

Abstract
Background and purpose:
Nitric oxide (NO) controls numerous physiological processes by activation of its receptor, guanylyl cyclase (sGC), leading to the accumulation of 3′-5′ cyclic guanosine monophosphate (cGMP). Ca2+-calmodulin (CaM) regulates both NO synthesis by NO synthase and cGMP hydrolysis by phosphodiesterase-1. We report that, unexpectedly, the CaM antagonists, calmidazolium, phenoxybenzamine and trifluoperazine, also inhibited cGMP accumulation in cerebellar cells evoked by an exogenous NO donor, with IC50 values of 11, 80 and 180 µM respectively. Here we sought to elucidate the underlying mechanism(s).
Experimental approach:
We used cerebellar cell suspensions to determine the influence of CaM antagonists on all steps of the NO-cGMP pathway. Homogenized tissue and purified enzyme were used to test effects of calmidazolium on sGC activity.
Key results:
Inhibition of cGMP accumulation in the cells did not depend on changes in intracellular Ca2+ concentration. Degradation of cGMP and inactivation of NO were both inhibited by the CaM antagonists, ruling out increased loss of cGMP or NO as explanations. Instead, calmidazolium directly inhibited purified sGC (IC50= 10 µM). The inhibition was not in competition with NO, nor did it arise from displacement of the haem moiety from sGC. Calmidazolium decreased enzyme Vmax and Km, indicating that it acts in an uncompetitive manner.
Conclusions and implications:
The disruption of every stage of NO signal transduction by common CaM antagonists, unrelated to CaM antagonism, cautions against their utility as pharmacological tools. More positively, the compounds exemplify a novel class of sGC inhibitors that, with improved selectivity, may be therapeutically valuable.
Keywords: guanylyl cyclase, nitric oxide, calmodulin, cGMP, calcium, cerebellum
Introduction
Accumulation of the second messenger, 3′-5′ cyclic guanosine monophosphate (cGMP), following activation of the nitric oxide receptor, soluble guanylyl cyclase (sGC), controls a wide range of physiological processes (Moncada et al., 1991; Garthwaite, 2008). Dysfunction of this signalling pathway is implied in several clinical conditions, including hypertension, erectile dysfunction and septic shock (Bredt, 1999; Vallance and Leiper, 2002). The pharmacology of this pathway is well developed (Rees et al., 1990; Morley and Keefer, 1993; Ko et al., 1994; Schrammel et al., 1996; Stasch et al., 2001) and continues to be of pharmaceutical benefit and interest (Moncada, 1999; Napoli and Ignarro, 2003; Evgenov et al., 2006).
Ever since the NO-cGMP pathway was first identified, its modulation by Ca2+ has been recognized (Palmer et al., 1988; Bredt and Snyder, 1990; Mayer et al., 1990). All isoforms of NO synthases (NOS) bind and are activated by calmodulin (CaM). In the case of the inducible isoform (iNOS, inducible nitric oxide synthase), CaM is bound even at resting Ca2+ concentrations, and the enzyme is therefore constitutively active after expression (Cho et al., 1992). In contrast, eNOS and nNOS bind CaM only after elevation of cytosolic Ca2+ concentration, and are thereby activated in a phasic manner, following, for example, opening of Ca2+-permeable NMDA receptors (Bredt and Snyder, 1990; Mayer et al., 1990; Stuehr, 1999). Similarly, Ca2+-CaM activates an isoform of phosphodiesterase (PDE1), the enzyme that hydrolyses both cGMP and 3′-5′ cyclic adenosine monophosphate (cAMP). This dual effect can lead to suppression of cGMP accumulation in those cells that are stimulated to release NO (Mayer et al., 1992).
A large number of compounds have been developed that disrupt CaM function. These ‘calmodulin antagonists’ are generally molecules with hydrophobic and positively charged groups, which occupy the hydrophobic pockets on CaM, preventing the interaction of these pockets with amphiphilic regions of target proteins, such as NOS and PDE1 (LaPorte et al., 1980; O'Neil and DeGrado, 1990). However, CaM regulates the activity of dozens of proteins (O'Neil and DeGrado, 1990; Jurado et al., 1999), and so antagonizing its binding to target proteins can have complex effects on a given signalling pathway. For example, CaM antagonists have been shown to relieve the CaM-dependent inhibition of phospholipase A2 (PLA2), opening store-operated or non-store-operated Ca2+ entry channels in several cell types (Peppiatt et al., 2004; Smani et al., 2004). This may be significant for NO signalling, as Ca2+ can directly inhibit the NO receptor (sGC) by binding to high and low affinity sites (Parkinson et al., 1999; Kazerounian et al., 2002), although the sensitivity of sGC to Ca2+ lies in the micromolar range (IC50∼ 2.6 µM), and evidence of direct inhibition of sGC (rather than activation of PDE1) in a physiological context is scarce. Conversely, cGMP has been shown to modulate Ca2+ signalling, mainly through the phosphorylation by cGMP-dependent protein kinase of several of the channels, pumps and buffers that comprise the Ca2+ signalling ‘toolkit’ (Clementi and Meldolesi, 1997; Berridge et al., 2003).
Given the potential for crosstalk, we investigated the effect of commonly used CaM antagonists – namely calmidazolium, trifluoperazine and phenoxybenzamine – on the NO-cGMP and Ca2+ signalling pathways in rat cerebellar cell suspensions. We found that in addition to the predicted effects due to CaM antagonism, these compounds also had pharmacological activity independent of Ca2+-CaM. Specifically, CaM antagonists inhibited NO inactivation in cell suspensions and inhibited purified sGC in an uncompetitive manner. Consequently, CaM antagonists could disrupt every step of the NO signalling pathway.
Methods
Preparation of cerebellar cells
All animal care and experimental procedures complied with UK Home Office and local ethics committee regulations. Cerebellar cell suspensions from 8 day old Wistar rats were prepared as previously described (Bellamy and Garthwaite, 2001a). Accumulation of cGMP was measured by inactivating an aliquot of the cell suspension in boiling buffer (containing 50 mM Tris and 4 mM EDTA) after a fixed period of NO exposure at 37°C. Total cGMP content was measured by standard radioimmunoassay or the ‘HitHunter’ enzyme fragment complementation chemiluminescence assay (GE Healthcare, Little Chalfont, UK), according to manufacturer instructions.
Ca2+ imaging
For Ca2+ imaging experiments, cell suspensions were prepared as above, and allowed to settle onto poly-l-lysine coated coverslips for 30 min. The coverslips were washed with imaging buffer containing (mM): NaCl 135, KCl 3, HEPES 10, glucose 15, MgSO4 2, CaCl2 2, and then incubated at room temperature with 3 µM Fluo-4 AM for 30 min. After washing in imaging buffer, the cells were left for a further 30 min to allow de-esterification of the indicator. Astrocytes were identified by their characteristic morphology – they adhere to the substrate more tightly than the abundant neurons and develop processes that gave them a ‘hairy’ appearance (Bellamy et al., 2000). Astrocytes readily took up Fluo-4 AM, as previously observed (Singaravelu et al., 2006). Cells were imaged in a static bath on the stage of an Olympus IX70 inverted microscope (objective 60×, NA 1.25; Olympus, Southend-on-Sea, UK), and images were acquired with a Hamamatsu ORCA-ER camera (exposure time, 100 ms; frame rate, 2 Hz; Hamamatsu, Welwyn Garden City, UK). Drugs were added by solution exchange in imaging buffer at the final concentration indicated.
Assay of NO and sGC activity
NO concentration in buffer and cell suspensions (1 mL) was measured in a sealed, stirred chamber at 37°C with an electrochemical probe (ISO-NO; World Precision Instruments, Stevenage, UK).
Purified α1β1 guanylyl cyclase (sGC) from bovine lung was purchased from Alexis Biochemicals (Nottingham, UK). The stock solution was diluted to a concentration of 5 µg·mL−1 in ice-cold buffer containing (mM): Tris 10, dithiothreitol (DTT) 1, and 0.05% bovine serum albumin (BSA), pH 7.4. Activity was measured after further dilution of the enzyme into reaction buffer to give final concentrations of (mM): Tris 50, MgCl2 3, DTT 0.01, guanosine triphosphate (GTP) 1, 0.05% BSA, and 50 ng·mL−1 of sGC at pH 7.4 and 37°C before addition of NO donor.
‘Clamped’ NO concentrations were achieved with the method of Griffiths et al. (2003). Briefly, sGC was incubated in a buffer containing (mM): Tris 50, MgCl2 3, ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) 0.1, DTT 0.01, uric acid 0.3, GTP 1, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide [Na+ salt] (carboxy-PTIO) 0.2 and 1000 U·mL−1 superoxide dismutase at pH 7.4 and 37°C. After pre-incubation with calmidazolium or dimethyl sulphoxide (DMSO), (Z)-1-{N-[3-aminopropyl]-N-[4-(3-aminopropylammonio)butyl]-amino}-diazen-1-ium-1,2-diolate (SPER/NO) was added at serial dilutions from 0.3 µM to 1 mM. After 2 min, an aliquot of reaction mixture was inactivated and assayed for cGMP content, as for cell suspensions. Free NO concentrations were calculated based on the values for kinetic parameters cited in Griffiths et al. (2003), and the EC50 for NO was ∼1 nM, closely similar to the previous report.
For experiments to test for haem dissociation, purified sGC (20 µL at 5 µg·mL−1) was incubated with Tween-20 (0.5%), calmidazolium Cl (30 µM) or DMSO (10%) for 15 min at 37°C. Thereafter, the samples were diluted 1:10 in buffer containing (mM): Tris 10, DTT 1, and 0.05% BSA, pH 7.4, and centrifuged at 12 500× g for 10 min at 4°C, through 30 kDa size-exclusion filters (Microcon YM-30; Millipore UK Ltd., Watford, UK). The samples were eluted from the filters in 20 µL of buffer by a further 5 min centrifugation, and then assayed for activity in the usual way.
Cerebellar homogenate was prepared from the cerebella of 16–20 day old Wistar rats using an Ultra-Turrax blade homogenizer (IKA, Staufen, Germany) in a buffer containing (mM): Tris 10, DTT 1, pH 7.4. Homogenate was diluted 1:4 into reaction buffer containing (mM): Tris 50, DTT 1, MgCl2 3, GTP 1, 3-isobutyl-1-methyl xanthine (IBMX) 1, phosphocreatine 5 and 150 µg·mL−1 creatine kinase, pH 7.4 at 37°C. Synthesis of cGMP was triggered by addition of 2-(N,N-diethylamino)-diazenolate-2-oxide [diethylammonium salt] (DEA/NO; 100 µM) for 10 min. Aliquots were inactivated and assayed for cGMP content as for cell suspensions.
Data analysis
Unless otherwise indicated, data are the means of three experiments ± SEM. Concentration–response curves were fitted with a logistic equation: y=c+[(a−c)/(1 + (x/x0)p)] to allow estimation of potency (x0= EC50 or IC50) using Origin 7.5 (Microcal, Northampton, MA, USA).
Materials
Superoxide dismutase, IBMX, phosphocreatine, creatine kinase, GTP, calmidazolium chloride, phenoxybenzamine, trifluoperazine and N-(6-aminohexyl)-5-chloro-1-naphthalenesulphonamide hydrochloride (W-7) were obtained from Sigma Aldrich (Poole, UK), Fluo 4-AM was obtained from Invitrogen (Paisley, UK), and 2-(N,N-diethylamino)-diazenolate-2-oxide [diethylammonium salt] (DEA/NO), (Z)-1-{N-[3-aminopropyl]-N-[4-(3-aminopropylammonio)butyl]-amino}-diazen-1-ium-1,2-diolate (SPER/NO), and 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide [Na+ salt] (carboxy-PTIO) were obtained from Alexis (Nottingham, UK). All other reagents were from VWR (Lutterworth, UK).
The NO donor DEA/NO was made up as a stock solution in ice-cold 10 mM NaOH, and diluted 1:100 into the cell suspension at t= 0, to give a final concentration of 1 µM. CaM antagonists were prepared as stock solutions in DMSO and diluted 1:100 to give final concentrations as indicated.
Drug target nomenclature conforms to the Guide to Receptors and Channels, 3rd edition (Alexander et al., 2008).
Results
Inhibition of cGMP accumulation by CaM antagonists
Addition of the nitric oxide donor, DEA/NO, to cerebellar cell suspensions caused an increase in intracellular cGMP concentration, which reached a plateau after ∼2 min (Figure 1A). This accumulation is largely confined to astrocytes (Southam et al., 1992; Bellamy and Garthwaite, 2001b). Preincubation of the cell suspension with the CaM antagonist, calmidazolium chloride (30 µM) for 10 min significantly inhibited both the amplitude of the NO-evoked cGMP response and the rate of onset of cGMP accumulation (Figure 1A). The concentration–response relationship for DEA/NO in the presence and absence of calmidazolium indicates that the antagonist decreases both the maximal cGMP concentration reached and the potency of the NO donor (EC50∼ 80 nM in the absence and 170 nM in the presence of antagonist; Figure 1B).
Figure 1.
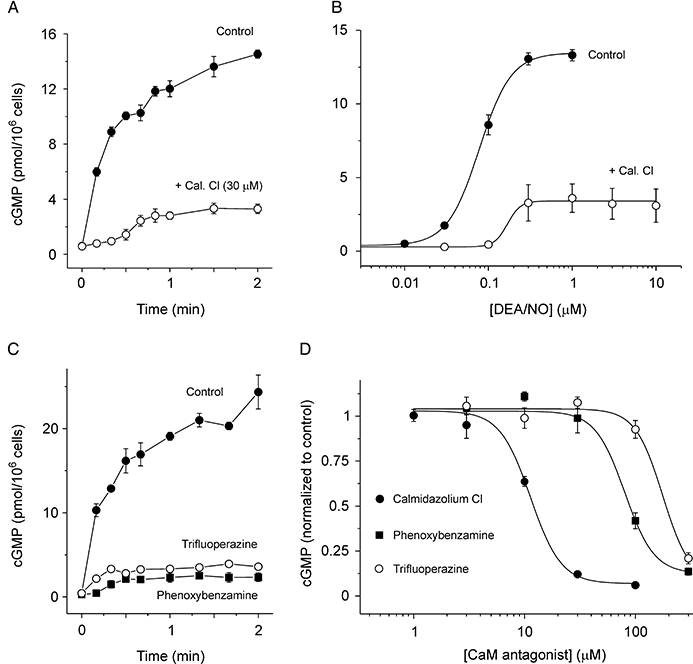
Effect of CaM antagonists on cGMP accumulation in cerebellar cells. (A) Time course of cGMP concentration in untreated cells (control) and cells preincubated for 10 min with calmidazolium chloride (Cal Cl; 30 µM). 1 µM DEA/NO was added at 0 s. (B) Concentration–response curves for 2 min exposure to DEA/NO in the presence and absence of 30 µM calmidazolium. Data were fitted with a logistic equation (Con: EC50= 80 nM, p= 2.3; Cal. Cl: EC50= 170 nM, p= 5.5; see Methods). (C) Time course of cGMP response to 1 µM DEA/NO in untreated cells and cells pre-incubated for 10 min with trifluoperazine (300 µM) or phenoxybenzamine (300 µM). (D) Concentration–response curves for inhibition of cGMP response to 2 min DEA/NO exposure by calmidazolium, phenoxybenzamine and trifluoperazine. Data normalized to control response in each case and fitted with a logistic equation (Cal. Cl: IC50= 11.3 µM, p= 3.5; phenoxybenzamine: IC50= 80 µM, p= 3.3; trifluoperazine: IC50= 177 µM; p= 3.5). CaM, calmodulin; cGMP, 3′-5′ cyclic guanosine monophosphate; DEA/NO, 2-(N,N-diethylamino)-diazenolate-2-oxide.
To explore whether this effect on cGMP accumulation was limited to calmidazolium, other CaM antagonists with different chemical structures were tested, namely trifluoperazine and phenoxybenzamine (Zimmer and Hofmann, 1987; Cimino and Weiss, 1988). Both of these compounds inhibited the accumulation of cGMP in cerebellar cells (Figure 1C), but the lag in cGMP accumulation caused by phenoxybenzamine was less pronounced than for calmidazolium (Figure 1A), and apparently absent for trifluoperazine. The comparative potencies of the CaM antagonists were tested by measuring cGMP accumulation after 2 min incubation with 1 µM DEA/NO (Figure 1D). The potencies were calmidazolium (IC50∼ 11 µM) > phenoxybenzamine (IC50∼ 80 µM) > trifluoperazine (IC50∼ 180 µM).
Effect of Ca2+ on cGMP accumulation and inhibition by calmidazolium
The inhibition of cGMP accumulation in cells by CaM antagonists could result from several mechanisms, but in the first instance, we investigated whether the known actions of calmidazolium on Ca2+-CaM signalling accounted for the inhibitory effect.
The interactions between Ca2+ and cGMP signalling pathways are complex (Clementi and Meldolesi, 1997). In addition to activating NO synthase and PDE1 via CaM, Ca2+ has been shown to directly inhibit purified sGC at concentrations in the micromolar range in an uncompetitive manner (Parkinson et al., 1999; Kazerounian et al., 2002). Calmidazolium has been shown to trigger Ca2+ entry in several cell types, including cerebellar astrocytes (Singaravelu et al., 2006), by relief of the CaM-dependent inhibition of PLA2. In principle, therefore, calmidazolium could inhibit sGC activation through elevation of cytosolic Ca2+ concentration. To test this possibility, we loaded cerebellar cell suspensions with the Ca2+ indicator Fluo 4-AM and measured the change in fluorescence intensity after exposing the cells to known Ca2+-mobilizing agonists (Verkhratsky et al., 1998) and calmidazolium, and tested the effect of these interventions on DEA/NO-evoked cGMP accumulation.
The excitatory neurotransmitter glutamate caused a large increase in Ca2+ concentration, as measured by Fluo-4 fluorescence, which, in many cells, became dysregulated after an initial peak (Figure 2A,B). This probably reflects the high concentration of glutamate used (1 mM), which can be toxic to astrocytes during prolonged exposure (Matute et al., 2006). Despite generating large Ca2+ signals, however, pre-incubation of cells with 1 mM glutamate for 30 s had no significant effect on the amplitude or time course of subsequent NO-evoked cGMP accumulation (Figure 2D). Agonists for metabotropic glutamate receptors (trans-ACPD, 100 µM) and AMPA/kainate receptors (kainate, 100 µM) also failed to inhibit cGMP formation (data not shown). Ca2+-dependent inhibition of sGC could be achieved, however, with the Ca2+ ionophore A23187 (10 µM), which reduced cGMP accumulation by approximately two-thirds (Figure 2E). Collectively, these results suggest that neurotransmitter receptors linked to Ca2+ elevation in astrocytes do not cause detectable inhibition of sGC – permeabilization of the cell membrane is required to reach sufficiently high concentrations of Ca2+ within the vicinity of cellular sGC.
Figure 2.
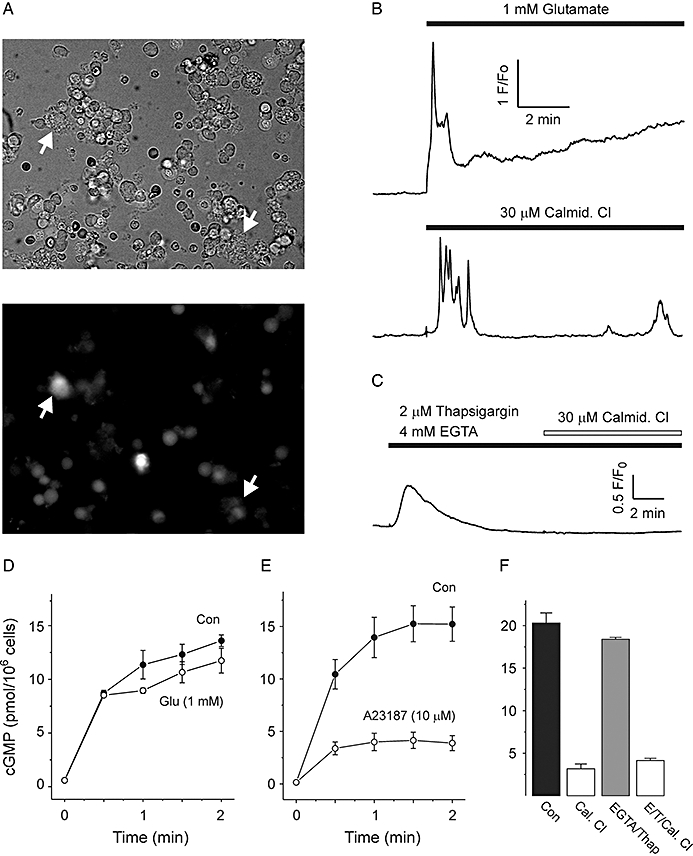
Effect of Ca2+ concentration changes on cGMP accumulation. (A) Brightfield (upper panel) and epifluorescent (lower panel) images of cerebellar cells loaded with 2 µM Fluo-4 AM. Astrocytes (indicated by arrows) adhere to the coverslip and send out disordered processes, and are readily distinguishable from smaller, more abundant granule neurons. (B) Representative time courses of Fluo-4 fluorescence intensity after addition of 1 mM glutamate and 30 µM calmidazolium (Calmid Cl) as indicated. (C) Representative time course from cells incubated with 2 µM thaspigargin and 4 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) for 10 min before addition of calmidazolium. Note that store depletion and removal of extracellular Ca2+ eliminates Ca2+ response to calmidazolium. (D) cGMP response to 2 min treatment with 1 µM DEA/NO in control cells (Con), and cells pre-incubated (30 s) with 1 mM glutamate (Glu). (E) Time course of cGMP accumulation in cells exposed to 10 µM A23187 compared with control cells. (F) cGMP response to 2 min treatment with 1 µM DEA/NO in untreated cells (Con), cells preincubated for 10 min with calmidazolium (Cal. Cl), cell preincubated for 10 min with 2 µM thapsigargin and 4 mM EGTA to deplete Ca2+ (EGTA/Thap), and cells pre-incubated with calmidazolium for 10 min after depletion of Ca2+ (E/T/Cal. Cl). cGMP, 3′-5′ cyclic guanosine monophosphate; DEA/NO, 2-(N,N-diethylamino)-diazenolate-2-oxide.
Calmidazolium (30 µM) triggered oscillatory increases in cytosolic Ca2+ (Figure 2B), confirming that the astrocytes express PLA2-stimulated Ca2+ entry channels, as previously noted in cerebellar slices (Singaravelu et al., 2006). Ca2+ elevation by calmidazolium could be eliminated by pre-incubation of cells for 10 min with the sarco/endoplasmic reticulum Ca2+ adenosine triphosphate (ATP)ase (SERCA) pump inhibitor thapsigargin (2 µM) and the Ca2+ chelator EGTA (4 mM), which transiently emptied internal stores and blocked subsequent Ca2+ entry (Figure 2C). This treatment had no effect on the magnitude of DEA/NO-evoked cGMP accumulation after 2 min (Figure 2F). Elimination of Ca2+ signalling in this way also failed to prevent the inhibition of DEA/NO-evoked cGMP accumulation by calmidazolium (Figure 2F), indicating that changes in Ca2+ concentration were neither necessary nor directly responsible for the inhibitory effect.
Effect of calmidazolium on cGMP degradation
In principle, the calmidazolium-dependent inhibition of cGMP accumulation could result from activation of PDEs, which hydrolyse cGMP to GMP (Conti and Beavo, 2007), although PDE1 would be inhibited by CaM antagonism and is not apparently expressed at significant levels in these cells (Bellamy and Garthwaite, 2001a).
Quenching free NO by addition of Hb after 2 min of DEA/NO stimulation abruptly halts cGMP synthesis, allowing cellular PDE activity to be measured from the subsequent decline in cGMP concentration (Bellamy and Garthwaite, 2001a). Addition of calmidazolium 5 s before Hb did not increase the rate of cGMP hydrolysis, but instead modestly inhibited the decline in cGMP over time (Figure 3). This result indicates that increased degradation of cGMP cannot account for the inhibitory effect of calmidazolium
Figure 3.
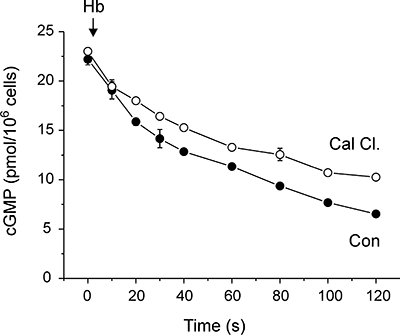
Effect of calmidazolium on cGMP degradation. Cells were pre-incubated with 2 µM DEA/NO for 2 min to allow cGMP accumulation. Calmidazolium (30 µM) or vehicle was added to cells 5 s before 10 µM Hb, which abruptly halts cGMP synthesis. Subsequent decay over 2 min allows cGMP hydrolysis to be measured. cGMP, 3′-5′ cyclic guanosine monophosphate; DEA/NO, 2-(N,N-diethylamino)-diazenolate-2-oxide.
Inhibition of NO inactivation by CaM antagonists
Another mechanism which could account for inhibition of cGMP accumulation is that CaM antagonists disrupt the kinetics of NO release from the donor, or enhance inactivation of NO by the cell suspension (Griffiths and Garthwaite, 2001), resulting in a decrease in free NO available to activate sGC.
Addition of calmidazolium to buffer before addition of DEA/NO had no effect on subsequent NO release kinetics (Figure 4A), indicating a lack of direct chemical interaction between the compounds. However, pre-incubation of cells with calmidazolium resulted in a lag in NO accumulation in the cell suspension (Figure 4B), the timing of which matched that of the lag in cGMP accumulation (Figure 1A). Thereafter, however, NO concentration rose with a similar time course to untreated cell suspensions, but overshot the maximal concentration achieved under control conditions (Figure 4B). Trifluoperazine and phenoxybenzamine did not result in a detectable lag in NO accumulation, but were even more effective than calmidazolium at increasing maximal NO concentration in the cell suspensions (Figure 4C). Furthermore, the relative potencies of the CaM antagonists for increasing free NO concentration were the opposite to that for inhibition of cGMP accumulation (Figure 1D).
Figure 4.
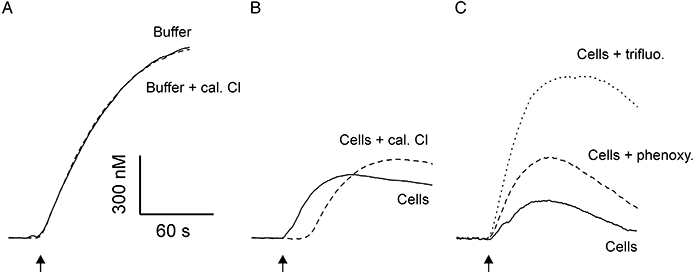
Effect of CaM antagonists on free NO concentration. Representative traces of NO concentration measured with an electrochemical probe in (A) buffer and buffer supplemented with 30 µM calmidazolium (Cal Cl), (B) control cells and cells preincubated with 30 µM calmidazolium, and (C) control cells and cells pre-incubated with 100 µM phenoxybenzamine (phenoxy) or cells pre-incubated with 300 µM trifluoperazine (trifluo). In all cases, 1 µM DEA/NO was added at arrow. CaM, calmodulin; cGMP, 3′-5′ cyclic guanosine monophosphate; DEA/NO, 2-(N,N-diethylamino)-diazenolate-2-oxide.
Direct inhibition of guanylyl cyclase by calmidazolium
A parsimonious explanation for the inhibition of cGMP accumulation by CaM antagonists is that these compounds directly inhibit sGC activation. In the first instance, we tested this possibility in whole tissue homogenate of rat cerebellum. By progressively diluting the concentration of the homogenate, any indirect inhibitory effect of calmidazolium (such as, for example, interaction with Ca2+-free apocalmodulin) should be reduced, whereas a direct effect on sGC should be unaffected. Unexpectedly, we found that 30 µM calmidazolium had no effect on cGMP synthesis when the homogenate was concentrated (1.5–0.5 mg protein·mL−1; Figure 5A), but as the homogenate was diluted, calmidazolium became progressively more effective at inhibiting NO-evoked cGMP accumulation (Figure 5A). The simplest explanation for this observation is that calmidazolium can be sequestered or metabolized by lipid or protein components of homogenized tissue, and that this influence is lost on dilution, revealing the inhibitory effect on sGC.
Figure 5.
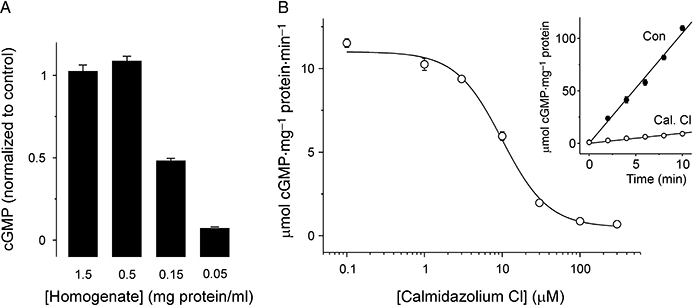
Effect of calmidazolium on homogenate and purified sGC. (A) cGMP accumulation in whole cerebellar homogenate at varying concentrations (expressed in mg protein·mL−1 as indicated), incubated with 100 µM DEA/NO for 10 min. Homogenate was pre-incubated for 10 min with 30 µM calmidazolium. At each concentration, cGMP response is normalized to the response in homogenate pre-incubated with vehicle alone. (B) Concentration–response curve for inhibition of purified bovine lung α1β1 sGC by calmidazolium. Data are fitted with a logistic equation (IC50= 9.9 µM; p= 1.5). Inset shows time course of cGMP synthesis in the presence (Cal. Cl) and absence (Con) of 30 µM calmidazolium chloride. cGMP, 3′-5′ cyclic guanosine monophosphate; DEA/NO, 2-(N,N-diethylamino)-diazenolate-2-oxide; sGC, soluble guanylyl cyclase.
To further test the hypothesis, we treated purified α1β1 sGC from bovine lung with the antagonists. Calmidazolium (30 µM) substantially inhibited cGMP synthesis by the purified enzyme exposed to a maximal concentration (100 µM) of DEA/NO (Figure 5B). The IC50 of calmidazolium for inhibition of purified sGC was ∼10 µM, indistinguishable from that for cell suspensions (cf. Figures 1D and 5B). Trifluoperazine and phenoxybenzamine were also tested against the purified enzyme at a concentration of 300 µM. Compared with enzyme treated with vehicle alone (1% DMSO; 114 ± 16 µmol cGMP·mg−1, n= 3), cGMP production over 10 min was inhibited by both compounds (trifluoperazine: 18.4 ± 2.1 µmol·mg−1, n= 3; phenoxybenzamine: 60.0 ± 8.2 µmol·mg−1, n= 3).
To investigate the mechanism of sGC inhibition, we first tested for a change in the potency of NO in activating sGC using clamped concentrations (Griffiths et al., 2003). Pre-incubation of sGC with 30 µM calmidazolium for 30 s under these conditions resulted in a ∼60% reduction in the maximal rate of cGMP synthesis, but no change in NO potency (EC50∼ 1.1 nM), indicating that calmidazolium does not act in a competitive manner at the sGC haem binding site (Figure 6A). This contrasts with the apparent increase in EC50 for DEA/NO observed in cells (Figure 1B).
Figure 6.
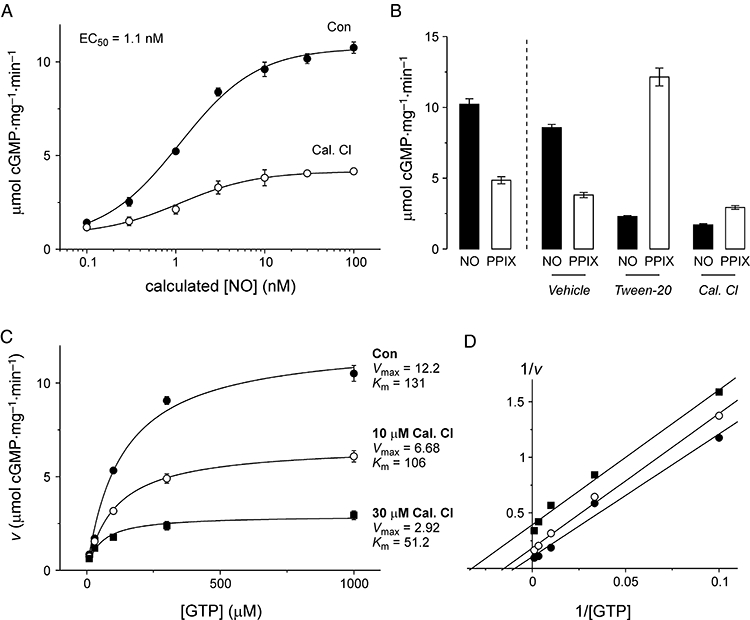
Mechanism of inhibition of purified sGC by calmidazolium. (A) Concentration–response of purified sGC to clamped nitric oxide (NO) concentrations (calculated from concentration of SPER/NO, as described in Griffiths et al., 2003), after 30 s pre-incubation with vehicle (Con) or 10 µM calmidazolium (Cal Cl). Both datasets were fitted with a logistic equation, with EC50= 1.1 nM, p= 1. (B) Response of sGC to 10 min incubation with DEA/NO (100 µM; NO) and protoporphyrin IX (1 µM; PPIX), under standard experimental conditions (columns to left of dashed line) and after 10 min pre-incubation with buffer + 1% DMSO (vehicle), 0.5% Tween-20 (Tween-20) or 30 µM calmidazolium chloride (Cal. Cl), followed by filtration and resuspension in normal buffer. (C) Concentration response of sGC activated by 100 µM DEA/NO to GTP, in the absence (Con) or presence of calmidazolium Cl (Cal. Cl) at 10 and 30 µM. Data were fitted with the Michaelis–Menten equation. Values for Vmax (in µmol·mg−1·min−1) and Km (in µM) were as indicated. (D) Data from panel c as a double reciprocal plot, to illustrate parallel lines indicative of uncompetitive inhibition. DEA/NO, 2-(N,N-diethylamino)-diazenolate-2-oxide; DMSO, dimethyl sulphoxide; sGC, soluble guanylyl cyclase; SPER/NO, (Z)-1-{N-[3-aminopropyl]-N-[4-(3-aminopropylammonio)butyl]-amino}-diazen-1-ium-1,2-diolate.
We next tested the hypothesis that as a hydrophobic molecule, calmidazolium could displace the haem moiety from sGC. Detergents such as Tween-20 can strip the haem from sGC in this manner (Roy et al., 2008), rendering the enzyme insensitive to NO. The haem mimetic protoporphyrin IX (PPIX) can substitute for haem and bind to sGC, making it more effective at activating haem-depleted sGC than the native enzyme.
PPIX (1 µM) activated untreated sGC (as supplied by the manufacturer) with around half the effectiveness of DEA/NO (Figure 6B). Pre-treatment of sGC with the detergent Tween-20 (0.5%), followed by filtration to remove the detergent, decreased the effectiveness of DEA/NO for activating cGMP synthesis by 73% (Figure 6B). In contrast, the effectiveness of PPIX for activating sGC was increased 3.2-fold by Tween-20 treatment (Figure 6B). Pre-treatment of sGC with calmidazolium, followed by filtration, caused an 80% inhibition of sGC activation by DEA/NO and a 23% inhibition of activation by PPIX (Figure 6B), indicating that the mechanism of action of calmidazolium does not involve displacement of the haem group. This result also indicates that the inhibitory effect of calmidazolium does not reverse (within the 10 min of this experiment) after filtration.
Finally, we tested calmidazolium for competition with the enzyme substrate, GTP. By measuring cGMP synthesis for 1 min after addition of NO, in the presence of various initial GTP concentrations, we gauged the ability of calmidazolium to alter the maximal rate of cGMP synthesis (Vmax) and concentration dependence of substrate binding (Km). As calmidazolium concentration increased from 10 to 30 µM, both the Vmax and the Km of sGC decreased (Figure 6C). This is indicative of an uncompetitive mechanism of enzyme inhibition – as illustrated by parallel lines in a double reciprocal plot (Figure 6D), which is reminiscent of the inhibitory mechanism of Ca2+ at sGC (Parkinson et al., 1999).
Discussion and conclusions
Regulation of the NO-cGMP signalling pathway by Ca2+-CaM is well established. CaM was first identified as an activator of PDE1 (Cheung, 1970), and activation of NO synthases by CaM was recognized from the earliest purifications of the enzyme (Bredt and Snyder, 1990; Mayer et al., 1990). Since then, CaM antagonists have been frequently used experimentally to intervene in NO signalling. The known actions of CaM would not, however, predict that its antagonism would lead to the inhibition of cGMP accumulation in cells exposed to an NO donor (Figure 1). This observation led us to investigate the action of common CaM antagonists, principally the widely adopted calmidazolium chloride, on each of the steps in the NO-cGMP signalling pathway in cerebellar astrocytes. Our results show that these compounds inhibit every step of the pathway, resulting in complex disruption of the timing and magnitude of cGMP accumulation.
The role of Ca2+ in regulating cellular cGMP dynamics has been much debated. Apart from the activation of PDE1, Ca2+ has also been shown to inhibit sGC directly by binding to high and low affinity allosteric sites (Parkinson et al., 1999, Kazerounian et al., 2002), suggesting that Ca2+ elevation may counter cGMP signalling by both accelerating degradation and retarding synthesis. This potential for crosstalk is of particular interest with regards to astrocytes, which are known to express numerous receptors coupled to Ca2+ elevation (Verkhratsky et al., 1998; Haydon, 2001). Furthermore, calmidazolium triggers Ca2+ entry in cerebellar astrocytes (Singaravelu et al., 2006) by antagonism of CaM-dependent inhibition of PLA2 (Harper and Daly, 2000; Jan and Tseng, 2000; Peppiatt et al., 2004; Smani et al., 2004), an observation we reproduced in freshly dissociated cerebellar cells (Figure 2B). We therefore explored the possibility that increased Ca2+ concentration could inhibit sGC, and that this was the means by which calmidazolium was acting. However, mobilization of Ca2+ by several neurotransmitter receptor agonists had no influence on the amplitude of cGMP responses evoked by DEA/NO – inhibition was only observed when cells were permeabilized with the Ca2+ ionophore A23187 (Figure 2E) – and depletion of Ca2+ from internal stores and the extracellular space did not alter the cGMP response to NO, nor prevent the inhibition of cGMP accumulation by calmidazolium (Figure 2C,F). These results indicate that the inhibitory effect of calmidazolium on cGMP accumulation does not depend on Ca2+ signalling, with or without CaM. The apparent insensitivity of NO-cGMP signalling to changes in Ca2+ also suggests that Ca2+ elevation by physiological mechanisms does not result in the inhibition of sGC activity in intact cerebellar astrocytes. It therefore seems unlikely that the dynamic Ca2+ signalling observed in astrocyte networks in vivo will alter the cells' responsiveness to NO.
Further investigation revealed little impact of calmidazolium on cGMP breakdown by PDEs, consistent with an earlier report that concluded these cells have negligible PDE1 activity (Bellamy and Garthwaite, 2001a). However, two CaM-independent, pharmacological effects of CaM antagonists on NO signalling were identified: inhibition of NO inactivation by the cell suspension, and direct inhibition of sGC.
NO inactivation under experimental conditions can result from numerous mechanisms, including generation of superoxide by buffers, reaction with cellular haemoproteins, reaction with cytochrome P450 and reaction with lipid peroxyl radicals (Martin et al., 1985; Crow and Beckman, 1995; Keynes et al., 2003; 2005; Hall et al., 2009). The latter mechanism has been found to predominate in cell suspensions and homogenates under the conditions adopted in this study (Keynes et al., 2005). CaM antagonists have previously been identified as inhibitors of lipid peroxidation (Hara et al., 1992), which could account for the reduction of NO inactivation observed in our experiments. How calmidazolium can generate a biphasic change in free NO concentration – an initial lag in release, followed by inhibition of inactivation – is not easily explained. The ‘NONOate’ donors release NO spontaneously in a temperature and pH-dependent manner (Morley and Keefer, 1993), and so inhibition of a metabolic process needed to trigger release (akin to that for nitroglycerin; Chen et al., 2002) can be ruled out. The generation of a saturable sink for NO by calmidazolium – by, for example, generation of radical species – could account for the lag in NO accumulation, although no obvious mechanism presents itself. Nevertheless, this peculiar effect does account for the disruption in the kinetics of cGMP accumulation caused by calmidazolium (Figure 1A), and probably also underlies the change in the shape and EC50 of the concentration–response curve for 2 min DEA/NO exposure (Figure 1B).
A previous study has reported that NO-dependent cGMP accumulation in pancreatic acinar cells was inhibited by several CaM antagonists (Gukovskaya and Pandol, 1995). In particular, the authors noted that W-7 inhibited cGMP synthesis in acinar cell lysates, an effect that varied with Ca2+ concentration (cf. our Figure 2F), but was not reversed by addition of excess CaM. The interpretation of Gukovskaya et al. was that the CaM antagonists may work through an alternative Ca2+ binding protein, which acted as an sGC inhibitor. The authors did not rule out direct inhibition of sGC, however, which would be consistent with our observation of inhibition of purified sGC by calmidazolium. Confusingly, we have been unable to reproduce the inhibition of cGMP synthesis by W-7 in cerebellar homogenate or with purified bovine lung sGC (up to 300 µM; data not shown). This tissue-specific discrepancy is surprising, but may result from variable potency of the drugs in different cellular contexts (see Figure 5A). In contrast, calmidazolium inhibited purified sGC with similar potency to its effect on cerebellar astrocytes (Figure 1B cf Figure 5B), and both phenoxybenzamine and trifluoperazine were also effective (at 300 µM).
The mechanism of inhibition by calmidazolium did not depend on competition with NO for the haem binding site (Figure 6A), or on the loss of haem from the receptor protein (Figure 6B). Calmidazolium also inhibited sGC that had been depleted of haem and stimulated by the haem mimetic protoporphyrin IX (Figure 6B). An analysis of the effect of calmidazolium on substrate (GTP) concentration dependence revealed that the CaM antagonist acts as an uncompetitive inhibitor of sGC – that is, the inhibitor acts selectively on the substrate-enzyme complex (Figure 6C,D). A straightforward interpretation of this result is that the conformational change in the active site associated with substrate binding enables the binding of calmidazolium, which decreases the rate of catalysis. The inhibition was found to be essentially irreversible, at least by filtration and resuspension of the receptor protein (Figure 6B), which may reflect a lasting disruption of protein structure, or a very tight association of calmidazolium with sGC, which survives filtration. The latter interpretation seems incompatible with the relatively modest potency of the drug (EC50∼ 0 µM), but detailed binding kinetics has not been determined.
CaM-independent effects of calmidazolium have been observed in other systems. For example, calmidazolium has been reported to facilitate steriodogenesis in Leydig and adrenocortical cells in a Ca2+-independent manner (Choi and Cooke, 1992), bind to peripheral benzodiazepine receptors (Zisterer and Williams, 1997), inhibit plasma membrane Ca2+ ATPases (Gietzen et al., 1982) and inhibit IP3 receptors (Khan et al., 2001). Of particular note, calmidazolium has a bimodal effect on some isoforms of Ca2+-CaM activated adenylyl cyclase (AC), being stimulatory at lower concentrations and inhibitory at higher concentrations (Haunso et al., 2003). The inhibitory effect was preserved in soluble recombinant fusion proteins of the catalytic subunits of AC and arose from direct binding, but in contrast to our findings with sGC, the inhibitory mechanism at AC was non-competitive (Haunso et al., 2003). It does not appear, therefore, that there is a common binding site for calmidazolium in the catalytic cores of AC and sGC, despite the similarities between the enzymes' catalytic mechanisms. The potency of calmidazolium for inhibition of these diverse targets, and for antagonising CaM, varies substantially between experimental preparations. Variation in the concentration of CaM, the binding of CaM to target proteins, the relative volumes of lipid and aqueous phases, and potential metabolism or sequestration of the drug (Figure 5A), can all influence the IC50 of a given target. As such, in the concentration range typically used (1–100 µM), calmidazolium is likely to act on numerous targets with overlapping concentration–response curves. Selective inhibition therefore appears difficult to achieve with confidence without detailed characterization of targets within the system of interest.
With respect to the binding site of calmidazolium on sGC, it is noteworthy that a low-affinity Ca2+ binding site has been described that also mediates uncompetitive inhibition of the enzyme (Parkinson et al., 1999). It will be worth exploring the potential for competition between Ca2+ and calmidazolium for binding at this site, as it may represent a new pharmacological target for small molecule inhibitors. Currently, the most widely applied inhibitor of sGC is 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), which binds to the receptor haem site and oxidizes the ferrous iron centre, greatly decreasing NO potency (Garthwaite et al., 1995; Bellamy and Garthwaite, 2002). Because of a similar interaction with haemoglobin, the bioavailability of ODQ in vivo is limited. Development of new compounds active at a different site, such as that putatively engaged by calmidazolium, would be advantageous both as experimental tools, and, in principle, as pharmaceutical agents.
In conclusion, these previously unrecognized pharmacological actions of calmidazolium, phenoxybenzamine and trifluoperazine highlight the need for caution in the use of CaM antagonists in a cellular context. For any physiological process in which NO signalling plays a role, clear interpretation of the impact of CaM antagonists on the experimental system will be challenging.
Acknowledgments
This work was supported by the Wellcome Trust, the Medical Research Council and the Biological and Biotechnological Research Council (grant number: BBB5009581). LRJ is a BBSRC CASE student, supported by Cairn Research Ltd. TCB is a BBSRC David Phillips Fellow.
Glossary
Abbreviations:
- CaM
calmodulin
- iNOS
inducible NOS
- NOS
nitric oxide synthase
- PDE
phosphodiesterase
- PLA2
phospholipase A2
- PPIX
protoporphyrin IX
- sGC
soluble guanylyl cyclase
Statement of conflicts of interest
None.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy TC, Garthwaite J. ‘cAMP-specific’ phosphodiesterase contributes to cGMP degradation in cerebellar cells exposed to nitric oxide. Mol Pharmacol. 2001a;59:54–61. doi: 10.1124/mol.59.1.54. [DOI] [PubMed] [Google Scholar]
- Bellamy TC, Garthwaite J. Sub-second kinetics of the nitric oxide receptor, soluble guanylyl cyclase, in intact cerebellar cells. J Biol Chem. 2001b;276:4287–4292. doi: 10.1074/jbc.M006677200. [DOI] [PubMed] [Google Scholar]
- Bellamy TC, Garthwaite J. Pharmacology of the nitric oxide receptor, soluble guanylyl cyclase, in cerebellar cells. Br J Pharmacol. 2002;136:95–103. doi: 10.1038/sj.bjp.0704687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy TC, Wood J, Goodwin DA, Garthwaite J. Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc Natl Acad Sci USA. 2000;97:2928–2933. doi: 10.1073/pnas.97.6.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res. 1999;31:577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WY. Cyclic 3′,5′-nucleotide phosphodiesterase. Demonstration of an activator. Biochem Biophys Res Commun. 1970;38:533–538. doi: 10.1016/0006-291x(70)90747-3. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Xie QW, Calaycay J, Mumford RA, Swiderek KM, Lee TD, et al. Calmodulin is a subunit of nitric oxide synthase from macrophages. J Exp Med. 1992;176:599–604. doi: 10.1084/jem.176.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MS, Cooke BA. Calmidazolium is a potent stimulator of steroidogenesis via mechanisms not involving cyclic AMP, calcium or protein synthesis. Biochem J. 1992;281(1):291–296. doi: 10.1042/bj2810291. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino M, Weiss B. Characteristics of the binding of phenoxybenzamine to calmodulin. Biochem Pharmacol. 1988;37:2739–2745. doi: 10.1016/0006-2952(88)90036-6. [DOI] [PubMed] [Google Scholar]
- Clementi E, Meldolesi J. The cross-talk between nitric oxide and Ca2+: a story with a complex past and a promising future. Trends Pharmacol Sci. 1997;18:266–269. doi: 10.1016/s0165-6147(97)01087-0. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Crow JP, Beckman JS. Reactions between nitric oxide, superoxide, and peroxynitrite: footprints of peroxynitrite in vivo. Adv Pharmacol. 1995;34:17–43. doi: 10.1016/s1054-3589(08)61079-0. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- Gietzen K, Sadorf I, Bader H. A model for the regulation of the calmodulin-dependent enzymes erythrocyte Ca2+-transport ATPase and brain phosphodiesterase by activators and inhibitors. Biochem J. 1982;207:541–548. doi: 10.1042/bj2070541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths C, Garthwaite J. The shaping of nitric oxide signals by a cellular sink. J Physiol. 2001;536:855–862. doi: 10.1111/j.1469-7793.2001.00855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths C, Wykes V, Bellamy TC, Garthwaite J. A new and simple method for delivering clamped nitric oxide concentrations in the physiological range: application to activation of guanylyl cyclase-coupled nitric oxide receptors. Mol Pharmacol. 2003;64:1349–1356. doi: 10.1124/mol.64.6.1349. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Pandol SJ. Dual regulation of cGMP formation by calcium in pancreatic acinar cells. Am J Physiol. 1995;268:G900–G907. doi: 10.1152/ajpgi.1995.268.6.G900. [DOI] [PubMed] [Google Scholar]
- Hall CN, Keynes RG, Garthwaite J. Cytochrome P450 oxidoreductase participates in nitric oxide consumption by rat brain. Biochem J. 2009;419:411–418. doi: 10.1042/BJ20082419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara S, Endo T, Kuriiwa F, Kano S, Iwata N. Antioxidant effect of calmodulin antagonists in rat brain homogenate. Res Commun Chem Pathol Pharmacol. 1992;76:367–370. [PubMed] [Google Scholar]
- Harper JL, Daly JW. Effect of calmidazolium analogs on calcium influx in HL-60 cells. Biochem Pharmacol. 2000;60:317–324. doi: 10.1016/s0006-2952(00)00349-x. [DOI] [PubMed] [Google Scholar]
- Haunso A, Simpson J, Antoni FA. Small ligands modulating the activity of mammalian adenylyl cyclases: a novel mode of inhibition by calmidazolium. Mol Pharmacol. 2003;63:624–631. doi: 10.1124/mol.63.3.624. [DOI] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Jan CR, Tseng CJ. Calmidazolium-induced rises in cytosolic calcium concentrations in Madin Darby canine kidney cells. Toxicol Appl Pharmacol. 2000;162:142–150. doi: 10.1006/taap.1999.8844. [DOI] [PubMed] [Google Scholar]
- Jurado LA, Chockalingam PS, Jarrett HW. Apocalmodulin. Physiol Rev. 1999;79:661–682. doi: 10.1152/physrev.1999.79.3.661. [DOI] [PubMed] [Google Scholar]
- Kazerounian S, Pitari GM, Ruiz-Stewart I, Schulz S, Waldman SA. Nitric oxide activation of soluble guanylyl cyclase reveals high and low affinity sites that mediate allosteric inhibition by calcium. Biochemistry. 2002;41:3396–3404. doi: 10.1021/bi0110894. [DOI] [PubMed] [Google Scholar]
- Keynes RG, Griffiths C, Garthwaite J. Superoxide-dependent consumption of nitric oxide in biological media may confound in vitro experiments. Biochem J. 2003;369:399–406. doi: 10.1042/BJ20020933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keynes RG, Griffiths CH, Hall C, Garthwaite J. Nitric oxide consumption through lipid peroxidation in brain cell suspensions and homogenates. Biochem J. 2005;387:685–694. doi: 10.1042/BJ20041431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SZ, Dyer JL, Michelangeli F. Inhibition of the type 1 inositol 1,4,5-trisphosphate-sensitive Ca2+ channel by calmodulin antagonists. Cell Signal. 2001;13:57–63. doi: 10.1016/s0898-6568(00)00140-6. [DOI] [PubMed] [Google Scholar]
- Ko FN, Wu CC, Kuo SC, Lee FY, Teng CM. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- LaPorte DC, Wierman BM, Storm DR. Calcium-induced exposure of a hydrophobic surface on calmodulin. Biochemistry. 1980;19:3814–3819. doi: 10.1021/bi00557a025. [DOI] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothianandan D, Furchgott RF. Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. J Pharmacol Exp Ther. 1985;232:708–716. [PubMed] [Google Scholar]
- Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006;53:212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- Mayer B, John M, Bohme E. Purification of a Ca2+/calmodulin-dependent nitric oxide synthase from porcine cerebellum. Cofactor-role of tetrahydrobiopterin. FEBS Lett. 1990;277:215–219. doi: 10.1016/0014-5793(90)80848-d. [DOI] [PubMed] [Google Scholar]
- Mayer B, Klatt P, Bohme E, Schmidt K. Regulation of neuronal nitric oxide and cyclic GMP formation by Ca2+ J Neurochem. 1992;59:2024–2029. doi: 10.1111/j.1471-4159.1992.tb10090.x. [DOI] [PubMed] [Google Scholar]
- Moncada S. Nitric oxide: discovery and impact on clinical medicine. J R Soc Med. 1999;92:164–169. doi: 10.1177/014107689909200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- Morley D, Keefer LK. Nitric oxide/nucleophile complexes: a unique class of nitric oxide-based vasodilators. J Cardiovasc Pharmacol. 1993;22(Suppl. 7):S3–S9. [PubMed] [Google Scholar]
- Napoli C, Ignarro LJ. Nitric oxide-releasing drugs. Annu Rev Pharmacol Toxicol. 2003;43:97–123. doi: 10.1146/annurev.pharmtox.43.100901.140226. [DOI] [PubMed] [Google Scholar]
- O'Neil KT, DeGrado WF. How calmodulin binds its targets: sequence independent recognition of amphiphilic alpha-helices. Trends Biochem Sci. 1990;15:59–64. doi: 10.1016/0968-0004(90)90177-d. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- Parkinson SJ, Jovanovic A, Jovanovic S, Wagner F, Terzic A, Waldman SA. Regulation of nitric oxide-responsive recombinant soluble guanylyl cyclase by calcium. Biochemistry. 1999;38:6441–6448. doi: 10.1021/bi990154v. [DOI] [PubMed] [Google Scholar]
- Peppiatt CM, Holmes AM, Seo JT, Bootman MD, Collins TJ, McDonald F, et al. Calmidazolium and arachidonate activate a calcium entry pathway that is distinct from store-operated calcium influx in HeLa cells. Biochem J. 2004;381:929–939. doi: 10.1042/BJ20040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Mo E, Vernon J, Garthwaite J. Probing the presence of the ligand-binding haem in cellular nitric oxide receptors. Br J Pharmacol. 2008;153:1495–1504. doi: 10.1038/sj.bjp.0707687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- Singaravelu K, Lohr C, Deitmer JW. Regulation of store-operated calcium entry by calcium-independent phospholipase A2 in rat cerebellar astrocytes. J Neurosci. 2006;26:9579–9592. doi: 10.1523/JNEUROSCI.2604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- Southam E, Morris R, Garthwaite J. Sources and targets of nitric oxide in rat cerebellum. Neurosci Lett. 1992;137:241–244. doi: 10.1016/0304-3940(92)90413-2. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, et al. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ. Mammalian nitric oxide synthases. Biochim Biophys Acta. 1999;1411:217–230. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- Vallance P, Leiper J. Blocking NO synthesis: how, where and why? Nat Rev Drug Discov. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiol Rev. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Zimmer M, Hofmann F. Differentiation of the drug-binding sites of calmodulin. Eur J Biochem. 1987;164:411–420. doi: 10.1111/j.1432-1033.1987.tb11073.x. [DOI] [PubMed] [Google Scholar]
- Zisterer DM, Williams DC. Calmidazolium and other imidazole compounds affect steroidogenesis in Y1 cells: lack of involvement of the peripheral-type benzodiazepine receptor. J Steroid Biochem Mol Biol. 1997;60:189–195. doi: 10.1016/s0960-0760(96)00189-6. [DOI] [PubMed] [Google Scholar]