

Abstract
Study of molecular actions of thyroid hormone receptor β (TRβ) mutants in vivo has been facilitated by creation of a mouse model (TRβPV mouse) that harbors a knockin mutant of TRβ (denoted PV). PV, which was identified in a patient with resistance to thyroid hormone, has lost T3 binding activity and transcription capacity. The striking phenotype of thyroid cancer exhibited by TRβPV/PV mice has allowed the elucidation of novel oncogenic activity of a TRβ mutant (PV) [PAS1]beyond nucleus-initiated transcription. PV was found to physically interact with the regulatory p85α subunit of phosphatidylinositol 3-kinase (PI3K) in both the nuclear and cytoplasmic compartments. This protein-protein interaction activates the PI3K signaling by increasing phosphorylation of AKT, mammalian target of rapamycin (mTOR), and p70S6K. PV, via interaction with p85α, also activates the PI3K-integrin-linked kinase-matrix metalloproteinase-2 signaling pathway in the extra-nuclear compartment. The PV-mediated PI3K activation results in increased cell proliferation, motility, migration, and metastasis.
In addition to affecting these membrane-initiated signaling events, PV affects [PAS2]the stability of the pituitary tumor-transforming gene (PTTG) product. PTTG (also known as securin), a critical mitotic checkpoint protein, is physically associated with TRβ or PV in vivo. Concomitant with T3-induced degradation of TRβ, PTTG is degraded by the proteasome machinery, but no such degradation occurs when PTTG is associated with PV. The degradation of PTTG/TRβ is activated by the direct interaction of the T3-bound TRβ with the steroid receptor coactivator-3 (SRC-3) that recruits a proteasome activator (PA28γ). PV that does not bind T3 cannot interact directly with SRC-3/PA28γ to activate proteasome degradation, and the absence of degradation results in an aberrant accumulation of PTTG. The PV-induced failure of timely degradation of PTTG results in mitotic abnormalities. PV, via novel protein-protein interaction and transcription regulation, acts to antagonize the functions of wild-type TRs and contributes to the oncogenic functions of this mutation.
Keywords: thyroid hormone receptors, phosphatidylinositol 3-kinase, pituitary tumor transforming gene, steroid hormone receptor coactivator-3, nongenomic actions, thyroid hormone receptor mutants, mouse model, thyroid cancer, carcinogenesis
1. Introduction
The thyroid hormone, T3, has diverse biological functions in growth, development, differentiation and metabolism. The cloning of cDNAs for thyroid hormone nuclear receptors (TRs) in 1986 [1, 2] ushered in an era of intensive study of target gene transcription regulation by TR in a T3-dependent manner. Two TR genes, α and β, located on two different chromosomes, encode four T3 binding receptors (α1, β1, β2, and β3) [3, 4]. The expression of these TR isoforms is tissue-dependent and developmentally regulated [5, 6]. They bind to the specific DNA sequences on the promoters of T3 target genes (TRE[PAS3]s). The TREs consist of consensus half site motifs of AGGTCA with two half sites in palindromic, direct repeat, and everted [PAS4]repeat arrangement [5]. The binding of liganded-TRs to TREs leads to the activation or repression of target genes [4]. However, binding of TRs to TREs is ligand-independent [5]. The TRE-bound unliganded TRs recruit corepressors such as the nuclear receptor corepressor (NCoR) [7], or its homolog, the silencing mediator of retinoid and thyroid hormone receptor (SMRT), together with other histone deacetylases [8, 9]. Binding of T3 induces conformational changes of TR, thereby releasing the corepressors and allowing the liganded TR to recruit coactivators such as p160 family members and their associated proteins [10, 11]. Several distinct multi-protein coactivator complexes have been identified, and these cofactor complexes could sequentially exchange to execute a series of enzymatic modifications required for regulated gene expression [11]. Thus, the transcription activity of TRs is tightly controlled at multiplelevels including the ligand, the type of TRE, the distribution of TR isoforms, and a host of corepressors and coactivators.
The cloning of cDNAs for TRs led to the discovery that mutations of the TRβ gene cause the genetic syndrome of thyroid hormone resistance (RTH) [PAS5][12, 13]. RTH is a syndrome characterized by reduced sensitivity of tissues to the actions of thyroid hormone and is inherited in an autosomal dominant manner. The hallmark of RTH is elevated thyroid hormone associated with nonsuppressible thyroid stimulating hormone (TSH). Other clinical signs are goiter, short stature, decreased weight, tachycardia, hearing loss, attention-deficit hyperactivity disorder, decreased IQ, and dyslexia [12, 13]. The clinical manifestations vary between families with different mutations, between families with the same mutation, and also between members of the same family with identical mutations. Most patients are heterozygous with only one mutated TRβ gene, and the clinical symptoms are mild [12, 13]. Only one patient homozygous for a mutant TRβ has been reported [14]. This patient, who died at a young age, displayed an extraordinary and complex phenotype of extreme RTH with very high levels of thyroid hormones and TSH [14].
Earlier work aimed at understanding how TRβ mutants mediate the clinical phenotype focused on study in the interfering with [PAS6]the transcription of target genes via dominant negative effects. These studies were mainly based on reporter activity in cultured cells, but a limitation in extrapolating findings to the physiological context was soon realized. To circumvent this limitation, Kaneshige et al. created a TRβ mutant knockin mouse, harboring a frame-shift mutation in the carboxyl-terminal 16 amino acids (TRβPV mouse) [15]. PV mutation was identified in a RTH patient [16] who had lost T3 binding activity and transcription capacity [17]. The TRβPV mouse faithfully reproduces RTH in humans with the loss of the feedback regulation in the pituitary-thyroid axis [15].
Consistent with phenotypes of RTH patients, TRβPV mice also exhibit growth retardation [15], abnormal regulation of serum cholesterol [18], hearing defects [19], thyrotoxic skeletal phenotype [20], and neurological dysfunction [21]. Through use of this mouse model of RTH, it was revealed that, in vivo, PV interferes with the transcription activity of TRs by competing with w-TRβ or w-TRα1 for binding to TREs and for heterodimerization with retinoid X receptors (RXRs) [22]. Such competition leads to repression of the positively T3-regulated target genes (e.g., S14, malic enzyme, and type 1 deiodinase) and activation of the negatively regulated genes (e.g., TSHβ subunit) [15]. Moreover, analysis of the altered expression gene profiles in three T3 target tissues of the TRβPV mouse (cerebellum, heart, and white adipocyte tissue) shows that “change-of-function” of PV could also contribute to the phenotypic changes via the transcription regulation [23].
Recent studies, however, indicate that the deleterious consequence of mutations of the TRβ gene extends beyond RTH. As TRβPV/PV mice age, they spontaneously develop follicular thyroid carcinoma similar to human cancer [24]. Delineation of the in vivo actions of PV in TRβPV/PV mice shows that its oncogenic functions could not be totally accounted for by the TRE-mediated transcription initiated in the nucleus. This article will highlight recent advances in the understanding of novel oncogenic functions of PV in vivo.
2. Extra-nuclear actions of TRβ mutants
2.1. TRβPV/PV mouse model of follicular thyroid carcinoma
TRβ mutants have long been implicated in the development and progression of human cancer [25, 26]. However, not until the discovery that TRβPV/PV mice spontaneously develop follicular thyroid carcinoma did it become possible to test the hypothesis that TRβ mutants could function as oncogenes. The thyroids of TRβPV/PV mice exhibit hyperplasia as early as 2 months old. Soon after that, TRβPV/PV mice develop capsular and vascular invasion, anaplasia, and eventually distant metastasis similar to human cancer [24]. Thus, the TRβPV/PV mouse provides an unusual opportunity to dissect gene alterations during carcinogenesis and to delineate the oncogenic functions of PV.
2.2. Activation of the phosphatidylinositol 3-kinase (PI3K) signaling by PV
Increasing evidence has shown that dysregulation of phosphatidylinositol 3-kinase (PI3K) signaling contributes to abnormal cell growth and cellular transformation in a variety of neoplasms [27]. Recent reports that AKT/protein kinase B is over-activated in human thyroid cancer, especially in follicular thyroid cancer [28, 29], prompted us to examine whether AKT is activated in the thyroid tumors of TRβPV/PV mice. Indeed, similar to human thyroid cancer, AKT is activated in the primary lesions of thyroid as indicated by increasing phosphorylation in an age-dependent manner (Fig. 1A). The ratio of phosphorylated-AKT (p-AKT) versus total AKT continues to increase during tumor progression (Fig. 1B) [30].
Fig. 1.
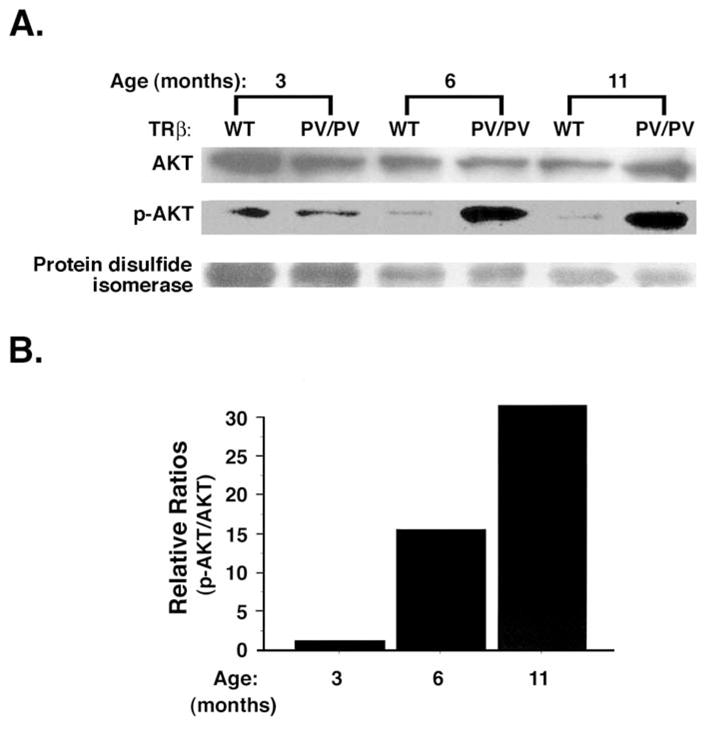
Activation of AKT by increased phosphorylation in the thyroids of TRβPV/PV mice in an age-dependent manner. (A). Thyroid extracts (50 μg) of TRβPV/PV mice and aged-matched wild-type mice were prepared and analyzed for total AKT, phosphorylated AKT (p-AKT), and protein disulfide isomerase (loading control) by Western blot analysis. (B). Densitometric analysis compared p-AKT to total AKT using a normalized ratio of TRβPV/PV mice (p-AKT/AKT)/wild-type mice (p-AKT/AKT) at each respective age [30].
To understand the molecular mechanisms by which the mutant PV increases the phosphorylation of AKT, the activity of its upstream kinase, PI3K, was compared between wild-type and TRβPV/PV mice. PI3K was immunoprecipitated from thyroid extracts by using antibody against the regulatory subunit p85α followed by kinase assays. Fig. 2 shows that at any given extract concentrations, PI3K activity is markedly increased in the thyroid of TRβPV/PV mice as compared with wild-type mice. Further biochemical analyses using GST-pull down assays indicated that the C-terminal SH2 domain of p85α physically interacts with the ligand-binding domain of TRβ1 or PV [31]. Importantly, p85α binds to PV 2–3 times stronger than to TRβ1. To identify the subcellular location at which such interaction takes place, thyroid extracts from TRβPV/PV mice were fractionated into nuclear and cytosolic fractions. Co-immunoprecipitation analysis of these fractions indicated that the association of p85α with TRβ1 or PV occurs in the nuclear compartment (lanes 3 and 4; Fig. 3) as well as in the extra-nuclear compartment (lanes 1 and 2; Fig. 3). It is important to note that p85α binds more to PV than to TRβ1 (lane 4 versus 3 and lane 2 versus 1; Fig. 3) in both compartments. Confocal microscopy also confirmed these subcellular interaction sites [31].
Fig. 2.
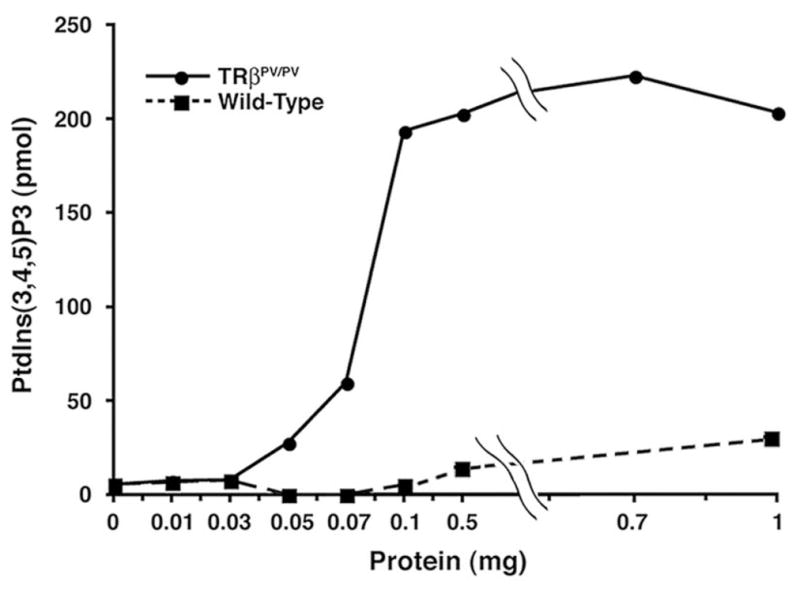
Activation of PI3K activity in the thyroids of TRβPV/PV mice. Increasing concentrations as marked of total thyroid extracts from wild-type mice (solid squares) and TRβPV/PV mice (solid circles) were immunoprecipitated with anti-p85α antibody. The PI3K activity of precipitates from each concentration was measured for the conversion of PtdIns(3,4,5)P2 to PtdIns(3,4,5)P3 by ELISA. The data are expressed as the relative production of PtdIns(3,4,5)P3 by each sample [31].
Fig. 3.
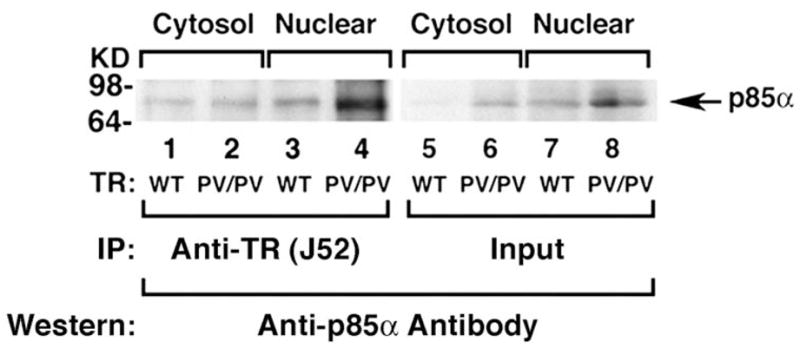
p85α interacts more avidly with PV than with TRβ1 in the cytosolic and nuclear compartments. The thyroid extracts of 12 wild-type mice or three TRβPV/PV mice were pooled and separated into nuclear or cytosolic fractions. The purity of each fraction was monitored by the respective markers, poly ADP-ribose polymerase for the nuclear fraction and α-tubulin for the cytosolic fraction. An equal amount of the nuclear or cytosolic fraction (100 μg proteins) were each immunoprecipitated with 5 μg of monoclonal anti-TR/PV antibody J52 followed by Western blot analysis using anti-p85α antibody. Lanes are as marked. Lanes 5 – 8 show the corresponding input of the p85α protein.
Emerging evidence has indicated the presence of PI3Ks, their lipid products, and AKT in the nuclear compartments of many cultured cells and some tissues [32, 33]. However, the control and actions of the nuclear PI3K remain unclear. Thus, the functional relevance of the interaction of p85α with PV in the nuclear compartment was assessed by PV-induced changes in the downstream signaling. Two signaling pathways were evaluated—AKT-mTOR-p70S6k and the integrin-linked kinase (ILK)-MMP (matrix metalloproteinase) pathways. The AKT-mTOR-p70S6k signaling is activated in both the nuclear and cytosolic compartments as shown by the increased ratios of p-AKT/total ATK (Fig. 4A-a), p-mTOR/total mTOR (Fig. 4A-b), and p-p70S6k (Fig. 4A-c). It is important to note that the PV-mediated AKT-mTOR-p70S6k signaling is markedly stronger in the nucleus than in the cytosolic compartment, as shown by a higher ratio in the nuclear compartment than in the cytosolic compartment (Fig. 4A). In contrast, the PV-mediated activation of the ILK-MMP signaling pathway occurs mainly in the extra-nuclear compartment (Fig. 4B) as no MMP2 was detected in the nuclear compartment. 00Therefore, the interaction of p85α with PV occurs in both the nuclear and cytosolic compartments and could activate the signaling that is subcellular site-dependent. The AKT-mTOR-p70S6k pathway is known to increase cell proliferation and suppress apoptosis [34], and the ILK-MMP pathway is involved in the degradation of the extra-cellular matrix that affects cancer cell invasion and metastasis [35, 36]. Given the critical role of PI3K, the activation of the PI3K activity [PAS7]via protein-protein interaction in both the nuclear and extra-nuclear compartments would be expected to affect the diverse downstream signaling cascades to mediate thyroid carcinogenesis.
Fig. 4.


Activation of the PI3K-AKT-mTOR-p70S6K and the integrin-linked kinase (ILK)-MMP2 pathways in the thyroids of TRβPV/PV mice. Pooled extracts from thyroids of 12 wild-type mice or three TRβPV/PV mice were separated into nuclear and cytosolic fractions. The purity of the fractions was confirmed by using respective markers, poly ADP-ribose polymerase for the nuclear fraction and α-tubulin for the cytosolic fraction. (A). Western blot analysis was carried out to determine cytosolic and nuclear abundance of the following proteins: total AKT, p-AKT(S473), total mTOR, p-mTOR, total p70S6K, and p-p70S6K. The band intensities were quantified and the ratios of p-AKT/total AKT (a), p-mTOR/total mTOR (b), p-p70S6K/total p70S6K (c) were expressed to indicate the activation of the respective effectors. (B). Western blot analysis of protein abundance of ILK (a) and MMP2 (b) in the cytosolic fraction of thyroid tumors of TRβPV/PV mice. The activation of this pathway occurs mainly in the cytosolic compartment.
2.3. Increase the stability of pituitary tumor-transforming gene (PTTG) by PV
PTTG is a securin and its association with separin (a protease) prevents the proteolytic activity of separin from acting on cohesions that hold the sister chromatids together before entering anaphase [37]. The timely degradation of securin by the proteasome-mediated pathway frees the separin to proteolyze cohesions for proper separation of sister chromatids at the anaphase. Thus, abnormally elevated PTTG disrupts proper separation of sister chromatids, resulting in aneuploidy [38, 39]. A strong correlation of the overexpressed PTTG with genetic instability has recently been demonstrated not only in a thyroid cancer mouse model (TRβPV/PV mice) [40], but also in human thyroid cancers [41]. PTTG was originally isolated from GH4 pituitary cells and shown to cause in vitro cell transformation and to induce tumor formation in vivo [42]. Overexpression of PTTG has been detected in human thyroid carcinomas [43, 44], colorectal carcinoma [45], pituitary adenomas [46], and hematopoietic neoplasms [47]. Despite the close association of overexpressed PTTG with carcinogenesis, very little is known about the mechanisms by which the cellular expression of PTTG is regulated.
One of significantly activated genes detected by cDNA microarray analysis during thyroid carcinogenesis of TRβPV/PV mice is PTTG [48]. In addition to an elevated mRNA expression, PTTG protein abundance is also markedly increased in the primary lesions of thyroid as well as the lung metastases [49]. The increase in PTTG mRNA in TRβPV/PV mice likely explains, at least partially, the increase in the PTTG protein, and it suggests some effect of PV on PTTG gene expression or mRNA stabilization. Since TRs [50] and PTTG [51] are known to participate in the proteasome-mediated degradation pathway, whether TRβ1 or PV could operate through such mechanisms to directly modulate the cellular abundance of PTTG proteins was explored.
Indeed, a series of studies by GST-pull down, confocal microscopy, and Gal4-reporter system showed that the DNA binding domain of TRβ1 or PV interacts with the amino-terminal region (amino acid 1–119) of PTTG [49]. Furthermore, the T3-induced proteasomal degradation of TRβ1 is tightly linked to the degradation of PTTG. As shown in Fig. 5, concomitant with the T3-induced degradation of transfected Flag tagged-TRβ1 (F-TRβ1; lane 6, Fig. 5A), transfected Flag tagged-PTTG was also degraded (F-PTTG). In contrast, in the presence of T3, but without F-TRβ1, no degradation of PTTG occurred (compare lane 2 to lane 6, Fig. 5A). That no degradation of liganded F-TRβ1 and F-PTTG occurred in the presence of the specific proteasome inhibitor, MG132, (lane 8, Fig. 5A) further supports the idea that the liganded TRβ1 and PTTG are degraded via the proteasomal machinery. Similar liganded TRβ1-facilitated degradation of the endogenous PTTG was also observed in cells [49].
Fig. 5.

T3-dependent proteasomal degradation of TRβ1 linked to the degradation of PTTG. Regulation of PTTG protein stability by the T3-bound TRβ1 via the proteasome-mediated pathway (A & B), but not by PV (C & D). [PAS10]CV1 cells were co-transfected with expression plasmids of F-PTTG, F-TRβ1 (A and B), F-PV (C and D) in the absence or presence (100 nM) of T3 and with or without MG132 as indicated. Antibodies used for Western blot analysis were: anti-Flag antibodies, anti-PTTG antibodies and for loading controls, anti-α-tubulin (B and D) [49].
In the presence of T3 and transfected Flag tagged-PV (F-PV), a different stability profile of PTTG emerged. In the presence of T3, PV remained at a level similar to that without T3 (compare lane 6 to lane 5, Figure 5C). Concomitant with the stability of PV, PTTG remained high, independent of T3 (lanes 5 and 6, Figure 5C). Similarly, accumulated endogenous PTTG was detected in cells stably expressing PV [49]. Taken together, these results indicate that the liganded TRβ1 regulates the stability of PTTG. The regulatory function is lost in PV that fails to bind to T3 due to mutation.
PTTG is a mammalian securin functioning to hold sister chromatids together during mitosis, and its overexpression has been shown to cause aneuploidy [37, 38, 41, 51]. The effect of PV-induced aberrant accumulation of PTTG on cell cycle progression was analyzed after arresting the FH-TRβ1 and FH-PV cells at the G2/M phase by thymidine/nocodazole block and releasing from [PAS8]the G2/M block by culturing in T3-containing growth media. FH-TRβ1 and FH-PV cells stably express TRβ1 and PV, respectively. Endogenous PTTG, F-TRβ1, F-PV, and the G2/M phase marker (cyclin B1) protein abundance were assessed in a time-dependent manner as cells were exiting from the G2/M phase (Fig. 6). Both phosphorylated (p-PTTG) and non-phosphorylated PTTG were detected in G2/M phase (lanes 2–5 and 8–12, Fig. 6). As shown in lanes 3–6 of Fig. 6, 14.8%, 60.8%, 71.6 %, and 74.2% of FH-TRβ1 had exited the G2/M phase to enter into the G0/G1 and S phase after 2, 4, 6, and 8 hours, respectively. However, significantly lower numbers of FH-PV cells had exited from the G2/M phase (14.9%, 30.3%, 38.2%, and 53.3% after 2, 4, 6, and 8 hours, respectively (lanes 9–12, Fig. 6). The severe delay in exiting from the G2/M phase was also shown by the delayed degradation of the G2/M phase marker, cyclin B1 (compare lanes 8–12 to lanes 2–6, Fig. 6). These results indicate that aberrant accumulation of PTTG induced by PV inhibits mitotic progression.
Fig. 6.
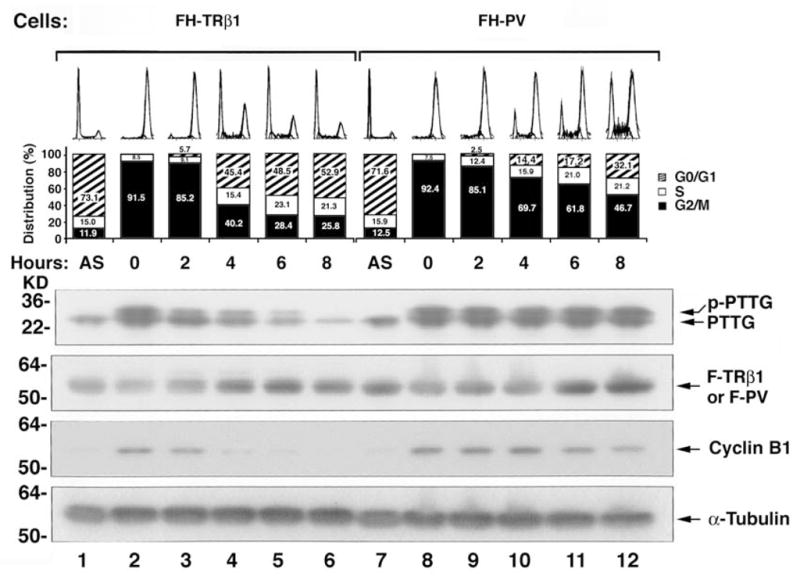
Delayed mitotic exit of cells stably expressing PV (FH-PV cells) as compared with cells stably expressing TRβ1 (FH-TRβ1 cells). Aberrant accumulation of PTTG induced by PV impedes mitotic progression. FH-TRβ1 and FH-PV cells were synchronized in prometaphase/metaphase by thymidine/nocodazole block. Synchronized cells were collected at time 0 hour and then released by changing to growth media containing 20% fetal bovine serum. Cells harvested at various time points after release were subjected to Western blot and FACS analyses. An asynchronous population of FH-TRβ1 and FH-PV cells was also examined (AS). The protein abundance of PTTG, F-TRβ1, F-PV, cyclin B1, and loading control, α-tubulin, was determined by Western blot analysis [49].
Recent reports have suggested that steroid receptor coactivator-3 (SRC-3) is physically associated with PA28γ, an activator of the trypsin-like activity of the proteasome and that this interaction results in the increased degradation of SRC-3 via 19S proteasome in an ubiquitin- and ATP-independent manner [52, 53]. These findings prompted us to ascertain whether the differential recruitment of TRβ1/PTTG and PV/PTTG complexes by SRC-3/PA28γ accounted for the functional differences in the regulation of the stability of PTTG. Using GST-pull down assays and cell-based coimmunoprecipitation analysis, we found that the liganded TRβ1/PTTG recruits SRC-3/PA28γ through the direct interaction of the liganded TRβ1 with SRC-3, whereas the unliganded TRβ1 and PV, that does not bind T3, fails [PAS9]to recruit SRC-3/PA28γ [49]. These findings are summarized in the molecular model shown in Fig. 7. PTTG physically associates with TRβ1 and its degradation is facilitated by T3-dependent recruitment of SRC-3/PA28γ complexes. The degradation of PTTG frees the separin to proteolyze cohesions to facilitate normal separation of sister chromatids during anaphase (Fig. 7A). The unliganded TRβ1, as well as PV (which does not bind T3), cannot recruit the SRC-3/PA28γ complexes to activate the proteasome degradation; therefore, the PV/PTTG complex remains stable as shown by the persistently elevated PTTG and PV (Fig. 7B; also see Fig. 6). The failure of timely degradation of PTTG severely inhibits the proper exit of cells from the G2/M phase that could lead to aneuploidy.
Fig. 7.
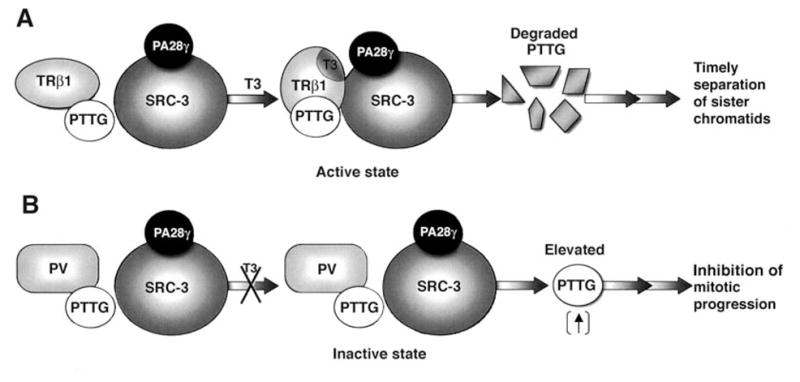
A proposed molecular model for the liganded TRβ1-dependent SRC-3/PA28γ activated degradation of PTTG (A). The direct interaction of TRβ1 with SRC-3 induced by the binding of T3 results in the formation of liganded TRβ1/PTTG/SRC-3/PA28γ complexes that activate the degradation of PTTG. The degradation of PTTG leads to timely separation of sister chromatids. (B). PV also forms complexes with PTTG. But because PV does not bind T3, PV cannot directly bind to SRC-3/PA28γ to activate degradation. The absence of degradation leads to elevated PTTG and results in inhibition of mitotic progression [49].
3. Conclusions and future challenges
Studies so far have shown that the deleterious effects of TRβ mutants in causing RTH are mediated via dominant negative actions at the transcriptional level [13, 54]. A prominent example is the persistently elevated serum TSH levels due to the loss of the negative regulation of TSHβ and the α-glycoprotein common subunit genes by the dominant negative effects of TRβ mutants at the transcription level [15]. The striking phenotype of thyroid cancer manifested by TRβPV/PV mice led to the identification of new modes of action of TRβ mutants that are beyond nucleus-initiated transcription. The direct protein-protein interaction of PV with p85α increases the activity of PI3K, leading to an activated cascade of downstream signaling to affect cell proliferation, apoptosis, migration, and metastasis. The direct protein-protein interaction of PV with PTTG derails the normal regulatory pathway, resulting in aberrant accumulation of cellular PTTG, leading to genetic instability. Thus, a complete manifestation of thyroid neoplasm in TRβPV/PV requires not only timely orchestration of transcription events, but also the active participation of actions of PV beyond nucleus- initiated transcription.
Currently, molecular details about the interaction sites/motifs of PV with p85α or with PTTG are unknown, nor is it known how these interactions are modulated by additional cellular factors. Similar to insulin receptor and IRS [55, 56], PV interacts with the SH2 domain of p85α. Insulin receptor and IRS bind to the SH2 domain due to their tyrosine phosphorylation via the motifs of YXXM. The activity of TR has been shown to be regulated by phosphorylation [57–60]. But it is presently unknown whether the activity of PV is modulated by tyrosine phosphorylation. In addition, very little is known about how PTTG interacts with TR or PV. These important questions have not yet been investigated.
A future challenge is to elucidate whether, in addition to thyroid cancer, these novel modes of action of PV also underscore the pathogenesis of other abnormalities such as RTH and pituitary tumors [61]. In view of the critical role of PI3K in diverse cellular functions, it is highly likely that the interaction of PV with p85α could also lead to aberrant signaling of some downstream pathways in a tissue-dependent manner. Growth retardation is apparent in patients with RTH caused by mutations of the TRβ gene [12, 13]. The accumulation of PTTG induced by PV leads to a marked delay for cells to exit the G2/M phase to advance to the next phase of the cell cycle, thereby impeding cell cycle progression and cell growth. The finding that TRβPV/PV mice, similar to RTH patients, exhibit severe growth retardation [15] further supports the idea that loss of the normal regulation of cellular PTTG could underlie the pathogenesis of RTH. The verification of these possibilities awaits future studies.
Acknowledgments
We regret any reference omissions due to length limitation. The work presented was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–6. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- 2.Sap J, Munoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennstrom B. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324:635–40. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- 3.Bassett JH, Harvey CB, Williams GR. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol Cell Endocrinol. 2003;213:1–11. doi: 10.1016/j.mce.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 4.Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X. Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol. 2006;246:121–7. doi: 10.1016/j.mce.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 5.Cheng SY. Multiple mechanisms for regulation of the transcriptional activity of thyroid hormone receptors. Rev Endocr Metab Disord. 2000;1:9–18. doi: 10.1023/a:1010052101214. [DOI] [PubMed] [Google Scholar]
- 6.Wondisford FE. Thyroid hormone action: insight from transgenic mouse models. J Investig Med. 2003;51:215–20. doi: 10.1136/jim-51-04-22. [DOI] [PubMed] [Google Scholar]
- 7.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, Rosenfeld MG. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 8.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–7. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 9.Tsai CC, Fondell JD. Nuclear receptor recruitment of histone-modifying enzymes to target gene promoters. Vitam Horm. 2004;68:93–122. doi: 10.1016/S0083-6729(04)68003-4. [DOI] [PubMed] [Google Scholar]
- 10.Moore JM, Guy RK. Coregulator interactions with the thyroid hormone receptor. Mol Cell Proteomics. 2005;4:475–82. doi: 10.1074/mcp.R500001-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–28. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 12.Weiss R, Refetoff S. Resistance to thyroid hormone. Rev Endocr Metab Disord. 2000;1:97–108. doi: 10.1023/a:1010072605757. [DOI] [PubMed] [Google Scholar]
- 13.Yen PM. Molecular basis of resistance to thyroid hormone. Trends Endocrinol Metab. 2003;14:327–33. doi: 10.1016/s1043-2760(03)00114-0. [DOI] [PubMed] [Google Scholar]
- 14.Ono S, Schwartz ID, Mueller OT, Root AW, Usala SJ, Bercu BB. Homozygosity for a dominant negative thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metab. 1991;73:990–4. doi: 10.1210/jcem-73-5-990. [DOI] [PubMed] [Google Scholar]
- 15.Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham NC, Barlow C, Cheng S-y. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA. 2000;97:13209–14. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parrilla R, Mixson AJ, McPherson JA, McClaskey JH, Weintraub BD. Characterization of seven novel mutations of the c-erbA beta gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest. 1991;88:2123–30. doi: 10.1172/JCI115542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meier CA, Dickstein BM, Ashizawa K, McClaskey JH, Muchmore P, Ransom SC, Menke JB, Hao EH, Usala SJ, Bercu BB. Variable transcriptional activity and ligand binding of mutant beta 1 3,5,3′-triiodothyronine receptors from four families with generalized resistance to thyroid hormone. Mol Endocrinol. 1992;6:248–58. doi: 10.1210/mend.6.2.1569968. [DOI] [PubMed] [Google Scholar]
- 18.Kamiya Y, Zhang XY, Ying H, Kato Y, Willingham MC, Xu J, O’Malley BW, Cheng S-y. Modulation by steroid receptor coactivator-1 of target-tissue responsiveness in resistance to thyroid hormone. Endocrinology. 2003;144:4144–53. doi: 10.1210/en.2003-0239. [DOI] [PubMed] [Google Scholar]
- 19.Griffith AJ, Szymko YM, Kaneshige M, Quinonez RE, Kaneshige K, Heintz KA, Mastroianni MA, Kelley MW, Cheng SY. Knock-in mouse model for resistance to thyroid hormone (RTH): an RTH mutation in the thyroid hormone receptor beta gene disrupts cochlear morphogenesis. J Assoc Res Otolaryngol. 2002;3:279–88. doi: 10.1007/s101620010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Shea PJ, Harvey CB, Suzuki H, Kaneshige M, Kaneshige K, Cheng SY, Williams GR. A thyrotoxic skeletal phenotype of advanced bone formation in mice with resistance to thyroid hormone. Mol Endocrinol. 2003;17:1410–24. doi: 10.1210/me.2002-0296. [DOI] [PubMed] [Google Scholar]
- 21.Siesser WB, Cheng SY, McDonald MP. Hyperactivity, impaired learning on a vigilance task, and a differential response to methylphenidate in the TRbetaPV knock-in mouse. Psychopharmacology (Berl) 2005;181:653–63. doi: 10.1007/s00213-005-0024-5. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X-y, Kaneshige M, Kamiya Y, Kaneshige K, McPhie P, Cheng SY. Differential expression of thyroid hormone receptor isoforms dictates the dominant negative activity of mutant β receptor. Mol Endocrinol. 2002;16:2077–92. doi: 10.1210/me.2002-0080. [DOI] [PubMed] [Google Scholar]
- 23.Miller LD, McPhie P, Suzuki H, Kato Y, Liu ET, Cheng SY. Multi-tissue gene-expression analysis in a mouse model of thyroid hormone resistance. Genome Biol. 2004;5:R31. doi: 10.1186/gb-2004-5-5-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor Beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–9. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- 25.Cheng SY. Thyroid hormone receptor mutations in cancer. Mol Cell Endocrinol. 2003;213:23–30. doi: 10.1016/j.mce.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 26.Cheng SY. Abnormalities of nuclear receptors in thyroid cancer. Cancer Treat Res. 2004;122:165–78. doi: 10.1007/1-4020-8107-3_9. [DOI] [PubMed] [Google Scholar]
- 27.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 28.Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, Bernet V, Burman KD, Kohn LD, Saji M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61:6105–11. [PubMed] [Google Scholar]
- 29.Miyakawa M, Tsushima T, Murakami H, Wakai K, Isozaki O, Takano K. Increased expression of phosphorylated p70S6 kinase and Akt in papillary thyroid cancer tissues. Endocr J. 2003;50:77–83. doi: 10.1507/endocrj.50.77. [DOI] [PubMed] [Google Scholar]
- 30.Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, Ringel MD. AKT activation promotes metastasis in a mouse model of folliculara thyroid carcinoma. Endocrinology. 2005;146:4456–63. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- 31.Furuya F, Hanover J, Cheng SY. Activation of the PI3K signaling by a mutant thyroid hormone β receptor. Proc Natl Acad Sci USA. 2006;103:1780–5. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neri LM, Borgatti P, Capitani S, Martelli AM. The nuclear phosphoinositide 3-kinase/AKT pathway: a new second messenger system. Biochim Biophys Acta. 2002;1584:73–80. doi: 10.1016/s1388-1981(02)00300-1. [DOI] [PubMed] [Google Scholar]
- 33.Irvine RF. Nuclear lipid signalling. Nat Rev Mol Cell Biol. 2003;4:349–60. doi: 10.1038/nrm1100. [DOI] [PubMed] [Google Scholar]
- 34.Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinease switching between life and death. Pharmacol Res. 2004;50:545–9. doi: 10.1016/j.phrs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2004;3:207–14. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 36.Turpeenniemi-Hujanen T. Gelatinases (MMP-2 and -9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie. 2005;87:287–97. doi: 10.1016/j.biochi.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H, McGarry TJ, Bernal T, Kirschner MW. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science. 1999;285:418–22. doi: 10.1126/science.285.5426.418. [DOI] [PubMed] [Google Scholar]
- 38.Yu R, Melmed S. Oncogene activation in pituitary tumors. Brain Pathol. 2001;11:328–41. doi: 10.1111/j.1750-3639.2001.tb00403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramos-Morales F, Dominguez A, Romero F, Luna R, Multon MC, Pintor-Toro JA, Tortolero M. Cell cycle regulated expression and phosphorylation of HPTTG proto-oncogene product. Oncogene. 2000;19:403–9. doi: 10.1038/sj.onc.1203320. [DOI] [PubMed] [Google Scholar]
- 40.Zimonjic DB, Kato Y, Ying H, Popescu NC, Cheng SY. Chromosomal aberrations in cell lines derived from thyroid tumors spontaneously developed in TRbetaPV/PV mice. Cancer Genet Cytogenet. 2005;161:104–9. doi: 10.1016/j.cancergencyto.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Kim D, Pemberton H, Stratford AL, Buelaert K, Watkinson JC, Lopes V, Franklyn JA, McCabe CJ. Pituitary tumour transforming gene (PTTG) induces genetic instability in thyroid cells. Oncogene. 2005;24:4861–6. doi: 10.1038/sj.onc.1208659. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Horwitz GA, Prezant TR, Valentini A, Nakashima M, Bronstein MD, Melmed S. Structure, expression, and function of human pituitary tumor-transforming gene (PTTG) Mol Endocrinol. 1999;13:156–66. doi: 10.1210/mend.13.1.0225. [DOI] [PubMed] [Google Scholar]
- 43.Heaney AP, Nelson V, Fernando M, Horwitz G. Transforming events in thyroid tumorigenesis and their association with follicular lesions. J Clin Endocrinol Metab. 2001;86:5025–32. doi: 10.1210/jcem.86.10.7886. [DOI] [PubMed] [Google Scholar]
- 44.Kim DS, McCabe CJ, Buchanan MA, Watkinson JC. Oncogenes in thyroid cancer. Clin Otolaryngol Allied Sci. 2003;28:386–95. doi: 10.1046/j.1365-2273.2003.00732.x. [DOI] [PubMed] [Google Scholar]
- 45.Heaney AP, Singson R, McCabe CJ, Nelson V, Nakashima M, Melmed S. Expression of pituitary-tumour transforming gene in colorectal tumours. Lancet. 2000;355:716–9. doi: 10.1016/S0140-6736(99)10238-1. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Horwitz GA, Heaney AP, Nakashima M, Prezant TR, Bronstein MD, Melmed S. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab. 1999;84:761–7. doi: 10.1210/jcem.84.2.5432. [DOI] [PubMed] [Google Scholar]
- 47.Dominguez A, Ramos-Morales F, Romero F, Rios RM, Dreyfus F, Tortolero M, Pintor-Toro JA. hPTTG, a human homologue of rat PTTG, is overexpressed in hematopoietic neoplasms. Evidence for a transcriptional activation function of hPTTG. Oncogene. 1998;17:2187–93. doi: 10.1038/sj.onc.1202140. [DOI] [PubMed] [Google Scholar]
- 48.Ying H, Suzuki H, Furumoto H, Walker R, Meltzer P, Willingham MC, Cheng SY. Alterations in genomic profiles during tumor progression in a mouse model of follicular thyroid carcinoma. Carcinogenesis. 2003;24:1467–79. doi: 10.1093/carcin/bgg111. [DOI] [PubMed] [Google Scholar]
- 49.Ying H, Furuya F, Zhao L, Araki O, West BL, Hanover JA, Willingham MC, Cheng SY. Inhibition of mitotic progression by aberrant accumulation of PTTG induced by a mutated thyroid hormone β receptor. J Clin Invest. 2006 doi: 10.1172/JCI28598. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dace A, Zhao L, Park KS, Furuno T, Takamura N, Nakanishi M, West BL, Hanover JA, Cheng S. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proc Natl Acad Sci USA. 2000;97:8985–9. doi: 10.1073/pnas.160257997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu R, Heaney AP, Lu W, Chen J, Melmed S. Pituitary tumor transforming gene causes aneuploidy and p53-dependent and p53-independent apoptosis. J Biol Chem. 2000;275:36502–5. doi: 10.1074/jbc.C000546200. [DOI] [PubMed] [Google Scholar]
- 52.Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai S, Tsai MJ, O’Malley B. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell. 2006;120:1123–34. doi: 10.1016/j.cell.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 53.Gao X, Li J, Pratt G, Wilk S, Rechsteiner M. Purification procedures determine the proteasome activation properties of REG gamma (PA28 gamma) Arch Biochem Biophys. 2004;425:158–64. doi: 10.1016/j.abb.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 54.Cheng SY. Thyroid hormone receptor mutations and disease: beyond thyroid hormone resistance. Trends Endocrinol Metab. 2005;16:176–82. doi: 10.1016/j.tem.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 55.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 56.Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J. 1998;333:471–90. doi: 10.1042/bj3330471. Erratum 1998;335:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhat MK, Ashizawa K, Cheng SY. Phosphorylation enhances the target gene sequence-dependent dimerization of thyroid hormone receptor with retinoid X receptor. Proc Natl Acad Sci USA. 1994;91:7927–31. doi: 10.1073/pnas.91.17.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ting YT, Cheng SY. Hormone-activated phosphorylation of human beta1 thyroid hormone nuclear receptor. Thyroid. 1997;7:463–9. doi: 10.1089/thy.1997.7.463. [DOI] [PubMed] [Google Scholar]
- 59.Ting YT, Bhat MK, Wong R, Cheng S. Tissue-specific stabilization of the thyroid hormone beta1 nuclear receptor by phosphorylation. J Biol Chem. 1997;272:4129–34. doi: 10.1074/jbc.272.7.4129. [DOI] [PubMed] [Google Scholar]
- 60.Davis PJ, Shih A, Lin HY, Martino LJ, Davis FB. Thyroxine promotes association of mitogen-activated protein kinase and nuclear thyroid hormone receptor (TR) and causes serine phosphorylation of TR. J Biol Chem. 2000;275:38032–9. doi: 10.1074/jbc.M002560200. [DOI] [PubMed] [Google Scholar]
- 61.Furumoto H, Ying H, Chandramouli GV, Zhao L, Walker RL, Meltzer PS, Willingham MC, Cheng SY. An unliganded thyroid hormone β receptor activates the cyclin D1/cyclin-dependent kinase/retinoblastoma/E2F pathway and induces pituitary tumorigenesis. Mol Cell Biol. 2005;25:124–35. doi: 10.1128/MCB.25.1.124-135.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]