ 158.8, 148.1, 146.9, 131.6, 122.3, 109.1, 108.8, 101.2, 73.9, 67.0, 64.6, 53.0, 50.0, 21.4; HRMS m/z (ESI) calcd for C15H21NO7Na (M + Na)+ 350.1216, found 350.1217.
Methyl N-[(1R,2R,3R,4R)-3,4-dihydroxy-1-(hydroxymethyl)-2-(7-methoxy-1,3-benzodioxol-5-yl)pentyl]carbamate (18b)
50%; Rf 0.51 (20% MeOH/CHCl3); [α]D22 +9.6 (c 0.062, CHCl3); 1H NMR (CDCl3) δ 6.34 (br s, 2H), 5.94 (s, 2H), 5.09 (m, 1H), 4.29 (m, 1H), 3.88 (s, 3H), 3.72 – 3.30 (m, 6H), 3.68 (s, 3H), 3.08 (m, 1H). 1.13 (m, 3H); 13C NMR (CDCl3) δ 158.8, 149.3, 143.8, 134.5, 132.4, 109.2, 102.5, 101.7, 73.9, 67.0, 64.3, 57.0, 53.0, 50.3, 21.2; HRMS m/z (ESI) calcd for C16H23NO8Na (M + Na)+ 380.1321, found 380.1326.
General Procedure for Acetyl Protection
To a mixture of corresponding triol (0.61 mmol) and DMAP (3 mg, 0.024 mmol) in dry pyridine (5 mL) at 0 °C was added acetic anhydride (1.87 g, 18.44 mmol). The reaction mixture was stirred at room temperature for 12 h and diluted with water (50 mL). The aqueous layer was extracted with ether (5 × 20 mL). The combined organic layers were washed with water (4 × 15 mL), brine, dried (MgSO4), and evaporated under reduced pressure. The oily residue was presorbed on silica gel and purified by column chromatography (2% - 5% MeOH/CHCl3) to yield corresponding triacetate.
(1R,2R,3R)-4-(Acetyloxy)-1-[(1R)-1-(acetyloxy)ethyl]-2-(1,3-benzodioxol-5-yl)-3-[(methoxycarbonyl)amino]butyl acetate (19a)
88%; Rf 0.47 (50% EtOAc/hexanes); [α]D22 +58.33 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 6.76 (d, J = 7.8 Hz, 1H), 6.58 (s, 1H), 6.52 (d, J = 7.8 Hz, 1H), 5.96 (s, 2H), 4.63 (m, 1H), 4.46 (m, 2H), 3.85 (dd, J = 11.0, 5.5 Hz, 1H), 3.65 (m, 3H), 3.19 (dd, J = 11.2, 0.4 Hz, 1H), 2.22 (s, 3H), 2.03 (s, 3H), 1.97 (s, 3H), 1.07 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 170.9, 169.8, 156.8, 148.6, 147.6, 128.6, 121.9, 109.2, 108.5, 101.5, 72.8, 69.4, 65.5, 52.6, 48.7, 46.6, 21.1, 20.8, 16.8; HRMS m/z (ESI) calcd for C21H27NO10Na (M + Na)+ 476.1538, found 476.1533.
(1R,2R,3R)-4-(Acetyloxy)-1-[(1R)-1-(acetyloxy)ethyl]-2-(7-methoxy-1,3-benzodi-oxol-5-yl)-3-[(methoxycarbonyl)amino]butyl acetate (19b)
96%; Rf 0.73 (8% MeOH/CHCl3); [α]D22 +60.0 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 6.26 (d, J = 1.6 Hz, 1H), 6.15 (d, J = 1.4 Hz, 1H), 5.93 (s, 2H), 5.32 (dd, J = 11.3, 1.6 Hz, 1H), 4.62 (qd, J = 6.3, 1.6 Hz, 1H), 4.52 (d, J = 11.0 Hz, 1H), 4.36 (m, Hz, 1H), 3.85 (m, 3H), 3.66 – 3.64 (m, 1H), 3.61 (s, 3H), 3.12 (dd, J = 11.3, 3.0 Hz, 1H), 2.19 (s, 3H), 2.00 (s, 3H), 1.95 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3) δ 170.8, 170.7, 169.7, 156.7, 149.8, 144.0, 135.2, 129.4, 109.1, 102.2, 101.7, 72.9, 69.5, 65.5, 57.2, 52.5, 46.9, 21.1, 20.9, 16.7; HRMS m/z (ESI) calcd for C22H29NO11Na (M + Na)+ 506.1638, found 506.1637.
Confirmation of Azide Stereochemistry
A mixture of azides 16a were subjected to the reaction conditions described in the preparation of compounds 18a,b to form lactones 20 and 21.
(3R,4R,5R)-3-Azido-4-(benzo[d][1,3]dioxol-5-yl)-5-((S)-1-hydroxyethyl)dihydrofuran-2(3H)-one (20)
Rf 0.59 (50% EtOAc/hexanes); [α]D22 +63.57 (c 0.03, CHCl3); 1H NMR (CDCl3) δ 6.68 – 6.73 (m, 3H), 6.02 (s, 2H), 4.41 (d, J = 11.6 Hz, 1H), 4.28 (dd, J = 9.91, 3.03 Hz, 1H), 3.74 (m, 1H), 3.47 (dd, J = 11.6 Hz, 1H), 1.24 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3) δ 171.5, 148.7, 147.9, 128.5, 121.7, 109.0, 107.6, 101.8, 85.6, 66.2, 64.8, 49.3, 19.9; HRMS m/z (ESI) calcd for C13H13N3O5Na 314.0752, found 314.0753.
(3S,4R,5R)-3-Azido-4-(benzo[d][1,3]dioxol-5-yl)-5-((S)-1-hydroxyethyl)dihydrofuran-2(3H)-one (21)
Rf 0.54 (50% EtOAc/hexanes); [α]D22 –62.75 (c 0.005, CHCl3); 1H NMR (CDCl3) δ 6.68 – 6.73 (m, 3H), 5.97 (s, 2H), 4.63 (d, J = 8.3 Hz, 1H), 4.50 (dd, J = 5.23, 2.48 Hz, 1H), 3.90 (m, 1H), 3.73 (dd, J = 8.1, 5.2 Hz, 1H), 1.33 (d, J = 6.6 Hz, 3H); HRMS m/z (ESI) calcd for C13H13N3O5Na 314.0752, found 314.0755.
General Procedure for Imidate Formation
Method 116
A 1.1 M solution of Tf2O (0.677 mL, 0.74 mmol) in anhydrous CH2Cl2 was added over period of 20 min to a solution of a corresponding carbamate (0.15 mmol) and DMAP (54.6 mg, 0.44 mmol) in CH2Cl2 (4.5 ml) at 0 °C. The reaction mixture was left to stir until complete consumption of the carbamate monitored by TLC. On average complete consumption of the carbamate appeared to be between 2 to 6 h. During this period of time the reaction was kept in the ice-bath but no further additions of ice were made. The reaction mixture was then diluted with EtOAc (20 mL) and poured into saturated aqueous NaHCO3. The aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with 0.5 M HCl (1 × 2 mL) and brine, dried over Na2SO4, and concentrated. The crude product was presorbed either on silica gel or TLC plate and separated chromatographically to yield the corresponding imidate as a colorless oil.
Method 220
Trifluoromethanesulfonic anhydride (64 μL, 0.39 mmol, 1.1 equiv) was added over period of 5 min to a solution of a corresponding carbamate (0.350 mmol, 1 equiv) and 2-chloropyridine (36 μL, 0.39 mmol, 1.1 equiv) in CH2Cl2 (1.8 ml) at -78 °C. The reaction mixture was left to stir for 5 min and then was placed in an ice-water bath for 15 min to warm up to 0 °C and stirred at that temperature for 1 h. The mixture was quenched with 1 M NaOH (0.5 mL), diluted with CH2Cl2 (5 mL). The aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (2 mL), dried over anhydrous Na2SO4, and concentrated. The crude product was separated chromatographically on alumina gel (5 - 30% EtOAc/hexanes) to yield a pale yellow fraction containing a corresponding imidate. The combined fractions were concentrated and additionally purified with preparative TLC (60 - 75% EtOAc/hexanes) to yield corresponding pure imidates as a colorless oil.
(1R,2R)-2-(Acetyloxy)-2-(7R,8R)-7-[(acetyloxy)methyl]-5-Methoxy-7,8-dihydro [1,3]dioxolo[4,5-g]isoquinolin-8-yl-1-methylethyl acetate (22)
54% (method 1), 71% (method 2); Rf 0.31 (75% EtOAc/hexanes); -24.0 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 7.13 (s, 1H), 6.49 (s, 1H), 5.98 (s, 2H), 4.95 (dd, J = 7.9, 3.0 Hz, 1H), 4.85 (dd, J = 6.3, 3.0 Hz, 1H), 4.13 (t, J = 5.5 Hz, 1H), 4.03 (td, J = 10.7, 5.2 Hz, 1H), 3.81 (s, 3H), 3.64 (dd, J = 10.7, 7.7 Hz, 1H), 3.00 (d, J = 7.9 Hz, 1H), 2.12 (s, 3H), 2.09 (s, 3H), 1.97 (s, 3H), 1.11 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 170.8, 170.2, 169.9, 160.2, 150.1, 147.6, 130.2, 119.0, 109.3, 105.7, 101.7, 75.1, 69.6, 64.3, 53.8, 53.2, 39.3, 21.3, 20.9, 16.9; HRMS m/z (ESI) calcd for C21H26NO9 (M + H)+ 436.1608, found 436.1602.
Mixture of (1R,2R)-2-(Acetyloxy)-2-(6R,7R)-6-[(acetyloxy)methyl]-4,9-dimethoxy-6,7-dihydro [1,3]dioxolo[4,5-h]isoquinolin-7-yl-1-methylethyl acetate (24) and (1R,2R)-2-(acetyloxy)-2-(7R,8R)-7-[(acetyloxy)methyl]-4,5-dimethoxy-7,8-dihydro[1,3]dioxolo-[4,5-g]isoquinolin-8-yl-1-methylethyl acetate (25)
18%, 1:2 (method 1), 59%, 1:2 (method 2); Rf 0.24 (75% EtOAc/hexanes); 1H NMR of minor regioisomer 24 (CDCl3): δ 6.20 (s, 1H), 6.05 (d, J = 15.1 Hz, 2H), 4.92 - 4.78 (m, 2H), 4.13 -3.95 (m, 2H), 3.89 (s, 3H), 3.85 (s, 3H), 3.73 – 3.59 (m, 1H), 2.95 (d, J = 9.1 Hz, 1H), 2.17 (s, 3H), 2.14 (s, 3H), 1.98 (s, 3H), 1.07 (m, 3H); 1H NMR of major regioisomer 25 (CDCl3): δ 6.25 (s, 1H), 5.97 (d, J = 3.8 Hz, 2H), 4.92 (dd, J = 9.1, 2.2 Hz, 1H), 4.81 (dd, J = 6.0, 2.5 Hz, 1H), 4.13 - 3.95 (m, 2H), 3.92 (s, 3H), 3.82 (s, 3H), 3.73 – 3.59 (m, 1H), 2.95 (d, J = 9.1 Hz, 1H), 2.15 (s, 3H), 2.13 (s, 3H), 1.98 (s, 3H), 1.09 (d, J = 6.0 Hz, 3H); 13C NMR of major regioisomer 25 (CDCl3): δ 170.8, 170.2, 169.9, 160.7, 151.0, 142.2, 138.9, 131.9, 108.5, 105.0, 102.5, 101.8, 74.0, 69.6, 63.8, 60.9, 53.3, 40.4, 21.3, 20.9, 16.8; HRMS m/z (ESI) calcd for C22H28NO10 (M + H)+ 466.1713, found 466.1722.
General Procedure for Conversion of Imidate to Amide
Method 1
A 1.3 M solution of trimethylsilylchloride (45.7 μL, 59.6 μmol) in dry acetonitrile was added to a stirred suspension of imidate (45.1 μmol) and anhydrous potassium iodide (45.1 μmol) in dry acetonitrile (1 mL). The resulting mixture was heated under reflux for 1 h. Water was added (2 mL) and the acetonitrile was evaporated under reduced pressure. The aqueous layer was extracted with EtOAc (3 × 5 mL), and the combined organic layers were washed with an aqueous solution of sodium thiosulphate (5%, 1 × 2 mL) and brine, dried over Na2SO4, and concentrated. The residue was purified by flash column chromatography (60% EtOAc/hexanes) to afford pure product as an oil.
Method 2
The corresponding imidate (82 μmol) was dissolved in THF (2 mL) and 2 M HCl (0.2 mL) and stirred at room temperature for 5 h. The reaction mixture was neutralized with saturated aqueous NaHCO3 and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel to afford pure product as a white solid.
(1R,2R)-2-(Acetyloxy)-2-(7R,8R)-7-[(acetyloxy)methyl]-5-Oxo-5,6,7,8-tetrahydro [1,3]dioxolo[4,5-g]isoquinolin-8-yl-1-methylethyl acetate (23)
84% (method 1), 78% (method 2); Rf 0.17 (75% EtOAc/hexanes); mp 194-196 °C (CH2Cl2/hexane); -40.0 (c 0.025, CHCl3); 1H NMR (CDCl3) δ 7.47 (s, 1H), 6.43 (s, 1H), 6.16 (s, 1H), 6.02 (s, 2H), 5.20 (dd, J = 8.8, 1.9 Hz, 1H), 4.70 (dd, J = 6.3, 2.5 Hz, 1H), 3.98 (dd, J = 10.7, 7.9 Hz, 1H), 3.89 (td, J = 11.3, 6.0 Hz, 1H), 3.67 (dd, J = 11.3, 5.8 Hz, 1H), 3.03 (d, J = 8.8 Hz, 1H), 2.15 (s, 3H), 2.14 (s, 3H), 2.02 (s, 3H), 1.00 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 170.6, 170.3, 170.0, 163.6, 151.5, 148.3, 129.9, 122.8, 108.6, 108.3, 102.1, 74.6, 69.4, 65.5, 50.0, 40.8, 21.4, 20.8, 16.8; HRMS m/z (ESI) calcd for C20H23NO9Na (M + Na)+ 444.1271, found 444.1278.
(1R,2R)-2-(Acetyloxy)-2-(7R,8R)-7-[(acetyloxy)methyl]-4-Methoxy-5-oxo-5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinolin-8-yl-1-methylethyl acetate (26)
83% (method 2); Rf 0.10 (75% EtOAc/hexanes); -62.3 (c 0.061, CHCl3); 1H NMR (CDCl3) δ 6.15 (s, 1H), 6.13 (d, J = 9.9 Hz, 2H), 6.05 (d, J = 5.2 Hz, 1H), 5.20 (dd, J = 9.4, 2.2 Hz, 1H), 4.76 (qd, J = 6.9, 6.6, 1.6, 1.4 Hz, 1H), 3.99 – 3.85 (br m, 2H), 3.90 (s, 3H), 3.64 (m, 1H), 3.00 (d, J = 9.6 Hz, 1H), 2.17 (s, 6H), 2.03 (s, 3H), 1.09 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 170.7, 170.2, 170.1, 161.9, 149.7, 146.3, 136.4, 129.1, 108.3, 105.7, 103.3, 74.2, 69.3, 65.6, 56.6, 50.2, 41.2, 21.5, 20.8, 16.8; HRMS m/z (ESI) calcd for C21H25NO10Na (M + Na)+ 474.1376, found 474.1381.
(1R,2R)-2-(Acetyloxy)-2-(7R,8R)-7-[(acetyloxy)methyl]-4-Hydroxy-5-oxo-5,6,7,8-tetrahydro[1,3]dioxolo[4,5-g]isoquinolin-8-yl-1-methylethyl acetate (27)
95% (method 1); Rf 0.4 (75% EtOAc/hexanes); -61.1 (c 0.085, CHCl3); 1H NMR (CDCl3) δ 12.09 (br s, 1H), 6.28 (m, 1H), 6.07 (s, 1H), 6.03 (d, J = 6.6 Hz, 2H), 5.15 (dd, J = 8.8, 2.2 Hz, 1H), 4.66 (qd, J = 6.3, 6.0, 2.2, 1.9 Hz, 1H), 4.01 (m, 2H), 3.65 (m, 1H), 3.03 (d, J = 9.1 Hz, 1H), 2.16 (s, 3H), 2.14 (s, 3H), 2.04 (s, 3H), 1.10 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3) δ 170.6, 170.4, 170.0, 168.2, 152.3, 146.2, 134.3, 130.0, 106.5, 102.6, 101.7, 74.5, 69.3, 65.2, 50.2, 40.8, 21.3, 20.8, 16.7; HRMS m/z (ESI) calcd for C20H23NO10Na (M + Na)+ 460.1220, found 460.1231.
General procedure for removing the acetyl protecting group
Potassium carbonate (50 mg, 0.36 mmol) was added to a solution of a corresponding protected ring C analog (0.045 mmol) in methanol (5 mL). The resulting suspension was stirred at room temperature until total consumption of the starting material (18 h). The reaction mixture was neutralized with acetic acid and concentrated to off-white residue, which was dissolved in 4:1 CH2Cl2 – MeOH, adsorbed on TLC plate, and separated chromatographically (40% MeOH/CH2Cl2) to yield corresponding pure product as a white powder.
(7R,8R)-8-[(1R,2R)-1,2-Dihydroxypropyl]-7-(hydroxymethyl)-7,8-Dihydro[1,3]di-oxolo[4,5-g]isoquinolin-5(6H)-one (6)
71%; Rf 0.3 (40% MeOH/CH2Cl2); -111.1 (c 0.045, MeOH); 1H NMR (DMSO-d6) δ 8.26 (br s, 1H), 7.21 (s, 1H), 6.92 (s, 1H), 6.03 (s, 2H), 3.83 (t, J = 7.4, Hz, 1H), 3.36 (dd, J = 12.9, 5.5 Hz, 1H), 3.27 (td, J = 10.5, 6.3 Hz, 1H), 3.16-3.04 (m, 3H), 1.01 (d, J = 6.3 Hz, 3H); 13C NMR (DMSO-d6) δ 163.4, 150.2, 146.9, 134.2, 123.6, 110.3, 106.7, 101.9, 74.6, 65.5, 63.3, 52.3, 41.8, 21.2; HRMS m/z (ESI) calcd for C14H17NO6Na (M + Na)+ 318.0954, found 318.0952.
(7R,8R)-8-[(1R,2R)-1,2-Dihydroxypropyl]-4-Hydroxy-7-(hydroxymethyl)-7,8-dihy-dro[1,3]dioxolo[4,5-g]isoquinolin-5(6H)-one (5)
83%; Rf 0.46 (20% MeOH/CH2Cl2); -52.9 (c 0.068, MeOH); 1H NMR (DMSO-d6) δ 8.35 (m, 1H), 6.50 (s, 1H), 6.01 (d, J = 3.3 Hz, 2H), 3.88 (m, 1H), 3.36 (m, 1H), 3.31 (dd, J = 10.5, 6.6 Hz, 1H), 3.14 (td, J = 10.5, 8.3 Hz, 1H), 3.07 (s, 2H), 1.73-1.54 (br s, 4H), 1.02 (d, J = 6.0 Hz, 3H); 13C NMR (DMSO-d6) δ 168.7, 151.7, 145.6, 134.9, 132.6, 107.1, 102.3, 102.1, 74.9, 65.5, 63.0, 52.3, 41.8, 21.2; HRMS m/z (ESI) calcd for C14H17NO7Na (M + Na)+ 334.0903, found 334.0911.
(1R,3aR,10bR)-1-Hydroxy-1,3a,4,10b-tetrahydro[1,3]dioxolo[4,5-g]furo[3,4-c]iso-quinolin-5(3H)-one (7)
To a solution of 6 (42 mg, 0.14 mmol) in methanol (2 mL) was added NaIO4 (36.4 mg, 0.17 mmol) in one portion at 0 °C. The resulting suspension was vigorously stirred for 1 h at room temperature. The precipitate was filtered and solution was concentrated under reduced pressure. The residue was purified by preparative TLC (20% MeOH/CH2Cl2) to afford pure product 7 (28.6 mg, 82%) as a white powder; Rf 0.59 (20% MeOH/CH2Cl2); -11.1 (c 0.018, MeOH); 1H NMR (DMSO-d6) δ 7.83 (s, 1H), 7.33 (s, 1H), 6.80 (s, 1H), 6.65 (br s, 1H), 6.08 (d, J = 8.81 Hz, 2H), 5.07 (t, J = 3.8, Hz, 1H), 4.26 (t, J = 3.8 Hz, 1H), 4.09 (dd, J = 9.1, 3.85 Hz, 1H), 3.88 (d, J = 8.81 Hz, 1H), 3.14 (t, J = 5.8 Hz, 1H); 13C NMR (DMSO-d6) δ 163.2, 151.0, 147.5, 132.3, 121.5, 108.3, 107.1, 104.5, 102.2, 72.8, 55.0, 48.8; HRMS m/z (ESI) calcd for C12H11NO5Na (M + Na)+ 272.0535, found 272.0548.
Cell Culture
Human cervical cancer cell line HeLa (ATCC S3) were cultured in RPMI-1640 (Gibco) and supplemented with 10% FBS (Gibco), 100 mg/L penicillin G, 100 mg/L streptomycin, 1.0 mM sodium pyruvate, 1.5 g/L sodium bicarbonate, and 4.5 g/L glucose (all from Sigma) at 37 °C in a humidified atmosphere with 10% CO2. MCF-7/AZ, a variant of the human mammary carcinoma cells MCF-7, were cultured in a mixture of DMEM and HAMF12 (50/50) (Invitrogen) supplemented with 250 IU/mL penicillin, 100 μg/mL streptomycin, and 10% FBS.
MTT Assay
Cells were transferred to microtiter plates in 100 μL of medium at a concentration of 2 × 104 cells/mL. HeLa and MCF-7/AZ cells were incubated for 24 h before treating with the drugs to allow proper adhesion. Cells were treated with the panel of the test compounds and incubated for 48 h. 20 μL of MTT reagent (5 mg/ mL) was added to each well and incubated further for 2 h. The resulting formazan crystals were dissolved in 100 μL of DMSO, and the OD was determined at a wavelength of 490 nm. The experiments were repeated at least three times for each compound per cell line.
Supplementary Material
Scheme 1.

Synthesis of carbamate 13.
Acknowledgments
This work is supported by the US National Institutes of Health (RR-16480 and CA-135579) under the BRIN/INBRE and AREA programs. A.A.Y. and M.Yu.A. are grateful to NSF/DMR (Grant 0420863) for the acquisition of X-ray single crystal diffractometer and to the Distributed Nanomaterials Characterization Network in the framework of New Mexico NSF EPSCoR Nanoscience initiative. W.A.L.v.O. gratefully acknowledges funding from the Research Office, University of the Witwatersrand, for supporting this collaboration. We thank Professor Tatiana Timofeeva for her kind assistance with X-ray crystallography.
Footnotes
Supporting Information Available: General experimental section, X-ray data of 23, details of the computational work, copies of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Hartwell JL. Lloydia. 1967;30:379. [Google Scholar]
- 2.Martin SF. The Amaryllidaceae alkaloids. In: Brossi AR, editor. The Alkaloids. Vol. 30. Academic Press; New York: 1987. pp. 251–376. [Google Scholar]
- 3.Hoshino O. The Amaryllidaceae alkaloids. In: Cordell GA, editor. The Alkaloids. Vol. 51. Academic Press; London: 1998. pp. 323–376. [Google Scholar]
- 4.Cook JW, Loudon JD. In: The Alkaloids. Manske RHF, Holmes HL, editors. Vol. 2. Academic Press; New York: 1952. p. 331. Chapter 11. [Google Scholar]
- 5.(a) Ceriotti G. Nature. 1967;213:595–596. doi: 10.1038/213595a0. [DOI] [PubMed] [Google Scholar]; (b) Kornienko A, Evidente A. Chem Rev. 2008;108:1982–2014. doi: 10.1021/cr078198u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Ingrassia L, Lefranc F, Dewelle J, Pottier L, Mathieu V, Spiegl-Kreinecker S, Sauvage S, El Yazidi M, Dehoux M, Berger W, Van Quaquebeke E, Kiss R. J Med Chem. 2009;52:1100–1114. doi: 10.1021/jm8013585. [DOI] [PubMed] [Google Scholar]; (b) Lefranc F, Ingrassia L, Van Quaquebeke E, Darro F, Kiss R. Neuro-oncology. 2008;10:1142–1142. [Google Scholar]
- 7.Pettit GR, Gaddamidi V, Cragg GM, Herald DL, Sagawa Y. J Chem Soc Chem Commun. 1984:1693–1694. [Google Scholar]
- 8.Ghosal S, Singh S, Kumar Y, Srivastava RS. Phytochemistry. 1989;28:611–613. [Google Scholar]
- 9.For recent reviews, see: Rinner U, Hudlicky T. Synlett. 2005:365–387.Chapleur Y, Chrétien F, Ibn-Ahmed S, Khaldi M. Curr Org Syn. 2006;3:341–378.Manpadi M, Kornienko A. Org Prep Proc Int. 2008;40:107–161. doi: 10.1080/00304940809458083.
- 10.For recent examples, see: Shnyder SD, Cooper PA, Millington NJ, Gill JH, Bibby MC. J Nat Prod. 2008;71:321–324. doi: 10.1021/np070477p.McNulty J, Nair JJ, Griffin C, Pandey S. J Nat Prod. 2008;71:357–363. doi: 10.1021/np0705460.Rinner U, Hillebrenner HL, Adams DR, Hudlicky T, Pettit GR. Bioorg Med Chem Lett. 2004;14:2911–2915. doi: 10.1016/j.bmcl.2004.03.032.Rinner U, Hudlicky T, Gordon H, Pettit GR. Angew Chem Int Ed. 2004;43:5342–5346. doi: 10.1002/anie.200460218.Hudlicky T, Rinner U, Finn KJ, Ghiviriga I. J Org Chem. 2005;70:3490–3499. doi: 10.1021/jo040292c.Hudlicky T, Moser M, Banfield SC, Rinner U, Chapuis JC, Pettit GR. Can J Chem. 2006;84:1313–1337.Hudlicky T, Rinner U, Gonzales D, Akgun H, Schilling S, Siengalewicz P, Martinot TA, Pettit GR. J Org Chem. 2002;67:8726–8743. doi: 10.1021/jo020129m.Ibn-Ahmed S, Khaldi M, Chrétien F, Chapleur Y. J Org Chem. 2004;69:6722–6731. doi: 10.1021/jo049153l.Chrétien F, Ibn-Ahmed S, Masion A, Chapleur Y. Tetrahedron. 1993;49:7463–7478.Kireev AS, Nadein ON, Agustin VJ, Bush NE, Evidente A, Manpadi M, Ogasawara MA, Rastogi SK, Rogelj S, Shors ST, Kornienko A. J Org Chem. 2006;71:5694–5707. doi: 10.1021/jo0607562.McNulty J, Mao J, Gibe R, Mo R, Wolf S, Pettit GR, Herald DL, Boyd MR. Bioorg Med Chem Lett. 2001;11:169–172. doi: 10.1016/s0960-894x(00)00614-4.McNulty J, Larichev V, Pandey S. Bioorg Med Chem Lett. 2005;15:5315–5318. doi: 10.1016/j.bmcl.2005.08.024.Beijnen JH, Flora KP, Halbert GW, Henrar REC, Slack JA. Br J Cancer. 1995;72:210–218. doi: 10.1038/bjc.1995.305.Torres-Labandeira JJ, Davignon P, Pitha J. J Pharm Sci. 1991;80:384–386. doi: 10.1002/jps.2600800421.Pettit GR, Freeman S, Simpson MJ, Thompson MA, Boyd MR, Williams MD, Pettit GR, III, Doubek DL. Anti-Cancer Drug Des. 1995;10:243–250.Pettit GR, Orr B, Ducki S. Anti-Cancer Drug Des. 2000;15:389–395.Pettit GR, Melody N, Simpson M, Thompson M, Herald DL, Knight JC. J Nat Prod. 2003;66:92–96. doi: 10.1021/np020225i.Pettit GR, Melody N, Herald DL. J Nat Prod. 2004;67:322–327. doi: 10.1021/np030299+.Pettit GR, Melody N. J Nat Prod. 2005;68:207–211. doi: 10.1021/np0304518.Shnyder SD, Cooper PA, Millington NJ, Gill JH, Pettit GR, Bibby MC. Clin Cancer Res. 2005;11(24 Suppl):8971s–8971s.Phung AN, Zannetti MT, Whited G, Fessner WD. Angew Chem Int Ed. 2003;42:4821–4824. doi: 10.1002/anie.200352023.Pettit GR, Melody N, Herald DL, Schmidt JM, Pettit RK, Chapuis JC. Heterocycles. 2002;56:139–155.
- 11.In the National Cancer Institute (NCI) in vitro 60-cell line screen, both pancratistatin and narciclasine displayed double digit nanomolar growth inhibitory potencies (mean GI50 = 91 and 16 nM, respectively). In addition, the differential cytotoxicity profiles of the two natural products were remarkably similar, with the correlation coefficient of 0.9. For details, see: Pettit GR, Gaddamidi V, Herald DL, Singh SB, Cragg GM, Schmidt JM, Boettner FE, Williams M, Sagawa Y. J Nat Prod. 1986;49:995–1002. doi: 10.1021/np50048a005.Pettit GR, Pettit GR, III, Backhaus RA, Boyd MR, Meerow AW. J Nat Prod. 1993;56:1682–1687. doi: 10.1021/np50100a004.
- 12.(a) McLachlan A, Kekre N, McNulty J, Pandey S. Apoptosis. 2005;10:619–630. doi: 10.1007/s10495-005-1896-x. [DOI] [PubMed] [Google Scholar]; (b) Pandey S, Kekre N, Naderi J, McNulty J. Artificial Cells, Blood Substitutes, and Biotechnology. 2005;33:279–295. doi: 10.1081/bio-200066621. [DOI] [PubMed] [Google Scholar]; (c) Kekre N, Griffin C, McNulty J, Pandey S. Cancer Chemother Pharmacol. 2005;56:29–38. doi: 10.1007/s00280-004-0941-8. [DOI] [PubMed] [Google Scholar]
- 13.Gabrielsen B, Monath TP, Huggins JW, Kefauver DF, Pettit GR, Groszek G, Hollingshead M, Kirsi JJ, Shannon WM, Shubert EM, Dare J, Ugarkar B, Ussery MA, Phelan MJ. J Nat Prod. 1992;55:1569–1581. doi: 10.1021/np50089a003. [DOI] [PubMed] [Google Scholar]
- 14.Quarzane-Amara M, Franetich JF, Mazier D, Pettit GR, Meijer L, Doerig C, Desportes-Livage I. Antimicrob Agents Chemother. 2001;45:3409–3415. doi: 10.1128/AAC.45.12.3409-3415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pettit GR, Eastham SA, Melody N, Orr B, Herald DL, McGregor J, Knight JC, Doubek DL, Pettit GR, III, Garner LC, Bell JA. J Nat Prod. 2006;69:7–13. doi: 10.1021/np058068l. [DOI] [PubMed] [Google Scholar]
- 16.Banwell MG, Bissett BD, Busato S, Cowden CJ, Hockless DCR, Holman JW, Read RW, Wu AW. Chem Commun. 1995:2551–2553. [Google Scholar]
- 17.For examples, see: Hanessian S, Wang W, Gai Y. Tetrahedron Lett. 1996;37:7477–7480.Hanessian S, Gai Y, Wang W. Tetrahedron Lett. 1996;37:7473–7476.
- 18.(a) Kireev AS, Manpadi M, Kornienko A. J Org Chem. 2006;71:2630–2640. doi: 10.1021/jo052383v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Manpadi M, Kornienko A. Tetrahedron Lett. 2005;46:4433–4437. [Google Scholar]
- 19.(a) Xu D, Crispino GA, Sharpless KB. J Am Chem Soc. 1992;114:7570–7571. [Google Scholar]; (b) Becker H, Soler MA, Sharpless KB. Tetrahedron. 1995;51:1345–1376. [Google Scholar]
- 20.Movassaghi M, Hill MD. Org Lett. 2008;10:3485–3488. doi: 10.1021/ol801264u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evidente A, Kireev AS, Jenkis AR, Romero AE, Steelant WFA, Van Slambrouck S, Kornienko A. Planta Med. 2009;75:501–507. doi: 10.1055/s-0029-1185340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettit GR, Meng Y, Herald DL, Knight JC, Day JFJ. Nat Prod. 2005;68:1256–1268. doi: 10.1021/np0402367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettit GR, Melody N, Herald DL. J Org Chem. 2001;66:2583. doi: 10.1021/jo000710n. [DOI] [PubMed] [Google Scholar]
- 24.Evidente A. Planta Med. 1991;57:293–295. doi: 10.1055/s-2006-960098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.