

Summary
Dystroglycan is a ubiquitously expressed cell adhesion protein. Its principal role has been determined as a component of the dystrophin-glycoprotein complex of muscle, where it constitutes a key component of the costameric cell adhesion system. To investigate more fundamental aspects of dystroglycan function in cell adhesion, we examined the role of dystroglycan in the dynamics and assembly of cellular adhesions in myoblasts. We show that β-dystroglycan is recruited to adhesion structures and, based on staining for vinculin, that overexpression or depletion of dystroglycan affects both size and number of fibrillar adhesions. Knockdown of dystroglycan increases the size and number of adhesions, whereas overexpression decreases the number of adhesions. Dystroglycan knockdown or overexpression affects the ability of cells to adhere to different substrates, and has effects on cell migration that are consistent with effects on the formation of fibrillar adhesions. Using an SH3 domain proteomic screen, we identified vinexin as a binding partner for dystroglycan. Furthermore, we show that dystroglycan can interact indirectly with vinculin by binding to the vinculin-binding protein vinexin, and that this interaction has a role in dystroglycan-mediated cell adhesion and spreading. For the first time, we also demonstrate unequivocally that β-dystroglycan is a resident of focal adhesions.
Keywords: Cell adhesion, Cell motility, Focal adhesions, Dystroglycan, Vinculin, Vinexin
Introduction
The adhesion of cells to each other and to their substrate is key to generating cell shape and cell organisation in tissues. Understanding how cells adhere is important in understanding disease processes such as cancer and some muscular dystrophies, both of which involve a failure in cell adhesion. The best studied molecules involved in adhesion are integrins. These are αβ heterodimeric transmembrane proteins formed by various combinations of 24 α-subunits and 9 β-subunits. Specific heterodimers are expressed in a tissue-specific manner, or in response to particular environmental cues.
In a recent review, 156 molecules are suggested to be involved in integrin-mediated cellular adhesion, forming 690 potential interactions (Zaidel-Bar et al., 2007). This averages almost nine interactions per molecule, and gives an immediate indication of the complexity of the cellular adhesion system. However, as with all studies of this type, spatiotemporal and organism-, tissue- or cell-specific factors play a part in determining which interactions can and do take place. In addition, there is a wide array of other proteins involved in cell-matrix and cell-cell adhesion. These will be involved in interactions that form part of the integrin adhesome, but in addition, they will also be involved in unique interactions. These include various cell adhesion molecules, cadherins and selectins, which along with integrins constitute the major classes of cell adhesion molecules.
Dystroglycan is unique amongst these proteins. It is widely expressed, but although it has a well-established adhesion and mechano-transductive role in skeletal muscle (Winder, 2001; Batchelor and Winder, 2006) its role in other tissues is not as well understood. Despite being a well-characterised general adhesion protein, the presence of both α- and β-dystroglycan in focal adhesions, and a specific role for them both in these structures has long been debated. It has been reported previously that α-dystroglycan is present in focal-adhesion-like structures (Belkin and Smalheiser, 1996; Spence et al., 2004a). Other dystroglycan-associated proteins such as utrophin, are present in focal adhesions in non-muscle cells (Belkin and Burridge, 1995). It is therefore assumed that β-dystroglycan is also likely to be present in these structures, although this has not been shown directly. We therefore examined the role of dystroglycan in the dynamics and assembly of cellular adhesions in conditionally immortalised myoblast cell lines. We investigated whether β-dystroglycan is recruited to adhesion structures, its role in the formation of cell adhesions during cell spreading and whether it can interact with proteins, such as vinculin, that are found in these cell adhesions.
Results
Dystroglycan localises to adhesions in myoblasts
Immunofluorescence staining has revealed that endogenous dystroglycan and vinculin co-localise in small adhesion puncta in the early stages of myoblast cell spreading (Thompson et al., 2008). The two proteins, however, appear less well co-localised in the more mature focal adhesion structures, but are closely localised in larger podosome structures, both during early spreading and in well spread cells treated with phorbol esters to stimulate podosome formation (Thompson et al., 2008).
Although α-dystroglycan has been demonstrated in focal adhesions in a variety of cell types (Belkin and Smalheiser, 1996; Spence et al., 2004a), robust detection of β-dystroglycan with either antibodies or fusion tags has proved elusive. It is thought that α-dystroglycan is an obligate heterodimer with β-dystroglycan, and the presence of α-dystroglycan on the extracellular face of the membrane infers the presence of the transmembrane β-dystroglycan. However, convincing evidence for β-dystroglycan, the transmembrane component of this heterodimeric adhesion receptor, in focal adhesions is lacking. It is assumed that β-dystroglycan must be present owing to its association with α-dystroglycan in focal adhesions. One possibility is that β-dystroglycan is present, but not accessible to antibodies because of molecular crowding or epitope-masking effects, but it is not clear why dystroglycan-GFP would not be visible, although molecular quenching of GFP has been described (Katz et al., 1998). β-dystroglycan can be easily detected in the less-dense podosome-like structures observed during the early stages of myoblast adhesion and spreading (supplementary material Fig. S1A,B) (Thompson et al., 2008). As cells spread and focal contacts and focal adhesions begin to form, however, the detection of β-dystroglycan in the plane of vinculin-delimited adhesions becomes less well defined and β-dystroglycan staining appears to be more apical to the vinculin of the adhesion (supplementary material Fig. S1A,C). The inability to detect β-dystroglycan directly in the adhesion has been attributed to epitope masking within the more dense focal adhesion structures, compared with the looser structure of podosomes (Thompson et al., 2008). We therefore used SDS antigen retrieval techniques (Brown et al., 1996; Ayscough and Drubin, 1998) to better visualise β-dystroglycan in classical focal adhesion structures (Fig. 1). β-dystroglycan was clearly and reliably localised with phosphorylated-tyrosine, talin or vinculin, in both peripheral focal complexes and more mature focal adhesions (Fig. 1A). Confocal sections taken through the basal aspect of cells confirmed the precise colocalisation of β-dystroglycan and vinculin within the adhesion plaque (Fig. 1B). Further examples of β-dystroglycan and vinculin staining are shown in supplementary material Fig. S2. GFP fluorescence or GFP antibody staining does not survive the SDS antigen retrieval treatment (Ayscough and Drubin, 1998), therefore to confirm the localisation and likely functionality of dystroglycan-GFP in adhesion we used a ventral membrane exposure technique (Avnur and Geiger, 1981) to improve the potential accessibility of β-dystroglycan to immunofluorescence microscopy. Although somewhat less reproducible than the SDS antigen-retrieval method, it did nonetheless allow us to confirm the presence of β-dystroglycan and dystroglycan-GFP in focal adhesions with vinculin or talin (supplementary material Fig. S3). Taken together, all of these observations tend to confirm the molecular crowding and epitope-masking hypothesis for β-dystroglycan in focal adhesions, and demonstrate for the first time that β-dystroglycan is a bona fide resident of focal adhesion structures.
Fig. 1.

Dystroglycan localises to focal adhesion in myoblasts. Epifluorescence and confocal images of β-dystroglycan in adhesions in myoblast cells. (A) Epifluorescence images of β-dystroglycan (green in merge) with either phospho-tyrosine, vinculin or talin (red in merged images). β-dystroglycan colocalises in focal adhesions and focal contacts with all three focal adhesion markers. Insets represent ×2.5 digital zooms of selected areas of the main panels. (B) Confocal images show a whole cell projection on the left with five consecutive confocal slices from basal left to apical right. Each optical section is 0.7 μm thick with 0.37 μm between sections. β-dystroglycan can be clearly seen to localise with vinculin in both the whole cell projection and in each individual section shown.
Effect of dystroglycan and substrate on cell spreading and polarisation
Given the clear presence of dystroglycan staining in focal adhesions and focal contacts, we next considered how altering the levels of cellular dystroglycan might affect cell spreading and polarisation in myoblasts plated on different substrates. In addition to wild-type H2k myoblasts, we used three stable myoblast cell lines: H2k myoblasts overexpressing dystroglycan-GFP to 200% of normal levels; H2k myoblasts depleted for dystroglycan by 50% using shRNA; and a control shRNA cell line expressing the sense sequence of dystroglycan used for the knockdown. These cell lines have been described previously (Thompson et al., 2008). Cells were seeded on laminin that could potentially engage both dystroglycan and integrins, fibronectin that would engage integrins but not dystroglycan, and a recombinant E3 fragment of laminin that would engage only dystroglycan (Ervasti and Campbell, 1993).
The different cellular levels of dystroglycan clearly affected the ability of the cells to spread on different substrates. On laminin, dystroglycan-GFP (DG-GFP)-expressing cells and control cells all spread reasonably well, but cells in which dystroglycan levels were reduced by 50% spread poorly. On rE3 laminin, DG-GFP-overexpressing cells spread well but control cells (sense and wild-type cells) and knockdown cells spread poorly. These results suggest that dystroglycan is important for spreading on ligands that engage dystroglycan (Fig. 2A,B). Finally, although DG-GFP expressing, and control cells had a very similar spreading response on fibronectin, all three types of cell spread less well than the dystroglycan knockdown cells (Fig. 2C). These results suggest that spreading on integrin engaging ligands might normally be inhibited by dystroglycan. None of the different types of substrate had an effect on initial cell attachment (supplementary material Fig. S4). In addition to examining the simple increase in cell area as an indication of spreading, we also monitored the change in circularity over time, which is a measure of polarisation. As can be seen from Fig. 2D-E, all cell lines plated on fibronectin achieved a reasonable degree of polarisation, whereas those plated on E3 laminin did not. The negative values for cell area and aspect ratio on rE3 laminin are a consequence of these cells becoming rounder and smaller during the course of the assay, and the sharp rise in spread area for wild-type cells on rE3 laminin at 30 minutes is probably a reflection of the associated change in aspect ratio from negative to positive at this time point. On whole laminin, however, which supported cell spreading in all but dystroglycan-depleted cells, both dystroglycan-depleted and dystroglycan-overexpressing cells failed to polarise well, whereas wild-type and shRNA control cells polarised very well (Fig. 2D). Dystroglycan has been described as a polarity determinant in a number of systems (see Discussion), therefore a lack of polarisation in dystroglycan-depleted cells is not surprising. However, one might assume that dystroglycan overexpression would induce better polarisation, which occurred to a certain extent on rE3 laminin, but not on fibronectin, and was worst of all on whole laminin (Fig. 2D-F). This might be due to ectopic localisation of excess dystroglycan also leading to a loss of a directed polarity signal, where cells could spread well, but continued to do so only in a radial manner.
Fig. 2.
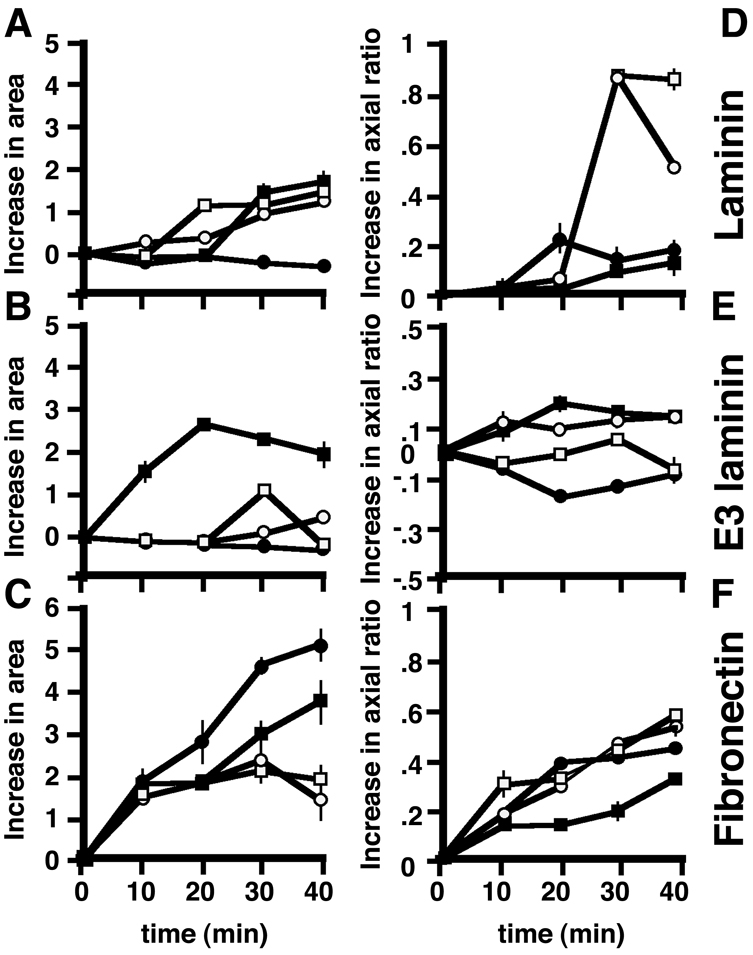
Effect of extracellular matrix and cellular dystroglycan content on cell spreading. Wild-type H2k myoblasts (white squares) or H2k myoblasts expressing control sense shRNA construct (white circles), an shRNA directed against dystroglycan (black circles) or dystroglycan-GFP (black squares) were allowed to adhere and spread for up to 40 minutes on whole laminin (A), rE3 fragment of laminin (B) or fibronectin (C). Spreading (cell area) was determined by measuring cell area using ImageJ. By 30 minutes, all cells have spread on laminin with the exception of the dystroglycan-knockdown cell line. On the rE3 fragment of laminin, only cells overexpressing dystroglycan are able to spread. Interestingly, on fibronectin, cells depleted for dystroglycan spread best of all. (D-F) Change in axial ratio with time for the same groups of cells as measured in A-C. Cells were assumed to be round at time 0 (whether they were or not), and all measurements of axial ratio calculated on that basis. Hence some cells, especially those on E3 show a negative value, because the cells became even rounder over time. All cells polarise well on fibronectin (F) and poorly on rE3 laminin (E). On laminin, cells with altered levels of dystroglycan do not polarise well, whereas controls on laminin are the most polarised of all (D).
Dystroglycan-dependent effects on adhesion type
We next determined whether dystroglycan levels affected the total number and subtype of cell adhesions formed in the various cell lines after they were fully spread. There is no universal marker of all cellular adhesion types; in most cell types vinculin is associated with focal complexes and focal adhesions, and not fibrillar adhesions. However, we have observed that vinculin can localise to structures in myoblasts that have all the hallmarks of fibrillar adhesions – longer thinner and more centrally located adhesions. We confirmed the identity of such structures as fibrillar adhesions by staining with parvin, a bona fide marker of these structures (Olski et al., 2001) (supplementary material Fig. S5). Based on this analysis, and for ease of quantification, we therefore chose to use vinculin as a marker of all three adhesion types in myoblasts. The cells were stained with vinculin as a marker for adhesion size and structure and the numbers of focal complexes, focal adhesions and fibrillar adhesions per cell were recorded.
The total number of adhesions per cell was reduced by 30% in cells that expressed higher levels of dystroglycan (DG-GFP cells; Fig. 3A) compared with wild-type or GFP-expressing cells. A drop in the number of focal complexes and fibrillar adhesions mostly accounted for the reduction in the total number of adhesions (Fig. 3C). However, although the absolute numbers decreased, when the number of focal complexes was expressed as a proportion of the total number, this value increased significantly (Fig. 3E). In dystroglycan-depleted cells, the total number of adhesions was also significantly decreased (Fig. 3B), but in contrast to DG-GFP cells, the numbers of focal complexes (both average numbers per cell and the proportion of focal complexes per cell) was reduced significantly in dystroglycan-depleted cells compared with controls (Fig. 3D,F). However, there was a dramatic compensatory increase in both absolute number and proportion of fibrillar adhesions (Fig. 3D,F). These data suggest that dystroglycan, possibly in concert with integrins, might have a role in modulating adhesion formation or maturation.
Fig. 3.

Dystroglycan-dependent effects on adhesion morphology. Wild-type H2k myoblasts, or cells expressing a control sense shRNA construct (sense), an shRNA directed against dystroglycan (knockdown), dystroglycan-GFP (DG-GFP) or GFP alone were allowed to adhere and spread and were then stained for vinculin. The number and type of adhesions formed were classified into focal complexes, focal adhesions and fibrillar adhesions. Altering dystroglycan levels either up (A) or down (B) reduces the total number of adhesions present in myoblasts. In dystroglycan-overexpressing cells, this is due to a decrease in absolute numbers of focal complexes and fibrillar adhesions (C), whereas in dystroglycan-depleted cells, although the number of focal complexes are markedly reduced, fibrillar adhesions increase (D). However, when expressed as a percentage of adhesion types for a given treatment, dystroglycan overexpression actually causes an increase in the proportion of focal complexes (E), whereas dystroglycan depletion reduces the proportion of focal complexes, but increases fibrillar adhesions (F). Data are mean ± s.e.m. of three independent experiments measuring adhesions from at least 30 cells. *P<0.05.
Altered dystroglycan levels affect cell motility
As altering dystroglycan levels affected size and types of adhesion, we also determined whether this had a concomitant affect on cell motility. Cell migration is a balance between the number and size of adhesion structures a cell can make, and the speed with which a cell can regulate or turn over these adhesions. Consequently, an increased number and size of focal-adhesion-like structures are associated with a less-motile phenotype, whereas a decrease in the number and size of adhesions is associated with a more motile phenotype (Ilic et al., 1995).
Altering the levels of dystroglycan altered both the velocity and persistence of cell migration. Cells that overexpressed dystroglycan migrated further than controls (Fig. 4A), probably because the average velocity of movement of these cells was increased by 40% (Fig. 4B). However, the persistence of these cells was decreased compared with wild-type cells (Fig. 4D). Persistence is a measure of the tendency of cells to continue to travel in the same direction, and these results show that although the velocity of the DG-GFP cells increased, they changed direction more frequently than control cells, which resulted in the reduction in the linear distance migrated from the starting point. This observation would also fit with the hypothesis that dystroglycan can act as a polarity determinant, and overexpression of dystroglycan could lead to ectopic localisation of polarity determinants. Cells expressing reduced levels of dystroglycan migrated much shorter distances than controls (Fig. 4A), and in this case, velocity was reduced by 40% (from 0.18 to 0.1 μm/minute), but persistence was similar to that in control cells. (Fig. 4C,E). Although migration was reduced, persistence was maintained because these cells still express ∼50% of normal levels of dystroglycan. The most likely explanation for the observed changes in velocity, is that fibrillar adhesions are reduced in dystroglycan-overexpressing cells, but increased in knockdown cells, as suggested by the data in Fig. 3. This results in the increase or decrease in velocity observed. Furthermore, if focal complexes were relatively more numerous in dystroglycan-overexpressing cells (Fig. 3) and/or were more dynamic, coupled with the potential ectopic localisation of a polarity determinant, this might also account for the decreased persistence.
Fig. 4.

Dystroglycan expression levels influence myoblast motility. (A) Migration of wild-type, knockdown, knockdown control (sense) and dystroglycan-overexpressing myoblast cells was analysed by time-lapse microscopy. Individual cell traces from 30 cells of each genotype are shown. Cells depleted in dystroglycan migrate shorter distances than the control, and cells overexpressing dystroglycan migrate more erratically and for greater distances. At least two independent isolates from each genotype were observed. (B,C) Mean velocity is decreased in dystroglycan-knockdown cells and increased in dystroglycan-overexpressing cells. (D,E) Cellular persistence is reduced in dystroglycan-overexpressing cells resulting in more erratic motion. (In B-E, results are mean ± s.e.m. of 30 cells from at least two independent isolates.)
β-dystroglycan interacts with vinexin and vinculin
To explore why β-dystroglycan affects focal adhesion numbers and how it might be recruited to focal adhesions, we searched for potential SH3-domain-containing proteins that might interact with the proline-rich dystroglycan cytoplasmic domain – a region that contains six potential PxxP SH3-binding motifs. We therefore carried out a human proteome-wide SH3 domain phage-display screen (Kärkkäinen et al., 2006) using the cytoplasmic domain of β-dystroglycan as an affinity ligand. We found a significant enrichment of clones from the SH3 domain phage library upon selection with β-dystroglycan (Thompson et al., 2008) (data not shown), confirming that the PxxP sites in β-dystroglycan can interact with SH3 domains.
By sequencing interacting clones, we identified that β-dystroglycan interacts with the third SH3 domain of vinexin. Vinexin contains three SH3 domains and was first identified as a vinculin-binding protein using a two-hybrid screen (Kioka et al., 1999). Vinexin has also been found to localise to focal adhesion structures in fibroblasts along with vinculin. The first and second SH3 domains of vinexin are important for its interaction with vinculin, and thus the third SH3 domain is free to interact with other binding partners, such as in this case, β-dystroglycan.
We verified this interaction between the vinexin SH3 domain and dystroglycan by three further approaches. First, we showed using a GST-SH3 domain affinity column (Fig. 5A), that β-dystroglycan bound to vinexin, whereas an unrelated SH3 domain (SH3 domain 1 or 5 from Tks5) (Thompson et al., 2008) used as control did not bind β-dystroglycan. Second, to examine the possible association of dystroglycan with vinexin in cells, we immunoprecipitated β-dystroglycan and used western blotting to determine whether vinexin, phosphorylated vinexin, vinculin or β-dystroglycan was present. Reciprocal immunoprecipitation revealed not only an interaction between dystroglycan and vinexin, as expected, but also revealed an interaction with phosphorylated vinexin (Fig. 5B). We also identified vinculin as a component of the dystroglycan-immunoprecipitated complex (Fig. 5C). We have no evidence for a direct interaction between vinculin and dystroglycan, and therefore presume this to be via the vinculin-binding protein vinexin. Control immunoprecipitations with non-immune IgG failed to immunoprecipitate dystroglycan, vinculin or vinexin. Coupled with the affinity column data (Fig. 5A), these experiments appear to substantiate the phage-display result. In addition, both vinculin and vinexin were localised to focal adhesion structures in myoblast cells expressing either GFP or dystroglycan-GFP; however, GFP-tagged dystroglycan did not localise strongly in focal adhesion structures, although some weak co-localisation with vinculin, vinexin and phosphorylated vinexin was apparent (supplementary material Fig. S6). To identify the potential vinexin SH3-domain-binding site in dystroglycan we used peptide-spot arrays to the entire cytoplasmic domain of β-dystroglycan (supplementary material Table S1). These arrays were probed with a biotinylated GST protein as a control and with a biotinylated GST fusion containing the third SH3 domain of vinexin. As can be seen from Fig. 6, the control GST did not bind to any peptide, with only the internal biotinylated peptide control being detected by the extravidin-HRP and ECL. However, the GST-SH3 fusion at two different stringencies clearly bound to peptides in the extreme C-terminus of the cytoplasmic domain of β-dystroglycan (Fig. 6BC). The peptide sequences delimited in these two incubations were NMTPYRSPPPYVPP and RSPPPYV, respectively.
Fig. 5.
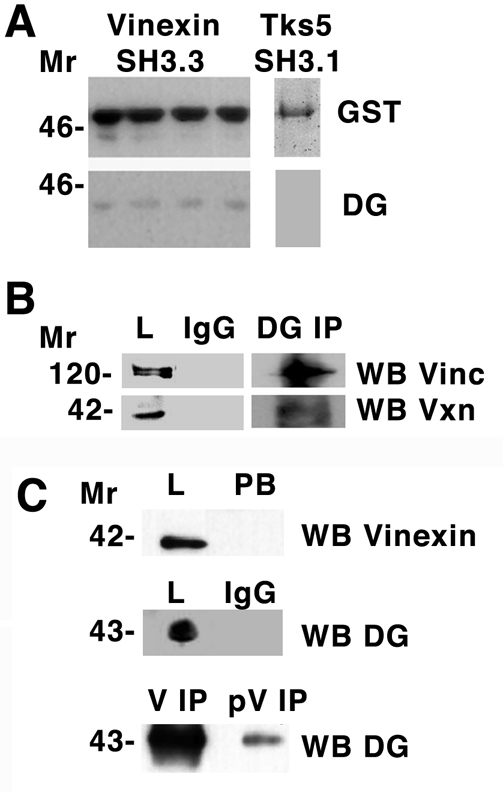
Dystroglycan interacts with vinculin and vinexin. (A) Total cell lysate from myoblasts was applied to a glutathione-Sepharose column with bound third SH3 domain of vinexin fused to GST. Following extensive washing, GST-SH3 was eluted with reduced glutathione and fractions western blotted (WB) for GST or dystroglycan (DG). Dystroglycan was specifically retained by the GST-SH3 column. A control SH3, the first SH3 domain of Tks5, did not interact with dystroglycan in a similar but batch bound experiment. (B) Immunoprecipitation (IP) of dystroglycan or control IgG from myoblast lysates followed by western blotting (WB) with antibodies that recognise vinculin (Vinc) or vinexin-β (Vxn). Vinculin or vinexin-β are recognised in the total lysate (L) fraction in the dystroglycan IP, but not when control IgG is used in the IP. (C) Dystroglycan is also identified by western blotting in immunoprecipitations carried out with either anti-vinexin-β (V) or anti-phosphorylated vinexin-β (pV); however, the amount of dystroglycan recovered in the anti-phosphorylated vinexin-β IP is considerably less. Anti-vinexin-β western blot of total lysate (L) and post binding (PB) fractions indicate that the anti-vinexin-β antibody completely depletes the lysate, whereas control IgG immunoprecipitations do not pull down any dystroglycan. Molecular size markers (in kDa) are shown on the left.
Fig. 6.

Vinexin SH3 binds the C-terminus of β-dystroglycan. Triplicate peptide spot arrays corresponding to the entire cytoplasmic domain of β-dystroglycan represented as a series 12-residue peptides, each different from the next by one amino acid in the sequence (see supplementary material Table S1 for exact peptide positions and sequences) were probed with GST alone (A) or with GST fused to the third SH3 domain of vinexin (B,C). Binding was detected using the endogenous biotin in the GST-pinpoint protein. In A, GST does not bind any peptide, and only the control biotinylated peptide was detected, whereas in B and C, the GST-SH3 binds peptides in the C-terminus of β-dystroglycan. In addition, the control biotinylated peptide was detected.
A dystroglycan-vinexin binding mutant fails to rescue spreading in dystroglycan-depleted cells
The vinexin SH3-domain-binding region in β-dystroglycan identified using the peptide spot arrays (Fig. 6) contained only one conventional PxxP SH3-domain-binding motif. This motif also overlapped with a previously described WW domain interaction motif: PPPY (James et al., 2000; Ilsley et al., 2001) (Fig. 7A). However, in addition to the minimum PxxP consensus for SH3-domain binding, many SH3-domain ligands also include basic residues as specificity determinants (Alexandropoulos et al., 1995). To avoid mutating the overlapping WW motif, we selected the N-terminal arginine and the C-terminal proline for mutagenesis to abolish the vinexin-SH3 interaction (Fig. 7A). Mutation of the putative vinexin-SH3-domain interaction site in the context of a full-length DG-GFP prevented the immunoprecipitation of vinexin with dystroglycan, indicating that this mutation could prevent vinexin binding to DG-GFP (Fig. 7B). DG-GFP or DG-GFP with a mutated vinexin-binding site (DGΔV-GFP) were expressed transiently in dystroglycan-depleted myoblast cells, immunoprecipitated with anti-GFP antiserum and blotted for GFP, vinexin and vinculin. The GFP blot clearly recognised both DG-GFP and DGΔV-GFP but vinexin was almost completely absent from the DGΔV-GFP immunoprecipitate, whereas vinculin was reduced significantly, although not completely. Nothing was detected in control lanes using an irrelevant IgG for immunoprecipitation (Fig. 7B). To test the functional relevance of the dystroglycan-vinexin interaction, we attempted to rescue the deficit in cell spreading observed in dystroglycan-depleted myoblast cells (Fig. 2) by re-expressing either DG-GFP or DGΔV-GFP. DG-GFP was able to rescue cell spreading, whereas cells expressing DGΔV-GFP spread significantly less (Fig. 7CD).
Fig. 7.

Mutagenesis of a putative vinexin binding site in β-dystroglycan. (A) Sequence of the peptides identified in Fig. 6 with the relative positions of SH3 (PxxP) and WW (PPxY) binding motifs, and the positions of the residues mutated to ablate the vinexin-binding site. (B) Immunoprecipitation of DG-DGP or DG-GFP with a mutated vinexin-binding site (DGΔV-GFP) using anti-GFP serum, indicates that unlike DG-GFP, DGΔV-GFP can no longer bind to vinexin, but still retains a weak interaction with vinculin. GFP, vinexin and vinculin were not immunoprecipitated with an irrelevant control IgG (lane C). (C,D) Myoblasts depleted of dystroglycan spread poorly on E3 laminin; re-expression of DG-GFP rescued the ability of cells to spread on E3 laminin, whereas DGΔV-GFP did not. These results substantiate the functionality of DG-GFP and also indicate a significant role for dystroglycan binding to vinexin in cell adhesion and spreading. Arrows in C point to GFP-marked transfected cells in upper panels and corresponding cells stained for vinculin in lower panels.
Discussion
We examined the role of dystroglycan in the dynamics and assembly of cellular adhesions in conditionally immortalised myoblast cell lines. In skeletal muscle, dystroglycan links the extracellular matrix (ECM) to the actin cytoskeleton via dystrophin; however, dystrophin is not expressed in most other tissues. Myoblasts have the ability to fuse and form syncitia, which in turn differentiate into contractile myotubes, an in vitro correlate of a myofibre. However, in their single cell state, they do not express muscle-specific genes such as skeletal muscle myosins and actin, or dystrophin. In this regard, a myoblast is little different from a fibroblast in terms of its resemblance to muscle. Expression profiles of muscle and non-muscle proteins in differentiating myoblasts demonstrate a shift from utrophin to dystrophin expression, whilst dystroglycan expression levels remain relatively constant (Ilsley et al., 2001; Rezniczek et al., 2007). β-vinexin is also expressed in many non-muscle cells, including C2C12 myoblasts, whereas α-vinexin is more predominantly expressed in differentiated C2C12 myoblasts (Kioka et al., 1999). The myoblast is therefore a useful cell type in which to examine the roles of adhesion proteins such as dystroglycan, in a non-muscle environment, and following differentiation of the myoblast, in a syncitial muscle fibre.
We determined that β-dystroglycan is recruited to adhesion structures, and, based on staining for vinculin, that overexpression or depletion of dystroglycan affects both the size and number of different adhesion types. Knockdown of dystroglycan increases the size and number of adhesions, whereas overexpression decreases their number. Knockdown or overexpression affects the ability of the cells to adhere to different substrates, and has effects on cell migration, consistent with their effects of the formation of fibrillar adhesions. Finally, we showed that dystroglycan can interact indirectly with vinculin by binding to the vinculin-binding protein vinexin. We demonstrated that a PxxP motif in the cytoplasmic domain of β-dystroglycan interacts with the third SH3 domain of vinexin, whereas the first and second SH3 domains of vinexin have been demonstrated previously to interact with vinculin (Kioka et al., 1999). Thus, dystroglycan could potentially form a ternary β-dystroglycan-vinexin-vinculin complex.
We found that levels of dystroglycan modulated the ability of cells to spread on different substrates. The ability of dystroglycan-overexpressing cells to spread on laminin was similar to wild-type cells, but it was substantially increased on substrates coated with a recombinant E3 fragment of laminin. Dystroglycan-knockdown cells spread poorly on laminin and E3 fragment, but spread better on a fibronectin substrate. Integrin-mediated affinity to E3 is low (Sonnenberg et al., 1990) and cell attachment to E3 is thought to be mediated by other adhesion molecules that do not belong to the integrin family. This suggests that dystroglycan is modulating cell adhesion to laminin, possibly through its interaction with E3. Our results are also broadly consistent with earlier work that shows that inhibiting dystroglycan function decreases cell attachment to laminin-1 (Hosokawa et al., 2002; Mehes et al., 2005).
During cell migration, the smallest and most dynamic focal complexes increase in size to become stress-fibre-associated focal adhesions, which further mature to fibrillar adhesions, which are involved in fibronectin fibril organisation. Fibrillar adhesions are large integrin-based adhesions located centrally within the cell, and increased numbers of these adhesions are characteristic of a less-motile state (Couchman et al., 1982; Duband et al., 1988). The maturation of adhesions from focal complexes is dependent on actomyosin-based contractility, or by external forces applied to the cells, and is also regulated by the rigidity of the extracellular matrix (Zamir and Geiger, 2001). We now show that the maturation of these fibrillar adhesions depends on the expression level of dystroglycan, because overexpressing or reducing levels of dystroglycan affects the number and size of these adhesions.
Therefore, an increase in number and/or size of fibrillar adhesions would be expected to affect cell motility. Consistent with this, we found that cells overexpressing dystroglycan had a reduction in fibrillar adhesions that was associated with an increase in velocity, but with a decrease in persistence, which resulted in an overall decrease in directed migration. By contrast, cells in which levels of dystroglycan expression were reduced had an increase in fibrillar adhesions, which was associated with a decrease in velocity, with little effect on persistence. These results suggest that fibrillar adhesions are important for persistence, and that unless cells form this type of adhesion they are more likely to change direction. Dystroglycan would therefore appear to be important for the maturation of adhesions into fibrillar adhesions and therefore in long-term adhesion and migration. Several studies have implicated dystroglycan in both vertebrate (Muschler et al., 2002) and invertebrate cell polarity (Deng et al., 2003). Overexpression of a polarity determinant might lead to loss of polarity, such as the more randomised movement pattern observed in dystroglycan-overexpressing cells. This might be a consequence of increased ectopic and depolarised Cdc42 activation owing to mislocalisation of a dystroglycan-Dbl complex (Batchelor et al., 2007), which leads to a loss of Cdc42 driven polarity. The cell spreading and polarisation data shown in Fig. 2 support this notion. Dystroglycan-overexpressing cells spread well, but maintain a circular shape, indicative of a loss of directed polarity. Dystroglycan-depleted cells spread less well, but also maintain a circular shape probably because of the lack of polarity determinants.
Focal complexes, focal adhesions and fibrillar adhesions contain varying amounts of vinculin. The stability and organisation of these adhesions might therefore depend wholly or partly on vinculin. We identified a PxxP motif in the extreme C-terminus of the cytoplasmic domain of dystroglycan that interacted with the third SH3 domain of vinexin. Mutation of a vinexin-binding site in dystroglycan (DGΔV) abolished the vinexin interaction, although a small amount of vinculin interaction remained, which was possibly mediated via other interactions. Dystroglycan with a vinexin binding site mutation could not rescue cell spreading in dystroglycan-depleted cells on E3 laminin, suggesting a possible role for the dystroglycan-vinexin-vinculin complex in cell spreading. A number of studies on vinculin-depleted and/or mutated cells have also documented a defect in cell spreading on laminin (Varnum-Finney and Reichardt, 1994; Volberg et al., 1995; Chandrasekar et al., 2005), and similarly to dystroglycan-depleted cells, these cells can still form adhesions because of compensatory changes in other adhesion proteins (Volberg et al., 1995). Furthermore, mutations in the regulatory PIP2-binding site in vinculin inhibit radial cell spreading and subsequent cell polarisation, and thus cell motility (Chandrasekar et al., 2005), a phenotype that is similar to that observed in dystroglycan-depleted myoblast cells.
It is possible that the dystroglycan-vinexin-vinculin interaction is important for either the maturation or de novo formation of focal complexes, focal adhesions and fibrillar adhesions. The number of fibrillar adhesions increased in dystroglycan-knockdown cells, and decreased in dystroglycan-overexpressing cells, such that overexpression of dystroglycan provides extra binding sites for vinexin. Through such a mechanism, dystroglycan could titrate vinculin away from other binding partners, which in turn has an adverse effect on vinculin-dependent adhesion structures. A similar phenomenon has been demonstrated for other dystroglycan binding partners involved in cell adhesion or adhesion-mediated signalling (Batchelor et al., 2007; Thompson et al., 2008). A dystroglycan-vinexin-vinculin complex could also link dystroglycan to integrins. Links between dystroglycan and integrin adhesion systems have been implied or hypothesised by others, both at a signalling level and a mechano-structural level (Ferletta et al., 2003; Jones et al., 2005; Colognato et al., 2007; Peng et al., 2008). Our preliminary data also suggest that vinculin-containing adhesion structures can form on E3 laminin, which is a non-integrin-engaging ligand, presumably mediated by dystroglycan. Dystroglycan is clearly present in focal adhesions, interacts indirectly with vinculin through its interaction with vinexin, and can contribute to focal adhesion stability and turnover, thus affecting cell migration. Our studies also provide a mechanism for a link via vinexin and vinculin between dystroglycan and integrin adhesion systems that could participate in dystroglycan-integrin crosstalk in both muscle and non-muscle cells.
Materials and Methods
Production of recombinant laminin E3 fragment
DNA coding for residues 2682-3060 of the mature mouse laminin α1 chain was obtained by PCR amplification from laminin α1 pCIS and cloned into pBluescript II KS+ as described (Harrison et al., 2007). The unpaired cysteine (C3014) was mutated to serine by strand-overlap extension PCR, and the sequence-verified mutated insert cloned into a modified pCEP-Pu vector (Kohfeldt et al., 1997). After cleavage of the BM-40 signal sequence, a vector-derived APLAHHHHHHALA sequence remains at the N-terminus of the secreted rE3 protein. The rE3 protein was purified from the conditioned medium of episomally transfected 293-EBNA cells (Harrison et al., 2007) using a 5 ml HisTrap column (GE Healthcare) and dialysed into PBS.
Cell culture
Mouse myoblast cells (H-2Kb-tsA58) were maintained as described previously (Morgan et al., 1994). Myoblast cell lines that stably expressed dystroglycan-GFP, GFP, an shRNA against DG and a sense strand control shRNA were generated using retroviral infection, as described previously (Batchelor et al., 2007; Higginson et al., 2008; Thompson et al., 2008). Where required, cells were plated onto glass coverslips coated overnight with 5 μg/ml in PBS of ECM protein, gelatin, laminin, fibronectin, poly-L-lysine (Sigma) or laminin rE3 fragment. Uncoated regions were blocked with 1% BSA for 1 hour at room temperature, rinsed once with PBS and used within 3 hours of preparation. Tissue culture cells were fixed and stained for immunofluorescence microscopy, as described previously (Spence et al., 2004b). For β-dystroglycan antigen retrieval, cells were fixed and permeabilised as above and then incubated in 1% SDS in PBS for 5 minutes, followed by three washes in PBS and immunolocalisation as above (Brown et al., 1996; Ayscough and Drubin, 1998). The number and type of adhesions in cells, where quantified, were estimated in at least 100 cells in three independent experiments per treatment.
The following antibodies were used for immunofluorescence microscopy (or western blotting or immunoprecipitation) at the indicated dilutions: MANDAG2 monoclonal anti-β-dystroglycan 1:25 (1:350 for western, 1:10 for IP) (Pereboev et al., 2001); 1709 polyclonal anti-β-DG 1:10 (Ilsley et al., 2001); vinculin, clone V9131, 1:400 and talin, clone 8D4, 1:100 (Sigma); phosphotyrosine PY20, 1:1000 and paxillin, 1:1000 (Transduction Labs); parvin 1:1 (Olski et al., 2001); GFP (1:5000 for western, 1:100 for IP); fluorescent phalloidins to detect F-actin 1 μg/ml (Molecular Probes); vinexin-β and phosphorylated vinexin-β, 1:500 (1:1000 for western, 1:50 for IP) (Ito et al., 2007) and fluorescent-species-specific secondary antibodies 1 μg/ml (Vector Labs).
Microscopy and image analysis
Epifluorescence images were captured using a Leica DMIRE2 fitted with a Leica DC350F monochrome CCD camera and Qflouro image acquisition software, or using an Olympus IX-81 inverted microscope using MAG Biosystems acquisition software version 7.5.2.0 with a Photometrics CoolsnapHQ2 Turbo 1394 camera. Confocal images were obtained using a Leica TCS SP1 confocal microscope fitted with Ar ion, Kr 568nm and Red HeNe 633nm lasers detected with Hamamatsu R6357 and R6358 photomultiplier tubes. Monochrome images were combined and processed using Adobe Photoshop and additionally as detailed below.
Fluorescence intensity models were created using ImageJ v.1.36b (NIH; http://rsb.info.nih.gov/ij/). The fluorophore channel of interest was first extracted using the `RGB Split' command, and the `Subtract background' tool applied to remove background noise. The Surface Plot 3D plugin (Kai Uwe Barthel Internationale Medieninformatik Berlin, Germany) was then applied and the images saved as TIFF files.
For measurement of whole cell area and circularity, or adhesion type and distribution, RGB images were split into their constituent channels using the ImageJ `RGB Split' command and the channel containing the fluorescently labelled cells or structures retained. Images were converted to 8-bit greyscale, the `Threshold' command was invoked and manually adjusted until all desired cells or structures were flood-filled. These settings were applied, and the `Analyse Particles' command selected. Settings were adjusted to exclude background particles and conjoined cells, and the `Measure' command implemented. In the case of adhesion types, this included size boundaries set to distinguish different classes of adhesion. In addition, visual inspection of the structures confirmed their presence in expected areas within the cell. See supplementary material Fig. S7. The classification of adhesions was based upon size, localisation, and morphological characteristics using vinculin staining as a marker. Focal complexes were classed as small punctate adhesions of 1-5 μm2, lying proximal to the cell periphery and at the leading edge of polarised cells. Focal adhesions were classed as larger structures (5-20 μm2); possessing a more linear morphology, with a broader distribution throughout the cell, both at the cell periphery and towards the cell body. Fibrillar adhesions were characterised as large, linear structures exceeding 20 μm2, and situated nearer to the centre of the cell. Data were curated manually to remove any values over 100 μm2 that resulted from areas of ambiguous, or apparently aberrant staining, an example of a cell imaged in this away is shown in supplementary material Fig. S7.
To determine cell velocity, time-lapse footage of cells in culture was analysed using the `Manual Tracker' plugin in Image J. Individual cells were tracked by selecting a point to monitor at the nucleus, then tracing the movement frame by frame. From the resulting data, average velocity per cell could be calculated, which was then averaged upon collection of 30 cell traces. This plugin also provided relative x-y coordinates for each cell trace. This data was normalised to start at x=0 and y=0 and the relative paths from this point plotted for each cell. Cellular persistence ratios, i.e. the tendency of a cell to maintain directionality during movement, were calculated by dividing the linear path length of each cell by the straight-line distance from start to finish. Cell diameters were measured after 45 minutes of cell respreading in at least 30 cells per treatment. The cell diameter was taken as the mean of two measurements through the centre of the cell and at right angles to each other.
Assays for protein-protein interaction
The cytoplasmic domain of mouse β-dystroglycan was expressed in E. coli, purified as described previously (Chen et al., 2003), and used to screen a phage-display library comprising an essentially complete collection (n=296) of human SH3 domains using a protocol described elsewhere (Kärkkäinen et al., 2006), and also specifically for β-dystroglycan (Thompson et al., 2008).
Pinpoint GST-fusion proteins corresponding to the third SH3 domains of vinexin were generated by PCR, expressed in E. coli and purified on glutathione-Sepharose according to the manufacturer's instructions (GE Healthcare). To evaluate binding of β-dystroglycan to the GST-SH3 domains, myoblast lysate prepared in RIPA buffer was passed over a GST-SH3 domain affinity column. Following extensive washing bound dystroglycan was eluted with the GST-SH3 domain using 20 mM glutathione. Fractions were separated by SDS-PAGE and analysed by western blotting, as described previously (James et al., 2000). Immunoprecipitation assays were carried out on myoblast extracts in RIPA buffer at 4°C overnight, following extensive washes and a final stringent wash in RIPA buffer containing 0.6 M LiCl2, bound proteins were analysed by SDS-PAGE and western blotting, as described above.
To identify the vinexin SH3-domain-binding site in dystroglycan, GST-SH3 (10 μg/ml) was incubated with a peptide CelluSpots array comprising the cytoplasmic domain of β-dystroglycan represented as 12 amino acid peptides with a one amino acid overlap (Intavis, Koln, Germany). Each array additionally contained a duplicate C-terminal peptide with a phospho-tyrosine at position 892, a negative control spot and a biotinylated peptide as a positive control for the streptavidin detection system used to detect the pinpoint-GST. Layout and full sequences are in supplementary material Table S1. Peptide spot arrays were developed essentially as described previously (James et al., 2000; Ilsley et al., 2001). Arrays were blocked in Tris-buffered saline Tween 20 (TBST) with 3% BSA for either 30 minutes or 1 hour, and incubated with vinexin SH3 (10 μg/ml) in TBST with or without 3% BSA for 4 hours. Following extensive washes in TBST, arrays were developed with Extravidin coupled to peroxidase and ECL. Dystroglycan-GFP and GST-dystroglycan with a point mutation in a putative vinexin SH3-domain-binding site was generated by QuikChange mutagenesis (Stratagene) according to the manufacturer's instructions and the presence of the desired mutation verified by sequencing.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/1/118/DC1
We are extremely grateful to Koh-ichi Nagata for antisera to vinexin-β and phosphorylated vinexin-β, Elena Korenbaum for anti-parvin antibody, Francis Barr for GFP antisera and to Mario Gimona for helpful discussions. O.T. was supported by a White Rose Studentship, the work was also supported by MRC grants G00000114 and G0701129 to S.J.W., grants from the Academy of Finland, the Sigrid Juselius Foundation, and Helsinki University Hospital EVO funds to K.S. K.R.A. is a senior non-clinical MRC Fellow (G0601600). S.H. and E.H. are supported by a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science to E.H. (ref 083942). The light microscopy imaging centre at the University of Sheffield was funded by a grant from the Wellcome Trust (GR077544AIA). Deposited in PMC for release after 6 months.
References
- Alexandropoulos, K., Cheng, G. and Baltimore, D. (1995). Proline-rich sequences that bind to Src homology 3 domains with individual specificities. Proc. Natl. Acad. Sci. USA 92, 3110-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avnur, Z. and Geiger, B. (1981). Substrate-attached membranes of cultured cells isolation and characterization of ventral cell membranes and the associated cytoskeleton. J. Mol. Biol. 153, 361-379. [DOI] [PubMed] [Google Scholar]
- Ayscough, K. R. and Drubin, D. G. (1998). Immunofluorescence methods in yeast. In Cell Biology: A Laboratory Handbook (ed. J. Celis), pp. 477-485. San Diego: Academic Press.
- Batchelor, C. and Winder, S. (2006). Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell Biol. 16, 198-205. [DOI] [PubMed] [Google Scholar]
- Batchelor, C. L., Higginson, J. R., Chen, Y.-J., Vanni, C., Eva, A. and Winder, S. J. (2007). Recruitment of Dbl by ezrin and dystroglycan drives membrane proximal Cdc42 activation and filopodia formation. Cell Cycle 6, 353-363. [DOI] [PubMed] [Google Scholar]
- Belkin, A. M. and Burridge, K. (1995). Localization of utrophin and aciculin at sites of cell-matrix and cell-cell adhesion in cultured cells. Exp. Cell Res. 221, 132-140. [DOI] [PubMed] [Google Scholar]
- Belkin, A. M. and Smalheiser, N. R. (1996). Localization of cranin (dystroglycan) at sites of cell-matrix and cell-cell contact: recruitment to focal adhesions is dependent upon extracellular ligands. Cell Adhesion Commun. 4, 281-296. [DOI] [PubMed] [Google Scholar]
- Brown, D., Lydon, J., McLaughlin, M., Stuart-Tilley, A., Tyszkowski, R. and Alper, S. (1996). Antigen retrieval in cryostat tissue sections and cultured cells by treatment with sodium dodecyl sulfate (SDS). Histochem. Cell Biol. 105, 261-267. [DOI] [PubMed] [Google Scholar]
- Chandrasekar, I., Stradal, T. E. B., Holt, M. R., Entschladen, F., Jockusch, B. M. and Ziegler, W. H. (2005). Vinculin acts as a sensor in lipid regulation of adhesion-site turnover. J. Cell Sci. 118, 1461-1472. [DOI] [PubMed] [Google Scholar]
- Chen, Y.-J., Spence, H. J., Cameron, J. M., Jess, T., Ilsley, J. L. and Winder, S. J. (2003). Direct interaction of β-dystroglycan with F-actin. Biochem. J. 375, 329-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato, H., Galvin, J., Wang, Z., Relucio, J., Nguyen, T., Harrison, D., Yurchenco, P. D. and ffrench-Constant, C. (2007). Identification of dystroglycan as a second laminin receptor in oligodendrocytes, with a role in myelination. Development 134, 1723-1736. [DOI] [PubMed] [Google Scholar]
- Couchman, J. R., Rees, D. A., Green, M. R. and Smith, C. G. (1982). Fibronectin has a dual role in locomotion and anchorage of primary chick fibroblasts and can promote entry into the division cycle. J. Cell Biol. 93, 402-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, W.-M., Schneider, M., Frock, R., Castillejo-Lopez, C., Gaman, E. A., Baumgartner, S. and Ruohola-Baker, H. (2003). Dystroglycan is required for polarizing the epithelial cells and the oocyte in Drosophila. Development 130, 173-184. [DOI] [PubMed] [Google Scholar]
- Duband, J. L., Nuckolls, G. H., Ishihara, A., Hasegawa, T., Yamada, K. M., Thiery, J. P. and Jacobson, K. (1988). Fibronectin receptor exhibits high lateral mobility in embryonic locomoting cells but is immobile in focal contacts and fibrillar streaks in stationary cells. J. Cell Biol. 107, 1385-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti, J. M. and Campbell, K. P. (1993). A role for the dystrophin glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 112, 809-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferletta, M., Kikkawa, Y., Yu, H., Talts, J. F., Durbeej, M., Sonnenberg, A., Timpl, R., Campbell, K. P., Ekblom, P. and Genersch, E. (2003). Opposing roles of integrin α6Aβ1 and dystroglycan in laminin-mediated extracellular signal-regulated kinase activation. Mol. Biol. Cell 14, 2088-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, D., Hussain, S.-A., Combs, A. C., Ervasti, J. M., Yurchenco, P. D. and Hohenester, E. (2007). Crystal structure and cell surface anchorage sites of laminin a-1LG4-5. J. Biol. Chem. 282, 11573-11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginson, J. R., Thompson, O. and Winder, S. J. (2008). Targeting of dystroglycan to the cleavage furrow and midbody in cytokinesis. Int. J. Biochem. Cell Biol. 40, 892-900. [DOI] [PubMed] [Google Scholar]
- Hosokawa, H., Ninomiya, H., Kitamura, Y., Fujiwara, K. and Masaki, T. (2002). Vascular endothelial cells that express dystroglycan are involved in angiogenesis. J. Cell Sci. 115, 1487-1496. [DOI] [PubMed] [Google Scholar]
- Ilic, D., Furuta, Y., Kanazawa, S., Takeda, N., Sobue, K., Akatsuji, N. N., Nomura, S., Fujimoto, J., Okada, M., Yamamoto, T. et al. (1995). Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377, 539-544. [DOI] [PubMed] [Google Scholar]
- Ilsley, J. L., Sudol, M. and Winder, S. J. (2001). The interaction of dystrophin with β-dystroglycan is regulated by tyrosine phosphorylation. Cell. Signal. 13, 625-632. [DOI] [PubMed] [Google Scholar]
- Ito, H., Usuda, N., Atsuzawa, K., Iwamoto, I., Sudo, K., Katoh-Semba, R., Mizutani, K., Morishita, R., Deguchi, T., Nozawa, Y. et al. (2007). Phosphorylation by extracellular signal-regulated kinase of a multidomain adaptor protein, vinexin, at synapses. J. Neurochem. 100, 545-554. [DOI] [PubMed] [Google Scholar]
- James, M., Nuttall, A., Ilsley, J. L., Ottersbach, K., Tinsley, J. N., Sudol, M. and Winder, S. J. (2000). Adhesion-dependent tyrosine phosphorylation of β-dystroglycan regulates its interaction with utrophin. J. Cell Sci. 113, 1717-1726. [DOI] [PubMed] [Google Scholar]
- Jones, J. C. R., Lane, K., Hopkinson, S. B., Lecuona, E., Geiger, R. C., Dean, D. A., Correa-Meyer, E., Gonzales, M., Campbell, K., Sznajder, J. I. et al. (2005). Laminin-6 assembles into multimolecular fibrillar complexes with perlecan and participates in mechanical-signal transduction via a dystroglycan-dependent, integrin-independent mechanism. J. Cell Sci. 118, 2557-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkkäinen, S., Hiipakka, M., Wang, J., Kleino, I., Vähä-Jaakkola, M., Renkema, G. H., Liss, M., Wagner, R. and Saksela, K. (2006). Identification of preferred protein interactions via phage-display of the human Src homology-3 proteome. EMBO Rep. 7, 186-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz, B., Krylov, D., Aota, S., Olive, M., Vinson, C. and Yamada, K. (1998). Green fluorescent protein labeling of cytoskeletal structures-novel targeting approach based on leucine zippers. Biotechniques 25, 298-302. [DOI] [PubMed] [Google Scholar]
- Kioka, N., Sakata, S., Kawauchi, T., Amachi, T., Akiyama, S. K., Okazaki, K., Yaen, C., Yamada, K. M. and Aota, S.-i. (1999). Vinexin: a novel vinculin-binding protein with multiple SH3 domains enhances actin cytoskeletal organization. J. Cell Biol. 144, 59-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohfeldt, E., Maurer, P., Vannahme, C. and Timpl, R. (1997). Properties of the extracellular calcium binding module of the proteoglycan testican. FEBS Letters 414, 557. [DOI] [PubMed] [Google Scholar]
- Mehes, E., Czirok, A., Hegedus, B., Szabo, B., Vicsek, T., Satz, J., Campbell, K. and Jancsik, V. (2005). Dystroglycan is involved in laminin-1-stimulated motility of Muller glial cells: combined velocity and directionality analysis. Glia 49, 492-500. [DOI] [PubMed] [Google Scholar]
- Morgan, J. E., Beauchamp, J. R., Pagel, C. N., Peckham, M., Ataliotis, P., Jat, P. S., Noble, M. D., Farmer, K. and Partridge, T. A. (1994). Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev. Biol. 162, 486-498. [DOI] [PubMed] [Google Scholar]
- Muschler, J., Levy, D., Boudreau, R., Henry, M., Campbell, K. and Bissell, M. J. (2002). A role for dystroglycan in epithelial polarization: loss of function in breast tumor cells. Cancer Res. 62, 7102-7109. [PubMed] [Google Scholar]
- Olski, T. M., Noegel, A. A. and Korenbaum, A. (2001). Parvin, a 42 kDa focal adhesion protein, related to the a-actinin superfamily. J. Cell Sci. 114, 525-538. [DOI] [PubMed] [Google Scholar]
- Peng, H., Shah, W., Holland, P. and Carbonetto, S. (2008). Integrins and dystroglycan regulate astrocyte wound healing: the integrin b1 subunit is necessary for process extension and orienting the microtubular network. Dev. Neurobiol. 68, 559-574. [DOI] [PubMed] [Google Scholar]
- Pereboev, A., Ahmed, N., Man, N. t. and Morris, G. (2001). Epitopes in the interacting regions of beta-dystroglycan (PPxY motif) and dystrophin (WW domain). Biochim. Biophys. Acta 1527, 54-60. [DOI] [PubMed] [Google Scholar]
- Rezniczek, G. A., Konieczny, P., Nikolic, B., Reipert, S., Schneller, D., Abrahamsberg, C., Davies, K. E., Winder, S. J. and Wiche, G. (2007). Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with β-dystroglycan. J. Cell Biol. 176, 965-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg, A., Linders, C. J. T., Modderman, P. W., Damsky, C. H., Aumailley, M. and Timpl, R. (1990). Integrin recognition of different cell-binding fragments of laminin (P1, E3, E8) and evidence that α6β1 but not α6β4 functions as a major receptor for fragment E8. J. Cell Biol. 110, 2145-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence, H. J., Dhillon, A. S., James, M. and Winder, S. J. (2004a). Dystroglycan a scaffold for the ERK-MAP kinase cascade. EMBO Rep. 5, 484-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence, H. J., Chen, Y.-J., Batchelor, C. L., Higginson, J. R., Suila, H., Carpen, O. and Winder, S. J. (2004b). Ezrin-dependent regulation of the actin cytoskeleton by β-dystroglycan. Hum. Mol. Genet. 13, 1657-1668. [DOI] [PubMed] [Google Scholar]
- Thompson, O., Kleino, I., Crimaldi, L., Gimona, M., Saksela, K. and Winder, S. J. (2008). Dystroglycan, Tks5 and Src mediated assembly of podosomes in myoblasts. PLoS One 3, e3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum-Finney, B. and Reichardt, L. F. (1994). Vinculin-deficient PC12 cell lines extend unstable lamellipodia and filopodia and have a reduced rate of neurite outgrowth. J. Cell Biol. 127, 1071-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberg, T., Geiger, B., Kam, Z., Pankov, R., Simcha, I., Sabanay, H., Coll, J. L., Adamson, E. and Ben-Ze'ev, A. (1995). Focal adhesion formation by F9 embryonal carcinoma cells after vinculin gene disruption. J. Cell Sci. 108, 2253-2260. [DOI] [PubMed] [Google Scholar]
- Winder, S. J. (2001). The complexities of dystroglycan. Trends Biochem. Sci. 26, 118-124. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar, R., Itzkovitz, S., Ma'ayan, A., Iyengar, R. and Geiger, B. (2007). Functional atlas of the integrin adhesome. Nat. Cell Biol. 99, 858-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir, E. and Geiger, B. (2001). Components of cell-matrix adhesions. J. Cell Sci. 114, 3577-3579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}