

Abstract
Dynamics of water are critically important to the energies of interaction between proteins and substrates and determine the efficiency of transport at the interface. The magnetic field dependence of nuclear spin-lattice relaxation rate constant 1/T1 of water protons provides a direct characterization of water diffusional dynamics at the protein interface. We find that the surface-average translational correlation time is 30–40 ps, and the magnetic field dependence of the water-proton 1/T1 is characteristic of 2-dimensional diffusion of water in the protein interfacial region. The reduced dimensionality substantially increases the intermolecular reencounter probability and the efficiency of the surface exploration by the small molecule, water in this case. We propose a comprehensive theory of the translational effects of a small diffusing particle confined in the vicinity of a spherical macromolecule as a function of the relative size of the two particles. We show that the change in the apparent dimensionality of the diffusive exploration is a general result of the small diffusing particle encountering a much larger particle that presents a diffusion barrier. Examination of the effects of the size of the confinement relative to the macromolecule size reveals that the reduced dimensionality characterizing the small molecule diffusion persists to remarkably small radius ratios. The experimental results on several different proteins in solution support the proposed theoretical model that may be generalized to other small particle-large body systems like vesicles and micelles.
Keywords: water-protein dynamics, MRD, proton spin-lattice relaxation, anisotropic water diffusion, surface dynamics, restricted diffusion
I. INTRODUCTION
Quantitative characterization of water molecule dynamics at protein interfaces is critical to understanding the energetic costs of intermolecular events such as molecular recognition and consequent molecular rearrangements [1–3]. Water dynamics also determines the effective interfacial viscosity. Experimental characterization of dynamics in the protein interfacial region has been challenging because it extends only a nanometer or so from the surface and represents a small volume fraction of the whole solution. Neutron scattering, magnetic relaxation spectroscopy, optical relaxation spectroscopy, and molecular dynamics simulations have partially addressed this region [4–20]. It is generally appreciated that rotational and translational correlation times for water in the interfacial region adjacent to a protein are short, i.e., motions are fast and not dynamically ice-like. However, characterization of how the macromolecule modifies the motion of small solutes or solvent in the interface is incomplete. Considerable insight is provided by simulations while experimental characterizations have derived from neutron scattering and magnetic resonance measurements. We focus here on nuclear 1H spin-lattice relaxation.
The magnetic field dependence of nuclear spin-lattice relaxation rate constants reports the frequency dependence of the noise that drives the spin relaxation which is determined by the nature of the motions that modulate the inter and intramolecular magnetic dipolar couplings. In the case that the relaxation is dominated by translational motions, the shape of magnetic relaxation dispersion profile (MRD) is sensitive to the dimensionality of the diffusion process and the correlation times for the relative motion of the spin-bearing molecules. The approach is well suited to characterization of interfacial dynamics. Here we apply this experimental approach to water in protein solutions and find that, at large Larmor frequencies, the water-proton-spin-lattice-relaxation-rate constants are logarithmic functions of the Larmor frequency. Analysis of these data using an original translational model yields a water translational correlation time at the protein surface of 30 ps for self diffusion and 15 ps for relative diffusion. The logarithmic field dependence implies that the 2-particle re-encounter probability density at the surface is strongly biased by the steric constraints of the surface and is characteristic of a 2-dimensional rather than a 3-dimensional exploration. This result is important in understanding how a small molecule samples the surface of a large molecule as a precursor to a binding event. The results here raise the fundamental question of how the surface creates the effects of reduced dimensionality and what the requirements may be for the spatial extent of the surface compared to the size of the observed molecule diffusing in this vicinity. Although motivated by the biophysical problem of water-protein interfaces, the question is general, and, to first approximation, independent of the origin of local molecular heterogeneity.
For a flat planar surface, such as a lamellar system created by liquid crystal environments or stacked plates or platelets as in clay minerals, it is well known that the dynamics of liquids are modified in a characteristic way [21]. One may expect that for spherical surface with a radius that is very large compared with the smaller explorer molecule, the interface may look locally planar. However, it is not clear how the dynamics should behave as a function of the relative size of the macromolecular or particle surface compared with the size of the diffusing small molecule that is observed. Even though most surfaces are not molecularly flat, we seek to address this question with a simple geometrically smooth spherical boundary. The spin-lattice relaxation depends on the power spectrum of the noise at the Larmor frequency, which may be deduced from the time correlation function characterizing the magnetic dipole-dipole couplings in the system. In this study we examine in detail the diffusion propagator P of the spin-bearing small explorer molecules moving in a layer of thickness δ in the immediate vicinity of a spherical molecule of radius R. We focus in particular on the time dependence of P (t), the probability of return to the spherical surface at various times. We show that P(t) ∝ 1/t with the time range tmin < t < tmax, where tmin = δ2/(πD) and tmax = L2/(2D) with L = R + δ for different ratios of δ/R compatible with experiments. These two times delimit the regime of diffusive behavior and predicts that the nuclear spin-lattice-relaxation-rate constant is a logarithmic function of the Larmor frequency in the corresponding frequency range.
The magnetic field dependence of the water-proton nuclear spin-lattice-relaxation-rate constant, 1/T1, in a protein system depends on the state of the protein. For pure water, the rotational and translational correlation times are less than 10 ps and the 1H2O spin-lattice relaxation rate constant is independent of field strength up to the highest magnetic fields currently available. In a protein solution, the water molecules may interact with the protein in several ways: 1) A few water molecules may bind to the protein and remain bound for times long compared to the rotational correlation time of the protein. While bound, the proton dipolar couplings are rotationally correlated with a correlation time equal to the rotational correlation time of the protein. The bulk water proton relaxation is sensitive to these rare binding interactions because the bound water molecules exchange with the bulk population in times short compared with the spin-lattice relaxation times in either environment. Therefore, the water relaxation dispersion reports the rotational correlation time of the protein [8, 10, 22–24] as shown in Fig. 1. 2) Water molecules may exchange protons with labile protein sites on the protein. This proton exchange adds a pH dependent contribution to the effects of water molecule exchange and is similarly sensitive to the rotational correlation time of the protein. 3) Water molecules at the protein surface may suffer transient couplings to the protein protons modulated by water molecule rotational and translational diffusion. Although the bulk of evidence suggests that both rotational and translational motion at the interface is slowed by a factor of 3 or more relative to pure water, the average surface motions of water are very rapid compared to the global rotational correlation time of the protein. It is these surface motions that contribute to the weak magnetic field dependence of the water spin-lattice relaxation rate constants investigated here [25–28]. For rotationally immobilized proteins, such as a cross-linked gel, the same water dynamics is important; however, the water-proton MRD is a power law in the Larmor frequency from 0.01–300 MHz and the relaxation rate constant is related to the structural fluctuations in the protein [29–31].
FIG. 1.
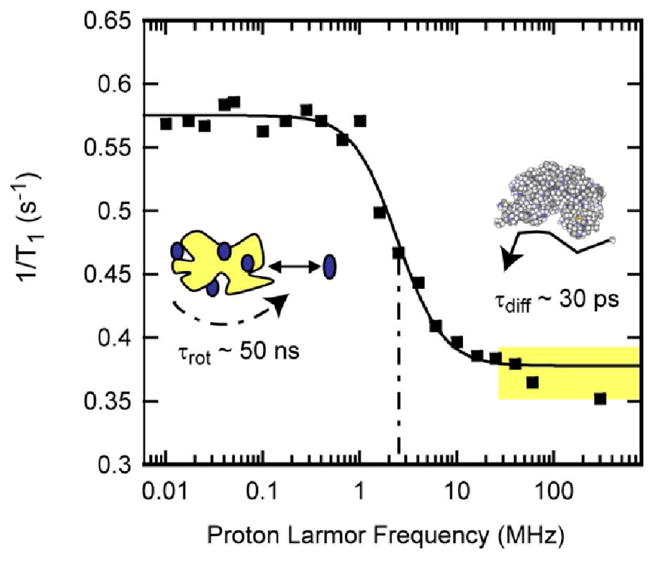
Proton spin-lattice relaxation rate constants as a function of magnetic field strength plotted as the Larmor frequency for 150 μM bovine serum albumin in H2O purged with nitrogen gas at laboratory temperature at 22°C. The solid line is a Lorentzian fit and reports the rotational correlation time of the protein at 45 ns [48]. The region marked in yellow above the rotational dispersion is dominated by relaxation events at the protein interface and is the focus of this work.
As noted earlier, the MRD measurement of the water-proton resonance reports on the spin-lattice-relaxation-rate constant that is averaged over the several environments among which the water molecules exchange. The observed relaxation-rate constant is a weighted average of relaxation-rate constants in each environment. At high magnetic field strengths, i.e., above the rotational dispersion shown in Fig. 1, the exchange process averages the water proton relaxation rate constants in the surface environments with those of the bulk. The bulk water has no magnetic field dependence in the field ranges studied; thus, the relaxation dispersion profile is determined by the dynamics suffered by the water in the immediate vicinity of the protein. An important aspect of spin relaxation and this averaging process is that the slower motions make often dominant contributions to the average rate constant. This bias is easiest to see in the low field limit where relaxation rate constants are approximately linear in the correlation time. If, for example, ten percent of spins in the interface suffer a correlation time that is ten times longer than the rest, the contribution of these slow moving spins is equal to that of the dominant population which moves more rapidly. Therefore, in the case of a distribution of effective translational or rotational correlation times, the observed spin relaxation rate constant is weighted more heavily by the slower dynamics.
II. MATERIALS AND METHODS
The NMR experiments for proton Larmor frequencies between 36 and 300 MHz were made using the fringe field of a 7.05 T magnet (Magnex Scientific, Oxford, UK) operating in conjunction with a Tecmag Apollo transceiver (Tecmag, Houston, TX), Miteq preamplifier (Miteq, Haupaug, NY, USA), an AMT power amplifier (American Microwave Technologies, Brea, CA, USA) and a probe constructed in this laboratory using a transmission line design. The 90° pulse length was 8.5 μs. The sample was polarized in the high-resolution isocenter or the resonance field of the superconducting magnet, after which the magnetization was inverted with a 180° pulse. The sample, then, was pneumatically shuttled to a calibrated position in the fringe field of the 7.05 T magnet for a variable relaxation delay and pneumatically returned to the resonance field, where the spectrum was promptly recorded using a 90° pulse. The residual water-proton resonance was easily detected in the presence of the background protein resonances. The integrated intensity measured as a function of the relaxation delay provided the relaxation rate at each calibrated fringe-field position. Within experimental error, all the decay/recovery curves of longitudinal magnetization were exponential. Fringe-field measurements were conducted at ambient laboratory temperature of ≃ 20–22° C.
Bovine Serum Albumin (BSA) (Fraction V), lysozyme from chicken egg white, ribonuclease A (type XII-A from bovine pancreas) were purchased from Sigma (St. Louis, MO, USA) as lyophilized powders. The proteins were dialyzed to remove buffer salts and lyophilized with a mechanical vacuum. Proteins used to prepare D2O solutions were dissolved in 99.9% D2O (Cambridge Isotope laboratories, Andover, MA, USA) and lyophilized several times to eliminate proton out-exchange from the protein into the D2O that causes drift in the proton T1 values. Lysozyme and bovine serum albumin solutions were 10% weight/volume and ribonuclease A was 5.5% weight/volume. Protein solutions were deoxygenated under a nitrogen stream for 45–60 minutes and transferred to the sample tube in a glove box flushed with flowing nitrogen gas.
III. RESULTS
Spin-lattice-relaxation rate constants are shown in Fig. 2 as a function of the proton Larmor frequency at magnetic field strengths above the protein rotational inflection for the water protons in H2O protein solutions and for the residual HOD protons in D2O protein solutions. In both cases, the spin-lattice-relaxation-rate constant for the water protons is proportional to the logarithm of the Larmor frequency. The water-proton-relaxation rate is dominated by dipolar interactions between water and protein protons that are modulated by both rotational and translational motions. The spin-lattice relaxation may be affected by the exchange of labile protein protons with water. Although the exchange times for some protein protons are short compared with the relaxation times measured, the exchange times are very long compared with the reciprocal of the Larmor frequency; thus, the exchange events make no contribution to the magnetic field dependence observed above the rotatinoal inflection. The water-water interactions may be separated from the water-protein coupling by exploiting the different magnetic properties of 1H and 2H (D).
FIG. 2.

The proton spin-lattice relaxation rate constants as a function of the magnetic field strength plotted as the proton Larmor frequency for B: 10% lysozyme solution in D2O (circles), 5% ribonuclease A solution in D2O (triangles up), A: 10% lysozyme solution in H2O (squares) and 10% BSA solution in H2O (triangles down) and 5.5% ribonuclease A (diamonds) at room temperature.
In D2O, the intramolecular contribution to the residual HOD proton dipolar coupling is diminished by a factor of approximately 20 relative to that of the proton-proton interaction in H2O; therefore, to within a small correction, the residual HOD proton relaxation in D2O is dominated by dipolar couplings between the rare water protons and the protein protons. Therefore, the proton-relaxation rates for ribonuclease A and lysozyme D2O solutions shown in Fig. 2B report the motion of the residual water protons relative to the interfacial protein protons. In the protein-free deoxygenated D2O, the background 1H relaxation rate constant is independent of magnetic field strength with a value between 0.03 and 0.04 s−1 depending on the concentration of residual protons. Therefore, the relaxation contribution from the interface dynamics is substantial even though the total relaxation-rate constant is not large. The lines are fits to Eq. 2 with the best-fit parameters (τdiff, A): (31 ± 2 ps, 0.55 ± 0.02) for lysozyme/D2O; (15 ± 2 ps, 3.4 ± 0.3) for lysozyme/H2O; (36 ± 4 ps, 0.68 ± 0.05) for RNaseA/D2O; (14 ± 11 ps, 1 ± 0.9) for ribonuclease A in H2O; (14 ± 3 ps, 4.7 ± 0.7) for BSA/H2O. In H2O, the magnetic field dependence of the water-proton-relaxation rate is logarithmic in the Larmor frequency as shown in Fig. 2A. In protein free deoxygenated water, the field independent rate constant is 0.28 s−1; thus, the contribution from the protein interface is again significant.
IV. DISCUSSION
The shape of the relaxation dispersion profile depends on the process that causes modulation of the coupling between the water and protein protons. A logarithmic dependence is characteristic of diffusive translational modulation of the spin-couplings in what is effectively a 2-dimensional space [32, 33]. We note that other dynamical models have been used to understand MRD profiles at surfaces. In particular, rotational motion modulated by translational jumps is a reasonable working hypothesis for the dynamics of water at a protein surface [34]. However, this model leads to an MRD profile that is a power law in the magnetic field strength or Larmor frequency that is not observed here. We note that a power law with a small exponent is difficult to distinguish from a logarithmic field dependence. In the present case, we examined the first derivative of the relaxation rate constant as a function of magnetic field strength, which should return a power law, if that is the appropriate model, and this test failed. Therefore, we accept the logarithmic dependence as the best description of the data. For proton-spin-lattice relaxation, rotational immobilization of the protein changes the relaxation profoundly because the protein-proton dipolar couplings are no longer averaged by rotation. Although surface effects make a contribution, the effects of cross-relaxation between water protons and the protein protons is also important over the Larmor frequency range studied here which we discuss elsewhere [35].
For a diffusive process, it is well known that the time dependence of the re-encounter probability density is mainly proportional to t−d/2 where d is the Euclidean dimensionality and t is time. The pair-wise dipolar correlation function is proportional to the re-encounter probability density; hence, it behaves at long time as the inverse of the volume explored or 1/(Dt)d/2, where D is the translational diffusion coefficient. In consequence, for d = 2, the time dependence of the correlation function is proportional to 1/t, and the spectral density, which is the Fourier transform of the time-correlation function, has a logarithmic dependence at low frequency. Thus, the dimensionality is critical in determining the re-encounter occurrence, which determines the shape of the spectral density functions that appear in the nuclear spin-relaxation equations. Because the 2-particle dipolar correlation is proportional to 1/r6, where r is the proton-proton distance, re-occurrence of first neighbor interactions dominate the time and spatial ensemble average.
The 1H-1H dipolar coupling is modulated by both rotational and translational motions. If we assume, for the moment, that rotational motion is isotropic, the rapid rotational reorientation of water, which is characterized by rotational correlation times of approximately 3 ps in pure water, will contribute a Lorentzian dispersion like that shown in Fig. 1 but at larger Larmor frequencies than achieved here. This would still be true if the rotational correlation time were longer by a factor of 5 or so as suggested by simulations and neutron scattering experiments, the Lorentzian dispersion would still be above the Larmor frequencies used here. The net effect of isotropic rotational motion is to add a field independent constant to the relaxation-rate constant. The translational spectral density functions yield a much broader MRD profile, and thus, appear to dominate the present observations. It is clear that in the present context, the water molecules at the protein surface may execute translational jumps in 3-dimensions because the protein surface is rough and strongly articulated on the length scale of a water molecule diameter. Nevertheless, the protein interface provides a steric barrier or excluded volume to uniform 3-dimensional averaging of proton-proton dipolar couplings between the diffusing water and the protein protons. This constraint may effectively create a solvent layering at the interface. Molecular dynamics simulations have reported anisotropy in the translational motion of water at the protein interface with motions parallel to the surface more rapid than motions perpendicular to the surface [19]. The logarithmic dependence of the spin-lattice-relaxation rates on Larmor frequency is consistent with this diffusion anisotropy and we proceed to examine a 2-dimensional model for relaxation. Spin-relaxation equations for 2-dimensional diffusion effects have been reported previously [32, 33, 36]. The spin relaxation at the interface is mixed with the bulk phase by exchange of molecules that is rapid compared with the relaxation rate constants in either environment.
| (1) |
The bulk contribution, 1/T1,Bulk is independent of magnetic field in the range studied [37]; thus, the field dependence is produced by the interfacial water molecule dynamics. The factor NS/N is the probability that the molecule samples the surface region and NS/N = δSpρliquid where δ is the surface layer thickness, Sp the specific surface area of the sphere, and ρliquid the density of the liquid. The second term depends on the Larmor frequency because of the reencounters between spins within this interfacial region where the dynamics are generally somewhat slower than in the bulk. If the interface residence time is τI, the relaxation equation is:
| (2) |
where A is a constant that includes the strength of the intermolecular coupling, and τdiff is the surface translational correlation time [36].
The interface residence time may provide a limit to the surface correlation time that results in a low frequency plateau. Although the residual high frequency tail of the Lorentzian rotational dispersion may obscure the plateau, none is apparent in Fig. 2. This result is consistent with several calculations that suggest some fraction of the interfacial water population experiences residence times of the order of ns [14, 15]. The lines in Fig. 2B result from best fits to Eqs. (1,2) with translational correlation times 31 ± 2 and 36 ± 4 ps for lysozyme and ribonuclease A in D2O respectively, which are similar to the values deduced from simulation [12–15]. Although relaxation by translational motion includes contributions from all intermoment distances, i.e., an integration over all space, the parameters of the relaxation equation are weighted strongly by nearest neighbor contacts because the pair-wise dipolar correlation falls as the sixth power of the distance. Thus, the diffusion constant extracted is dominated by water-protein contacts.
For protein solutions in H2O, the proton relaxation rate constant is larger but also logarithmic in the Larmor frequency as shown in Fig. 2A. In H2O, both intra and intermolecular contributions to spin relaxation are important. If rotation of water is isotropic, the intramolecular proton-proton coupling as noted above is modulated by rapid rotation at frequencies larger than those directly sampled in the present experiments and adds a constant to the relaxation rate. The intermolecular contribution consists of parts resulting from water-protein and water-water magnetic dipolar couplings. The water-protein contribution is well represented by the measurements made in D2O, and is small compared with the water-water contribution which is dominated by the relative translational motion of diffusing water molecules at the interface. The fitted lines in Fig. 2A for lysozyme, ribonuclease A, and BSA in H2O were computed using Eq. 1 with correlation times of 15 ± 2, 14 ± 11 and 14 ± 3 ps respectively. The error in the ribonuclease case is large because of the low concentration. For the water-water coupling, the interacting spins are both mobile and characterized by identical translational diffusion constants for the water molecules. Therefore, the relative diffusion constant is twice that for the diffusion of a single water molecule and the translational correlation time for the pair-wise interaction is half that for the individual particle. The single molecule translational correlation time is, then, twice the correlation time for the relative interaction or 30 ps, which is in good agreement with result from D2O solutions (Fig. 3).
FIG. 3.

Schematic representation of water molecule diffusion at the surface of ribonuclease A. The scale of the water molecules on the left side is enlarged to clarify self and relative diffusion.
Simulations have suggested that water molecule rotational motion near the protein surface is anisotropic perhaps biased by electrostatic coupling of the water electric dipole moment to the asymmetric charge distribution of the protein [11, 14, 16, 18]. If the orientation of water molecules in the interfacial region is biased, then the intramolecular proton dipolar coupling is not completely averaged at short times, and the rotational contribution to the spin relaxation may be field dependent in the observed range of Larmor frequencies. Nevertheless, the observed magnetic field dependence is logarithmic in the magnetic field strength. Therefore, if the rotational anisotropy makes an important contribution to the spin relaxation, the averaging of any residual rotational anisotropy in the water molecule dynamics at the interface creates the same magnetic field dependence as translational modulation of the dipolar couplings. If the origin of the putative water-molecule-orientation bias is coupling of the water-molecule electric dipole moment to the nonuniform charge distribution on the protein [16], eliminating this source of orientation bias would require translational displacement of the water molecule from one region of particular charge density to another; i.e., a surface translational displacement and the correlation time would approximate that for translation. While this contribution is physically reasonable, we have no direct test of this hypothesis and note that the analysis of the relaxation data in terms of the translational model alone is self consistent.
We note that analysis of relaxation data is necessarily dependent on the model employed. The analysis presented above is simple, internally consistent, but not necessarily unique. Intramolecular dipolar coupling modulated by rotational diffusion results in a spin-relaxation equation that is a sum of Lorentzian functions which lead to a MRD like that shown in Fig. 1. Historically, a common approach to spin relaxation in heterogeneous systems is to neglect translational contributions and presume that the water-molecule-rotational correlation times are distributed, perhaps widely distributed. Such a model may be utilized here to achieve a similar magnetic field dependence to that shown in Fig. 1. For example, a Pareto distribution of correlation times may yield a power law in the Larmor frequency and a small exponent is difficult to distinguish from a logarithmic dependence as already noted. Distributions of surface dynamics are likely and supported by simulations [11, 12, 15–18]. Several have suggested that surface molecule residence times are distributed by a power law, a suggestion consistent with 1H MRD on paramagnetic hemoglobin [38]. However, the distribution required in such approaches is both broad and strongly asymmetric. By comparison, the translational models employed here are far simpler and the distribution of effective correlation times implicit in the volume averaging required in building the model. This is clear by considering that the effective correlation time when particles are far apart is much longer than when they are close together yet the translational jumps are the same. While we cannot eliminate such rotational models and the associated very broad correlation-time-distribution functions, the internal consistency of the translational analysis provides a simpler view that is consistent with other approaches.
V. THEORETICAL MODEL FOR THE TWO-DIMENSIONAL EXPLORATION
We examine here the origin of 2-dimensional effects in spectral density functions, and therefore, the time correlation functions that characterize diffusive modulation of dipole-dipole couplings at the interface of a rotating spherical protein. A fundamental question is how large the spherical macromolecule must be to affect the dimensionality of the diffusional exploration of the small molecule.
A. Relevance of the probability of return
We consider an ensemble of a large number N of mobile small molecule explorer spins, I = 1/2, in the vicinity of a spherical macromolecule (protein) uniformly distributed in solution. The explorer spin diffuses within a surface layer of finite thickness, δ, in the proximity of a sphere of radius R (Fig. 4). To a first approximation, this weak confinement can be incorporated by setting a fictitious reflecting boundary at distance δ (an outer sphere of radius R + δ). We explore the consequences of the diffusion on the spin-lattice relaxation rate constant for the system placed in a constant magnetic field, B0. The layered geometry in proximity of protein interface is sufficient to create the logarithmic dependence of nuclear spin relaxation on magnetic field strength. Of course, in a solution, the total explorer molecule magnetic resonance signal is observed, not just that from the interfacial region as shown in Fig. 4. However, the relaxation rate constants in both the interface and the bulk are small compared with the exchange rates between these regions of the solution; therefore, a rapid mixing equation is appropriate, justifying Eq. (1).
FIG. 4.

“Thick” and “thin” spherical layers (gray regions) representing confinement of water molecules near protein’s surface.
The dynamics of the bulk liquid are very rapid and the contribution 1/T1,Bulk, is independent of magnetic field strength at presently achievable field strengths. Here, we focus on the layer contribution 1/T1,Layer(ωI). This term depends on the Larmor frequency ωI/(2π) because of the re-encounters between spins within this interfacial region where the dynamics are generally somewhat slower than in the bulk. From the first principles of proton magnetic relaxation in a confined or bulk liquid [21, 37, 39], 1/T1,Layer(ωI) can be written as the linear combination of spectral densities:
| (3) |
where A is a constant that contains the usual NMR parameters and is the spectral density in the laboratory frame L (associated with the constant magnetic field B0) are the exponential Fourier transforms,
| (4) |
of the stationary pair-wise dipolar correlation functions (with m = −2, −1, 0, 1, 2) given by:
| (5) |
Equation (5) describes the persistence of correlation of the second order irreducible spherical spatial dipolar tensor, , between the magnetic moments associated with the water-proton spins I and protein-protons I′ and modulated by the translational diffusion of spins I in the layer δ relative to the fixed spins I′ during a short time interval τ. The notation < … > stands for the ensemble average over all the positions of the spins I at times 0 and τ for a given density of spins I′. For a diffusive process, this ensemble average can be expressed as a stationary integral average over the diffusive propagator P (r0, r, τ):
| (6) |
In Eq. (6), p(r0) is the initial water density in the layer and P (r0, r, τ ) is the conditional probability for the spin I to move from a position r0 to another position r (relatively to proton I′ at the protein surface) in the layer in a given time τ. Here we consider a restricted diffusion inside a spherical layer Ω = {r ∈ ℝ3: R < |r| < R + δ} between two reflecting boundaries.
Eq. (6) can be greatly simplified because we are only interested in the short time dependence (high frequency) of these pair-wise dipolar correlations. At short times, the water-proton spins I have no time to explore the whole confining domain Ω. Moreover, the rotational invariance of the problem implies that the probability of return to an initial position r0 within the confining domain Ω in a time t, P (r0, r0, τ ), is independent of the angular coordinates of the starting point r0: P (r0, r0, τ ) = P (τ ). In consequence, one has the following approximation
| (7) |
This means that the time dependence of is equivalent to that for the probability to return to the origin. The only loss in this approximation is some numerical constants coming from the integration. The role of the probability of return is outlined in [39–41].
B. Asymptotic behavior of diffusive propagators
In mathematical terms, restricted diffusion in a domain Ω with a reflecting boundary ∂Ω is governed by the diffusive propagator P (r0, r, t) satisfying
| (8) |
where ∂/∂n is the normal derivative to the boundary, and δ(r − r′) the Dirac distribution.
The diffusive propagators are thoroughly investigated in mathematics and physics [42–44]. For a bounded diffusion-confining domain, a spectral decomposition over the eigenvalues λm and eigenfunctions um(r) of the Laplace operator Δ provides an explicit though formal dependence of P (r0, r, t) on time t:
| (9) |
where the asterisk denotes complex conjugate. The two asymptotic limits for the probability of return P (r0, r0, t) are well understood:
-
In the short-time limit (t → 0), the molecules have no time to explore the diffusion-confining domain, they only “see” a small neighborhood of the starting point. If the molecules start from an interior point of the domain, P (r0, r, t) is close to the Gaussian propagator for free (unrestricted) diffusion. If the molecules start from a boundary point, the propagator P (r0, r, t) is close to the propagator for the half-space which is obtained by mirror reflection principle [42, 43]. The short-time behavior is therefore summarized as
(10) d being the dimension of space. In particular, one obtains P (r0, r0, t) ∝ t−3/2 in three dimensions.
-
In the opposite long-time limit (t → 8), the molecules explore the whole diffusion-confining domain many times so that the probability of return becomes independent of the exploration time, being inversely proportional to the volume V of the domain. More precisely, the spectral decomposition (9) yields for a domain with a reflecting boundary
(11) where λ1 is the lowest strictly positive eigenvalue of the Laplace operator (the first term is the ground eigenmode with λ0 = 0 and |u0|2 = 1/V ). The eigenvalue λ1 sets a typical time tmax = (Dλ1)−1 for approaching a constant.
For intermediate times t, the behavior of the probability of return P (r0, r0, t) depends on the geometry of the diffusion-confining domain through Eq. (9). If the confining domain is thin, diffusion in one spatial direction is much more restricted than in the other ones. If δ is the thickness of the domain, δ2/(2D) is the typical time for a random walker to cross the diffusion-confining domain from one side to the other. One can therefore expect an intermediate region of times δ2/(2D) ≪ t ≪ tmax for which diffusion is effectively two-dimensional with P (r0, r0, t) ∝ t−1.
The simplest example of a thin bounded domain is a parallelepiped of size L×L×δ with L ≫ δ. In this case, the displacements along the three spatial directions are independent from each other, and
so that the problem is reduced to one-dimensional diffusion in the interval with reflecting endpoints. When δ2/2D ≪ t ≪ L2/2D, the first two propagators obey the short-time behavior (10), while the last propagator follows Eq. (11). For the molecules started from the center of the bottom face, one therefore gets
| (12) |
Here, the denominator can be interpreted as a typical diffusion-explored volume (the volume of a cylinder of radius and of height δ, located between the top and bottom faces of the parallelepiped).
The crossover time tmin between the short-time asymptotic behavior (10) with d = 3 and the intermediate behavior (12) can be found by equating the right-hand-sides of these expressions:
| (13) |
This crossover time is close to the earlier qualitative estimate δ2/(2D).
It is worth stressing that the emergence of the intermediate region is related to the separation of times scales δ2/2D and L2/2D for transverse and longitudinal diffusions in a thin parallelepiped. Since diffusion is weakly sensitive to small geometrical details at long enough times, one can expect an intermediate regime for any “thin” confinement, not necessarily a flat one. In the next Section, we demonstrate this result for a thin spherical layer, for which we retrieve exactly Eq. (12). What is more striking is that this result remains accurate even for not-too-thin layers.
C. Probability of return for thin spherical layers
We consider restricted diffusion inside a spherical layer Ω = {r ∈ ℝ3: R < |r| < R + δ} between two reflecting boundaries: the inner sphere of radius R (the surface of a protein) and the outer sphere of radius R + δ (a fictitious boundary that implements confinement of water molecules by interaction potential). The rotational invariance implies that the probability of return P (r0, r0, t) is independent of the angular coordinates of the starting point. Since we are interested in water molecules that start from the protein’s surface (the inner sphere), the probability of return can be written as
| (14) |
where all the starting points are equivalent and thus averaged over the inner surface.
For spherical layers, the Laplace operator eigenfunctions are known explicitly [42, 43] which permits efficient analytical and numerical computation of the probability of return. In Appendix, we have shown that P (t) naturally splits in two parts, Plong(t) and Ptran(t), which represent “contributions” from longitudinal (effectively two-dimensional) and transverse diffusions in thin spherical layers (Fig. 4b). We derived the leading terms of the asymptotic behaviors for each part in the short-time limit (t → 0):
| (15) |
| (16) |
The crossover time tmin at which these two contributions are equal is given by Eq. (13) as for a parallelepiped.
For very short times (t ≪ tmin), the contribution of Plong(t) can be neglected so that one retrieves the expected short-time behavior P (t) ∝ t−3/2 in agreement with Eq. (10). For longer times t ≫ tmin, Plong(t) is in turn the determinant contribution, yielding P (t) ∝ t−1. This effectively two-dimensional behavior is limited to times t smaller than tmax. When t exceeds tmax, both asymptotic relations (15) and (16) become invalid, and P (t) approaches a constant according to Eq. (11). The time tmax is determined by the lowest strictly positive eigenvalue λ1 of the Laplace operator. Its value lies between approximately 4.3330/δ2 for the “thickest” spherical layer (a ball of radius δ between the spheres of radii R = 0 and R + δ) and 2/R2 for the “thinnest” spherical layer (an infinitely thin layer between two spheres of radius R). If 2L is the diameter of a spherical layer (L = δ and L = R for the two ultimate cases mentioned above), one can use an estimate tmax ≈ L2/(2D). We summarize the asymptotic results for the probability of return in thin spherical layers:
| (17) |
It is worth stressing a technical difference in how the probability of return was treated in a thin parallelepiped and in a thin spherical layer. In the parallelepiped, the probability of return was a product of three probabilities of return for each spatial direction. Each of these factors behaved as t−1/2 for short times and approached a constant for long times. When the sizes of the parallelepiped in each spatial direction were comparable, the asymptotic behaviors of three factors were similar that yielded the probability of return to be t−3/2 in the short-time limit. When one size was much smaller than the others, an intermediate region emerged for which P (r0, r0, t) ∝ t−1. For a spherical layer, the same asymptotic behavior was derived, but the “mechanism” of the crossover between two regime was different. In fact, we split the probability of return into the sum of two parts formally corresponding to longitudinal and transverse diffusions. In this case, P (t) is not a product of terms, but a sum.
D. Numerical results for spherical layers
The asymptotic result (17) was derived for very thin spherical layers, for which the Laplace operator eigenvalues can be determined explicitly [45]. Strictly speaking, it is valid for spherical layers of vanishing thickness δ → 0. In this Subsection, we present numerical computations that extend the applicability of this result to not-too-thin layers.
The numerical computation is based on the exact spectral decomposition Eq. (21) of the probability of return. As described in the Appendix, the computation is reduced to finding zeros of some explicit equations involving spherical Bessel functions. The only technical difficulty of this computation is that one needs more and more eigenvalues for smaller and smaller times.
Figure 5 shows the probability of return for three spherical layers with normalized thickness δ = 0.01, δ = 0.1, and δ = 0.5 (with L = R + δ = 1). In all three cases, one can clearly distinguish three asymptotic regimes, in agreement with our theoretical result (17). The crossover times tmin = δ2/(πD) and tmax = L2/(2D) accurately delimit the intermediate regime with an effectively two-dimensional behavior P (t) ∝ t−1. Even for a “thick” layer with δ = R = 0.5, the intermediate regime is still visible, although its time range between tmin and tmax is quite narrow.
FIG. 5.

The probability density P(t) for a walker to return to the starting boundary point on the inner boundary of a spherical layer of inner radius R, outer radius R + δ = 1, and thickness δ: δ = 0.01 (squares), δ = 0.1 (circles), and δ = 0.5 (triangles). The corresponding dashed lines show the t−1 behavior in the intermediate regime, while the black solid line shows the very short-time behavior t−3/2. The first three vertical dotted lines indicate the crossover times tmin = δ2/π for each spherical layer, the last one gives tmax = 1/2 (with D = 1). For very small times, one can notice an unphysical deviation from the very short-time asymptotic behavior (for δ = 0.1 and δ= 0.5) because of an insufficient number of the eigenvalues used in numerical computation.
E. Comparison with experiments
The data in Fig. 2 demonstrate a logarithmic dependence on the Larmor frequency from 36 to 300 MHz corresponding to values of 1/(2π(2ν)) of 2.2 ns and 0.26 ns respectively. For the smallest protein we may estimate the radius R of about 1.5 nm and estimate the thickness δ of the surface water regime as 0.9 nm or three water molecules thick. If the interfacial diffusion constant is approximately one third that of the bulk as shown by the present and previous work, D = Dbulk/3 ≃ 0.77 · 10−9 m2/s, then the values of tmin and tmax are approximately 0.33 ns and 3.7 ns respectively. This range is consistent with the data of Fig. 2 where deviations from logarithmic behavior are not detected.
VI. CONCLUSION
The high field MRD profiles observed here report on water molecule dynamics that are faster than the rotational reorientation of the protein. Therefore, the same dynamics obtain if the protein does not rotate and these surface water dynamics will make a contribution to water-proton-spin relaxation in dynamically heterogeneous systems such as tissues. Although this dependence may be masked by the effects of proton coupling to the internal dynamics of the protein [30], it must become important at high magnetic fields. The logarithmic dependence derives from translational motions at the interface that are practically two dimensional because of the geometric barriers to diffusion imposed by the large protein adjacent to a small diffusing water molecule as shown schematically in Fig. 3.
The translational correlation times for self diffusion at the surface deduced from the measurements are weighted averages over the interfacial heterogeneity; however, different measurements in H2O and D2O return the same translational correlation times of 30–36 ps, which are longer than that for pure bulk water by only a factor of about 3. This rapid motion at the interface is consistent with earlier measurements as well as simulations, and guarantees that the effective dielectric constant at the water side of the interface is attenuated little from the value in the bulk. The 2-dimensional character of the water-protein interface dynamics increases profoundly the re-encounter probability that a particular site on the protein will be visited or revisited by the diffusive exploration of the interface, whether the explorer molecule is water or a substrate molecule.
The reduced dimensionality for diffusive exploration at the protein interface is supported by a theoretical investigation of the effects of spherical diffusion barriers that approximate the protein. We calculate the probability of return P (t) first analytically for very thin spherical layers and then extend the result to thicker layers by numerical computation. We observe three asymptotic regimes for P (t): very short-time limit P (t) ∝ t−3/2 as expected in three dimensions; intermediate regime with effectively two-dimensional behavior P (t) ∝ t−1; and exponential approach to a constant at long times. For thin spherical layers, it is not surprising that we have retrieved the same behavior as for a flat surface of a thin parallelepiped because the radius of curvature is large. What is more striking is that the derived asymptotic relations agree quite well with the MRD data and remain accurate even for thick layers.
Acknowledgments
This work was supported by the National Institutes of Health, RO1 NIBIB 2805, the University of Virginia, the CNRS, Ecole Polytechnique and the ANR “DYOPTRI”, France. Helpful discussions with Pierre Levitz are gratefully acknowledged.
APPENDIX: COMPUTATIONS FOR THIN SPHERICAL LAYERS
In this Appendix, we provide some mathematical details for calculation of the probability of return for a spherical layer Ω = {r ∈ ℝ3: R < |r| < R + δ}. To simplify the expressions, we take D = 1 and normalize the distances to have R + δ = 1. The dimensional units can later be incorporated into the derived formulas by simple dimensional analysis.
The eigenfunctions of the Laplace operator in the spherical layer O are conventionally enumerated by triple index nkl, with n = 0, 1, 2, 3, …, k = 0, 1, 2, 3, …, and l = −n, −(n − 1), …, (n − 1), n. In spherical coordinates r = (r, θ, ϕ), these eigenfunctions can be written as [42, 43]:
| (18) |
where Pn(z) are Legendre polynomials, βnk the normalization constants, and
| (19) |
jn(z) and yn(z) are the spherical Bessel functions of the first and second kinds, respectively (the prime denoting derivative). This particular form of the radial dependence υnk(r) automatically ensures the Neumann boundary condition on the outer sphere: ∂υnk(r)/∂r = 0 at r = 1. In turn, this equality for the inner sphere r = R yields the conditions
| (20) |
For each n, this equation has an infinite set of positive solutions αnk enumerated by the index k. Finally, the normalization constants βnk are set to get the integral of |unkl(r)|2 over Ω to be 1:
where
Note that the eigenvalue λnk associated to the eigenfunction unkl(r) is (2n + 1) times degenerate.
Given that the spherical Bessel functions are known explicitly, the eigenfunctions and eigenvalues can be fully determined by solving Eq. (20) to get αnk. In particular, the probability of return P (t) from Eq. (14) can be written in terms of these solutions
| (21) |
(the summation over the last index l yields the factor (2n + 1) that is the degeneracy of the corresponding eigenvalue λnk). This is an exact result that can be used for a numerical computation of P (t) for any spherical layer.
Although Eq. (21) is explicit, the analysis of its asymptotic behavior is still difficult because of the implicit form (20) for the solutions αnk. For thin layers (when the normalized thickness δ = 1 − R is much smaller than 1), asymptotic formulas for λnk and ξnk were derived in [45]
| (22) |
The smallness of the thickness δ implies that the eigenvalues λn0 describing longitudinal (effectively two-dimensional) diffusion are much smaller than the eigenvalues λnk (with k > 0) describing transverse diffusion. It is therefore convenient to split the spectral decomposition (21) in two parts, longitudinal (with k = 0) and transverse (k > 0):
| (23) |
For the first sum, the asymptotic relations for λn0 and ξn0 imply
| (24) |
In the short-time limit t → 0, the sum behaves as t−1 yielding the asymptotic behavior (15) for Plong(t). This theoretical estimate relies on the fact that the function e−x is close to 1 for small x and close to 0 for large x. When t is small enough, there are many terms for which n(n + 1)t ≪ 1. At the same time, there is always an infinity of terms for which n(n + 1)t ≫ 1. The time t sets a border index N between these two cases: N(N + 1)t ~ 1 so that . In a first approximation, e−n(n+1)t can be replaced by 1 for n ≤ N, and by 0 otherwise. This crude approximation yields
| (25) |
This rough estimate has also been verified numerically (see Fig. 5).
An accurate analysis of the second sum Ptran(t) in Eq. (23) is more complicated. But we know that the sum Plong(t) + Ptran(t) should behave as t−3/2 in the short-time limit according to the general formula (10). Given that the first sum behaves as t−1, the second one necessarily behaves as t−3/2, yielding Eq. (16).
References
- 1.Daniel RM, Dunn RV, Finney JL, Smith JC. Annu Rev Biophys Biomolec Struct. 2003;32:69. doi: 10.1146/annurev.biophys.32.110601.142445. [DOI] [PubMed] [Google Scholar]
- 2.Frauenfelder H, McMahon B. Proc Natl Acad Sci U S A. 1998;95:4795. doi: 10.1073/pnas.95.9.4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frauenfelder H, McMahon BH, Austin RH, Chu K, Groves JT. Proc Natl Acad Sci U S A. 2001;98:2370. doi: 10.1073/pnas.041614298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pal SK, Peon J, Zewail AH. Proc Natl Acad Sci U S A. 2002;99:15297. doi: 10.1073/pnas.242600399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pal SK, Zewail AH. Chem Rev. 2004;104:2099. doi: 10.1021/cr020689l. [DOI] [PubMed] [Google Scholar]
- 6.Peon J, Pal SK, Zewail AH. Proc Natl Acad Sci U S A. 2002;99:10964. doi: 10.1073/pnas.162366099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellissent-Funel MC, Dellerue S. Chem Phys Lett. 2000;258:315. [Google Scholar]
- 8.Bryant RG. Annu Rev Biophys Biomol Struct. 1996;25:29. doi: 10.1146/annurev.bb.25.060196.000333. [DOI] [PubMed] [Google Scholar]
- 9.Halle B, Denisov VP, Venu K. Multinuclear Relaxation Dispersion Studies of Protein Hydration. In: Krishna NR, LJB, editors. Biological Magnetic Resonance. Vol. 17. Kluwer Academic/Plenum; New York: 1999. p. 419. [Google Scholar]
- 10.Halle B, Davidovic M. Proc Natl Acad Sci U S A. 2003;100:12135. doi: 10.1073/pnas.2033320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchi M, Sterpone F, Ceccarelli M. J Amer Chem Soc. 2002;124:6787. doi: 10.1021/ja025905m. [DOI] [PubMed] [Google Scholar]
- 12.Dastidar SG, Mukhopadhyay C. Phys Rev E. 2003;68:021921. doi: 10.1103/PhysRevE.68.021921. [DOI] [PubMed] [Google Scholar]
- 13.Rocchi C, Bizzarri AR, Cannistraro S. Phys Rev E. 1998;57:3315. [Google Scholar]
- 14.Pizzitutti F, Marchi M, Sterpone F, Rossky PJ. J Phys Chem B. 2007;111:7584. doi: 10.1021/jp0717185. [DOI] [PubMed] [Google Scholar]
- 15.Hua L, Huang X, Zhou R, Berne BJ. J Phys Chem B. 2006;110:3704. doi: 10.1021/jp055399y. [DOI] [PubMed] [Google Scholar]
- 16.Bizzarri AR, Cannistraro S. J Phys Chem B. 2002;106:6617. [Google Scholar]
- 17.Bandyopadhyay S, Chakraborty S, Balasubramanian S, Pal S, Bagchi B. J Phys Chem B. 2004;108:12608. [Google Scholar]
- 18.Bandyopadhyay S, Chakraborty S, Bagchi B. J Am Chem Soc. 2005;127:16660. doi: 10.1021/ja054462u. [DOI] [PubMed] [Google Scholar]
- 19.Bizzarri AR, Cannistraro S. Europhys Lett. 1997;37:201. [Google Scholar]
- 20.Dellerue S, Bellissent-Funel MC. Chemical Physics. 2000;258:315. [Google Scholar]
- 21.Kimmich R, Weber HW. Phys Rev B. 1993;47:11788. doi: 10.1103/physrevb.47.11788. [DOI] [PubMed] [Google Scholar]
- 22.Koenig SH, Schillinger WE. J Biol Chem. 1969;244:3283. [PubMed] [Google Scholar]
- 23.Hallenga K, Koenig SH. Biochemistry. 1976;15:4255. doi: 10.1021/bi00664a019. [DOI] [PubMed] [Google Scholar]
- 24.Koenig SH, Brown RD, Ugolini R. Magn Reson Med. 1993;29:77. doi: 10.1002/mrm.1910290114. [DOI] [PubMed] [Google Scholar]
- 25.Bottomley PA, Foster TH, Argersinger RE, Pfeifer LH. Med Phys. 1984;11:425. doi: 10.1118/1.595535. [DOI] [PubMed] [Google Scholar]
- 26.Koenig SH, Brown RD. NMR Encycl. 1996;6:4108. [Google Scholar]
- 27.Fischer HW, Rinck PA, Van Haverbeke Y, Muller RN. Magn Reson Med. 1990;16:317. doi: 10.1002/mrm.1910160212. [DOI] [PubMed] [Google Scholar]
- 28.Shirley WM, Bryant RG. J Amer Chem Soc. 1982;104:2910. [Google Scholar]
- 29.Korb JP, Bryant RG. J Chem Phys. 2001;115:10964. [Google Scholar]
- 30.Korb JP, Bryant RG. Magn Reson Med. 2002;48:21. doi: 10.1002/mrm.10185. [DOI] [PubMed] [Google Scholar]
- 31.Korb JP, Bryant RG. Biophys J. 2005;89:2685. doi: 10.1529/biophysj.105.060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korb JP, Torney DC, McConnell HM. J Chem Phys. 1983;78:5782. [Google Scholar]
- 33.Korb JP, Winterhalter M, McConnell HM. J Chem Phys. 1984;80:1059. [Google Scholar]
- 34.Levitz PE. J Phys: Cond Matter. 2005;17:S4059. [Google Scholar]
- 35.Goddard YA, Diakova G, Korb J-P, Bryant RG. Biophys J. 2009 submitted. [Google Scholar]
- 36.Godefroy S, Korb JP, Fleury M, Bryant RG. Phys Rev E. 2001;64:021605–1. doi: 10.1103/PhysRevE.64.021605. [DOI] [PubMed] [Google Scholar]
- 37.Abragam A. Principles of Nuclear Magnetism. Oxford University; Oxford: 1961. [Google Scholar]
- 38.Victor K, Van-Quynh A, Bryant RG. Biophys J. 2005;88:443. doi: 10.1529/biophysj.104.046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korb JP, Whaley-Hodges M, Bryant RG. Phys Rev E. 1997;56:1934. [Google Scholar]
- 40.Mitra PP, Latour LL, Kleinberg RL, Sotak CH. J Magn Reson A. 1995;114:47. [Google Scholar]
- 41.Schwartz LM, Hurlimann MD, Dunn KJ, Mitra PP, Bergman DJ. Phys Rev E. 1997;55:4225. [Google Scholar]
- 42.Carslaw HS, Jaeger JC. Conduction of Heat in Solids. 2. Oxford: Clarendon; 1959. [Google Scholar]
- 43.Crank J. The Mathematics of Diffusion. 2. Oxford: Clarendon; 1975. [Google Scholar]
- 44.Grebenkov DS. Rev Mod Phys. 2007;79:1077. [Google Scholar]
- 45.Grebenkov DS. J Chem Phys. 2008;128:134702. doi: 10.1063/1.2841367. [DOI] [PubMed] [Google Scholar]
- 46.Fripiat JJ, Letellier M, Levitz PE. Philos Trans R Soc London A. 1984;311:287. [Google Scholar]
- 47.Polnaszek CF, Bryant RG. J Am Chem Soc. 1984;106:428. [Google Scholar]
- 48.Kiihne S, Bryant RG. Biophys J. 2000;78:2163. doi: 10.1016/S0006-3495(00)76763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]