

Abstract
Metals are the most commonly encountered protein cofactors, and they play important structural and functional roles in biology. In many cases, metal-binding provides a major driving force for a polypeptide chain to fold. While there are many studies on the structure, stability, and function of metal-binding proteins, there are few studies focusing on the understanding of the kinetic mechanism of metal-induced folding. Herein, the Zn2+-induced folding kinetics of a small zinc-binding protein are studied; the CH11 peptide is derived from the first cysteine/histidine-rich region (CH1 domain) of protein interaction domains of the transcriptional coregulator CBP. Computational design is used to introduce tryptophan and histidine mutations that are structurally consistent with CH11; these mutants are studied using stopped-flow tryptophan fluorescence experiments. The Zn2+-induced CH11 folding kinetics are consistent with two parallel pathways, where the initial binding of Zn2+ occurs at two sites. However, the initially formed Zn2+-bound complexes can proceed either directly to the folded state where zinc adopts a tetrahedral coordination or to an off-pathway, misligated intermediate. While elimination of those ligands responsible for misligation simplifies the folding kinetics, it also leads to a decrease in the zinc binding constant. Therefore, these results suggest why these non-native zinc ligands in the CH11 domain are conserved in several distantly related organisms and that the requirement for function can lead to kinetic frustration in folding. In addition, the loop closure rate of the CH11 peptide is determined based on the proposed model and temperature dependent kinetic measurements.
Keywords: Protein folding, computational design, stopped-flow fluorescence, zinc-binding peptides, folding intermediate
Introduction
The study of the folding mechanism of single domain proteins that lack cofactors has reached an advanced stage wherein the sequence of events along the course of refolding after denaturation begins to be understood.1–10 In several cases, it has been even possible to describe in detail the energetics, position, and structure of the folding transition state ensembles and/or intermediate states involved.2,3,7–9 In contrast, there are far fewer studies on cofactor-induced folding kinetics, even though a large number of proteins require cofactors for proper folding and/or function.11,12 While the effect of cofactor-binding on the structure and stability may differ for different proteins, many polypeptides can only attain a well-folded structure when associated with their respective cofactors. For example, cytochrome c, an important electron transfer protein, cannot fold without incorporation of its heme cofactor.12 Similarly, many small metal-binding peptides, such as the zinc finger motif,13,14 also require their respective cofactors to maintain the integrity of their native fold. For such systems, cofactor-binding thus provides not only stabilization but also a major driving force directing the folding of an otherwise unstructured protein to a well-defined structure. Nonetheless, the prerequisite of a cofactor for folding may complicate the folding free-energy landscape and thus the kinetic mechanism,15 because cofactor-binding often requires the formation of a precisely defined cofactor-binding environment in the native state. In addition, a protein may also transiently associate with a cofactor in a non-native fashion, leading to kinetic frustration in folding. For example, cytochrome c exhibits rather complex folding kinetics arising from non-native iron ligation during or prior to folding.16–20 Herein, the folding of designed variants of a small Zn2+-binding protein are examined in an effort to probe the roles of native and non-native interactions in the folding kinetics of metal-binding proteins.
Metal ions, such as Zn2+, Mg2+ and Ca2+, are among the most commonly encountered cofactors. Metal-binding not only plays many important structural and functional roles in cells,21 but it is also increasingly used in de novo protein designs to generate or stabilize a specific fold.22–32 For example, when coordinated with a metal ion, polypeptides composed of several heptad repeats are observed to undergo a transition from a disordered conformation to an α-helical coiled-coil structure.24,32 Despite the increase in the number of metal-binding peptides (or domains) designed and discovered in recent years, studies on metal-induced folding kinetics are scarce.25,33–35 Moreover, small miniproteins have led to advances in our understanding of elementary events in protein folding and detailed comparisons between theory and experiment.36–39 Similarly, it is useful to identify and develop small cofactor-containing miniproteins that lend themselves to kinetic folding studies. To provide further insight into the understanding of the mechanism of metal-mediated protein folding processes, herein we have carried out detailed stopped-flow studies of the Zn2+-induced folding kinetics of computationally-designed variants of a zinc-binding motif, the CH11 peptide.
The CH11 peptide (sequence: 1 EVRACSLPHC-11 RTMKNVLNHM-21 THCQAGK) is a highly conserved motif found in the first cysteine/histidine-rich (Cys/His-rich) region (CH1 domain) of protein interaction domains of the transcriptional coregulator CBP (CREB binding protein).40 NMR studies have revealed that the CH11 peptide binds a single Zn2+ ion through coordination with the side chain of C5, C10, H19, and C23 residues, and that the folded structure is composed of a 310-helix and a short α-helical stretch packing around the zinc (Figure 1). The CH11 peptide has also been termed as CHANCE finger (or a Cys/His-rich peptide exhibiting an unexpected conformational ensemble) as it constitutes a new protein fold.40 Previous studies40, 41 have shown that this CHANCE fold is not only thermally stable (no substantial secondary structural changes occur up to 85 °C as judged by CD), but is also tolerant to both multiple-alanine (Ala) mutations and changes in the sequence spacing of the zinc-ligating ligands. In addition, it is found that the two non-native ligands, especially His22, are conserved in the CH11 sequence in several distantly related organisms,40 suggesting that they may be functionally important, for example, by facilitating ligand binding via non-native interactions. However, such interactions are expected to occur at the expense of introducing frustration in the folding kinetics. Thus, the advantages of studying the folding kinetics of the CH11 peptide are multi-fold: (a) the robustness of the peptide sequence renders multiple mutations feasible, a necessity for elucidating the folding mechanism; (b) the small size of the peptide also makes it feasible to achieve a mechanistic description of the folding process; (c) it allows one to examine the kinetic role of those non-native ligands, thus providing a rationalization of their possible functional role; and (d) because of its small size, this miniprotein is amenable and should provide a rich model system for further computer simulation studies.
Figure 1.

The NMR structures of CH11 Zn-binding domain (PDB code: 1liq).40 Zn coordinating residues (gray) and residues of triple mutant L7W/H9V/H22I (magenta) are shown.
To study the zinc-induced folding process of the CH11 peptide, it is useful to introduce a probe that is sensitive to local structure and to mutate potential nonnative ligands, H9 and H22, while retaining the parent structure. For the CH11 variants studied herein, a single tryptophan (Trp) residue was introduced to serve as a fluorescent probe of structure, since both cysteine (Cys) and histidine (His) side chains are efficient quenchers of Trp fluorescence.42 Though the peptide is tolerant of multiple alanine mutations, mutations involving larger residues may not be compatible with folding to the native structure. Computational design methods facilitate the identification of viable mutations. Four structurally-consistent mutants (L7W, T21W, L7W/H9V, and L7W/H9V/H22I) were designed using a statistical, computationally-assisted design strategy (SCADS).43,44 The folding process was initiated by mixing a Zn2+ solution with a CH11 peptide solution using a stopped-flow technique, and the folding kinetics were monitored by following changes in Trp fluorescence intensity. The Zn2+-induced folding kinetics of the CH11 peptide are complex and involve at least three kinetic phases. Based on extensive concentration and temperature dependent studies, a model that is capable of describing the observed stopped-flow kinetics of these mutants is proposed. This model suggests that the binding of Zn2+ to the unfolded CH11 peptide involves parallel pathways and that the initially formed Zn2+-bound peptide can proceed either directly to the native state or to an off-pathway misligated state.
Results
Identification of Trp-tag sites
Native fluorescence, especially that arising from Trp, is often used in protein folding and binding studies. However, the native sequence of the CH11 peptide does not contain a suitable fluorophore for monitoring folding or cofactor binding. Therefore, we have employed a statistical computational design method (i.e., SCADS) to identify Trp-containing mutants consistent with the known structure of CH11.40 While the calculations suggest that several sites may be tolerant toward Trp-mutation, two single mutants, L7W and T21W, were targeted because in both cases the Trp residue is in proximity to two zinc ligands: C5/C10 for L7W and H19/C23 for T21W. The probabilities of these two Trp residues at their respective positions are shown in Figure 2. As shown in the predicted model structures (Figure 2), Trp7 in L7W is located close to the metal binding pocket, thus, it is expected to be sensitive to Zn2+-ligand coordination. On the other hand, while the side chain of Trp21 in T21W is highly exposed to solvent (Figure 2), it is near the zinc-binding site and the location (i.e., sandwiched between H19 and C23) makes it a good candidate for the observation of local environmental changes associated with the transition from a heterogeneous ensemble of conformations in the unfolded state to a well organized structure upon folding.
Figure 2.

Site-specific amino acid probability profiles for introduction of Trp-mutation on L7 and T21 (left) and His-replacement for H9 and H22 (right). Teff = 0.5 kcal/mol. For H9, probabilities at two different sampling temperatures of 2.0 and 0.5 kcal/mol are presented with gray squares and red solid bars, respectively. Mutated residue appears in yellow. Zn-coordinating residues (gray) and residues of triple mutant L7W/H9V/H22I (magenta) are shown.
Identification of mutations for His9 and His22
To elucidate the roles of the two non-binding His residues (i.e., H9 and H22) in CH11 folding, the site-specific amino acid probability profiles for sites 9 and 22 were calculated. At each position, 19 amino acids were permitted (i.e., Cys was excluded), and the modeled structure of the mutant L7W was used. As shown (Figure 2), glutamic acid (Glu) shows slightly higher probability for site 9 than several other amino acids at Teff = 2 kcal/mol. However, at lower energies (Teff = 0.5 kcal/mol), the aliphatic nonpolar amino acid valine (Val) is the most probable amino acid for this site due to the stabilizing short ranged van der Waals interactions. With the goal of testing the function of metal-reactive His at position 9, it would be desirable to replace His with an amino acid that is neutral and unlikely to coordinate the metal ion. Thus, Val was identified to replace His9, resulting in the double mutant L7W/H9V.
Further calculations were performed to replace the His22 based on L7W/H9V. As shown (Figure 2), isoleucine (Ile) and Val are highly probable amino acids, in addition to His, and have similar average conformational energies as compared to that of His. However, taking into consideration that Ile has a higher helical propensity than Val for an α-helical motif,45 Ile was selected to substitute His22, resulting in the triple mutant L7W/H9V/H22I.
CD studies
The CD spectra of the CH11 mutants in aqueous solution, as that shown in Figure S1 (Supporting Information) for apo L7W, show characteristics of unfolded peptides. Addition of ZnCl2 to the peptide solution, however, leads to a distinct change in the CD spectrum: the development of the bands around 208 and 222 nm, respectively, consistent with the formation of a partial helical structure (Supporting Information, Figure S1). These results are in agreement with the study of Sharpe et al.40 who have shown that upon association with Zn2+, the CH11 peptide folds into a structure that contains a short 310-helix and a short α-helical stretch. Addition of 5 mM EDTA into the peptide-zinc solution results in a loss of the helical CD signal (data not shown) due to the chelation of Zn2+ by EDTA,46 further demonstrating the important structural role of Zn2+ in the folding of these CH11 mutants. In addition, one-dimensional (1D) proton NMR measurements also indicate that the zinc-bound peptide adopts a folded conformation (see Figure S2 in Supporting Information for an example).
Moreover, the CD spectra of the CH11 mutants considered here indicate that the helical content of each peptide is somewhat different. This is likely due to a combination of effects arising from the difference in helix-forming propensities of different amino acids45 and/or structural constrains induced by the presence or absence of H9 and/or H22.47 Indeed, Sharpeet al. 40 have shown that the mean residual helicity of the CH11 peptide increases when 15 native residues are replaced with Ala.
Fluorescence titrations
The Trp fluorescence intensity of these CH11 mutants increases upon association with Zn2+ and is therefore used to determine their apparent Zn2+-binding constants. As an example, the fluorescence spectra of L7W obtained at various Zn2+ concentrations are shown (Figure 3a). Interestingly, the fluorescence spectra of all mutants exhibit a common maximum centered at 354 ± 1 nm that is independent of the concentration of Zn2+, indicating that the Trp side chain is largely exposed to solvent in both the folded and unfolded states. Thus, the increase in Trp fluorescence upon Zn2+ binding must arise from an alleviation of intramolecular quenching of the Trp fluorescence by various amino acids,42,48–50 most likely Cys and His residues in the current case. The study of Chen et al.42 has shown that Cys and protonated His are among the strongest quenchers of Trp fluorescence existing in natural proteins. As described above, the single Trp in all of these CH11 mutants is deliberately introduced through design at a position that is in close proximity to, at least, a pair of putative zinc coordinating residues: C5/C10 or H19/C23. Therefore, it is expected that the Trp fluorescence of these peptides increases upon association with Zn2+ because coordination with a metal ion greatly reduces the quenching efficiency of these residues.48
Figure 3.

(a) Representative Trp fluorescence spectra of L7W at various Zn2+ concentrations, as indicated. The peptide concentration was 10 μM (in 10 mM Tris buffer, pH 6.5). (b) Integrated area of the Trp fluorescence of CH11 mutants versus Zn2+ concentration. Lines are fits of these data to the binding model described in the text.
As indicated (Figure 3b), the total Trp fluorescence intensity (i.e., the integrated area of the fluorescence spectrum) of these CH11 mutants as a function of Zn2+ concentration can be described by a 1:1 binding model. The apparent dissociation constants (Kd) determined from the best fit of these data to this model (i.e., eqs. 1 and 4) are Kd (L7W) = 3.8 ± 0.5 μM, Kd (T21W) = 0.52 ± 0.35 μM, and Kd (L7W/H9V) = 4.6 ± 0.8 μM. Because the triple mutant L7W/H9V/H22I was found to form aggregates at relatively high Zn2+ concentrations, we did not attempt to determine its Kd.
Stopped-flow kinetics
The Zn2+-induced folding kinetics of these CH11 mutants were investigated by a stopped-flow fluorescence technique.51 Consistent with the equilibrium measurements, rapid mixing of a Zn2+ solution with a peptide solution at pH 6.5 leads to the Trp fluorescence to increase as a function of time. However, the stopped-flow kinetics thus obtained for these mutants are quite complex. As shown (Figure 4a), the stopped-flow traces of L7W, T21W, and L7W/H9V are adequately described by a tri-exponential function with well-separated rate constants (Table 1). While such phenomenological kinetic analyses do not provide molecular insights into the Zn2+-induced folding mechanism of the CH11 peptide, they do show that the CH11 folding is not a simple two-state cooperative process. This is in marked contrast to results obtained for other Zn2+-binding peptides,35, 52 which only show pseudo-first-order stopped-flow kinetics even under different experimental conditions (e.g., different zinc concentrations and pH values). In addition, concentration dependent measurements show that the rate constant of the initial or fastest kinetic phase (i.e., k1obs in Table 1) is linearly proportional to the Zn2+ concentration (Figure 4b), typical of a bimolecular reaction taking place under pseudo-first-order reaction conditions. Therefore, we attribute this kinetic phase to the initial binding of Zn2+ to the peptide. Moreover, the second-order Zn2+-binding rate constant for L7W and T21W is determined to be about 1.8 × 106 M−1 s−1, which is comparable to that observed for other zinc-binding proteins.52–55 To further verify this assignment, additional stopped-flow mixing experiments were carried out at different pH conditions. As expected (Figure 5), taking T21W as an example, the rate of the first stopped-flow kinetic phase increases approximately 4 times when the pH of the final mixed solution is increased from 6.5 to 7.5, due to an increase of the population of deprotonated ligands (especially deprotonated His). Interestingly, the Zn2+-induced folding kinetics of L7W/H9V/H22I can be well described by a bi-exponential function (Figure 6), suggesting that replacement of H9 and H22 with non-ligating amino acids simplifies the folding process.
Figure 4.

(a) Representative stopped-flow kinetics of L7W, T21W, and L7W/H9V (these data have been offset for easy comparison) at 15 °C and pH 6.5, which were initiated by mixing equal volumes of a peptide solution and a ZnCl2 solution. The final peptide and Zn2+ concentrations were 10 μM and 0.15 mM, respectively. Smooth lines represent the best fits to the following tri-exponential function, F(t) = B − C × [A1%·exp(−k1obst) + A2%·exp(−k2obst) + A3%·exp(−k3obst)], and the resultant best-fit parameters are listed in Table 1. (b) The observed rate constant of the first or fastest kinetic phase (i.e., k1obs) versus Zn2+ concentration. Linear regressions to these data yield those straight lines with the following slopes: 1.8×106 M−1 s−1 (L7W), 1.8×106 M−1 s−1 (T21W), and 1.2×106 M−1 s−1 (L7W/H9V).
Table 1.
Phenomenological rate constant (kiobs) and relative amplitude (Ai%) of each kinetics phase obtained by fitting the stopped-flow kinetics presented in Figure 4a and Figure 5 to a tri-exponential function.
| k1obs | k2obs | k3obs | A1% | A2% | A3% | |
|---|---|---|---|---|---|---|
| L7W (pH 6.5) | 282 ± 29 | 11 ± 1 | 2.0 ± 0.1 | 56 ± 5 | 10 ± 2 | 34 ± 2 |
| T21W (pH 6.5) | 217 ± 10 | 25 ± 7 | 1.4 ± 0.1 | 49 ± 6 | 31 ± 4 | 20 ± 2 |
| L7W/H9V (pH 6.5) | 174 ± 10 | 8 ± 1 | 2.2 ± 0.6 | 31 ± 1 | 11 ± 2 | 58 ± 3 |
| T21W (pH 7.5) | 956 ± 50 | 39 ± 3 | 1.3 ± 0.2 | 54 ± 6 | 30 ± 4 | 16 ± 2 |
Figure 5.

Stopped-flow kinetics of T21W obtained at pH 6.5 and 7.5, as indicated. These data have been offset for easy comparison.
Figure 6.

Stopped-flow kinetics obtained by mixing equal volumes of a L7W/H9V/H22I peptide solution and a ZnCl2 solution at 15 °C. The final peptide and Zn2+ concentrations were 10 μM and 0.15 mM, respectively. The smooth line is the best fit of these data to a bi-exponential function, and the resultant rate constants are k1obs = 92 s−1 and k2obs = 6.8 s−1.
Finally, temperature dependent measurements were carried out in the temperature range of 5 to 30 °C to further characterize the apparent activation energy (Ea) encountered by each kinetic phase. As shown (Figure 7 and Table 2), all the rate constants show Arrhenius temperature dependence with an Ea ranging from 6 – 14 kcal/mol.
Figure 7.

Arrhenius plots of the rate constants obtained by fitting the stopped-flow kinetics of the CH11 mutants (as indicated) to the tri-exponential function discussed in Figure 4 caption. Lines are fits to the Arrhenius equation, , where is the apparent activation energy for kinetic phase i (i = 1, 2, or 3) and its value is listed in Table 2.
Table 2.
Apparent activation energy of each stopped-flow kinetic phase.
| L7W | T21W | L7W/H9V | |
|---|---|---|---|
| (kcal/mol) | 6.2±0.5 | 7.1±1.7 | 8.4±1.5 |
| (kcal/mol) | 14.1±1.2 | 10.2±0.5 | 12.8±0.3 |
| (kcal/mol) | 9.3±0.6 | 12.9±0.3 | 12.0±1.5 |
Discussion
The CH11 peptide is a novel zinc-binding motif found in the first Cys/His-rich region of CREB binding protein.40 It is known that this peptide binds to zinc through a consensus CCHC motif (i.e., Cys-X4-Cys-X8-His-X3-Cys wherein Cys and His residues are the ligands for Zn2+) and forms a thermostable, well-defined tertiary structure that is extremely tolerant to sequence mutations at non-ligating positions.40,41 In other words, Zn2+-binding provides a major driving force for CH11 folding. Therefore, this peptide provides an ideal model system for understanding the mechanism of metal-induced protein folding processes.
Since the native sequence of CH11 peptide does not contain a suitable fluorophore (e.g., Trp) that can be used to follow the kinetic process of binding or folding using fluorescence-based techniques, we have employed a statistical computational design method43,44 to identify structurally consistent, Trp-containing mutants of CH11. First, two single mutants, L7W and T21W, were identified as the best targets because in both cases the Trp residue is sandwiched between two zinc ligands, i.e., C5/C10 for L7W and H19/C23 for T21W. Thus, their fluorescence is likely sensitive to Zn2+-binding. Second, a double mutant, L7W/H9V, and a triple mutant, L7W/H9V/H22I, were designed to investigate the role of the non-ligating His residues (i.e., His9 and His22) in the kinetics of CH11 folding. Indeed, both equilibrium and stopped-flow fluorescence measurements show that upon coordination with Zn2+ the Trp fluorescence of these mutants increases significantly, indicative of the sensitivity of this fluorescent probe to metal-induced folding processes. Furthermore, the structural integrity of these mutants was verified by low-resolution methods (i.e., CD and 1D NMR).
A simple sequential model is inadequate to describe the folding mechanism of the CH11 peptide
Compared to other metal-binding motifs,34,35,54–56 these CH11 mutants exhibit rather complex and slow cofactor-induced folding kinetics. As shown (Figure 4a), the stopped-flow kinetics of L7W consist of (at least) three exponential phases, indicative of a non-two-state folding scenario. While this result is not surprising, it is nevertheless interesting as many proteins of similar size that lack cofactors have been shown to fold in a two-state manner37, 57–59 and on a much faster time scale.37, 60–62 Therefore, the slower and more complex folding kinetics of L7W must arise from the fact that the folding of CH11 necessitates Zn2+ ligation in a tetrahedral geometry.
The fastest phase in the stopped-flow kinetics of L7W is characteristic of a bimolecular reaction, as its rate constant (i.e., k1obs in Table 1) depends linearly on the Zn2+ concentration (Figure 4b). Therefore, we attribute this kinetic phase to the initial association of the peptide with zinc wherein a Zn2+-peptide complex (I1) is formed. Given the fact that the four native ligands in CH11 are segregated into two pairs, one (C5 and C10) close to the N-terminus and the other (C23 and H19) close to the C-terminus, it is likely that only two ligands in I1 are ligated. Indeed, Bombarda et al.35 have recently shown that the first step in the Zn2+-induced folding kinetics of the distal CCHC finger motif of the HIV-1 nucleocapsid protein corresponds to the formation of a bidentate state wherein only C36 and H44 are coordinated with the cofactor. Therefore, the simplest interpretation of the tri-exponential stopped-flow fluorescence kinetics of L7W is that this initially formed Zn2+-peptide complex, I1, further evolves to produce the final folded state (F) via a sequential mechanism involving an on-pathway folding intermediate (I2), as indicated in Scheme 1. In fact, zinc coordination has been proposed to proceed in an intrinsically multistep and sequential manner for several peptides.35, 54, 63 While Scheme 1 can indeed fit the stopped-flow kinetics of L7W obtained at different Zn2+ concentrations, the microscopic rate constants recovered from the best fit (Supporting Information, Table S1) however give rise to an equilibrium constant that is not overwhelmingly favorable for the folded state. Instead, this model indicates that the folding intermediate states are significantly populated at equilibrium (e.g., [U]:[I1]:[I2]: [F] = 1:9:9:14 at 300 μM Zn2+), which is inconsistent with previous40 and current equilibrium results (Figure 3b). This inconsistency suggests that a simple sequential model, such as Scheme 1, is inadequate to describe the Zn2+-induced folding mechanism of the CH11 peptide.
Scheme 1.

A sequential folding mechanism involving an on-pathway folding intermediate.
CH11 folding involves parallel pathways
The inadequacy of Scheme 1 in describing the folding mechanism of the CH11 peptide promoted us to design and study a second mutant (i.e., T21W) wherein the Trp is placed near the second group of Zn2+ ligands, H19 and C23. The rationale is to explore whether or not the Zn2+-induced CH11 folding involves parallel pathways. In principle, the initial binding of Zn2+ to the peptide could occur at both the N- and C-terminal regions, leading to the formation of N-Zn2+ and C-Zn2+ (Figure 8). Since Trp fluorescence quenching by various amino acids is strongly distance dependent, we expect that both L7W and T21W will exhibit similar fluorescence intensity changes upon the initial binding of Zn2+ if both N-Zn2+ and C-Zn2+ are formed. Indeed, the relative amplitude of the first stopped-flow kinetic phase of T21W (Table 1), arising from the initial Zn2+-peptide association, is 49%, similar to that (i.e., 56%) of L7W. Taken together, these results thus suggest that the Zn2+-induced CH11 folding involves at least two parallel pathways, as shown in Scheme 2, which represents the simplest parallel folding mechanism wherein N-Zn2+ (I1) and C-Zn2+ (I2) are directly converted to the folded state. Similar to Scheme 1, this folding mechanism possesses sufficient number of (fitting) parameters that can be adjusted to fit the observed stopped-flow kinetics of both L7W and T21W. However, the microscopic rate constants recovered from the best fits (Supporting Information, Table S2) suggest that this folding scheme is also unlikely to be correct. For example, the values of both k3 and k−3 for T21W nearly approach zero, suggesting that T21W can only fold to its native state via I2, whereas in the case of L7W k3 is comparable to k4. While it is possible that the folding mechanism of T21W is different from that of L7W, it seems more plausible that a more complicated kinetic scheme is required.
Figure 8.

Two putative Zn2+-peptide complexes formed after the initial binding of zinc, where H9 and H22 are the two non-native ligands which may also participate in binding.
Scheme 2.

A parallel folding mechanism involving two on-pathway folding intermediates.
CH11 folding involves a misligated intermediate
Since the CH11 peptide contains two ‘extra’ zinc ligands (i.e., H9 and H22), it is therefore possible that its complex folding kinetics upon association with zinc arise from the formation of a misligated state involving either H9 or H22 or both. To test this possibility, we have further studied a double mutant, L7W/H9V, and a triple mutant, L7W/H9V/H22I. If misligation involving both H9 and H22 indeed occurs during CH11 folding, we would expect that only the stopped-flow kinetics of the triple mutant, wherein all non-native zinc ligands are replaced by non-ligating amino acids, become simpler. Indeed, the Zn2+-induced folding kinetics of L7W/H9V remain tri-exponential (Figure 4a), whereas those of L7W/H9V/H22I can be well described by a bi-exponential function (Figure 6). Taken together, these results hence support the idea that the Zn2+-induced CH11 folding process involves misligation of Zn2+ to H9 and/or H22.
While other mechanisms are possible, Scheme 3 represents the simplest scenario wherein such a misligated state (i.e., I3) is populated. As shown (Supporting Information, Figure S3), Scheme 3 not only adequately fits the stopped-flow kinetics of L7W and T21W obtained under different conditions, but also yields physically meaningful fitting parameters (Table 3). For example, the values of k1 and k2 are comparable for both mutants, consistent with the aforementioned picture that the initial binding of Zn2+ to the peptide proceeds in two parallel pathways wherein N-Zn2+ and C-Zn2+ are formed. In addition, the overall bimolecular rate constant for zinc binding is determined to be ~1.5 × 106 M−1 s−1 (i.e., the sum of k1 and k2 in Table 3), which is similar to that determined according to the Eigen-Wilkins mechanism (i.e., 5 × 106 M−1 s−1 for Zn2+ ligation).54 Furthermore, both k5 and k6 are larger than k−5 and k−6, indicating that I3 is only transiently populated (same as I1 and I2). More importantly, the overall equilibrium constant for each peptide (i.e., Kd (L7W) = 20.8 μM, Kd (T21W) = 3.2 μM, and Kd (L7W/H9V) = 30.3 μM) calculated based on the recovered microscopic rate constants is comparable to that determined from the equilibrium titration experiment (Figure 3b), which assumes a simpler binding model.
Scheme 3.

A parallel folding mechanism involving two on-pathway folding intermediates and also a misligated state (I3).
Table 3.
Microscopic rate constants (ki) and relative fluorescence intensities (Q) obtained from globally fitting the stopped-flow kinetics obtained at different Zn2+ concentrations for each mutant to Scheme-3 or Scheme-2 (for L7W/H9V/H22I only).
| L7W | T21W | L7W/H9V | L7W/H9V/H22I | |
|---|---|---|---|---|
| k1 (M−1 s−1) | (5.8 ± 0.5) × 105 | (6.2 ± 1.2) × 105 | (2.0 ± 0.4)× 105 | (2.4 ± 0.6) ×105 |
| k−1 (s−1) | 76 ± 10 | 49 ± 21 | 77 ± 4 | 5 ± 3 |
| k2 (M−1 s−1) | (8.6 ± 0.4) × 105 | (8.5 ± 1.6) × 105 | (6.9 ± 0.7) × 105 | (3.5 ± 0.2) ×105 |
| k−2 (s−1) | 56 ± 2 | 12 ± 1 | 156 ± 10 | 0.2 ± 0.1 |
| k3 (s−1) | 2.6 ± 0.8 | 3.0 ± 0.9 | 7.9 ± 3 | 3.9 ± 0.5 |
| k−3 (s−1) | 0.1 ± 0.1 | (3 ± 1) × 10−4 | 0.6 ± 0.2 | 1.8 ± 0.4 |
| k4 (s−1) | 1.4 ± 0.1 | 1.3 ± 0.4 | 0.1 ± 0.1 | 2.1 ± 0.9 |
| k−4 (s−1) | 0.7 ± 0.3 | 0.3 ± 0.1 | 0.02 ± 0.01 | 1.2 ± 0.5 |
| k5 (s−1) | 22 ± 8 | 9 ± 2 | 7 ± 0.3 | -- |
| k−5 (s−1) | 0.2 ± 0.1 | 7 ± 3 | 0.5 ± 0.3 | -- |
| k6 (s−1) | 4 ± 2 | 7 ± 2 | ~ 0 | -- |
| k−6 (s−1) | 0.4 ± 0.2 | 5 ± 1 | 0.5 ± 0.05 | -- |
| Q(U) | 8.2 ± 0.6 | 11.6 ± 0.5 | 12 ± 3 | 9.9 ± 0.5 |
| Q(I1) | 15 ± 3 | 13 ± 1 | 19 ± 3 | 12.0 ± 0.2 |
| Q(I2) | 11 ± 2 | 14.9 ± 0.5 | 14 ± 2 | 11.6 ± 0.2 |
| Q(I3) | 80 ± 20 | 18 ± 4 | 49 ± 5 | -- |
| Q(F) | 16 ± 1 | 16 ± 1 | 20 ± 3 | 12.1 ± 0.2 |
The relative fluorescence intensity determined from the fitting for every species provides a more stringent test of the current model. Since the major Trp fluorescence quenchers in both L7W and T21W are Cys and His residues, it is possible to rationalize, albeit in a qualitative manner, the relative fluorescence intensities of the folded, unfolded and intermediate states involved in a specific folding mechanism. For Scheme 3, it is expected that the fluorescence intensity of F is higher than those of U, I1 and I2 because more Trp fluorescence quenchers are bound to Zn2+ in the folded state (F). Similarly, if I1 and I2 represent N-Zn2+ and C-Zn2+ in Scheme 3, respectively, one would expect that for L7W (T21W) the fluorescence intensity of I1 (I2) is larger than that of I2 (I1) because in this case the Trp fluorophore is located near the N-terminal (C-terminal) ligands. As indicated (Table 3), the relative fluorescence intensities of F, I1 and I2 recovered from the best fits of the stopped-flow data of L7W and T21W to Scheme 3 indeed meet these expectations. Moreover, for both mutants the off-pathway intermediate I3 exhibits the largest relative fluorescence intensity, suggesting that it adopts a conformation wherein the Trp fluorescence quenching by all potential quenchers (i.e., all Cys and His residues) is maximally reduced. Thus, this result corroborates the idea that the misligated state involves H9 and/or H22.
While the structure of the misligated state cannot be exclusively determined from the current study, a possible Zn2+ coordination is presented in Figure 9, where Zn2+ not only coordinates with the native ligands, but is also associated with one or both of the non-native ligands (i.e., H9 and H22). Another possibility is that both H9 and H22 are in rapid exchange with one of the native ligands.55, 64 In addition, the more pronounced increase in the relative fluorescence intensity of I3 (compared to F) for L7W (and L7W/H9V) than that for T21W (Table 3) likely arises from difference in the orientations of the indole rings of Trp7 and Trp21. As shown in the model structures provided by the design calculations (Figure 2), the side chain of Trp7 is oriented towards the zinc and is thus expected to be more sensitive than Trp21 to changes in the coordination number of Zn2+.
Figure 9.
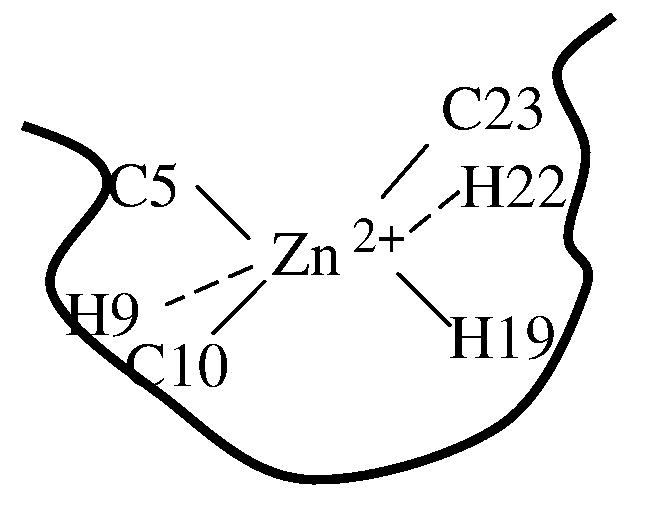
A possible zinc coordination in the misligated state.
Moreover, fitting the stopped-flow kinetics of L7W/H9V to Scheme 3 and those of L7W/H9V/H22I to Scheme 2 (where I3 is no longer needed because H9 and H22 are deleted) also yields a set of microscopic rate constants and relative fluorescence intensities (Table 3) that meet the aforementioned structural characteristics of I1, I2, and I3. For example, k1 of L7W/H9V is smaller than that of L7W as a result of H9 deletion, and both k1 and k2 of L7W/H9V/H22I are decreased as compared to those of L7W as a result of H9 and H22 deletion. In addition, the resultant microscopic rate constants for L7W/H9V/H22I (i.e., k3, k−3, k4, and k−4) indicate that deleting non-native ligands results in a less stable CH11 fold. This is consistent with the fact that both H9 and H22 are conserved in the CH11 sequence40 to some extent and also similar to that observed in a recent study47 on the NZF-1 zinc-binding domain, which showed that an ‘extra’ histidine is not only important for maintaining the tertiary structure of the peptide, but it can also coordinate with the Zn2+ when the native histidine is absent.
Taken together, our results show that Scheme 3 is capable of providing a comprehensive description of the zinc-induced folding kinetics of four computationally designed mutants of the CH11 peptide as monitored by Trp fluorescence. While earlier studies have shown that binding of Zn2+ to a peptide could proceed in a multistep and sequential manner35,54,65 and that misligation of nearby non-native ligands to Zn2+ could occur in the later stage of the folding process,65 the current model highlights the importance of parallel, binding-coupled folding pathways, as well as the non-native ligands, for achieving a higher metal binding affinity. Furthermore, the finding that the two non-native ligands (i.e., H9 and H22) stabilize the CH11 fold even though they complicate the folding kinetics provides strong evidence suggesting that in certain cases functional requirements for folding could produce significant local frustrations in the folding energy landscape, thus slowing down folding. Similar conclusions regarding the divergent requirements for folding and function have also been reached in other studies.66–69
Estimating the loop closure rate
Temperature-dependent measurements indicate that all three stopped-flow kinetic phases exhibit Arrhenius temperature dependence with an apparent activation energy ranging from 6 – 14 kcal/mol (Table 2), depending on the kinetic phase and mutant. While it is difficult to rationalize these apparent activation energies and also the subtle differences among them, they likely arise from one of the following events:70 (a) dehydration of Zn(H2O)62+, (b) deprotonation of His and Cys ligands, (c) peptide chain motions required to bring the corresponding ligands to a suitable position for coordination, and (e) disruption of misligation for correct folding. Regardless of the origin of those energetic barriers, however, the value of obtained for each CH11 mutant could be used to estimate the loop closure rate for short peptides. According to Scheme 3, in order to form the final folded and closed conformation of CH11 from either N-Zn2+ (I1) or C-Zn2+ (I2) the opposite end of the peptide chain must be brought to a suitable distance to the peptide-bound Zn2+, so that further coordination can take place. Therefore, using the apparent rate constant and the activation energy obtained for the third kinetic step, which approximately measures the total rate leading to the formation of the native and closed conformation of CH11 from both N-Zn2+ and C-Zn2+, we can estimate the free diffusion rate of the CH11 peptide chain in aqueous solution. In other words, the prefactor in the Arrhenius equation provides an upper limit for the rate of the peptide chain diffusion. At 15 °C, this rate was estimated to be 2.1 × 107 s−1 for L7W, 6.6 × 106 s−1 for L7W/H9V, and 7.0 × 109 s−1 for T21W. Despite its simplicity, this simple analysis yields peptide chain diffusion rates that are in good agreement with those determined by measuring the end-to-end collision rate in short peptides (e.g., 7.1 × 106 to 1.5 × 108 s−1),71,72 which provides further support for the proposed zinc-induced folding mechanism of CH11.
Conclusions
In summary, the Zn2+-induced CH11 folding can be described by a parallel pathway wherein Zn2+ first binds to either the N-terminal group of ligands (C5/C10/H9) or C-terminal group of ligands (H19/H22/C23). This initial binding event is followed by a rearrangement of the peptide chain, leading to the closure of peptide loop and the concomitant formation of the native zinc tetrahedral coordination. During folding, however, a misligated Zn2+-peptide complex is also transiently populated which may have a higher coordination number. While the existence of non-native Zn2+ ligands is responsible for the formation of this off-pathway folding intermediate, these ‘extra’ ligands nevertheless facilitate the binding of Zn2+ to CH11 peptide and also enhance the stability of the final fold. In addition, the loop closure rate of this peptide is estimated to be in the range of 6.6 × 106 to 7.0 × 109 s−1.
MATERIALS AND METHODS
Materials and sample preparation
All peptides were synthesized using standard Fmoc-based solid-phase method on a PS3 peptide synthesizer (Protein Technologies, MA) and purified by reverse-phase high performance liquid chromatography. The identity of each peptide was further verified by MALDI-TOF mass spectrometry. All peptide solutions were prepared by directly dissolving lyophilized peptide solid in 10 mM deoxygenated Tris buffer (pH 6.5 or 7.5, depending on the experiment). To prevent cysteine from oxidation, each peptide solution also contains a 10-fold excess of tris(2-carboxyethyl) phosphine hydrochloride (TCEP·HCl) (Pierce Biotechnology, IL). The final peptide concentration of each solution was determined optically using Trp absorbance at 280 nm and a molar extinction coefficient of 5600 M−1 cm−1. The Zn2+ solution was freshly prepared before each experiment by dissolving ZnCl2 (Sigma, MO) in 10 mM deoxygenated Tris buffer (pH 6.5 or 7.5).
Equilibrium CD measurement
Far-UV CD spectra at 25 °C were collected on an AVIV 62DS spectropolarimeter (Aviv Associates, NJ) using a 1 mm quartz cell. For each case, the concentrations of the peptide and Zn2+ were ~30 μM and 1 mM, respectively.
Equilibrium fluorescence measurement
Trp fluorescence spectra were collected under a nitrogen atmosphere on a Fluorolog 3.10 spectrofluorometer (Jobin Yvon Horiba, NJ) using a 1 cm quartz sample holder. The excitation wavelength was 285 nm and the spectral resolution was 2 nm (for both excitation and emission).
Equilibrium binding experiment
The Zn2+-binding affinity of the CH11 mutants at 15 °C was determined by Trp fluorescence titration. The Zn2+-peptide solution (10 μM peptide) for each measurement was prepared by adding an appropriate aliquot of a 4 mM ZnCl2 stock solution to a peptide solution, which was allowed to equilibrate for 5 minutes before fluorescence measurement. The total fluorescence of each Zn2+-peptide solution was taken as the sum of those of the free and Zn2+-bound peptides, i.e.,
| (1) |
where Fint is the integrated area of the fluorescence spectrum, Ff and Fb represent the molar fluorescence intensities of the free and Zn2+-bound peptides, respectively, and [P]f and [P]b denote the molar concentrations of the free and Zn2+-bound peptides, respectively. For 1:1 binding, [P]f and [P]b follow the following relationship,
| (2) |
where Kd is the dissociation constant of the Zn2+-peptide complex. Since Zn2+ also binds to Tris, the Zn2+ concentration ([Zn2+]) in the above equation should also satisfies the following relationship,
| (3) |
where Kd′ is the dissociation constant of the Zn2+-Tris complex (Kd′= 11.5 mM),73 and [Tris]f and [Tris]b are the molar concentrations of the free and Zn2+-bound Tris molecules. Since Kd′ is relatively large and the total concentration of Tris (i.e., [Tris]T = [Tris]f + [Tris]b = 10 mM) is much larger than that of [Zn2+] (in μM range), we have assumed that [Tris]f = [Tris]T in the derivation of the following equation,
| (4) |
where [P]T is the total concentration of the peptide. For each experiment, the total concentrations of the peptide, Zn2+, and Tris were known. Thus, eqs. (1) and (4) allow one to determine the dissociation constant of the Zn2+-peptide complex via fitting the equilibrium fluorescence binding curve (i.e., Figure 3b) by varying Kd, Fb and Ff.
Stopped-flow measurement
The zinc-induced CH11 folding kinetics were monitored via Trp fluorescence using a SFM-300 stopped-flow module (Bio-logic, Claix, France) equipped with homebuilt optics, the detail of which has been described elsewhere.51 In the current study, a microcuvette (μFC-08) with an optical pathlength of 0.8 mm was used and the dead time of the system was ~600 μs determined by the method of N-acetyl-tryptophanamide fluorescence quenching by N-bromo-succinimide. The stopped-flow kinetics were initiated by mixing equal volumes of a peptide solution and a ZnCl2 solution. The final peptide concentration was 10 μM and the final Zn2+ concentration was 0.1–0.3 mM, depending on the experiment. The Trp fluorophore was excited at 285 nm and the resultant fluorescence (for λem > 315 nm) was measured by a photomultiplier tube. Sample temperature (5–30 °C) was controlled by a Thermo Neslab RTE-7 circulator (Thermo, NH). The stopped-flow traces shown in the Figures correspond to an average of 6–10 shots. The stopped-flow kinetics thus obtained were fit to either a multi-exponential function using a nonlinear least squares method or a kinetic model, i.e., Scheme 1, 2 or 3. For the latter case, all individual kinetic steps were assumed to be first-order with respect to each species and the corresponding rate equations were solved numerically using the program Scientist (Micromath Research, LC) to yield the best fit.
Mutant design
Four structurally consistent mutants of CH11 were identified using a statistical, computationally assisted design strategy (SCADS), the details of which have been described elsewhere.30,43,44,74 Briefly, the site-specific probabilities of the amino acids at variable positions in a given structure are calculated, allowing identification of structurally consistent mutations. These probabilities are estimated by optimizing an effective sequence-conformation entropy subject to constraints involving the total average energy of the structure, as measured using an atom-based energy function. The conformational energy is represented as an average over sequence and side chain conformational (rotamer) states of the amino acids,30,43,44 where all interatomic interactions are quantified using the AMBER force field75 with a modified hydrogen bonding energy76 and a distance dependent electrostatic dielectric constant (ε = 4rij, where rij is the distance between atoms i and j). The nonbonding parameters for Zn2+ were obtained from Hoops et al. (Lennard-Jones parameters: R* = 1.1 Å, ε = 0.012 kcal/mol; partial charge: qZn2+=0.866).77 The protein backbone coordinates were constrained to those in the experimentally determined NMR structure (PDB code: 1liq).40 Side chain variability was addressed using a backbone-dependent rotamer library with maximum 81 rotamers per amino acid.78
The sequence design was divided into two components. The first involved identifying sites for introducing a Trp as an intrinsic fluorescent probe. The residue positions in the protein were scanned for possible mutations, allowing Trp, Tyr and wild type amino acid, while fixing the remaining residues at their wild type amino acids. For all calculations, the four metal-binding residues (Cys5, Cys10, His19 and Cys23) were fixed to their wild type conformations, while other residues were allowed to take on rotamer states contained in the library. The second part of the design identified suitable mutations for the two non-ligating histidines (His9 and His22), to inform the roles of these non-ligating histidines in folding. All 19 natural amino acids except Cys were allowed for the site-directed mutagenesis of these two sites. The amino acid probabilities were obtained at medium (Teff = 2.0 kcal/mol) and low (Teff = 0.5 kcal/mol) effective temperatures. Teff represents an effective temperature conjugate to the configurational energy of the sequences; lower temperatures correspond to lower average energy of the sequence when in the target structure.30,44 In addition, mutations between the two sites 9 and 22 were not correlated with each other, presumably due to the relatively large distance between them (dCβ−Cβ = 14.1 Å).
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the NIH (GM-065978 and GM-61267) and the NSF (DMR05-20020). Support for computational resources was provided in part by the NSF (CHE-0131132).
Abbreviation used
- CD
Circular dichroism
- SCADS
Statistical computationally-assisted design strategy
- NMR
nuclear magnetic resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferguson N, Fersht AR. Early events in protein folding. Curr Opin Struct Biol. 2003;13:75–81. doi: 10.1016/s0959-440x(02)00009-x. [DOI] [PubMed] [Google Scholar]
- 2.Fersht AR, Sato S. ψ-value analysis and the nature of protein-folding transition states. Proc Natl Acad Sci USA. 2004;101:7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krishna MM, Lin Y, Englander SW. Protein misfolding: Optional barriers, misfolded intermediates, and pathway heterogeneity. J Mol Biol. 2004;343:1095–1009. doi: 10.1016/j.jmb.2004.08.098. [DOI] [PubMed] [Google Scholar]
- 4.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38:245–88. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 5.Deechongkit S, Nguyen H, Jager M, Powers ET, Gruebele M, Kelly JW. β-sheet folding mechanisms from perturbation energetics. Curr Opin Struct Biol. 2006;16:94–101. doi: 10.1016/j.sbi.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 6.Naganathan AN, Doshi U, Fung A, Sadqi M, Munoz V. Dynamics, energetics, and structure in protein folding. Biochemistry. 2006;45:8466–8475. doi: 10.1021/bi060643c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrovich M, Jonsson AL, Ferguson N, Daggett V, Fersht AR. ψ-analysis at the experimental limits: Mechanism of beta-hairpin formation. J Mol Biol. 2006;360:865–881. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 8.Sosnick TR, Krantz BA, Dothager RS, Baxa M. Characterizing the protein folding transition state using φ analysis. Chem Rev. 2006;106:1862–1876. doi: 10.1021/cr040431q. [DOI] [PubMed] [Google Scholar]
- 9.Beck DA, White GW, Daggett V. Exploring the energy landscape of protein folding using replica-exchange and conventional molecular dynamics simulations. J Struct Biol. 2007;157:514–523. doi: 10.1016/j.jsb.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dyer RB. Ultrafast and downhill protein folding. Curr Opin Struct Biol. 2007;17:38–47. doi: 10.1016/j.sbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Gray HB. Biological inorganic chemistry at the beginning of the 21st century. Proc Natl Acad Sci USA. 2003;100:3563–3568. doi: 10.1073/pnas.0730378100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson CJ, Apiyo D, Wittung-Stafshede P. Role of cofactors in metalloprotein folding. Q R ev Biophys. 2004;37:285–231. doi: 10.1017/S003358350500404X. [DOI] [PubMed] [Google Scholar]
- 13.Frankel AD, Berg JM, Pabo CO. Metal-dependent folding of a single zinc finger from transcription factor IIIA. Proc Natl Acad Sci USA. 1987;84:4841–4845. doi: 10.1073/pnas.84.14.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krizek BA, Amann BT, Kilfoil VJ, Merkle DL, Berg JM. A consensus zinc finger peptide: Design, high-affinity metal-binding, a pH-dependent structure, and a His to Cys sequence variant. J Am Chem Soc. 1991;113:4518–4523. [Google Scholar]
- 15.Zong CH, Wilson CJ, Shen TY, Wittung-Stafshede P, Mayo SL, Wolynes PG. Establishing the entatic state in folding metallated Pseudomonas aeruginosa azurin. Proc Natl Acad Sci USA. 2007;104:3159–3164. doi: 10.1073/pnas.0611149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones CM, Henry ER, Hu Y, Chan CK, Luck SD, Bhuyan A, Roder H, Hofrichter J, Eaton WA. Fast events in protein-folding initiated by nanosecond laser photolysis . Proc Natl Acad Sci USA. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shastry MCR, Sauder JM, Roder H. Kinetic and structural analysis of submillisecond folding events in cytochrome c. Acc Chem Res. 1998;31:717–725. [Google Scholar]
- 18.Englander SW, Sosnick TR, Mayne LC, Shtilerman M, Qi PX, Bai YW. Fast and slow folding in cytochrome c. Acc Chem Res. 1998;31:737–744. [Google Scholar]
- 19.Guidry J, Wittung-Stafshede P. Cytochrome c(553), a small heme protein that lacks misligation in its unfolded state, folds with rapid two-state kinetics. J Mol Biol. 2000;301:769–773. doi: 10.1006/jmbi.2000.3993. [DOI] [PubMed] [Google Scholar]
- 20.Chen EF, Goldbeck RA, Kliger DS. The earliest events in protein folding: A structural requirement for ultrafast folding in cytochrome c. J Am Chem Soc. 2004;126:11175–11181. doi: 10.1021/ja0400077. [DOI] [PubMed] [Google Scholar]
- 21.Bertini I, Gray HB, Lippard SJ, Valentine JS. Bioinorganic Chemistry. 2. Mill Valley; California: 1994. [Google Scholar]
- 22.Cerasoli E, Sharpe BK, Woolfson DN. ZiCo: a peptide designed to switch folded state upon binding zinc. J Am Chem Soc. 2005;127:15008–15009. doi: 10.1021/ja0543604. [DOI] [PubMed] [Google Scholar]
- 23.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. De novo design and structural characterization of proteins and metalloproteins. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 24.Kohn WD, Kay CM, Sykes BD, Hodges RS. Metal ion induced folding of a de novo designed coiled-coil peptide. J Am Chem Soc. 1998;120:1124–1132. [Google Scholar]
- 25.Kharenko OA, Ogawa MY. Metal-induced folding of a designed metalloprotein. J Inorg Biochem. 2004;98:1971–1974. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 26.Maglio O, Nastri F, Calhoun JR, Lahr S, Wade H, Pavone V, DeGrado WF, Lombardi A. Artificial di-iron proteins: Solution characterization of four helix bundles containing two distinct types of inter-helical loops. J Biol Inorg Chem. 2005;10:539–549. doi: 10.1007/s00775-005-0002-8. [DOI] [PubMed] [Google Scholar]
- 27.Shiraishi Y, Imanishi M, Morisaki T, Sugiura Y. Swapping of the β-hairpin region between Sp1 and GLI zinc fingers: Significant role of the β-hairpin region in DNA binding properties of C2H2-type zinc finger peptides. Biochemistry. 2005;44:2523–2538. doi: 10.1021/bi047797h. [DOI] [PubMed] [Google Scholar]
- 28.Iranzo O, Ghosh D, Pecoraro VL. Assessing the integrity of designed homomeric parallel three-stranded coiled coils in the presence of metal ion. Inorg Chem. 2006;45:9959–9973. doi: 10.1021/ic061183e. [DOI] [PubMed] [Google Scholar]
- 29.Papworth M, Kolasinska P, Minczuk M. Designer zinc-finger proteins and their applications. Gene. 2006;17:27–38. doi: 10.1016/j.gene.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J Mol Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, DeGrado WF. De novo design of a redox active minimal rubredoxin mimic. J Am Chem Soc. 2005;127:5804–5805. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki K, Hiroaki H, Kohda D, Nakamura H, Tanaka T. Metal ion induced self-assembly of a designed peptide into a triple-stranded α-helical bundle: A novel metal binding site in the hydrophobic core. J Am Chem Soc. 1998;120:13008–13015. [Google Scholar]
- 33.Farrer BT, Pecoraro VL. Hg(II) binding to a weakly associated coiled coil nucleates an encoded metalloprotein fold: A kinetic analysis. Proc Natl Acad Sci USA. 2003;100:3760–3765. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pozdnyakova I, Wittung-Stafshede P. Biological relevance of metal binding before protein folding. J Am Chem Soc. 2001;123:10135–10136. doi: 10.1021/ja016252x. [DOI] [PubMed] [Google Scholar]
- 35.Bombarda E, Grell E, Roques B, Mely Y. Molecular mechanism of the Zn2+-induced folding of the distal CCHC finger motif of the HIV-1 nucleocapsid protein. Biophys J. 2007;93:208–217. doi: 10.1529/biophysj.106.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du DG, Zhu YJ, Huang CY, Gai F. Understanding the key factors that control the rate of beta-hairpin folding. Proc Natl Acad Sci USA. 2004;101:15915–15920. doi: 10.1073/pnas.0405904101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bunagan MR, Yang X, Saven JG, Gai F. Ultrafast folding of a computationally designed Trp-cage mutant: Trp2-cage. J Phys Chem B. 2006;110:3759–3763. doi: 10.1021/jp055288z. [DOI] [PubMed] [Google Scholar]
- 38.Xu Y, Purkayastha P, Gai F. Nanosecond folding dynamics of a three-stranded β-sheet. J Am Chem Soc. 2006;128:15836–15842. doi: 10.1021/ja064865+. [DOI] [PubMed] [Google Scholar]
- 39.Sadqi M, de Alba E, Pérez-Jiménez R, Sanchez-Ruiz JM, Muñoz V. A designed protein as experimental model of primordial folding. Proc Natl Acad Sci USA. 2009;106:4127–4132. doi: 10.1073/pnas.0812108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharpe BK, Matthews JM, Kwan AH, Newton A, Gell DA, Crossley M, Mackay JP. A new zinc binding fold underlines the versatility of zinc binding modules in protein evolution. Structure. 2002;10:639–648. doi: 10.1016/s0969-2126(02)00757-8. [DOI] [PubMed] [Google Scholar]
- 41.Sharpe BK, Liew CK, Kwan AH, Wilce JA, Crossley M, Matthews JM, Mackay JP. Assessment of the robustness of a serendipitous zinc binding fold: Mutagenesis and protein grafting. Structure. 2005;13:257–266. doi: 10.1016/j.str.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 43.Zou J, Saven JG. Statistical theory of combinatorial libraries of folding proteins: Energetic discrimination of a target structure. J Mol Biol. 2000;296:281–294. doi: 10.1006/jmbi.1999.3426. [DOI] [PubMed] [Google Scholar]
- 44.Kono H, Saven JG. Statistical theory for protein combinatorial libraries. Packing interactions, backbone flexibility, and the sequence variability of a main-chain structure. J Mol Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 45.Oneil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino-acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 46.Nyborg JK, Peersen OB. That zincing feeling: The effects of EDTA on the behaviour of zinc-binding transcriptional regulators. Biochem J. 2004;381:e3–e4. doi: 10.1042/BJ20041096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berkovits-Cymet HJ, Amann BT, Berg JM. Solution structure of a CCHHC domain of neural zinc finger factor-1 and its implications for DNA binding. Biochemistry. 2004;43:898–903. doi: 10.1021/bi035159d. [DOI] [PubMed] [Google Scholar]
- 48.Mély Y, De Rocquigny H, Piemont E, Demene H, Jullian N, Fournie-Zaluski MC, Roques B, Gerard D. Influence of the N-terminal and C-terminal chains on the zinc-binding and conformational properties of the central zinc-finger structure of moloney murine leukemia-virus nucleocapsid protein: A steady-state and time-resolved fluorescence study. Biochim Biophys Acta. 1993;1161:6–18. doi: 10.1016/0167-4838(93)90189-x. [DOI] [PubMed] [Google Scholar]
- 49.Mély Y, De Rocquigny H, Morellet N, Roques BP, Gerad D. Zinc binding to the HIV-1 nucleocapsid protein: A thermodynamic investigation by fluorescence spectroscopy. Biochemistry. 1996;35:5175–5182. doi: 10.1021/bi952587d. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Liu B, Yu HT, Barkley MD. The peptide bond quenches indole fluorescence. J Am Chem Soc. 1996;118:9271–9278. [Google Scholar]
- 51.Tucker MJ, Tang J, Gai F. Probing the kinetics of membrane-mediated helix folding. J Phys Chem B. 2006;110:8105–8109. doi: 10.1021/jp060900n. [DOI] [PubMed] [Google Scholar]
- 52.Buchsbaum JC, Berg JM. Kinetics of metal binding by a zinc finger peptide. Inorg Chim Acta. 2000;297:217–219. [Google Scholar]
- 53.Keeble AH, Hemmings AM, James R, Moore GR, Kleanthous C. Multistep binding of transition metals to the H-N-H endonuclease toxin colicin E9. Biochemistry. 2002;41:10234–10244. doi: 10.1021/bi020174o. [DOI] [PubMed] [Google Scholar]
- 54.Bombarda E, Roques BP, Mély Y, Grell E. Mechanism of zinc coordination by point-mutated structures of the distal CCHC binding motif of the HIV-1NCp7 proteins. Biochemistry. 2005;44:7315–7325. doi: 10.1021/bi047349+. [DOI] [PubMed] [Google Scholar]
- 55.Heinz U, Kiefer M, Tholey A, Adolph HW. On the competition for available zinc. J Biol Chem. 2005;280:3197–3207. doi: 10.1074/jbc.M409425200. [DOI] [PubMed] [Google Scholar]
- 56.Dutta SJ, Liu J, Mitra B. Kinetic analysis of metal binding to the amino-terminal domain of ZntA by monitoring metal-thiolate charge-transfer complexes. Biochemistry. 2005;44:14268–14274. doi: 10.1021/bi050761k. [DOI] [PubMed] [Google Scholar]
- 57.Bunagan MR, Cristian L, DeGrado WF, Gai F. Truncation of a cross-linked GCN4-p1 coiled-coil leads to ultrafast folding. Biochemistry. 2006;45:10981–10986. doi: 10.1021/bi0606142. [DOI] [PubMed] [Google Scholar]
- 58.Du DG, Gai F. Understanding the folding mechanism of an α-helical hairpin. Biochemistry. 2006;45:13131–13139. doi: 10.1021/bi0615745. [DOI] [PubMed] [Google Scholar]
- 59.Qiu LL, Pabit SA, Roitberg AE, Hagen SJ. Smaller and faster: The 20-residue Trp-cage protein folds in 4 μs. J Am Chem Soc. 2002;124:12952–12953. doi: 10.1021/ja0279141. [DOI] [PubMed] [Google Scholar]
- 60.Williams S, Causgrove TP, Gilmanshin R, Fang KS, Callender RH, Woodruff WH, Dyer RB. Fast events in protein folding: Helix melting and formation in a small peptide. Biochemistry. 1996;35:691–697. doi: 10.1021/bi952217p. [DOI] [PubMed] [Google Scholar]
- 61.Du DG, Tucker MJ, Gai F. Understanding the mechanism of β-hairpin folding via φ-value analysis. Biochemistry. 2006;45:2668–2678. doi: 10.1021/bi052039s. [DOI] [PubMed] [Google Scholar]
- 62.Wang T, Zhu YJ, Getahun Z, Du DG, Huang CY, DeGrado WF, Gai F. Length dependent helix-coil transition kinetics of nine alanine-based peptides. J Phys Chem B. 2004;108:15301–15310. doi: 10.1021/jp037272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bombarda E, Morellet N, Cherradi H, Spiess B, Bouaziz S, Grell E, Roques BP, Mely Y. Determination of the pKa of the four Zn2+-coordinating residues of the distal finger motif of the HIV-1 nucleocapsid protein: Consequences on the binding of Zn2+ J Mol Biol. 2001;310:659–672. doi: 10.1006/jmbi.2001.4770. [DOI] [PubMed] [Google Scholar]
- 64.Siemann S, Badiei HR, Karanassios V, Viswanatha T, Dmitrienko GI. 68Zn isotope exchange experiments reveal an unusual kinetic lability of the metal ions in the di-zinc form of IMP-1 metallo-b-lactamase. Chem Commun. 2006:532–534. doi: 10.1039/b514227j. [DOI] [PubMed] [Google Scholar]
- 65.Li WF, Zhang J, Wang J, Wang W. Metal-coupled folding of Cys2His2 zinc-finger. J Am Chem Soc. 2007;130:892–900. doi: 10.1021/ja075302g. [DOI] [PubMed] [Google Scholar]
- 66.Jager M, Zhang Y, Bieschke J, Nguyen H, Dendle M, Bowman ME, Noel JP, Gruebele M, Kelly JW. Structure-function-folding relationship in a WW domain. Proc Natl Acad Sci USA. 2006;103:10648–10653. doi: 10.1073/pnas.0600511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferreiro DU, Hegler JA, Komives EA, Wolynes PG. Localizing frustration in native proteins and protein assemblies. Proc Natl Acad Sci USA. 2007;104:19819–19824. doi: 10.1073/pnas.0709915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hills RD, Brooks CL. Coevolution of function and the folding landscape: Correlation with density of native contact. Biophys J. 2008;95:L57–L59. doi: 10.1529/biophysj.108.143388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ivarsson Y, Travaglini-Allocatelli C, Brunori M, Gianni S. Folding and misfolding in a naturally occurring circularly permuted PDZ domain. J Biol Chem. 2008;283:8954–8960. doi: 10.1074/jbc.M707424200. [DOI] [PubMed] [Google Scholar]
- 70.Blasie CA, Berg JM. Structure-based thermodynamic analysis of a coupled metal binding-protein folding reaction involving a zinc finger peptide. Biochemistry. 2002;41:15068–15073. doi: 10.1021/bi026621h. [DOI] [PubMed] [Google Scholar]
- 71.Lapidus LJ, Eaton WA, Hofrichter J. Measuring the rate of intramolecular contact formation in polypeptides. Proc Natl Acad Sci USA. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roccatano D, Sahoo H, Zacharias M, Nau WM. Temperature dependence of looping rates in a short peptide. J Phys Chem B. 2007;111:2639–2646. doi: 10.1021/jp066418m. [DOI] [PubMed] [Google Scholar]
- 73.Magyara JS, Godwin HA. Spectropotentiometric analysis of metal binding to structural zinc-binding sites: accounting quantitatively for pH and metal ion buffering effect. Anal Biochem. 2003;320:39–54. doi: 10.1016/s0003-2697(03)00281-1. [DOI] [PubMed] [Google Scholar]
- 74.Zhu YJ, Fu XR, Wang T, Tamura A, Takada S, Saven JG, Gai F. Guiding the search for a protein’s maximum rate of folding. Chem Phys. 2004;307:99–109. [Google Scholar]
- 75.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. A new force-field for molecular mechanical simulation of nucleic-acids and proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 76.Kono H, Doi J. A new method for sidechain conformation prediction using a Hopfield network and reproduced rotamers. J Comput Chem. 1996;17:1667–1683. [Google Scholar]
- 77.Hoops SC, Anderson KW, Merz KM. Force-field design for metalloproteins. J Am Chem Soc. 1991;113:8262–8270. [Google Scholar]
- 78.Dunbrack RL, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.