

Abstract
Molecular mechanisms for cell migration, especially how signaling and cytoskeletal systems are integrated, are not understood well. Here, we examined the role of CARMIL (capping protein, Arp2/3, and Myosin-I linker) family proteins in migrating cells. Vertebrates express three conserved genes for CARMIL, and we examined the functions of the two CARMIL genes expressed in migrating human cultured cells. Both isoforms, CARMIL1 and 2, were necessary for cell migration, but for different reasons. CARMIL1 localized to lamellipodia and macropinosomes, and loss of its function caused loss of lamellipodial actin, along with defects in protrusion, ruffling, and macropinocytosis. CARMIL1-knockdown cells showed loss of activation of Rac1, and CARMIL1 was biochemically associated with the GEF Trio. CARMIL2, in contrast, colocalized with vimentin intermediate filaments, and loss of its function caused a distinctive multipolar phenotype. Loss of CARMIL2 also caused decreased levels of myosin-IIB, which may contribute to the polarity phenotype. Expression of one CARMIL isoform was not able to rescue the knockdown phenotypes of the other. Thus, the two isoforms are both important for cell migration, but they have distinct functions.
INTRODUCTION
Cell migration is an essential element of many aspects of animal cell biology, such as morphogenesis during development, immune response to disease, and chemotaxis (Ridley et al., 2003; Vicente-Manzanares et al., 2005). In some settings, cell migration is a prominent component of disease, involved in the progression of malignant cancers and autoimmune syndromes. Cell migration requires proper function of the cytoskeleton, with integration of the actin and microtubule and intermediate filament cytoskeletons.
A migrating cell is generally polarized with broad actin-rich lamellipodia at its leading edge. Lamellipodia contain dense meshworks of actin filaments with their fast-growing, barbed ends of actin filaments oriented toward the direction of migration, and polymerization at barbed ends provides the driving force pushing the plasma membrane forward (Le Clainche and Carlier, 2008). Protrusions at the leading edge also include long thin structures termed filopodia or microspikes, which are composed of bundles of actin filaments that often appear to arise from the lamellipodial actin network (Svitkina et al., 2003). Lamellipodia are often accompanied by ruffles, which are wave-like structures that form by protruding upward and then moving rearward, sometimes resulting in macropinocytotic engulfment of extracellular fluid. The actin network of the leading edge contains many proteins, including Arp2/3 complex, cofilin, and capping protein (CP). In vitro, a synthetic mix of these proteins can form branched networks of filaments, and the assembly of those networks can produce movement (Pollard, 2007). The structure, molecular nature, and dynamics of these network in cells is not understood well, with considerable controversy as to their assembly, function, and turnover (Koestler et al., 2008; Lai et al., 2008).
Regulation of barbed ends, their creation and capping, is considered to be a key element controlling the architecture and force production of actin filament networks. Biochemically, free barbed ends can be created by nucleation from actin subunits de novo, by uncapping capped ends or by severing existing filaments. To create free barbed ends, the dendritic nucleation model proposes that activated Arp2/3 complex binds to an existing mother filament, which nucleates the formation of a new daughter filament with a free barbed end (Pollard, 2007). Other models propose that activation of cofilin to sever filaments is a primary event that creates free barbed ends (van Rheenen et al., 2007) or that inhibition of capping by proteins such as formins or Ena/VASP is critical (Applewhite et al., 2007; Le Clainche and Carlier, 2008).
In this study, we investigated how CARMIL family proteins function in cell migration. In particular, we compared the functions of the human CARMIL1 and 2 proteins, which are expressed together in many cells and tissues. In migrating cancer cells, we found both proteins to be important but with distinct nonoverlapping roles. CARMIL2 controls cell polarity and associates with vimentin intermediate filaments, whereas CARMIL1 controls actin dynamics in lamellipodia, possibly through regulation of Rac1 via interaction with the guanine nucleotide exchange factor (GEF) Trio.
MATERIALS AND METHODS
Antibodies and Reagents
Reagents and materials were from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA) unless stated otherwise.
For CP, mouse mAb 3F2 specific for the C-terminus of beta2 and 5B12 recognizing alpha1 and alpha2 were used for immunoblots as described (Developmental Studies Hybridoma Bank, University of Iowa; Schafer et al., 1996). Rabbit pAb R26 against the C-terminus of beta2 was used for immunostaining (Schafer et al., 1994). For CARMIL, chicken antibodies against a fragment of human CARMIL1 (538–1371) were produced by Dr. Ilgu Kang in our lab. Other antibodies and sources were as follows: VASP (rabbit pAb) from Dr. Frank Gertler (MIT); Myosin 1E (rabbit pAb) from Drs. Mira Krendel and Mark Mooseker (Yale University); ARPC2/p34 (rabbit pAb) and cortactin (mouse mAb 4F11) from Upstate Millipore (Lake Placid, NY); VASP (rabbit, pAb) from Calbiochem (La Jolla CA); giantin (rabbit, pAb) from Covance (Madison, WI); Myosin-IIA and Myosin-II B (rabbit, pAbs) from Covance and Dr. Paul Bridgman (Washington University); paxillin (mouse, mAb) from BD Bioscience (San Jose, CA); Trio (goat pAb) from Biotechnology (Santa Cruz, CA); and actin (mouse mAb C4), a gift from Dr. James Lessard (University of Cincinnati). Antibodies to α-tubulin (mouse, mAb), acetylated tubulin (mouse, mAb), γ-tubulin (mouse, mAb), and FLAG (mouse, mAb) were from Sigma-Aldrich, as was FLAG M2 affinity beads. Anti-green fluorescent protein (GFP; rabbit, pAb), Dynabeads M-280 sheep anti-rabbit IgG, and HRP- and Alexa-conjugated secondary antibodies were from Invitrogen (Carlsbad, CA).
cDNA Cloning and RT-PCR
Human total RNA was purified from HeLa cells using an RNeasy kit (QIAGEN, Valencia, CA), and 30 ng was reverse-transcribed to first-strand cDNA using Superscript (Invitrogen). Using this cDNA as template, PCR amplification with specific primer pairs (Supplemental Table S1) produced cDNA clones for full-length human CARMIL1 and 2.
For CARMIL1, the primers were chosen based on the National Center for Biotechnology Information (NCBI) sequence NM_017640, extending from the initiating ATG to the stop codon of the CDS. Several independent clones were obtained, and their sequences agreed with that of NM_017640, except for an insertion of 135 bases between 3906 and 3907. The sequence of the clones we obtained was submitted to NCBI with accession number FJ009082. We refer to this novel longer form as CARMIL1a and the shorter form, predicted by NM_017640, as CARMIL1b. Our CARMIL1a cDNA was used for all experiments in this study.
For CARMIL2, primers were chosen based on NM_001013838, from initiating ATG to stop codon, which we call CARMIL2a. We obtained several independent full-length ORF clones all corresponding to a slightly shorter variant that we call CARMIL2b, and we made a new NCBI accession FJ026014 for this variant. In CARMIL2b, exon 37 is missing. Furthermore, the intronic sequence between exons 14 and 15 in the RLTPR gene is not excised but is translated in-frame. Variations such as these were among ones described previously for partial clones (Matsuzaka et al., 2004). Our CARMIL2b full-length ORF cDNA was used for all experiments in this study.
For expression and localization, the full-length cDNAs encoding HsCARMIL1a and HsCARMIL2b were subcloned into the EcoRI/BamHI and HindIII/SpeI sites of pEYFP-C1 (Clontech, Palo Alto, CA), respectively. To express FLAG-tagged CARMIL fragments, the corresponding cDNA regions of HsCARMIL1 and HsCARMIL2 were subcloned into pUHDF30 (Liang et al., 2004). All constructs were sequenced fully.
To assay expression from cultured cell lines, 30 ng of total RNA was used for first-strand cDNA synthesis with platinum Taq DNA polymerase (Invitrogen). β-Actin was used as an internal control. PCR amplification was performed with primers pairs listed in Supplemental Table S1.
Sequence Alignment and Phylogenetic Tree Analysis
An unrooted phylogenetic tree was generated from a multiple sequence alignment of CARMIL family members. The sequences were retrieved from NCBI with the following accession numbers: human1a FJ009082, human2b FJ026014, human3 NP_612369, mouse1 NP_081101.3, mouse2 NP_001028492.1, mouse3 NP_001019816.1, rat1 XP_225336.4, rat3 Q5XHY1 (UniProt), stickleback ENSGACG00000006707 (ENSEMBL), Danio rerio XP_684159.3, Anopheles gambiae XP_314353.3, Ades egypti AAEL000228-RA, Drosophila melanogaster NP_610316.2, Dictyostelium XP_629656.1, Acanthamoeba castellanii AAB57739, chimpanzee1 XP_511754, chimpanzee2 XP_523395.2, chimpanzee3 XP_509860.2, dog1 XP_545371.2, dog2 XP_536814.2, dog3 Q8ND23 (UniProt), chicken1 XP_419088, chicken2 XP_414033, cow1 XP_617473, cow2 XP_587484.3, cow3 XP_595353.3, Caenorhabditis elegans Q21301 (UniProt) and C. briggsae CAP37466.1, Tetraodon1 CAF93839, and Apis mellifera XP_624410. The initial list of CARMIL family members was obtained from the TREEFAM database (www.treefam.org) as family TF316381. Each family member was verified by BLAST searching against human CARMIL homologues. Only sequences containing a consensus CARMIL homology domain (CHD), a leucine-rich repeat (LRR) region, and a CP-binding region (CBR) were included in the alignment. The multiple sequence alignment and unrooted phylogenetic tree were produced using ClustalW (Larkin et al., 2007) and Njplot Unrooted (Saitou and Nei, 1987). The reliability of the tree structure was tested by bootstrapping with 1000 trials. All nodes on the tree were present in >90% of the trials.
Sequence alignments for the CHD and CBR regions were made with ClustalW and MegAlign (DNASTAR, Madison, WI), with database sequences from NCBI as listed above. The percentage identities between various regions of human CARMILs 1, 2, and 3 (listed here as C1, C2 and C3 for brevity) were calculated using MegAlign. The boundaries of the regions were as follows: N-terminus (start to CHD): C1 1-155, C2 1-154, C3 1-152. CHD: C1 156-181, C2b 155-180, C3 153-178. LRR: C1 214-595, C2b 213-597, C3 211-593. Central region containing putative verprolin-homology and acidic regions: C1 596-964, C2b 598-963, C3 594-954. CBR: C1 964-1078, C2b 964-1072, C3 955-1063. C-terminal region: C1 1079-1371, C2b 1073-1372, C3 1064-1372. The acidic regions are located between residues 878-889, 899-911, and 937-955 in C1, 874-905 and 929-945 in C2 and 889-903 in C3. The verprolin-like sequence is located between residues 702-735 of C1, 705-738 in C2 and residues 700-733 of C3.
Cell Culture, Transfection, and Knockdown
Human HT-1080 cells (ATCC, Manassas, VA) and human embryonic kidney HEK293T cells were grown in DMEM (GIBCO BRL, Rockville, MD) supplemented with 10% calf serum (GIBCO) in an atmosphere containing 5% CO2. Cells were transfected with FuGENE6 (Roche, Indianapolis, IN) or Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For overexpression, cells were fixed 48 h after transfection.
To knock down human CARMIL1 and 2, we targeted the coding region sequences ATGCCATTGTTCATCTGGAT and GCAAAGATGGCGAGATCAAG, respectively. BLAST searches against the human genome revealed no other targets. A scrambled sequence, CAGTCGCGTTTGCGACTGG, was used as a control. Pairs of complementary oligonucleotides were annealed and inserted into an short hairpin RNA (shRNA)-expression vector, pSUPER, according to the manufacturer's instructions (OligoEngine, Seattle, WA). The resulting plasmids were transfected into HT-1080 cells using Lipofectamine, and cells were fixed or harvested at 48–72 h after transfection. Red fluorescent protein (RFP) or GFP-actin served as a cotransfection marker. To obtain a knockdown-positive cell population, the target sequences were subcloned into the plasmid pFLRu-FH-GFP (a gift from Dr. Yunfeng Feng in the Longmore lab, Washington University). GFP-positive cells were sorted at 24 h after transfection and maintained in media with puromycin (3 μg/ml) for up to 1 wk. Phenotypes were analyzed on day 3–4 after transfection. For rescue by expression, site-directed mutagenesis was used to construct a pFLRu-CARMIL1shRNA plasmid with three codon-neutral mutations (GCC to GCT, GTT to GTG, and CTG to CTC), and a pFLRu-CARMIL2shRNA plasmid with three codon-neutral mutations (AAA to AAG, GAT to GAC, and GGC to GGG).
To knock down YFP-HsCARMIL1 or YFP-HsCARMIL2, HEK293 cells or HT-1080 cells were cotransfected with plasmids expressing an shRNA, YFP-HsCARMIL1, or YFP-HsCARMIL2 and mRFP using Lipofectamine. Cells were processed 48 h after transfection for fluorescence microscopy. To knockdown Arp2/3, an siRNA for human ArpC4 (p20), AAGGAGATCAGTGAGATGAAG, was used, with the scrambled sequence, CAGTCGCGTTTGCGACTGG, as control. HT-1080 cells were transfected with siRNA using Lipofectamine2000 (Invitrogen), and cells were processed for immunoblot and immunofluorescence at 72 h.
The vimentin-knockdown plasmid was a gift from Dr. Yunfeng Feng (Washington University). The target sequence GGCACGTCTTGACCTTGAAC against human vimentin was subcloned into the plasmid pFLRu-FH. Cells were transfected with pFLRu-Vimentin shRNA using Lipofectamine. For YFP-CARMIL2 localization, YFP-CARMIL2 was introduced into cells transfected with pFLRu-Vimentin shRNA 72 h after transfection.
Microscopy of Cells
Cells were plated onto glass coverslips coated with 30 μg/ml fibronectin (Sigma-Aldrich), fixed in paraformaldehyde or methanol, and processed as described (Mejillano et al., 2004). Immunostaining was performed with appropriate antibodies. Free barbed ends in permeabilized cells were visualized by addition of fluorescent actin, essentially as described (Bryce et al., 2005). Cells were permeabilized and labeled with 0.4 μM Alexa-568-actin (Molecular Probes, Eugene, OR) in saponin buffer for 30 s and then fixed with 4% paraformaldehyde. Cells were imaged with 10× or 60× objectives on an Olympus IX70 inverted microscope (Olympus, Melville, NY) equipped with a Coolsnap HQ camera (Photometrics, Woburn, MA). Images were collected and initially process with QED In Vivo software (Media Cybernetics, Silver Spring, MD). Images were converted to pseudocolor using Adobe Photoshop (San Jose, CA). Polarity was scored by “blind” observers as described (Sidani et al., 2007).
For wound-healing assays, shRNA-transfected cells were grown to a monolayer, serum-starved for 12 h, wounded with a pipette tip, and fed fresh medium with 10% FBS. Cells were allowed to recover for 30 min, and then time-lapse images were captured every 1.5 min for 2 h.
For time-lapse movies, cells were grown on glass-bottom culture dishes (MatTek, Ashland, MA) coated with fibronectin (30 μg/ml). Cells were adapted to L-15 medium (Invitrogen) supplemented with 10% FBS (GIBCO) at 37°C. For long-term movies, phase-contrast images were captured every 2 min for 2 h. For short-term movies, images were captured every 1 s for 5 min.
For cell spreading assays, trypsinized cells were seeded onto a glass-bottom culture dish coated with fibronectin. After 1–5 min, images were captured every 1 min for 30 min. For quantitative analysis of lamellipodial motility parameters, phase-contrast time-lapse movies were viewed. Each experimental group contained at least 13 cells. Images were taken every second for 5 min. A cell with one or more broad lamellipodia or phase-dark ruffles was scored as positive for lamellipodia or ruffles, respectively. A cell with no obvious protrusion or having a smooth edge without protrusion and withdrawal was scored as a negative for lamellipodia or ruffles. Persistence was defined as the continuous presence of protrusion or ruffling activity for 5 min. Macropinocytosis was scored as positive if more than two macropinosomes were observed during the time course of the movie. Two “blind” observers evaluated the movies independently, and the data were quantified for each parameter by averaging the percent of cells determined by each observer. Two-tailed t statistical tests were performed for statistical analysis, and p < 0.05 was considered significant.
Disassembly of vimentin filaments or microtubules. For withaferin A (WFA) experiments, cells were treated with 0.5–10 μM WFA (Chromadex, Irvine, CA) from a stock solution in DMSO or the appropriate volume % of DMSO as a control for 1 h and then fixed with 4% paraformaldehyde (Bargagna-Mohan et al., 2007). Nocodazole treatment was at 5 μM for 60 min, with a 60-min washout.
Coimmunoprecipitations and Immunoblots
Coimmunoprecipitation with anti-FLAG M2 affinity gel was performed as described (Liang et al., 2004). Coimmunoprecipitation with anti-GFP (Invitrogen) was performed as instructed by the manufacturer. Briefly, cells were lysed with 20 mM Tris/HCl, pH 7.5, 100 mM NaCl, 0.5% NP-40, 10% glycerol, and protease cocktail inhibitor (Calbiochem). The lysates were centrifuged to remove debris, and supernatants were incubated for 2 h at 4°C with Dynabeads precoated with anti-GFP antibody. Beads were washed three times with lysis buffer, boiled with 2× SDS loading buffer, and then subjected to SDS-PAGE, followed by immunoblotting. For the Rac1 activity assay, 2 × 106 cells were allowed to adhere to fibronectin-coated culture dishes for 0, 15, and 30 min. The Rac1 activity was measured by pulling down with PAK-PBD as described by the manufacturer (Upstate). Immunoblots were developed with ECL (Perkin Elmer-Cetus, Boston, MA) and exposed to autoradiography film.
RESULTS
The CARMIL Protein Family: Cloning, Phylogeny, and Expression
To study CARMIL function, we first asked how many forms of CARMIL exist in mammalian systems. Early after the discovery of CARMIL in Acanthamoeba as Acan125, a C. elegans gene predicted to encode a protein of similar sequence was identified (Xu et al., 1997). A Dictyostelium protein, p116, was then found to have sequence similarity to Acan125 (Jung et al., 2001). At that time, a CARMIL family protein was defined as a polypeptide of >1000 aa with a long LRR domain near the N-terminus and a proline-rich region capable of binding the SH3 domain of myosin-I (Xu et al., 1995, 1997; Zot et al., 2000; Jung et al., 2001). In vertebrates, CARMIL genes and proteins have been described in mice, rats, and humans (Yang et al., 2005; Uruno et al., 2006), and evidence for three isoforms has been uncovered (Uruno et al., 2006).
To determine the extent to which different organisms express CARMIL family members, we searched genome databases. First, BLAST searches of the human genome using human and mouse forms of CARMIL1 protein, encoded by genes on human chromosome 6 and mouse chromosome 13, respectively (Yang et al., 2005), identified two additional human genes, predicted to encode proteins that we refer to as CARMIL2 and 3. The three predicted proteins are similar in size and have sequence similarity across their entire length (Figure 1A).
Figure 1.

CARMIL protein family sequence and expression analysis. (A) Domain architecture and sequence similarity among human CARMILs. CHD, CARMIL-homology domain; LRR, leucine-rich repeat; V, verprolin homology; CBR, capping protein-binding region (Bruck et al., 2006; Canton et al., 2006). (B) Alignment of CHD sequences. Residues identical to the majority in red, and similar ones in blue. (C) CARMIL family phylogenetic tree. Included proteins possess all three major domains: CHD, LRR, and CBR. Bootstrap analysis showed the branch points to be significant. Scale bar, the number of amino acid substitutions per site. Sequence accession numbers are in Materials and Methods. (D) Alignment of CBR sequences, performed as for the CHD sequences of B. (E) Expression of CARMIL and CP-alpha genes in cultured cells, determined by RT-PCR. Primers are listed in Supplemental Table S1. β-Actin was a positive control for loading. For CARMIL1, the primers flank a region present in CARMIL1a but not CARMIL1b, and the position of the single band is consistent with the predicted size of 443 base pairs for CARMIL1a. No band is seen at the position of 308 base pairs, predicted for CARMIL1b. For CARMIL2, the primers flank exon 37, which is present in CARMIL2a but not in CARMIL2b (Matsuzaka et al., 2004). The position of the upper band is consistent with the predicted size of 592 base pairs for CARMIL2a and that of the lower band with 511 base pairs, predicted for CARMIL2b. HT-1080 cells have more CARMIL2b than 2a. For the CP alpha subunit, isoforms alpha1 and alpha2 were detected, consistent with previous studies (Hart et al., 1997).
We cloned cDNAs for human CARMIL1 from HeLa cells by RT-PCR and obtained a single isoform, which we call CARMIL1a (described in detail in Materials and Methods). The GenBank accession number for this cDNA is FJ026014. CARMIL1a is predicted to contain 1371 aa residues and have a molecular weight of 151.6 kDa. The gene is LRRC16A, GeneID 55604, located at chromosome 6p22.2.
Human CARMIL2 is encoded by GeneID 146206 at chromosome 16q22.1. The gene name is RLTPR (RGD motif, leucine-rich repeats, tropomodulin domain, and proline-rich containing). The gene was discovered on the basis of its transcript being down-regulated in the skin of patients with psoriasis vulgaris (Matsuzaka et al., 2004). That study described sequence similarity to Acanthamoeba CARMIL, expression in all of 30 different tissues examined, and evidence for alternative splicing in three regions. The principal transcript is predicted to encode a protein of 1436 aa and Mr 154.7 kDa, which we refer to here as CARMIL2a. We cloned cDNAs for CARMIL2 from HeLa cells by RT-PCR. We found a single splicing variant, which we call CARMIL2b and which has GenBank accession number FJ009082. This variant lacks exon 37 and intronic sequence between exons 14 and 15. CARMIL2b is predicted to contain 1372 aa and have an Mr of 148.2 kDa.
The human gene predicted to encode CARMIL3 is GeneID 90668, named LRRC16B, for “leucine-rich repeat containing 16B” and located at chromosome 14q11.2-q12. The predicted protein, NP_612369, is 1372 aa with Mr 150.3 kDa.
Next, we searched for CARMIL family proteins across eukaryotes with BLAST sequence searches. Predicted proteins similar to CARMIL were found encoded in most eukaryotic genomes, including amoebas, nematodes, insects, fish, birds, and mammals. No genes predicted to encode CARMIL-like proteins were found in plants or fungi, including Saccharomyces cerevisiae and Schizosaccharomyces pombe. Phylogenetic tree analysis showed five clusters of sequences (Figure 1C). CARMIL families 1, 2, and 3 of vertebrates each defined a cluster, as did the CARMILs of insects. For fish, well-characterized genomes had single genes for CARMIL, which clustered with vertebrates. Nematode genomes contained one CARMIL-like protein, which clustered near Acanthamoeba and Dictyostelium CARMIL.
Near their N-terminus, all three of the predicted human proteins contain a region of high sequence similarity, specific for CARMIL, which we refer to as the CARMIL homology domain (CHD; Figure 1, A and B). Following the CHD, the proteins contain a conserved LRR region, a verprolin-like sequence, an acidic region, and a C-terminal proline-rich domain. Within the N-terminal part of the proline-rich region, all three contain a conserved region similar to a region of mouse CARMIL1 that was shown to be necessary and sufficient for binding CP (Yang et al., 2005). We refer here to this region, previously designated C-1 (Yang et al., 2005) or CAH-3 (Uruno et al., 2006), as the CBR.
CARMIL1 and 3 family members are slightly more similar to each other than they are to CARMIL2, based primarily on the CHD (Table 1, Figure 1A). The LRRs, defined by the consensus sequence LxxLxLxxN/CxxL (Enkhbayar et al., 2004), are also more similar to those of other CARMILs than they are to LRRs of non-CARMIL proteins.
Table 1.
Sequence similarity among CARMIL family proteins
| Dicty | Human1a | Human2b | Human3 | Mouse1 | Mouse2 | Mouse3 | |
|---|---|---|---|---|---|---|---|
| Acan125 | 33 | 23 | 21 | 24 | 22 | 13 | 24 |
| Dicty | 23 | 22 | 23 | 23 | 15 | 23 | |
| Human1a | 29 | 39 | 90 | 18 | 39 | ||
| Human2b | 33 | 29 | 86 | 33 | |||
| Human3 | 38 | 21 | 95 |
Percent identity determined with DNASTAR MegAlign and ClustalW.
Verprolin WH2 domains can bind actin subunits and promote nucleation of polymerization (Paunola et al., 2002). Alignment of the sequences of CARMIL with WH2 sequences shows some limited sequence similarity (Supplemental Figure S1A), and this includes some of the conserved residues characteristic of WH2 domains (Chereau et al., 2005). In WASp family proteins, the WH2 domain combines with an acidic Arp2/3-binding region to nucleate actin polymerization. The acidic region of Dictyostelium CARMIL is one continuous region of 72 residues, and that of Acanthamoeba CARMIL is 55 residues; both exhibit strong negative charges (Supplemental Figure S1B). Although the acidic region of CARMIL2 shows a very strong negative charge, those of CARMIL1 and 3 are less strongly negative. The acidic regions of all three human CARMIL proteins are not continuous, but are made up of two or three short pieces (Figure 1A). We looked for evidence of interaction of CARMIL1 with actin or Arp2/3 complex by immunoprecipitation of epitope-tagged CARMIL from HT-1080 cells, but no such evidence was found (Supplemental Figure S2). This result, indicating lack of interaction with actin or Arp2/3, is consistent with previous actin polymerization data for mouse CARMIL1 (Yang et al., 2005).
The CBR is relatively well conserved among the three human CARMIL proteins (Figure 1D). The CBR includes a motif, LxHxTxxRPK, identified as necessary for binding to CP in previous studies with CARMIL1, CD2AP, and CKIP-1; the latter two are otherwise unrelated to CARMIL (Bruck et al., 2006; Canton et al., 2006; Uruno et al., 2006). The CBR is located within a long proline-rich region at the C-terminus of the three human CARMIL proteins. In a separate study, we examined the role of CP-binding by CARMIL (Liang, Y., Kim, T., Niederstrasser, H., Edwards, M., Jackson, C. E., Butler, B., and Cooper, J. A., unpublished data). Purified CARMIL2 CBR had biochemical activities similar to those of CARMIL1, in that it bound CP, prevented capping, and caused uncapping.
The C-terminal region of Dictyostelium and Acanthamoeba CARMIL is able to bind the SH3 domain of certain class-I myosins (Xu et al., 1997; Jung et al., 2001). We found that human CARMIL1a can bind to myosin 1E, an SH3-containing form of myosin 1 in humans (Krendel et al., 2007). Anti-Flag antibodies precipitated endogenous myosin 1E from lysates of HT-1080 cells expressing Flag-tagged CARMIL1 but not CARMIL2 (Supplemental Figure S2).
To determine the expression profile of the three CARMIL genes in cultured cell lines, RT-PCR was performed. CARMIL1 and 2 were expressed in all the cell lines tested (Figure 1E). For CARMIL1 and 2, we chose PCR primers designed to detect the splice forms mentioned above. For CARMIL1, we detected CARMIL1a, the form that we cloned (NCBI FJ026014), but not CARMIL1b (NCBI NM_017640). Differences between CARMIL1a and 1b are described further in Materials and Methods section. For CARMIL2, the sizes of the PCR products were consistent with alternative splicing described previously (Matsuzaka et al., 2004). The major band corresponded to CARMIL2b, the cDNA that we cloned above and use for expression below. CARMIL3 expression was more restricted, detected primarily in immune cells (Figure 1E). In this study, to address the role of CARMIL in cell migration, we chose the human fibrosarcoma cell line HT-1080. Therefore, based on the expression results, we examined the functions of CARMIL1 and 2, but not CARMIL3, in the HT-1080 cells.
Distinct Roles for CARMIL1 and 2 in Cell Migration and Spreading
To test the roles of CARMIL1 and 2 in cell migration, isoform-specific shRNA-expression plasmids, called CARMIL1i and 2i, were constructed. Their efficacy and specificity were tested in HT-1080 and HEK293 cells expressing yellow fluorescent protein (YFP) fusions of CARMIL1 or 2 (Supplemental Figure S3). Each shRNA successfully lowered the level of its target protein isoform without affecting the level of the other isoform. As a negative control, a scrambled shRNA sequence had little to no effect on the level of either isoform. In addition, CARMIL1i decreased the level of endogenous CARMIL1 protein in HT-1080 cells (Figure 2A), and CARMIL2i decreased the level of endogenous CARMIL2 RNA in HT-1080 cells (Figure 2B).
Figure 2.

Knockdown of CARMIL1 or 2 inhibits cell migration during wound healing. (A) Knockdown of endogenous CARMIL1 protein. HT-1080 cells were transfected with vector, scrambled shRNA or shRNA targeting CARMIL1. Immunoblots of whole cell lysates were probed with anti-CARMIL1, as well as anti-CP and anti-actin. (B) Knockdown of endogenous CARMIL2 RNA. Cells were treated as in A, except with shRNA targeting CARMIL2, and RT-PCR was performed to assess the level of RNA expression. Serial twofold dilutions of the control RNA sample showed that the level of knockdown was between two- and fourfold (data not shown). (C) Cell migration during wound healing, monitored by time-lapse phase-contrast microscopy of HT-1080 cells. Images are frames from Movies 1–3. A monolayer of HT-1080 cells expressing scrambled, CARMIL1i, or CARMIL2 shRNA was scratched 3 d after transfection. Yellow lines show the boundary of the wound, the white arrow points in the direction of cell migration, and black arrowheads indicate protrusions and ruffles at the leading edge of cells. (D) Cell speed comparison, based on tracking nuclear position from frame to frame in time-lapse movies. At least 15 cells from two to three independent experiments were analyzed using Image J (http://rsb.info.nih.gov/ij/). Results plotted are mean and SEM; **p <0.001 and ***p < 0.0001.
To assess the effect of CARMIL knockdown on cell migration, a scratch wound-healing assay was performed with HT-1080 cells and time-lapse video microscopy. This human fibrosarcoma cell line grows as a monolayer, and cells migrate rapidly to fill wounds. Based on time-lapse movies, cells expressing either CARMIL1i or 2i shRNA filled the wound gap more slowly than did control scramble-expressing cells (Figure 2C, Movies 1–3). In CARMIL1 knockdown cells at the wound edge, the movies revealed decreases in lamellipodia, ruflling, and macropinosomes, compared with control cells. In CARMIL2 knockdown cells, the leading edge was smooth and flat, with no ruffles or macropinosomes. Also, the leading edge changed direction over time.
To quantitate the results from the wound-healing assay in another way, we calculated cell speed by tracking the displacements of individual cell nuclei over time, from each frame to the next (Figure 2D). The average speed of CARMIL1i cells was greatly decreased (to 29% of control), and the average speed of CARMIL2i cells was also decreased (to 52% of control), by a smaller amount. Both differences were highly statistically significant.
To investigate why depletion of CARMIL1 or 2 impaired cell migration in wound healing, we next examined the actin-rich leading edge of single cells. HT-1080 cells have prominent lamellipodia and filopodia, which are highly dynamic. Cells were cotransfected with an shRNA-knockdown plasmid and an RFP expression plasmid, in order to identify transfected cells and compare them with nontransfected cells. The RFP fluorescence provided a sense of the level of transfection for each cell, allowing one to compare low, medium, and high expressors. This approach provided some indication of whether effects were primary versus secondary, when multiple effects were observed. Time-lapse phase-contrast movies of living cells were collected, and cells were fixed and stained with fluorescent phalloidin to visualize filamentous actin.
In the phalloidin-stained fixed-cell images, control cells were well polarized with multiple lamellipodia at a single broad leading edge (Figure 3A.) Actin filament bundles, which might be called filopodia or microspikes depending on their protrusion length, were embedded in the lamellipodia (Figure 3A). Cells displayed tail retraction at the trailing edge. This overall morphology, which we refer to as unipolar, was seen in 83 ± 0.9% of cells expressing scrambled shRNA (n = 576, Figure 3B).
Figure 3.

Differential effects of CARMIL1 versus CARMIL2 knockdown on lamellipodium formation and cell polarity in HT-1080 cells. (A) Effects on lamellipodia revealed by fluorescence images of cells stained with coumarin-phalloidin. Lamellipodial regions are enlarged in insets. Arrowheads point to the leading edge of migrating cells. An RFP expression plasmid was cotransfected to identify transfected cells. Scale bar, 20 μm on all panels. (B) Scoring of the degree of polarity in cells, as a percentage of total cells. Polarity scored by blind observers as described (Sidani et al., 2007). n = >300 cells per group. Results plotted are mean and SEM from three experiments. Representative cells are shown. (C) Profile of fluorescent phalloidin intensity at the cell edge. The average fluorescence pixel intensity at each distance from the cell edge was determined by an Image J macro as described (Cai et al., 2007). The values were normalized to the highest value of the scrambled group. *p < 0.05 and **p < 0.001. n = >14 cells per group. (D) Effects on cell polarity revealed by time-lapse phase-contrast images, in frames from Movies 4–7. Black arrowheads indicate the leading edge of migrating cells, white arrowheads indicate sites of constriction, and white arrows indicate macropinosomes.
In CARMIL1 knockdown cells, the shape of the cell outline was properly polarized (Figure 3, A and B, 86 ± 1.5% of cells, n = 649), but the intensity of lamellipodial staining by phalloidin was greatly decreased, as illustrated by representative images (Figure 3A). Quantitation of the intensity of staining at the leading edge confirmed that the staining intensity was less, and the curve of intensity versus position was flat, lacking a peak behind the leading edge (Figure 3C). Actin filament bundles resembling short stress fibers were scattered about the cytoplasm (Figure 3A).
The CARMIL2 knockdown cells exhibited phenotypes distinct from those of CARMIL1i cells. Many CARMIL2i cells (44 ± 17%, n = 563) displayed a multipolar morphology, with lamellipodia at several locations on the cortex (Figure 3, A, B, and D). A substantial fraction (20 ± 7%) of CARMIL2i cells had apolar shapes without a distinguishable leading edge or retracting tail (Figure 3B), and only 17 ± 7% of cells had a unipolar shape (Figure 3B). At high magnification, some CARMIL2i lamellipodia were relatively normal, with actin bundles in lamellipodia (Figure 3A), whereas others had decreased lamellipodial actin staining and lacked lamellipodial bundles (Figure 3A). In the quantitation of actin staining at the leading edge (Figure 3C), CARMIL2i cells showed an overall moderate decrease of staining intensity, but the shape of the intensity versus position curve was similar to that of control cells, in contrast to the result for CARMIL1i cells. CARMIL2i cells did not show increased stress fiber-like structures, also in contrast to CARMIL1i cells. Off-target silencing was excluded by rescue expression with RNAi-resistant forms of CARMIL1 or 2, discussed further below in the context of cross-rescue. Thus, CARMIL1 appeared to be required for the assembly of lamellipodial actin, and CARMIL2 appeared necessary primarily for cell polarity, with some contribution to lamellipodial actin assembly.
We followed these observation of fixed cells with movies of living cells migrating on a fibronectin-coated surface. Again, cells were plated at low density to allow observations of individual cells (Figure 3D, Movies 4–7). To quantitate effects objectively, “blind” observers of the movies scored defined parameters. In terms of cell polarity, control scrambled-shRNA cells remained well polarized, with a unipolar shape, for 2 h (89%, 16/18, Figure 3D). During this time, cells moved and changed direction; this was accomplished by retracting existing lamellipodia and extending new lamellipodia in a concerted manner (Figure 3D, Movie 4). Characteristically, new lamellipodia showed prominent ruffling, which led to the formation of phase-bright macropinosomes. For CARMIL1 knockdown cells, the unipolar morphology was maintained (86%, 12/14, Figure 3B); however, lamellipodia and peripheral ruffles were less frequent, short-lived, or not formed at all, whereas macropinosomes were rarely seen (Figure 3D, Movie 5). In contrast, CARMIL2 knockdown cells exhibited with multiple leading edges and frequent changes in directions; individual lamellipodia appeared to pull the cell in different directions (76% of cells, 16/21; Figure 3D, Movies 6 and 7). Some of these leading edges ultimately became long tails. At the leading edge, some CARMIL2 knockdown cells had regions deficient in lamellipodia formation, membrane ruffling, and macropinosome formation.
To compare the effects on lamellipodia in CARMIL1 knockdown versus CARMIL2 knockdown cells in detail, we viewed the leading edge at high magnification with high time resolution (Figure 4A). Control scrambled cells exhibited cycles of protrusion and withdrawal, accompanied by ruffling and macropinosome formation (Figure 4A, Movie 8). At the leading edge of CARMIL1i cells, membrane protrusions were smaller and extended more slowly and less frequently over time (Figure 4A, Movie 9). Ruffles were small and infrequent, and no macropinosomes appeared to form. Focusing on the abnormal lamellipodial regions of CARMIL2i cells, we saw persistent protrusions at the leading edges, but they were smooth, slow and free of ruffles and macropinosomes (Figure 4A, Movie 10).
Figure 4.

Effects of CARMIL1 versus 2 knockdown on lamellipodial dynamics and cell spreading. (A) Lamellipodial dynamics in frames from phase-contrast time-lapse Movies 8–10. Arrowheads indicate the cell edge. Scale bar, 20 μm. (B) Cell spreading, in frames from phase-contrast time-lapse Movies 11–13. White arrowheads, lamellipodia with features described in the text.
To gain additional insight into the roles of CARMIL1 and 2, we examined how individual cells spread on a surface, with time-lapse movies, with observations scored by blind observers (Figure 4B, Table 2, Movies 11–13). Control scrambled cells extended and retracted membrane protrusions, with prominent ruffling, all along the cell perimeter (Figure 4B, inset, Movie 11). At ∼18 min, cells became polarized with a major lamellipodial protrusion on one side of the cell and retraction fibers on the other side (Table 2). This morphology persisted until the end of the 30-min movie, with few cells (14%) changing direction. In CARMIL1 knockdown cells, cells initially spread well, with intense ruffling. However, after ∼10 min, the edge of most cells (75%) consisted of aberrant structures that were small, phase-dark, and shaped like blebs or fingers (Figure 4B, inset, Movie 12). CARMIL1i cells became polarized at 17 min and did not change direction, similar to control cells (Table 2). Spreading of CARMIL2 knockdown cells differed from that of control or CARMIL1i cells (Figure 4B, inset, Table 2, Movie 13). As CARMIL2i cells spread, the cell edge was smooth without ruffles. Retraction fibers appeared early, and the cells did not adopt a well-polarized shape until late. Moreover, most cells changed direction. Thus, consistent with the observations on migrating cells, spreading CARMIL2i cells showed decreased protrusion/retraction activity, impaired ruffle formation, and disrupted polarity.
Table 2.
Effect of CARMIL knockdown on cell spreading
| Maintenance ofprotrusion/retraction(% of cells) | Change of direction(% of cells) | Smooth edgewithout ruffles(% of cells) | Appearance ofretractionfibers (min) | Onset ofpolarization (min) | No. ofcells | |
|---|---|---|---|---|---|---|
| Scrambled | 100 | 14 | 0 | 18.3 ± 1.9 | 18.3 ± 1.9 | 7 |
| CARMIL1i | 25 | 13 | 0 | 17.4 ± 1.8 | 17.4 ± 1.8 | 8 |
| CARMIL2i | 13 | 63 | 75 | 12.8 ± 2.0 | 21.5 ± 3.7 | 8 |
Time-lapse phase-contrast microscope movies of living cells were observed. Cells that maintained protrusion and retraction of lamellipodia over a 30-min time course were scored, as were cells that changed the direction of their protrusions. The times when retraction fibers appeared and polarization began were determined. Values are mean ± SEM.
Effects of CARMIL1 on Lamellipodial Actin
To understand more fully the role of CARMIL1 in lamellipodia formation and dynamics, we localized molecular markers and free barbed ends in the cells. For free barbed ends, we used a permeabilized-cell in situ assay (Symons and Mitchison, 1991; Bryce et al., 2005). We found incorporation of fluorescent actin at the leading edge, reflecting the number of free barbed ends, to be much less in CARMIL1 knockdown cells than in control scrambled cells (Figure 5A). We stained cells with antibodies against Arp2/3, cortactin and VASP, proteins that function at the leading edge to control lamellipodial architecture and dynamics (Bear et al., 2002). CARMIL1 knockdown cells showed decreased Arp2/3 and cortactin staining at the leading edge (Figure 5B). VASP staining was enriched at the leading edge in control cells, especially at tips of actin filament bundles (Figure 5C). In CARMIL1 knockdown cells, the intensity of VASP staining at the leading edge was greatly decreased. Taken together, these results show that the loss of CARMIL1 leads to a decrease of each of all the molecular components of lamellipodia that were examined. In contrast, knockdown of CARMIL2 often had little to no effect on Arp2/3 and cortactin staining at the leading edge, as described more fully below (see Figure 7B and data not shown).
Figure 5.

Effect of CARMIL1 knockdown on molecular markers and GTP-Rac1 activity. (A) Free barbed ends of actin filaments, assessed by incorporation of fluorescent actin in permeabilized cells. Scale bar, 20 μm on all panels. (B and C) Cells stained with antibodies to Arp2/3, cortactin, or VASP and costained with coumarin-phalloidin. Asterisks indicate transfected cells. Arrows in C indicate tips of filopodia. (D) Levels of GTP-Rac1 activity in cells spreading on fibronectin, over time in minutes. GTP-Rac1 was pulled down with PAK-PBD, and precipitates were analyzed by immunoblot.
Figure 7.

Effect of CARMIL knockdown on Myosin-II. (A) Cells stained with anti-Myosin-IIA heavy chain and anti-paxillin Abs. Arrowheads, lamellipodia structures; arrows, focal adhesions. GFP-actin is a transfection marker. (B) Cells stained with anti-Myosin-IIB heavy chain and anti-Cortactin Abs. Arrows, Myosin-IIB localization; arrowheads, the leading edge of cells. Scale bar, 20 μm. (C) Expression of Myosin-II heavy chain isoforms in cells, analyzed by immunoblot.
To gain insight into the molecular mechanism of CARMIL action in lamellipodia formation, we measured the activation of Rac1 over time in spreading cells (Figure 5D). In control scrambled and CARMIL2i cells, the level of active Rac1 was increased at 15 and 30 min. In contrast, in CARMIL1i cells, the level of active Rac1 did not change. A recent study in C. elegans found biochemical and genetic interactions between CARMIL and Trio/UNC-73, a large protein with both Rac- and Rho-GEF domains (Vanderzalm et al., 2009). We looked for a biochemical interaction between CARMILs and Trio in our cells, by coimmunoprecipitation. We observed coprecipitation of human Trio with CARMIL1, but not with CARMIL2 (Supplemental Figure S2).
Overall, these results indicate that CARMIL1 contributes to the dendritic actin network assembly of lamellipodia and ruffles, apparently upstream of Rac1 with the possible involvement of Trio. CARMIL2 appears to have some role in lamellipodial actin assembly, but the role appears to be smaller and less important than that of CARMIL1.
Effects of CARMIL2 on Cell Polarity: Microtubules and Myosin-II
Cell polarity, including the polarity of migrating cells, often involves the position of the microtubules and the microtubule-organizing center (MTOC; Gomes et al., 2005; Siegrist and Doe, 2007). Because loss of CARMIL2 conferred an apparent multipolar phenotype, we examined cell polarity. First, we asked whether the orientation of the microtubule cytoskeleton was affected. Cells were costained with antibodies against microtubules and the Golgi complex, which often colocalizes with the MTOC. Control scrambled cells displayed a unipolar morphology, with microtubules and the Golgi located between the nucleus and the leading edge (Figure 6, A and B.) CARMIL2 knockdown cells showed poor polarity, with the MTOC and Golgi situated either behind the nucleus or off to one side, but not in the direction of the leading edge (Figure 6, A and B). γ-Tubulin, which marks the MTOC, and acetylated tubulin, which marks stable microtubules, were present but not polarized properly (Figure 6C). In contrast to CARMIL2, CARMIL1 knockdown cells showed a well-polarized microtubule morphology, indistinguishable from control scrambled cells (Figure 6B and data not shown).
Figure 6.

Effect of CARMIL2 knockdown on polarity in migrating cells. (A) Fluorescence images of CARMIL2 knockdown cells stained for giantin of the Golgi complex and for microtubules. GFP-actin is a transfection marker. Arrows indicate the MTOC/Golgi. Insets show the relationship of microtubules with actin at the cell cortex. (B) MTOC polarization scored from images like those in A. Data are plotted as percent of cells ± SE of proportion; n = >100 cells per sample. (C) Cells stained with fluorescent antibodies for acetylated tubulin and γ-tubulin. Arrowheads indicate the centrosome. Scale bar, 20 μm.
Examining the interface between the microtubule cytoskeleton and the leading edge at high magnification, many distal ends of microtubules appeared to contact the actin-rich lamellipodium in control cells (Figure 6A, inset). In CARMIL2 knockdown cells, the density of microtubule ends was less. Also, as noted above, bundles of actin filaments were absent in the lamellipodial actin.
Conventional class-II myosin has been implicated in cell polarity during cell migration, and migrating fibroblasts express two of the three genes for myosin-II, A and B (Lo et al., 2004; Vicente-Manzanares et al., 2007). To examine the role of myosin-II in cells lacking CARMIL, we stained cells with antibodies to myosin-IIA and myosin-IIB. Costaining with antibodies to paxillin and cortactin provided markers for focal adhesions and lamellipodial actin, respectively. In control cells, myosin-IIA and -IIB were mainly concentrated in the central part of the cell, with little to no staining of the leading edge (Figure 7, A and B), consistent with previous findings (Vicente-Manzanares et al., 2007).
In CARMIL2 knockdown cells, the most notable change was a decrease in the overall intensity of staining for myosin-IIB (Figure 7B), with no apparent change in the intensity of myosin-IIA staining (Figure 7A). These results were confirmed by immunoblots (Figure 7C). In addition, in CARMIL2 knockdown cells, the anti-paxillin-stained focal adhesions were more numerous and less intense (Figure 7A). For CARMIL1 knockdown cells, the staining intensity for myosin-IIA and -IIB was relatively normal, confirmed by immunoblot (Figure 7C). The number and staining intensity of stress-fiber-like structures was increased, consistent with the phalloidin staining described above (Figures 3A and 5), and the intensity of anti-paxillin staining at the ends of the stress fibers was increased.
Thus, the polarity defect of CARMIL2-deficient cells is accompanied by defects in the microtubule cytoskeleton and myosin-II, none of which have been previously implicated in the function of CARMIL.
Localization of CARMIL1 and 2
To gain further insight into the roles of CARMIL1 and 2 in cell migration, we performed subcellular localization studies. We localized YFP-fusions of full-length CARMIL1 and 2, expressed at low level (Figure 8, A and B; see Materials and Methods for details of the cDNAs used). Endogenous CARMIL1 and 2 were difficult to visualize by immunofluorescence because of weak staining. In a parallel study, we documented the functionality of these YFP fusions (Liang, Y., Kim, T., Niederstrasser, H., Edwards, M., Jackson, C. E., Butler, B., and Cooper, J. A., unpublished data). Each fusion rescued shRNA-knockdown phenotypes fully, based on qualitative and quantitative assays of their characteristic traits, and each fusion immunoprecipitated endogenous CP.
Figure 8.

Localization of CARMIL1 and 2. (A) HT-1080 cells expressing YFP, YFP-CARMIL1, or YFP-CARMIL2, stained with phalloidin-coumarin. Scale bar, 20 μm. (B) Cells cotransfected with RFP-CARMIL1 and YFP-CARMIL2 and then stained with anti-cortactin Abs. Arrowheads indicate areas of the leading edge where CARMIL1 and 2 localizations differ. Merge includes RFP-CARMIL1 and YFP-CARMIL2. (C) Sequential images of live cells expressing YFP-CARMIL1 or YFP-CARMIL2. Arrowheads point to macropinosome formation. Boxed regions are enlarged to show the detail. See Movies 14–15.
For YFP-CARMIL1, fluorescence was enriched at the leading edge of migrating HT-1080 cells (Figure 8, A and B), consistent with its role in lamellipodial formation. Similar results were obtained with migrating SNB-19 and B16-F1 cells (Supplemental Figure S4), consistent with previous findings (Yang et al., 2005). As a negative control, YFP alone was evenly distributed throughout the cytoplasm (Figure 8A).
Migrating HT1080 cells often form macropinosomes (Figures 2C and 3D). We found that YFP-CARMIL1 was enriched in newly formed macropinosomes (Figure 8C, Movie 14), as previously reported for Dictyostelium (Jung et al., 2001). The association of CARMIL1 macropinosomes was transient, disappearing as the vesicles moved into the cell (Movie 14). Taken together, the localization of CARMIL1 to lamellipodia and macropinosomes confirms its role in actin assembly dynamics at the plasma membrane.
To investigate how CARMIL1 was recruited to the leading edge, we depleted Arp2/3 complex by siRNA knockdown of the p20 subunit (Supplemental Figure S5, A and B). YFP-CARMIL1 localization was lost, along with the cortactin and phalloidin staining characteristic of lamellipodial actin (Supplemental Figure S5, B and C). In contrast, in a concurrent set of experiments described in another study, depletion of CP, which enhances actin polymerization at the leading edge, did not impair YFP-CARMIL1 localization there (Liang, Y., Kim, T., Niederstrasser, H., Edwards, M., Jackson, C. E., Butler, B., and Cooper, J. A., unpublished data). Thus, CARMIL1 appears to be recruited to lamellipodia and to be important for their formation, suggesting its participation in a positive feedback cycle.
In contrast, YFP-CARMIL2 fluorescence was not concentrated at the actin-rich leading edge (Figure 8A) or at macropinosomes (Figure 8C, Movie 15). This difference was seen most clearly in a direct comparison of RFP-CARMIL1 with YFP-CARMIL2 in doubly transfected cells (Figure 8B). Instead, most CARMIL2 fluorescence was distributed throughout the cytoplasm in a coarse fibrillar pattern reminiscent of intermediate filaments (Figure 8A). Indeed, staining of YFP-CARMIL2 cells with antibodies to vimentin revealed extensive colocalization (Figure 9A and Supplemental Figure S6A). Treatment of these cells with WFA, a specific inhibitor of vimentin filament assembly (Bargagna-Mohan et al., 2007), led to collapse of the vimentin filament network, with relatively little effect on microtubules or actin. YFP-CARMIL2 colocalized with the collapsed vimentin filaments (Figure 9A). The effect of WFA on vimentin and CARMIL2 depended on the concentration and time of WFA treatment.
Figure 9.

CARMIL2 colocalization with vimentin filaments. (A) Cells were treated with withaferin A (WFA) under conditions chosen for maximal collapse of vimentin filaments with minimal effects on microtubules. Fixed cells were stained for vimentin, microtubules, and filamentous actin. Scale bar, 20 μm. (B) Efficacy of vimentin knockdown determined by immunoblot. (C) Effects of vimentin knockdown on CARMIL2 localization. Control scrambled and vimentin-knockdown cells were cotransfected with YFP-CARMIL2 and stained with anti-vimentin antibody. Scale bar, 20 μm. (D) Fluorescence images showing colocalization of YFP-CARMIL2 with vimentin but not keratin in A549 cells. Control and WFA-treated cells are shown.
Vimentin intermediate filaments are also known to collapse toward the nucleus when microtubules are depolymerized, based on a dynamic association between the two filament systems (Gurland and Gundersen, 1995; Ho et al., 1998). In our cells, nocodazole treatment completely depolymerized microtubules and also caused the vimentin intermediate filaments to collapse (Supplemental Figure S6B). The distribution of YFP-CARMIL2 mirrored that of vimentin, during nocodazole treatment and washout.
In addition, we examined the localization of CARMIL2 in vimentin-depleted cells, using shRNA to knock down vimentin. Vimentin depletion was nearly complete (Figure 9, B and C). In the absence of vimentin, CARMIL2 had a diffuse pattern, not a fibrillar one (Figure 9C). Conversely, we examined the distribution of vimentin in CARMIL2 knockdown cells, by immunofluorescence staining, and we observed no gross effects (data not shown).
We investigated the specificity of the association of CARMIL2 with vimentin by localizing CARMIL2 in the A549 cell line, which expresses both vimentin and keratin intermediate filament proteins. Vimentin and keratin staining showed distinct patterns, and the pattern of CARMIL2 localization was similar to that of vimentin and different from that of keratin (Figure 9D). Moreover, the collapse of vimentin induced by WFA in these cells caused a collapse of CARMIL2 staining with little effect on the distribution of keratin (Figure 9D). Thus, CARMIL2 appears to have a specific association with vimentin intermediate filaments, which has not been reported for any CARMIL protein.
Failure of Cross-Rescue between CARMIL1 and 2
To address directly the question of overlapping versus distinct functions for CARMIL1 and 2, we asked whether expression of one isoform was able to rescue or suppress the knockdown phenotypes of the other. Using shRNA plasmids, we knocked down each isoform and then expressed an RNAi-resistant cDNA, constructed as a YFP-CARMIL fusion, which allowed us to identify transfected cells and assess the relative level of expression. In control experiments in a parallel study (Liang, Y., Kim, T., Niederstrasser, H., Edwards, M., Jackson, C. E., Butler, B., and Cooper, J. A., unpublished data), wild-type CARMIL1 expression rescued CARMIL1 knockdown phenotypes, and wild-type CARMIL2 rescued CARMIL2 knockdown phenotypes. Here, neither isoform was able to rescue the knockdown of the other one in any noticeable manner (Figure 10), showing that the cellular functions of CARMIL1 and 2 are distinct.
Figure 10.
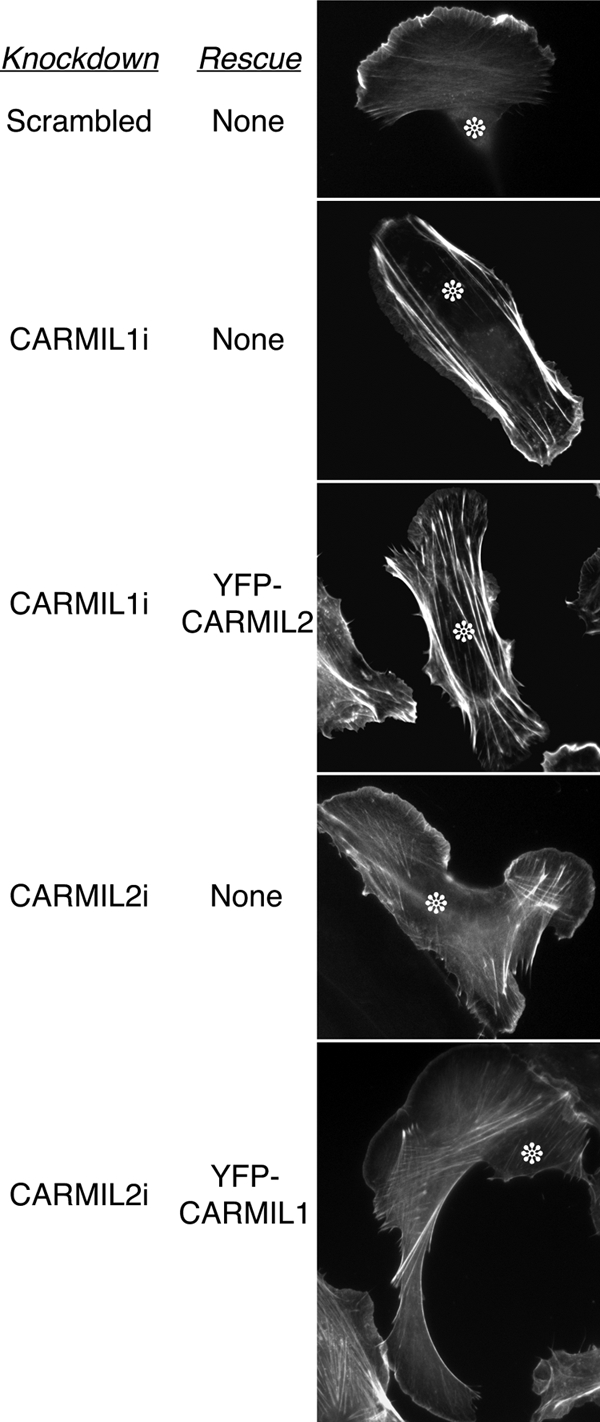
Cross-rescue expression analysis. CARMIL1 knockdown cells expressing CARMIL2, and vice versa. Cells are stained with coumarin-phalloidin. The characteristic phenotypes were not rescued in either case. In controls, each expression plasmid rescues its own knockdown phenotypes (Liang et al., 2009).
Overexpression
To gain further insight into the functions of CARMILs, we asked how overexpression of CARMILs might affect cell motility and cell migration by expressing YFP-CARMIL fusions in otherwise normal HT-1080 cells, which had normal levels of endogenous CARMILs. First, we observed that YFP-CARMIL1 expression caused alterations in lamellipodia (Figure 8A, Supplemental Figure S7; Liang, Y., Kim, T., Niederstrasser, H., Edwards, M., Jackson, C. E., Butler, B., and Cooper, J. A., unpublished data). At low expression levels, the intensity of phalloidin staining in the lamellipodium was slightly decreased. At higher expression levels, normal lamellipodia were not seen; instead, abnormal small, contorted protrusions were present. Movies (not shown) confirmed that these structures were protrusions and not the end-product of retractions. With further increases in the expression levels of YFP-CARMIL1, the leading edge adopted an irregular shape, with long narrow protrusions, rather than the single broad sheet seen normally. These irregular protrusions contained Arp2/3, CP, and cortactin along their periphery, by immunofluorescence (Supplemental Figure S7). In contrast, VASP was not detected at the tips of these structures. Thus, these protrusions appear to be abnormal forms of lamellipodia, not filopodia.
For CARMIL2, expression of YFP-CARMIL2 did not result in a detectable effect on the shape of the cell or the distribution of cytoskeletal filaments, including vimentin intermediate filaments, microtubules or actin, even at high levels of expression (Figures 8A and 9A, Supplemental Figure S6, A and B, and data not shown).
DISCUSSION
The most important conclusions from our study relate to the function of CARMIL in cells. First, the CARMIL1 and 2 isoforms have distinct functions, consistent with their conservation across vertebrates. Second, CARMIL2 is necessary for cell polarity, a novel finding for a CARMIL family protein, and CARMIL2 is associated with vimentin filaments.
The conclusion that CARMIL1 and 2 have distinct functions is based on observations of different knockdown phenotypes, protein localizations, and overexpression phenotypes. Furthermore, expression of CARMIL2 did not rescue knockdown phenotypes of CARMIL1-deficient cells and vice versa. In addition, we found that CARMIL1 homodimerized with itself, as Acanthamoeba CARMIL does (Remmert et al., 2004), but CARMIL1 did not heterodimerize with CARMIL2, based on immunoprecipitation with pairs of different epitope tags (data not shown).
Phylogenetic analysis revealed conserved CARMIL-encoding genes in a wide variety of metazoans, including insects and amoebae. Most interesting, we found that vertebrates have three conserved isoforms of CARMIL, expressed from different genes. On this basis alone, one might expect CARMILs to have distinct roles in animal cells.
As cell migration is a defining feature of animals, we focused our studies of CARMIL function on cell migration, using predominantly the human fibrosarcoma cell line HT-1080. We found that these cells and most other cell lines express CARMIL1 and 2, and we cloned full-length cDNAs to avoid relying on genome-based predictions. CARMIL3 showed very low expression in HT-1080 cells and in most cells and tissues, so we did not include it in this analysis. CARMIL3 appears to be expressed in immune and other hematopoietic cells, where its function remains to be explored.
For CARMIL1, our results show that it functions in lamellipodia in the control of actin assembly, consistent with previous studies in Dictyostelium and cultured human cells (Jung et al., 2001; Yang et al., 2005). We found that CARMIL1 localized to lamellipodia, and that RNAi-mediated knockdown of CARMIL1 led to inhibition of lamellipodial actin assembly. Expression of CARMIL1 at low levels over the endogenous level also caused inhibition of lamellipodial actin. At high levels of expression, CARMIL1 caused extreme distortion of lamellipodial structures. Together, the results suggest a complex nonlinear role for CARMIL. In CARMIL1 knockdown cells, Rac1 activity was decreased, consistent with inhibition of lamellipodia. In addition, we found that CARMIL1 associates with the Rac-GEF Trio, as recently observed in C. elegans (Vanderzalm et al., 2009). Thus, CARMIL1 may control lamellipodial actin assembly via effects on Trio and Rac1.
For CARMIL2, one might have hypothesized a priori that it would function in a manner similar to CARMIL1, being a closely related isoform. This was not the case. First, CARMIL2 localized to vimentin intermediate filaments, not to lamellipodia or the leading edge. This conclusion is based on knocking down vimentin and on treatment with a vimentin pharmacologic inhibitor, WFA. In addition, in cells expressing both keratin and vimentin, CARMIL2 colocalized with vimentin but not keratin filaments, providing evidence for specificity.
Second, cells depleted of CARMIL2, by RNAi knockdown, showed a distinctive multipolar phenotype not observed in CARMIL1 knockdown cells. Snapshots of migrating cells showed that one cell would often have two to four leading edges, and movies showed that the multiple leading edges would form, move forward, and disassemble with no apparent coordination. In particular, the knockdown cells seemed to suffer from the inability to disassemble or suppress the minor leading edges, and cells often stretched themselves thin as their different parts attempted to walk away from each other. Sometimes, the leading edges showed flat and smooth protrusions, with decreased actin and little ruffling or macropinocytosis, whereas sometimes the leading edges were relatively normal. These effects may be a consequence of having multiple leading edges; actin assembly is a cooperative process, so its occurrence at one location might decrease the availability of subunits for assembly at some of them.
How might a protein associated with vimentin intermediate filaments affect cell migration? In previous studies, the cellular level of vimentin was found to be proportional to the ability of cells to migrate, based on expression, antisense and knockout approaches (Eckes et al., 1998; Gilles et al., 1999). Microtubules and myosin-II are both important for cell migration (Gomes et al., 2005). Here, we observed the microtubule cytoskeleton to be improperly polarized and myosin-IIB levels to be decreased in CARMIL2-knockdown cells. In a previous study, the phenotype of embryonic fibroblasts from myosin-IIB knockout mice included evidence for multipolarity (Lo et al., 2004), reminiscent of the CARMIL2- knockdown phenotype here. The molecule basis of the connections between CARMIL2, vimentin, and myosin-IIB remain to be explored.
In addition, we found that vimentin filaments depend on microtubules for assembly, but not vice versa, based on pharmacologic inhibitor results, consistent with previous studies (Shohat et al., 1976a,b; Gurland and Gundersen, 1995). However, we found that simple drug treatments alone, affecting either microtubules or vimentin, did not cause the cells to become multipolar in the manner seen with loss of CARMIL2. Multiple previous studies have found that vimentin filament assembly depends on microtubules, but there has been little evidence for the converse (Gyoeva and Gelfand, 1991; Chou et al., 2007; Shim et al., 2008). In one study, inhibition of vimentin assembly decreased the formation of cell “microtentacles,” which contain microtubules (Whipple et al., 2008). On the other hand, in many cell systems complete collapse of vimentin intermediate filaments has had no effect on microtubules (Klymkowsky, 1981, 1983; Lin and Feramisco, 1981; Gyoeva and Gelfand, 1991). To understand how CARMIL2 may link vimentin filaments with other elements of the cytoskeleton and cell polarity will require further study.
The molecular phylogeny of CARMILs shows conservation in multiple domains of this large protein, raising the possibility of multiple interactions and functions. Initial studies with amoeba CARMILs suggesting binding to CP, Arp2/3, actin, and myosin-I as potential bases of function. Previous work with the CARMIL1 isoform of vertebrates suggested a role in lamellipodial actin assembly (Yang et al., 2005). Our results here confirm that idea and show that the function of the CARMIL2 isoform, also expressed in migrating cells, is distinct. Further work will be required to identify the molecular basis of action of the CARMIL isoforms. Our identification here of the high conservation of CHD focuses attention on this region, to be considered with the multiple other regions defined previously by sequence analysis and biochemical studies. In addition, one other feature shared by the two isoforms is that both interact with some form of myosin: CARMIL1 with myosin-IE and CARMIL2 with myosin-IIB.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Drs. Mira Krendel and Mark Mooseker (Yale University) for antibodies to myosin 1E, Dr. Ilgu Kang of our lab for anti-CARMIL1, Dr. Frank Gertler (MIT) for anti-VASP antibody, Dr. Paul Bridgman (Washington University) for rabbit anti-Myosin-IIB antibody, Dr. James Lessard (University of Cincinnati) for C4 monoclonal anti-actin antibody, Dr. Narashimhan Gautam (Washington University) for the A549 cell line, and Dr. Yunfeng Feng of the Longmore lab (Washington University) for the vimentin knockdown plasmid and for assistance with construction of shRNA plasmids. We thank our lab colleagues for their comments and assistance with the project and the manuscript. This work was supported by National Institutes of Health (NIH) R01 Grant GM 38542. M.E. was supported by NIH training grant T90 DA022871.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-10-1071) on October 21, 2009.
REFERENCES
- Applewhite D. A., Barzik M., Kojima S., Svitkina T. M., Gertler F. B., Borisy G. G. Ena/VASP proteins have an anti-capping independent function in filopodia formation. Mol. Biol. Cell. 2007;18:2579–2591. doi: 10.1091/mbc.E06-11-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargagna-Mohan P., et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 2007;14:623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear J. E., et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Bruck S., Huber T. B., Ingham R. J., Kim K., Niederstrasser H., Allen P. M., Pawson T., Cooper J. A., Shaw A. S. Identification of a novel inhibitory actin-capping protein binding motif in CD2-associated protein. J. Biol. Chem. 2006;281:19196–19203. doi: 10.1074/jbc.M600166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryce N. S., Clark E. S., Leysath J. L., Currie J. D., Webb D. J., Weaver A. M. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr. Biol. 2005;15:1276–1285. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Cai L., Marshall T. W., Uetrecht A. C., Schafer D. A., Bear J. E. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128:915–929. doi: 10.1016/j.cell.2007.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton D. A., Olsten M. E., Niederstrasser H., Cooper J. A., Litchfield D. W. The role of CKIP-1 in cell morphology depends on its interaction with actin-capping protein. J. Biol. Chem. 2006;281:36347–36359. doi: 10.1074/jbc.M607595200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereau D., Kerff F., Graceffa P., Grabarek Z., Langsetmo K., Dominguez R. Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc. Natl. Acad. Sci. USA. 2005;102:16644–16649. doi: 10.1073/pnas.0507021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y. H., Flitney F. W., Chang L., Mendez M., Grin B., Goldman R. D. The motility and dynamic properties of intermediate filaments and their constituent proteins. Exp. Cell Res. 2007;313:2236–2243. doi: 10.1016/j.yexcr.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Eckes B., et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 1998;111:1897–1907. doi: 10.1242/jcs.111.13.1897. [DOI] [PubMed] [Google Scholar]
- Enkhbayar P., Kamiya M., Osaki M., Matsumoto T., Matsushima N. Structural principles of leucine-rich repeat (LRR) proteins. Proteins. 2004;54:394–403. doi: 10.1002/prot.10605. [DOI] [PubMed] [Google Scholar]
- Gilles C., Polette M., Zahm J. M., Tournier J. M., Volders L., Foidart J. M., Birembaut P. Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci. 1999;112:4615–4625. doi: 10.1242/jcs.112.24.4615. [DOI] [PubMed] [Google Scholar]
- Gomes E. R., Jani S., Gundersen G. G. Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell. 2005;121:451–463. doi: 10.1016/j.cell.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Gurland G., Gundersen G. G. Stable, detyrosinated microtubules function to localize vimentin intermediate filaments in fibroblasts. J. Cell Biol. 1995;131:1275–1290. doi: 10.1083/jcb.131.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoeva F. K., Gelfand V. I. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature. 1991;353:445–448. doi: 10.1038/353445a0. [DOI] [PubMed] [Google Scholar]
- Hart M. C., Korshunova Y. O., Cooper J. A. Vertebrates have conserved capping protein alpha isoforms with specific expression patterns. Cell Motil. Cytoskelet. 1997;38:120–132. doi: 10.1002/(SICI)1097-0169(1997)38:2<120::AID-CM2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Ho C. L., Martys J. L., Mikhailov A., Gundersen G. G., Liem R. K. Novel features of intermediate filament dynamics revealed by green fluorescent protein chimeras. J. Cell Sci. 1998;111:1767–1778. doi: 10.1242/jcs.111.13.1767. [DOI] [PubMed] [Google Scholar]
- Jung G., Remmert K., Wu X., Volosky J. M., Hammer J. A. The Dictyostelium CARMIL protein links capping protein and the Arp2/3 complex to type I myosins through their SH3 domains. J. Cell Biol. 2001;153:1479–1497. doi: 10.1083/jcb.153.7.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymkowsky M. W. Intermediate filaments in 3T3 cells collapse after intracellular injection of a monoclonal anti-intermediate filament antibody. Nature. 1981;291:249–251. doi: 10.1038/291249a0. [DOI] [PubMed] [Google Scholar]
- Klymkowsky M. W., Miller R. H., Lane E. B. Morphology, behavior, and interaction of cultured epithelial cells after the antibody-induced disruption of keratin filament organization. J. Cell Biol. 1983;96:494–509. doi: 10.1083/jcb.96.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koestler S. A., Auinger S., Vinzenz M., Rottner K., Small J. V. Differentially oriented populations of actin filaments generated in lamellipodia collaborate in pushing and pausing at the cell front. Nat. Cell Biol. 2008;10:306–313. doi: 10.1038/ncb1692. [DOI] [PubMed] [Google Scholar]
- Krendel M., Osterweil E. K., Mooseker M. S. Myosin 1E interacts with synaptojanin-1 and dynamin and is involved in endocytosis. FEBS Lett. 2007;581:644–650. doi: 10.1016/j.febslet.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F. P., Szczodrak M., Block J., Faix J., Breitsprecher D., Mannherz H. G., Stradal T. E., Dunn G. A., Small J. V., Rottner K. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 2008;27:982–992. doi: 10.1038/emboj.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Le Clainche C., Carlier M. F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 2008;88:489–513. doi: 10.1152/physrev.00021.2007. [DOI] [PubMed] [Google Scholar]
- Liang Y., Yu W., Li Y., Yang Z., Yan X., Huang Q., Zhu X. NUDEL functions in membrane traffic mainly through association with Lis1 and cytoplasmic dynein. J. Cell Biol. 2004;164:557–566. doi: 10.1083/jcb.200308058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. J., Feramisco J. R. Disruption of the in vivo distribution of the intermediate filaments in fibroblasts through the microinjection of a specific monoclonal antibody. Cell. 1981;24:185–193. doi: 10.1016/0092-8674(81)90514-6. [DOI] [PubMed] [Google Scholar]
- Lo C. M., Buxton D. B., Chua G. C., Dembo M., Adelstein R. S., Wang Y. L. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration. Mol. Biol. Cell. 2004;15:982–989. doi: 10.1091/mbc.E03-06-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaka Y., Okamoto K., Mabuchi T., Iizuka M., Ozawa A., Oka A., Tamiya G., Kulski J. K., Inoko H. Identification, expression analysis and polymorphism of a novel RLTPR gene encoding a RGD motif, tropomodulin domain and proline/leucine-rich regions. Gene. 2004;343:291–304. doi: 10.1016/j.gene.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Mejillano M. R., Kojima S., Applewhite D. A., Gertler F. B., Svitkina T. M., Borisy G. G. Lamellipodial versus filopodial mode of the actin nanomachinery; pivotal role of the filament barbed end. Cell. 2004;118:363–373. doi: 10.1016/j.cell.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Paunola E., Mattila P. K., Lappalainen P. WH2 domain: a small, versatile adapter for actin monomers. FEBS Lett. 2002;513:92–97. doi: 10.1016/s0014-5793(01)03242-2. [DOI] [PubMed] [Google Scholar]
- Pollard T. D. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- Remmert K., Olszewski T. E., Bowers M. B., Dimitrova M., Ginsburg A., Hammer J. A., 3rd. CARMIL is a bona fide capping protein interactant. J. Biol. Chem. 2004;279:3068–3077. doi: 10.1074/jbc.M308829200. [DOI] [PubMed] [Google Scholar]
- Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schafer D. A., Jennings P. B., Cooper J. A. Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J. Cell Biol. 1996;135:169–179. doi: 10.1083/jcb.135.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer D. A., Korshunova Y. O., Schroer T. A., Cooper J. A. Differential localization and sequence analysis of capping protein beta-subunit isoforms of vertebrates. J. Cell Biol. 1994;127:453–465. doi: 10.1083/jcb.127.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S. Y., Samuels B. A., Wang J., Neumayer G., Belzil C., Ayala R., Shi Y., Shi Y., Tsai L. H., Nguyen M. D. Ndel1 controls the dynein-mediated transport of vimentin during neurite outgrowth. J. Biol. Chem. 2008;283:12232–12240. doi: 10.1074/jbc.M710200200. [DOI] [PubMed] [Google Scholar]
- Shohat B., Ben-Bassat M., Shaltiel A., Joshua H. The effect of withaferin A on human peripheral blood lymphocytes. An electron-microscope study. Cancer Lett. 1976a;2:63–70. doi: 10.1016/s0304-3835(76)80013-4. [DOI] [PubMed] [Google Scholar]
- Shohat B., Shaltiel A., Ben-Bassat M., Joshua H. The effect of withaferin A, a natural steroidal lactone, on the fine structure of S-180 tumor cells. Cancer Lett. 1976b;2:71–77. doi: 10.1016/s0304-3835(76)80014-6. [DOI] [PubMed] [Google Scholar]
- Sidani M., et al. Cofilin determines the migration behavior and turning frequency of metastatic cancer cells. J. Cell Biol. 2007;179:777–791. doi: 10.1083/jcb.200707009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist S. E., Doe C. Q. Microtubule-induced cortical cell polarity. Genes Dev. 2007;21:483–496. doi: 10.1101/gad.1511207. [DOI] [PubMed] [Google Scholar]
- Svitkina T. M., Bulanova E. A., Chaga O. Y., Vignjevic D. M., Kojima S., Vasiliev J. M., Borisy G. G. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 2003;160:409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons M. H., Mitchison T. J. Control of actin polymerization in live and permeabilized fibroblasts. J. Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uruno T., Remmert K., Hammer J. A. CARMIL is a potent capping protein antagonist: identification of a conserved CARMIL domain that inhibits the activity of capping protein and uncaps capped actin filaments. J. Biol. Chem. 2006;281:10635–10650. doi: 10.1074/jbc.M513186200. [DOI] [PubMed] [Google Scholar]
- van Rheenen J., Song X., van Roosmalen W., Cammer M., Chen X., Desmarais V., Yip S. C., Backer J. M., Eddy R. J., Condeelis J. S. EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J. Cell Biol. 2007;179:1247–1259. doi: 10.1083/jcb.200706206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderzalm P. J., Pandey A., Hurwitz M. E., Bloom L., Horvitz H. R., Garriga G. C. elegans CARMIL negatively regulates UNC-73/Trio function during neuronal development. Development. 2009;136:1201–1210. doi: 10.1242/dev.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M., Webb D. J., Horwitz A. R. Cell migration at a glance. J. Cell Sci. 2005;118:4917–4919. doi: 10.1242/jcs.02662. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M., Zareno J., Whitmore L., Choi C. K., Horwitz A. F. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 2007;176:573–580. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple R. A., Balzer E. M., Cho E. H., Matrone M. A., Yoon J. R., Martin S. S. Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Res. 2008;68:5678–5688. doi: 10.1158/0008-5472.CAN-07-6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P., Mitchelhill K. I., Kobe B., Kemp B. E., Zot H. G. The myosin-I-binding protein Acan125 binds the SH3 domain and belongs to the superfamily of leucine-rich repeat proteins. Proc. Natl. Acad. Sci. USA. 1997;94:3685–3690. doi: 10.1073/pnas.94.8.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P., Zot A. S., Zot H. G. Identification of Acan125 as a myosin-I-binding protein present with myosin-I on cellular organelles of Acanthamoeba. J. Biol. Chem. 1995;270:25316–25319. doi: 10.1074/jbc.270.43.25316. [DOI] [PubMed] [Google Scholar]
- Yang C., Pring M., Wear M. A., Huang M., Cooper J. A., Svitkina T. M., Zigmond S. H. Mammalian CARMIL inhibits actin filament capping by capping protein. Dev. Cell. 2005;9:209–221. doi: 10.1016/j.devcel.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zot H. G., Bhaskara V., Liu L. Acan125 binding to the SH3 domain of Acanthamoeba myosin-IC. Arch. Biochem. Biophys. 2000;375:161–164. doi: 10.1006/abbi.1999.1648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.