

Abstract
Background and purpose:
Mesenteric and carotid arteries from the α1B/D-adrenoceptor knockout (α1B/D-KO) were employed to isolate α1A-adrenoceptor pharmacology and location and to reveal these features in the wild-type (WT) mouse.
Experimental approach:
Functional pharmacology by wire myography and receptor localization by confocal microscopy, using the fluorescent α1-adrenoceptor ligand BODIPY FL-Prazosin (QAPB), on mesenteric (an ‘α1A-adrenoceptor’ tissue) and carotid (an ‘α1D-adrenoceptor’ tissue) arteries.
Key results:
α1B/D-KO mesenteric arteries showed straightforward α1A-adrenoceptor agonist/antagonist pharmacology. WT had complex pharmacology with α1A- and α1D-adrenoceptor components. α1B/D-KO had a larger α1A-adrenoceptor response suggesting compensatory up-regulation: no increase in fluorescent ligand binding suggests up-regulation of signalling. α1B/D-KO carotid arteries had low efficacy α1A-adrenoceptor responses. WT had complex pharmacology consistent with co-activation of all three subtypes. Fluorescent binding had straightforward α1A-adrenoceptor characteristics in both arteries of α1B/D-KO. Fluorescent binding varied between cells in relative intracellular and surface distribution. Total fluorescence was reduced in the α1B/D-KO due to fewer smooth muscle cells showing fluorescent binding. WT binding was greater and sensitive to α1A- and α1D-adrenoceptor antagonists.
Conclusions and implications:
The straightforward pharmacology and fluorescent binding in the α1B/D-KO was used to interpret the properties of the α1A-adrenoceptor in the WT. Reduced total fluorescence in α1B/D-KO arteries, despite a clear difference in the functionally dominant subtype, indicates that measurement of receptor protein is unlikely to correlate with function. Fewer cells bound QAPB in the α1B/D-KO suggesting different cellular phenotypes of α1A-adrenoceptor exist. The α1B/D-KO provides robust assays for the α1A-adrenoceptor and takes us closer to understanding multi-receptor subtype interactions.
Keywords: vascular smooth muscle, adrenoceptor, α1B/D-adrenoceptor knockout, wire myography, fluorescent ligand binding, confocal microscopy
Introduction
Three genetically defined α1-adrenoceptor subtypes (α1A, α1B, α1D; nomenclature follows Alexander et al., 2008) can produce similar cellular responses, including contraction of smooth muscle (Guimaraes and Moura, 2001). Previous pharmacological work on α1-adrenoceptor subtypes suggests that multiple subtypes are present in arterial preparations, causing the analysis of one subtype to be contaminated by the presence of the others (Daly et al., 2002; Deighan et al., 2005).
Little is known of the functional pharmacology of the three subtypes due to the similarity of their signalling, their widespread co-expression and the incomplete availability of selective pharmacological agents (Alexander et al., 2008). This explains the conflicting pA2 or pKB estimates calculated by different groups using various assays for what is ostensibly the same subtype.
Little is known also of the location of the subtypes in native tissues. In cell culture, using epitope-tagged recombinant receptors, it has been proposed that through heterodimerization, the α1-adrenoceptor subtypes might influence each other's cellular location and functional capabilities (Uberti et al., 2003; Stanasila et al., 2003; Hague et al., 2004a,b;). Through such approaches it has also been proposed that these receptor–receptor interactions might result in a different cellular distribution for each subtype. This relates to the concept that the α1B-adrenoceptor is located mainly on the cell surface, while the α1A-adrenoceptor and α1D-adrenoceptor are mainly located at intracellular sites (Hirasawa et al., 1997; Hrometz et al., 1999; McCune et al., 2000; Sugawara et al., 2002). This is not a universal finding as, even in cultured cells, all three subtypes have been shown to be located both on the surface and at intracellular locations (Daly et al., 1998; Hrometz et al., 1999; Pediani et al., 2000; Morris et al., 2004).
There is, however, an alternative approach to studying α1-adrenoceptor location, using the fluorescent ligand BODIPY FL-Prazosin (QAPB). This ligand has affinity for all three α1-adrenoceptor subtypes (Daly et al., 1998; Mackenzie et al., 2000; Daly and McGrath, 2003; McGrath et al., 2005). Because the α1A-adrenoceptor spontaneously internalizes, when QAPB binds it is taken inside the cell and labels the receptor population in intracellular organelles; the receptors continuously recycle so that equilibrium is established between extracellular free ligand and the receptor population (Pediani et al., 2005). This process can be blocked by subtype-selective competitor antagonists to identify subtypes in particular locations (Mackenzie et al., 2000) and enables experiments in native tissue that are normally possible only in cell culture.
Normally in native tissues the presence of more than one receptor subtype complicates both pharmacological analysis and localization of receptors. However, mouse strains are available in which genetic knockouts (KO) of each α1-adrenoceptor are present [α1A-KO (Rokosh and Simpson, 2002); α1B-KO (Cavalli et al., 1997); α1D-KO (Tanoue et al., 2002b; Turnbull et al., 2003)]. We have therefore pursued a strategy of isolating the α1A-adrenoceptor in the double KO of the other subtypes (α1B/D-adrenoceptor knockout; α1B/D-KO). We employed a small artery, mesenteric first order, in which the α1A-adrenoceptor is the major mediator of α1-adrenoceptor-mediated vasoconstriction (Daly et al., 2002; Martinez-Salas et al., 2007) and where the other subtypes are expected to be modulatory (Daly et al., 2002). A conductance artery, the carotid, was also examined to study a system in which the α1D-adrenoceptor is the major mediator of α1-adrenoceptor contractility but where an additional functional α1A-adrenoceptor may exist (Deighan et al., 2005).
The aim of this study was to use the α1B/D-KO to allow the study in native tissue of the remaining subtype, the α1A-adrenoceptor, in respect of its pharmacological properties and cellular distribution and whether these change when it is isolated from the other α1-adrenoceptor subtypes. We subsequently used this knowledge to try to understand the greater complexity that occurs when the other subtypes are present in the wild-type (WT) and to elucidate the possible interactions between subtypes.
Methods
Animals used and vessel preparation
All animal care and this investigation conformed to the provisions of the UK Animals (Scientific procedures) Act 1986. WT mice of C57BL6J/129Sv mixed background were used as controls for single knockouts of the α1B-adrenoceptor (α1B-KO), α1D-adrenoceptor (α1D-KO) and the α1B/D-KO. Mice were bred at the University of Glasgow from breeding pairs kindly provided by Professor Susanna Cotecchia (University of Lausanne, Lausanne, Switzerland; α1B-KO) and Professor Gozoh Tsujimoto (National Children's Medical Research Center, Tokyo, Japan; α1D-KO). α1B/D-KO were generated by cross-breeding the homozygous single KO mice at the University of Glasgow. The generation and background of the KO have previously been described in detail [α1B-KO (Cavalli et al., 1997); α1D-KO (Tanoue et al., 2002b); α1B/D-KO (Hosoda et al., 2005)].
Mice were male weighing between 26 and 55 g [WT (26–47 g; n= 74); α1B-KO (27–48 g; n= 12); α1D-KO (26–42 g; n= 12); α1BD-KO (26–55 g; n= 80)]. An earlier study showed that the predominant subtype present in hepatocytes could change between the commonly employed ages of 2–3 months (Deighan et al., 2004), therefore the more mature 4–5-month-old mice were used. Mice were maintained on a 12:12 hour light/dark schedule at 22–25°C with 45–65% humidity and fed ad libitum on a standard rodent diet and provided drinking water. Mice were killed by carbon dioxide asphyxiation, and first order mesenteric and carotid arteries were isolated using a dissection microscope.
Experimental protocols for functional pharmacology
Two millimetre sections of artery were mounted in physiological salt solution on a four-chamber wire myograph (Danish Myotechnology, Aarhus, Denmark). Following a 30 min equilibration period, passive tension of 250 mg was applied to both types of arteries, and vessels were allowed to equilibrate for a further 45 min. Data were recorded by using Powerlab and Chart (version 5.0) (ADInstruments, Chalgrove, UK). Vessels were challenged with 10 µM of one of three agonists [the standard α1-adrenoceptor agonist, phenylephrine; an agonist selective for the α1A-adrenoceptor, A-61603 (N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide) (Knepper et al., 1995); or 5-hydroxytryptamine]. Acetylcholine (3 µM) was used to test the viability of the endothelium following preconstriction.
Cumulative concentration–response curves were produced for each agonist in half-log increments from 1 nM to 30 µM or 100 µM. Subtype-selective antagonists were incubated for 30 min: prazosin 1 nM, 10 nM and 100 nM (non-selective α1-adrenoceptor antagonist); 5-methylurapidil 10 nM and 100 nM [α1A-adrenoceptor-selective antagonist (Gross et al., 1988)]; RS100-329 (N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride) 1 nM, 10 nM and 100 nM [α1A-adrenoceptor-selective antagonist (Williams et al., 1999)]; BMY 7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione) 1 nM, 10 nM and 100 nM [α1D-adrenoceptor-selective antagonist (Goetz et al., 1995; Saussy et al., 1996)]; or rauwolscine 10 nM (non-selective α2-adrenoceptor antagonist).
Experimental protocols for visualization of smooth muscle cell α1-adrenoceptors
QAPB incubations
3–5 millimetre segments of mesenteric and carotid arteries were incubated with QAPB (100 nM) for 120 min. Live unfixed tissue was used to remain as faithful as possible to the in vitro state used to assess functional pharmacology. All incubations were performed at room temperature (21°C), and solutions were replaced every 30 min to maintain pH conditions.
Additional experiments using non-fluorescent antagonists to compete for QAPB binding sites were performed in segments from the same animals as QAPB controls (see Figure S1). Based on the timescale reported by Pediani et al. (2005), segments were incubated with QAPB for 60 min to establish an equilibrium, then co-incubated for a further 60 min with QAPB and an antagonist at 100 nM: prazosin, rauwolscine, RS100-329 or BMY 7378. The concentration of each antagonist was selected to be capable of competing effectively with QAPB at a specific α1-adrenoceptor subtype but not at other potential binding sites based on pKi estimates from the literature at the cloned α1-adrenoceptor subtypes (Table 1). For instance, at α1A-adrenoceptors, RS100-329 has higher affinity than QAPB, but at the α1B-adrenoceptor or α1D-adrenoceptor, its affinity is lower. Thus, used at the same concentration as QAPB (100 nM), RS100-329 should be capable of displacing QAPB from α1A-adrenoceptors but not from other binding sites.
Table 1.
Ligand binding at α1-adrenoceptors (AR) (mean pKi for antagonists at cloned receptors)
| Antagonist | α1A-AR | α1B-AR | α1D-AR | References | |
|---|---|---|---|---|---|
| Prazosin | Mean | 9.6 | 9.8 | 9.7 | 1–3 |
| Range | 9.0–9.9 | 9.0–10.3 | 9.0–10.0 | ||
| 5-Methylurapidil | Mean | 8.7 | 7.0 | 7.4 | 1, 4–10 |
| Range | 8.3–9.2 | 6.1–7.4 | 6.1–7.9 | ||
| RS100-329 | Mean | 9.5 | 7.6 | 7.9 | 11–12 |
| Range | 9.3–9.6 | 7.3–7.8 | 7.9–7.9 | ||
| BMY 7378 | Mean | 6.4 | 6.6 | 8.5 | 1, 4, 8, 12–17 |
| Range | 6.0–7.1 | 6.2–7.5 | 8.1–9.3 | ||
| QAPB | 8.7 | 8.4 | 8.1 | 1 |
1 MacKenzie et al. (2000); 2 Knepper et al. (1995); 3 Michel and Goepel (1998); 4 Saussy et al. (1996); 5 Goetz et al. (1994); 6 Leonardi et al. (1997); 7 Garcia-Sainz et al. (1995); 8 Yoshio et al. (2001); 9 Buckner et al. (1996); 10 Meyer et al. (1996); 11 Williams et al. (1999); 12 Deighan et al. (2004); 13 Goetz et al. (1995); 14 Kenny et al. (1995); 15 Testa et al. (1997); 16 Hieble et al. (1995); 17 Piascik et al. (1997).
BMY 7378, 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione; QAPB, BODIPY FL-Prazosin; RS100-329, N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride.
Slide preparation and image capture
Arteries were mounted in the incubation solutions with a coverslip (No. 1.5) on top. The thin-walled mesenteric artery was mounted intact, while the carotid artery was sliced open with a single-edged razor blade and laid flat (Miquel et al., 2005) to optimize visualization through the luminal side of the thick arterial wall.
Optical sections were collected on a Bio-Rad Radiance 2100 Confocal Laser Scanning System at an excitation/emission of 488/515 nm for QAPB. Arteries were visualized by using ×40 oil immersion objective (NA 1.0). Optimal laser, gain and offset (contrast and brightness) were determined and then kept constant for each artery. Photomultiplier sensitivities were set such that all pixels fell within the range 0 (black) to 255 (white). Carotid arteries were imaged at high power (zoom 8) to include only smooth muscle cells and avoid areas containing elastic lamina in the media. Mesenteric artery images were collected at low power (zoom 3), which allows coverage of a wider area of exclusively smooth muscle cells. Arteries were scanned as 0.5 µm slices from the internal elastic lamina through the media producing Z-series in stacks of approximately 20–30 µm in depth. Each procedure was carried out in triplicate on four different mice per strain.
Data analysis and statistical procedures
All graphical and statistical analysis was performed by using Graph Pad Prism (version 5.0). Mean data were compared across mouse strains and also in the presence and absence of the selected antagonists using one-way anova and Bonferroni's post test or Student's t-test.
Functional pharmacology
Data were expressed as a percentage of the control curve maximum and represents the mean value ± standard error of the mean. When a maximum response was achieved, pEC50 values were calculated for control curves and agonist curves in the presence of antagonists.
Quantitative confocal microscopy
Image analysis was based on the 2D image analysis as described by Miquel et al. (2005) and performed by using Metamorph software (version 4). In cell cultures fluorescent ligand binding follows similar kinetics to radioligand binding (Mackenzie et al., 2000). However, the relationship between receptor number and fluorescence is not quantitatively established. Therefore, the measurement of ligand binding can be interpreted to identify which receptor subtypes are present but cannot be interpreted in terms of precise numbers of receptors. A single plane at a depth of 4–6 µm from the internal elastic lamina of each Z-series that captured only smooth muscle cells was selected for analysis. Average pixel intensity was measured for the entire plane (mesenteric artery 96 × 96 µm; carotid artery 36 × 36 µm). In addition, individual smooth muscle cells from each image were outlined as a Region of Interest to measure average pixel intensity (see Figure S2). It should be noted that this analysis was performed only on smooth muscle cells that showed fluorescent binding. The average pixel intensity correlated to a greyscale range of 0 (black) to 255 (white), with intermediate intensities being assigned an appropriate grey level.
Materials
Physiological salt solution composition (in mM): 119 NaCl, 4.7 KCl, 1.2 MgSO4.H2O, 1.2 KH2PO4, 24.9 NaHCO3, 2.5 CaCl2 and 11.1 glucose.
All analytical grade drugs were dissolved and diluted to the required concentration in distilled water (unless otherwise stated). Drugs supplied by Sigma Aldrich (Poole, UK) were phenylephrine, acetylcholine, 5-methylurapidil, prazosin, 5-hydroxytryptamine, BMY 7378; by Tocris(Bristol, UK) were A-61603, RS100-329, rauwolscine; and by Invitrogen (Paisley, UK) was QAPB (dissolved in DMSO and diluted in freshly gassed physiological salt solution).
Results
Functional pharmacology in mesenteric arterial rings
In the WT and all three KO strains, the maximum response and sensitivity to 5-hydroxytryptamine was similar (Table 2). This eliminates generic structural or smooth muscle sensitivity changes as a basis for differences subsequently found using drugs acting via α1-adrenoceptors.
Table 2.
Agonist responses in mouse mesenteric arteries
| Mouse |
Phenylephrine |
A-61603 |
5-Hydroxytryptamine |
|||
|---|---|---|---|---|---|---|
| Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | |
| WT | 0.52 ± 0.06 | 6.2 ± 0.08 | 0.41 ± 0.05 | 8.3 ± 0.11*** | 0.18 ± 0.04 | 6.9 ± 0.18 |
| α1B-KO | 0.42 ± 0.07 | 5.8 ± 0.14 | – | – | 0.17 ± 0.04 | 6.9 ± 0.16 |
| α1D-KO | 0.48 ± 0.05 | 5.9 ± 0.09 | – | – | 0.17 ± 0.02 | 6.8 ± 0.12 |
| α1B/D-KO | 0.49 ± 0.08 | 6.4 ± 0.17 | 0.66 ± 0.04** | 8.0 ± 0.09*** | 0.19 ± 0.02 | 7.0 ± 0.13 |
Maximum responses and pEC50 values are expressed as mean ± SEM.
A-61603, N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide; α1B/D-KO, α1B/D-adrenoceptor knockout mouse; WT, wild-type.
P < 0.01 compared with WT (one-way anova, Bonferroni's post test).
P < 0.001 compared with phenylephrine-induced response (one-way anova, Bonferroni's post test).
Cumulative concentration–response curves to the non-selective α1-adrenoceptor agonist phenylephrine were obtained in all mouse strains [Figure 1A (data not shown for α1B-KO and α1D-KO)], with no significant differences between the WT and KO mice (Table 2).
Figure 1.
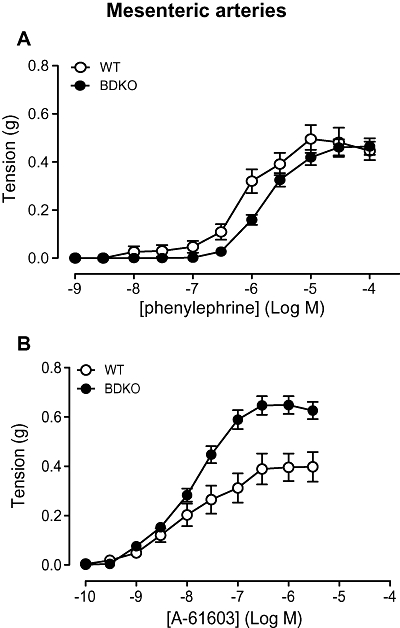
Contractile responses to (A) phenylephrine (n= 6) and (B) A-61603 (N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide) (n= 7) in mesenteric arteries of wild-type (WT) and α1B/D-adrenoceptor knockout (BDKO) mice. Data points are expressed in g tension as mean ± SEM.
The more selective agonist A-61603 was used to study the α1A-adrenoceptor response. Concentration-dependent contractions were obtained to A-61603 in the WT and α1B/D-KO (Figure 1B). The maximum response was significantly higher in the α1B/D-KO than in the WT (Table 2). In both the WT and α1B/D-KO, sensitivity to A-61603 was significantly higher than to phenylephrine (Table 2) consistent with the α1A-adrenoceptor being the main functional α1-adrenoceptor in the mesenteric artery.
In both the WT and α1B/D-KO tissues, prazosin competitively antagonized the responses to phenylephrine (Figure 2A,B). The high pA2 of 9.6 calculated for the α1B/D-KO (Table 3) is consistent with α1-adrenoceptor pharmacology at recombinant α1A-adrenoceptor (Table 1). However, in the WT arteries, prazosin showed only moderate potency as indicated by the pKB of 8.8 (Table 3). Prazosin was also tested against A-61603 in the α1B/D-KO (Table 3), producing a pKB indicative of an α1-adrenoceptor-mediated response.
Figure 2.
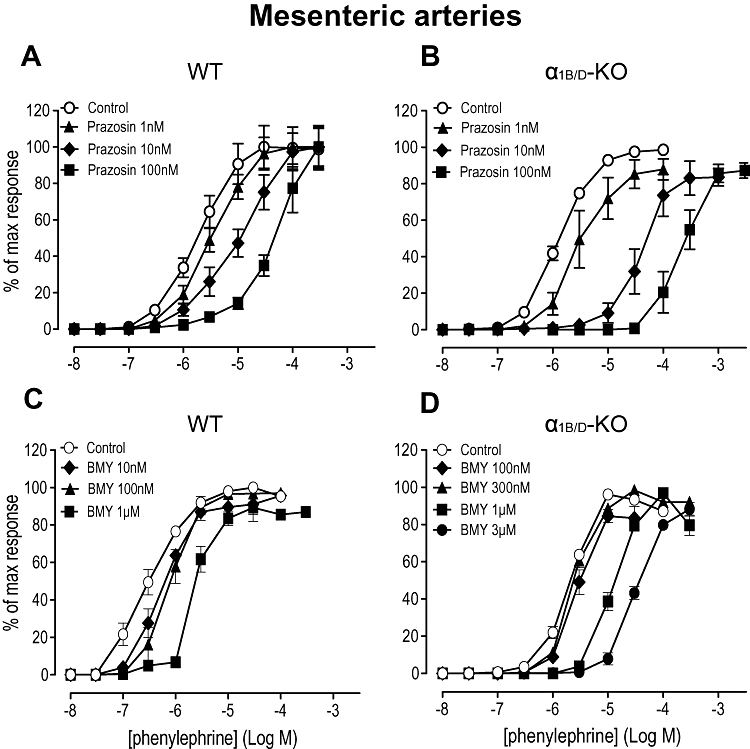
Effects of prazosin (A) and (B), and BMY 7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione) (C) and (D) on contractile response to phenylephrine in mesenteric arteries of the wild-type (WT) (n= 6) and α1B/D-adrenoceptor knockout (α1B/D-KO) mice (n= 7). Data points are expressed as percentage of control curve maximum as mean ± SEM.
Table 3.
Potency values of antagonists of responses to phenylephrine and A-61603 in mouse mesenteric arteries
| Antagonist |
Phenylephrine |
A-61603 |
||||
|---|---|---|---|---|---|---|
|
WT |
α1B/D-KO |
WT |
α1B/D-KO |
|||
| pA2/pKB | Slope (95% CI) | pA2/pKB | Slope (95% CI) | pKB | pKB | |
| Prazosin | 8.8 | – | 9.6 | 1.0 (0.8–1.2) | – | 9.3 |
| RS100-329 | – | – | – | – | 9.0 | 9.9 |
| 5-Methylurapidil | 9.0 | – | 8.9 | – | 8.8 | 8.9 |
| BMY 7378 | 8.3 (at 10 nM) | – | 6.5 | 1.1 (0.8–1.4) | – | 6.5 |
| 7.0 (at 1 µM) | – | |||||
A-61603, N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide; α1B/D-KO, α1B/D-adrenoceptor knockout mouse; BMY 7378, 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione; RS100-329, N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride; WT, wild-type.
The effect of the selective α1D-adrenoceptor antagonist BMY 7378 was studied to examine the contractile role of the α1D-adrenoceptor in the mesenteric artery. In WT arteries BMY 7378 caused parallel shifts of the phenylephrine concentration–response curve but the relationship to antagonist concentration was not clearly dose-related (Figure 2C). Thus, at the lowest BMY 7378 concentration (10 nM), the concentration–response curve was shifted significantly to the right (P < 0.05). An intermediate concentration (100 nM) had little further effect but the highest concentration (1 µM) produced a further significant rightward shift (P < 0.01). pKB values were calculated for BMY 7378 at the lowest and highest concentrations (Table 3), and these were comparable with α1D-adrenoceptor and α1A-adrenoceptor values respectively (Table 1). In the α1B/D-KO, BMY 7378, at high concentrations (300 nM, 1 µM and 3 µM), caused concentration-related, rightward shifts of the phenylephrine response curve (Figure 2D). In the tissues from this mouse strain, the pA2 value (6.5) (Table 3) was identical to that obtained against the A-61603 response and indicates the potency of BMY 7378 at the α1A-adrenoceptor.
5-Methylurapidil was employed as much of the existing literature uses this antagonist as being ‘α1A-adrenoceptor-selective’. In WT arteries, 5-methylurapidil caused a parallel rightward shift of the phenylephrine response at 10 nM (Figure 3A), which produced a pKB (Table 3) indicative of an α1A-adrenoceptor-mediated response (Table 1). However, 100 nM shifted only the upper part of the curve, creating a biphasic concentration–response curve, suggestive of a component of the response being attributable to either α1D-adrenoceptor or α1B-adrenoceptor. Consequently, 5-methylurapidil (100 nM) was studied in the α1B-KO, to reduce the possibilities to the α1D-adrenoceptor and α1A-adrenoceptor subtypes. In this strain, 5-methylurapidil (100 nM) also revealed a biphasic response, indicating that the additional response was α1D-adrenoceptor-mediated (Figure 3B). The position was further clarified by using 5-methylurapidil in combination with BMY 7378: BMY 7378 eliminated the minor component. This was quantified by showing that, while the pEC50 for phenylephrine in the presence of 5-methylurapidil (100 nM) was unaffected by BMY 7378 treatment (4.5 ± 0.1), the pEC25, which was more representative of the first ‘phase’, was shifted significantly rightward by BMY 7378 from 6.4 ± 0.3 to 4.8 ± 0.1 (P < 0.001). The response after 5-methylurapidil plus BMY 7378 was thus monophasic (lacking the first phase) (Figure 3B). In the α1B/D-KO, a monophasic response to phenylephrine was observed in the presence of 5-methylurapidil (100 nM) (Figure 3C). The phenylephrine concentration–response curve was shifted rightward in parallel by 5-methylurapidil, producing a pKB value (Table 3) comparable to published values for α1A-adrenoceptors (Table 1).
Figure 3.
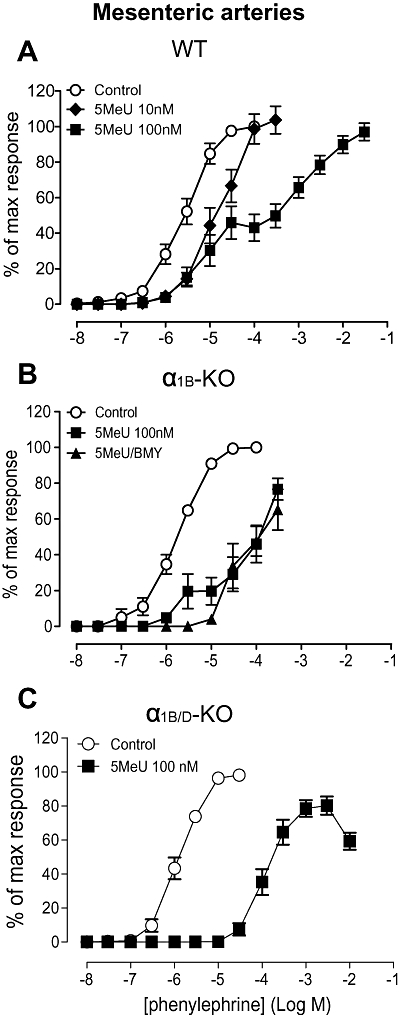
Effects of 5-methylurapidil (5MeU) on contractile response to phenylephrine in mouse mesenteric arteries. (A) Antagonizing effect of 5-methylurapidil in wild-type (WT) (n= 6). (B) Antagonizing effect of 5-methylurapidil and elimination of biphasic response by BMY 7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione) in α1B-adrenoceptor knockout (α1B-KO) (n= 6). (C) Elimination of biphasic response in α1B/D-KO (n= 6). Data points are expressed as percentage of control curve maximum as mean ± SEM.
As phenylephrine was found to be acting on more than one subtype, hence complicating assessment, antagonist potency for the α1A-adrenoceptor was tested against a selective α1A-adrenoceptor agonist, A-61603. Both 5-methylurapidil and a second α1A-adrenoceptor antagonist, RS100-329, caused a concentration-related, parallel, rightward shift of the A-61603 response curve in both the WT and α1B/D-KO arteries (Table 3). This verifies that A-61603 activated only the α1A-adrenoceptor in mesenteric arteries from both the WT and α1B/D-KO.
Functional pharmacology in carotid arterial rings
In the WT and all three KO strains the maximum response and sensitivity to 5-hydroxytryptamine was similar (Table 4). This eliminates generic structural or smooth muscle sensitivity changes as a basis for differences subsequently found using drugs acting via α1-adrenoceptors.
Table 4.
Agonist responses in mouse carotid arteries
| Mouse |
Phenylephrine |
A-61603 |
5-Hydroxytryptamine |
|||
|---|---|---|---|---|---|---|
| Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | |
| WT | 0.27 ± 0.01 | 6.6 ± 0.05 | 0.28 ± 0.02 | 6.0 ± 0.06 | 0.27 ± 0.04 | 6.9 ± 0.08 |
| α1B-KO | 0.32 ± 0.01 | 6.9 ± 0.10 | 0.26 ± 0.02 | 6.3 ± 0.04* | 0.27 ± 0.05 | 6.9 ± 0.08 |
| α1D-KO | 0.17 ± 0.01*** | 5.6 ± 0.06*** | 0.14 ± 0.01*** | 6.7 ± 0.08*** | 0.26 ± 0.06 | 6.8 ± 0.04 |
| α1B/D-KO | 0.06 ± 0.01*** | 4.9 ± 0.07*** | 0.13 ± 0.01*** | 6.6 ± 0.09*** | 0.26 ± 0.03 | 6.8 ± 0.07 |
Maximum responses and pEC50 values are expressed as mean ± SEM.
A-61603, N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide; αIB-KO, α1B-adrenoceptor knockout; αID-KO, α1D-adrenoreceptor knockout; α1B/D-KO, α1B/D-adrenoceptor knockout; WT, wild-type.
P < 0.05;
P < 0.001 compared with WT (one-way anova, Bonferroni's post test).
Concentration-dependent contractions to phenylephrine were produced in all mouse strains (Figure 4A; Table 4). Maximum response and sensitivity to phenylephrine were significantly reduced in the α1D-KO compared with the WT. This suggests that the WT response can be attributed predominantly to α1D-adrenoceptors and the small response in the α1B/D-KO suggests a minor contribution from α1A-adrenoceptors. The effect of knocking out the α1B-adrenoceptor differed according to the other receptors present. Its elimination from the WT (α1B-KO) resulted in an increase in maximum response and sensitivity to phenylephrine indicating either an up-regulation of the α1D- or α1A-adrenoceptor-mediated response or loss of an inhibitory response. In contrast, elimination of the α1B-adrenoceptors from the α1D-adrenoceptor strain (α1B/D-KO) resulted in a reduction in response. This suggests the existence of an α1B-adrenoceptor-mediated contraction or a down-regulation of an α1A-adrenoceptor-mediated response.
Figure 4.
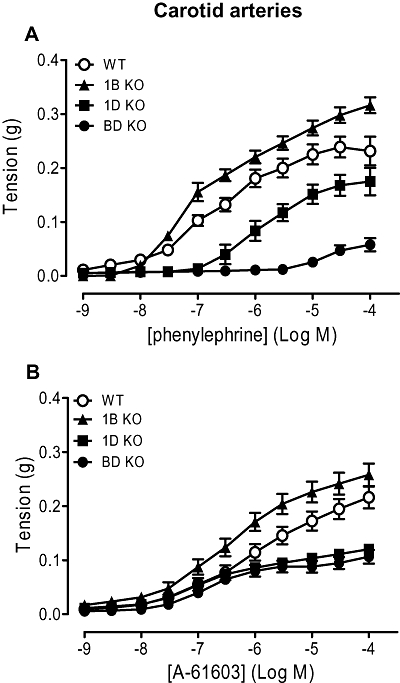
Contractile responses to (A) phenylephrine (n= 8) and (B) A-61603 (N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide) (n= 8) in carotid arteries of wild-type (WT), α1B-adrenoceptor knockout (1B KO), α1D-adrenoceptor knockout (1D KO) and α1B/D-adrenoceptor knockout (BD KO) mice. Data points are expressed in g tension as mean ± SEM.
The α1A-adrenoceptor agonist A-61603 induced concentration-related contractions in all mouse strains (Figure 4B; Table 4). The maximum responses to A-61603 in the α1D-KO and α1B/D-KO were significantly lower compared with the WT. pEC50 values for the A-61603-induced response were significantly higher in the α1B-KO, α1D-KO and α1B/D-KO than in the WT, although the pEC50 value to A-61603 in the α1B-KO was significantly lower than in the α1D-KO and α1B/D-KO (P < 0.001 and P < 0.05 respectively). This can be explained by the presence of the α1D-adrenoceptors: there is an additional component at high concentrations of A-61603 that has the effect of reducing the apparent pEC50 value; this is eliminated after knocking out the α1D-adrenoceptors, revealing the true, higher, sensitivity of A-61603 at α1A-adrenoceptors. Also, for this reason, sensitivity, but not maximum response, to A-61603 appears to be significantly lower than for phenylephrine in the WT (Table 4). In the α1B/D-KO both maximum response and sensitivity to A-61603 were significantly higher than for phenylephrine, reflecting the true situation at α1A-adrenoceptors, with a difference in pEC50 values of 1.7, similar to the value of 1.6 in the mesenteric artery.
In the carotid arteries from WT mice, prazosin competitively antagonized the phenylephrine concentration–response curve (Figure 5A; Table 5). In the α1B/D-KO the phenylephrine concentration–response curve was significantly shifted rightward by prazosin 10 nM, although a true maximum response was not achieved within the agonist concentration used (Figure 5B; Table 5).
Figure 5.
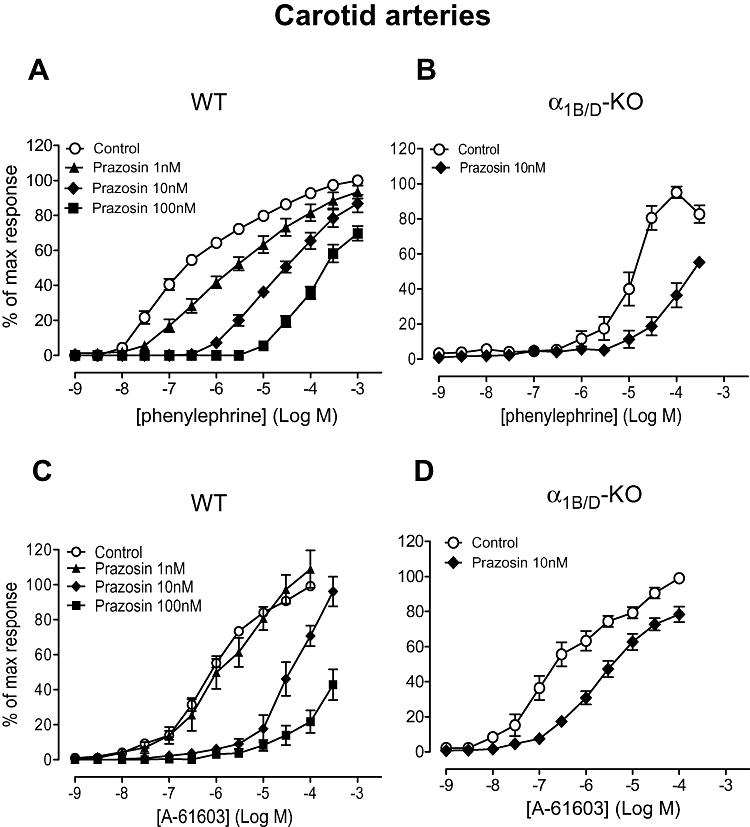
Effects of prazosin on contractile response to phenylephrine (A) and (B), and A-61603 (N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide) (C) and (D) in carotid arteries of wild-type (WT) (n= 7) and α1B/D-adrenoceptor knockout (α1B/D-KO) (n= 6) mice. Data points are expressed as percentage of control curve maximum as mean ± SEM.
Table 5.
Potency values of antagonists of responses to phenylephrine and A-61603 in mouse carotid arteries
| Antagonist |
Phenylephrine |
A-61603 |
||||||
|---|---|---|---|---|---|---|---|---|
|
WT |
α1B/D-KO |
WT |
α1B/D-KO |
|||||
| pA2/pKB | Slope | pA2/pKB | Slope | pA2/pKB | Slope | pA2/pKB | Slope | |
| Prazosin | 9.6* | 0.9 (0.8–1.1) | 9.0 at 10 nM | – | 9.6 at 10 nM | – | 9.1 at 10 nM | – |
| BMY 7378 | 8.3* | – | – | – | 8.3 | – | No shift at 10 nM and 100 nM | – |
| 5-Methylurapidil | 7.5* | 1.1 (0.7–1.5) | Response abolished | – | 8.3 | 1.0 (0.6–1.0) | 8.3 at 10 nM | – |
| RS100-329 | 7.9 | – | – | – | 8.7 | – | Non-competitive block (at 10 nM) | |
A-61603, N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide; α1B/D-KO, α1B/D-adrenoceptor knockout; BMY 7378, 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione; RS100-329, N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride; WT, wild-type.
Denotes data from Deighan et al. (2005).
In the WT, only 10 nM prazosin produced a parallel shift in the A-61603 curve, enabling a pKB to be calculated (Figure 5C; Table 5). Prazosin (10 nM) also produced a parallel shift in the A-61603-induced response in the α1B/D-KO (Figure 5D) producing a pKB (9.1), comparable to published values for α1A-adrenoceptors.
In the α1B/D-KO, in the presence of the selective α1A-adrenoceptor antagonist 5-methylurapidil the contractile response to phenylephrine was obliterated (Table 5). No further antagonist studies were performed against phenylephrine in the α1B/D-KO due to the low potency of the agonist in this artery.
The potency estimates for 5-methylurapidil against A-61603 were similar in the α1B/D-KO and WT (Figure S3; Table 5). Therefore, another selective α1A-adrenoceptor antagonist, RS100-329, was also employed to investigate the role of the α1A-adrenoceptor in the carotid artery. RS100-329 10 nM acted competitively in the WT (Figure 6A; Table 5) but blocked the A-61603-induced response in a non-competitive manner in the α1B/D-KO (Figure 6B). The pKB for 5-methylurapidil against A-61603 in the WT (8.3) was higher than that obtained against phenylephrine (7.5) and closer to published values for an α1A-adrenoceptor-mediated response (average pKi 8.7, Table 1).
Figure 6.
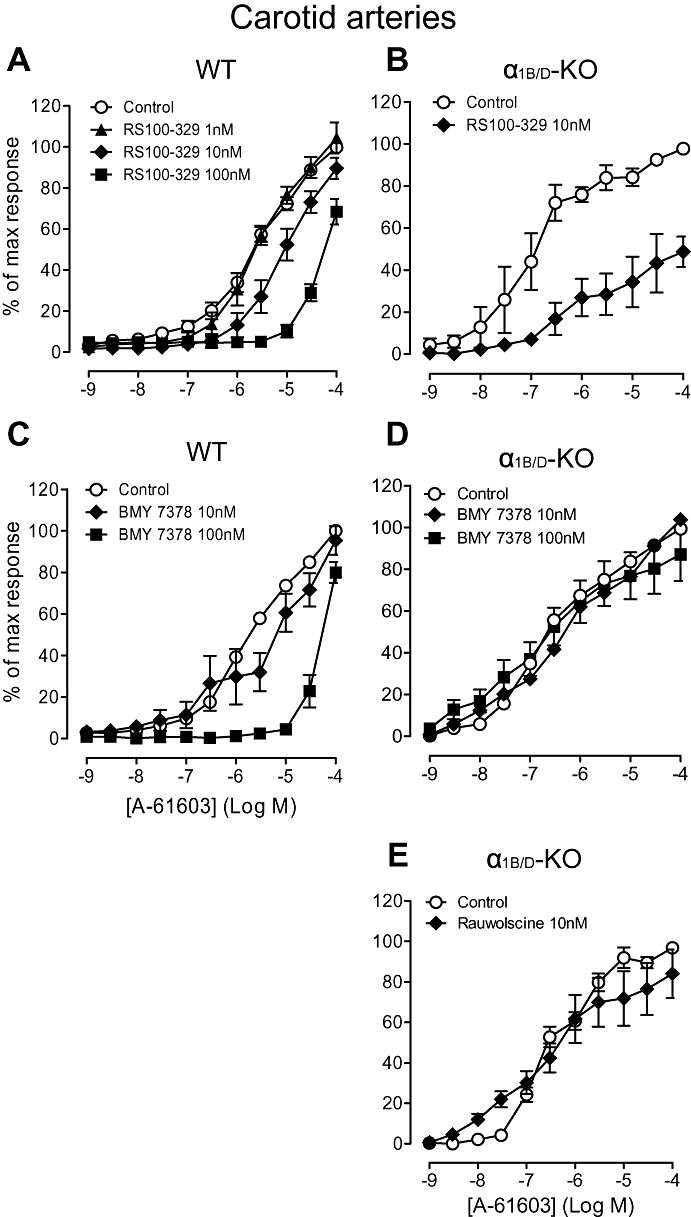
Effects of RS100-329 (N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride) (A) and (B), BMY 7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione) (C) and (D), and rauwolscine (E), on contractile response to A-61603 (N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide) in carotid arteries of wild-type (WT) (n= 7) and α1B/D-adrenoceptor knockout (α1B/D-KO) mice (n= 7). Data points are expressed as percentage of control curve maximum as mean ± SEM.
In the WT, a biphasic response was produced in the presence of BMY 7378 10 nM (Figure 6C), highlighting the involvement of two subtypes mediating the A-61603 response, as noted above. BMY 7378 100 nM produced a rightward displacement of the A-61603 concentration–response curve producing a pKB (Figure 6C; Table 5) in line with published values for α1D-adrenoceptors (average pKi 8.5, Table 1). This indicates α1D-adrenoceptor agonism by A-61603 when an efficient α1D-adrenoceptor system is present. To assess its potency versus α1A-adrenoceptors and, hence, assess its selectivity, BMY 7378 was tested against A-61603 in the α1B/D-KO carotid arteries: BMY 7378 (10 nM and 100 nM) had no effect on the concentration–response curve to A-61603 (Figure 6D).
In the α1B/D-KO, the α2-adrenoceptor antagonist, rauwolscine did not produce a rightward shift of the A-61603 concentration–response curve (Figure 6E; Table 5), suggesting that this agonist-induced response was not α2-adrenoceptor-mediated.
Visualization of α1-adrenoceptors using the fluorescent ligand QAPB
Mesenteric artery
In both strains, smooth muscle cells were clearly defined throughout the medial layer although the amount of fluorescence varied between individual cells (Figure 7). In the WT, QAPB appeared to bind all smooth muscle cells, while in the α1B/D-KO, some smooth muscle cells did not appear to show fluorescent ligand binding. When measured quantitatively, the intensity of fluorescence of the 96 × 96 µm image was significantly reduced in the α1B/D-KO compared with the level in the WT (Table 6). However, in the α1B/D-KO tissues, the intensity of the fluorescence of those smooth muscle cells that QAPB did bind was not significantly different to that of WT smooth muscle cells (Table 6). These data suggest that there was a reduction in the number of smooth muscle cells that bound QAPB in the α1B/D-KO mesenteric artery. Binding was present both on the cell surface and intracellularly in punctate compartments, showing that the α1A-adrenoceptor is non-uniformly distributed.
Figure 7.
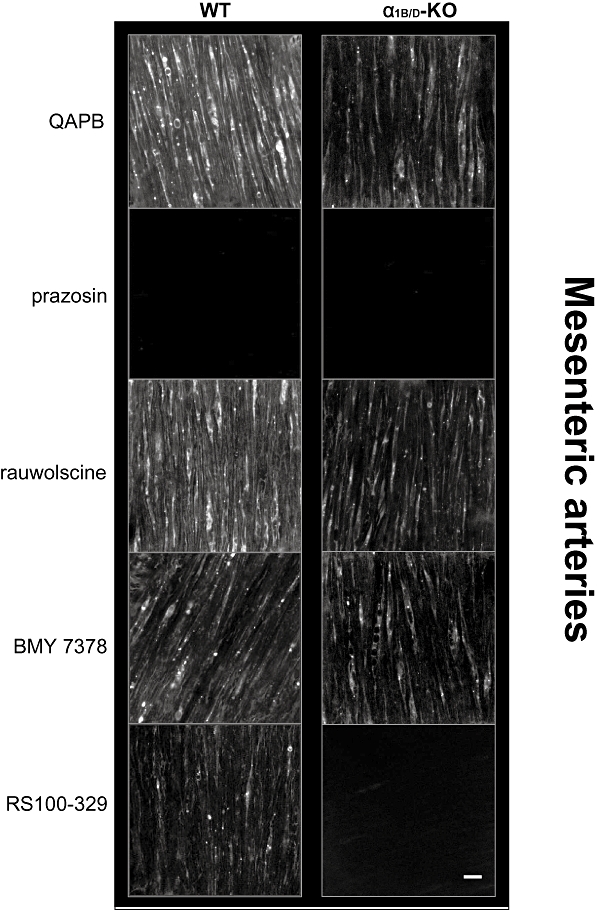
Comparison of QAPB fluorescent ligand binding in mesenteric arteries of WT and α1B/D-KO mice. Non-fluorescent antagonists (100 nM) were used to compete with the fluorescent ligand QAPB (100 nM) after QAPB equilibrium was established. All images are collected under identical conditions of laser intensity, photomultiplier gain (contrast) and offset (brightness). Imaging was performed at low power (zoom 3). An image size of 512 × 512 pixels produced a field size of 96 × 96 µm. Calibration bar indicates 10 µm. Each image is representative of those generated from four mice. α1B/D-KO, α1B/D-adrenoceptor knockout mouse; BMY 7378, 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione; QAPB, BODIPY FL-Prazosin; RS100-329, N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride; WT, wild-type.
Table 6.
Comparison of average pixel intensity of entire image and smooth muscle cells (SMC) in wild-type (WT) and α1B/D-adrenoceptor knockout mouse (α1B/D-KO)
| Mouse strain |
Mesenteric arteries |
Carotid arteries |
||
|---|---|---|---|---|
| Average pixel intensity of 96×96 µm image | Average pixel intensity of SMC | Average pixel intensity 36×36 µm image | Average pixel intensity of SMC | |
| WT | 79.9 ± 4.1 | 84.8 ± 2.6 | 41.2 ± 9.2 | 48.8 ± 2.5 |
| α1B/D-KO | 24.3 ± 2.3*** | 75.7 ± 3.7 | 20.6 ± 2.9* | 50.4 ± 3.6 |
Data are expressed as mean ± SEM.
Mesenteric arteries imaged at low power (zoom 3); carotid arteries imaged at high power (zoom 8).
Only smooth muscle cells showing fluorescent ligand binding were outlined as a Region of Interest to measure average pixel intensity.
P > 0.05 for smooth muscle cells, α1B/D-KO compared with WT (Student's t-test).
P < 0.05;
P < 0.001 compared with WT (Student's t-test).
Effective competition from the non-selective α1-adrenoceptor antagonist prazosin but not from the non-selective α2-adrenoceptor antagonist rauwolscine confirmed that the fluorescent ligand was binding to α1-adrenoceptors and not α2-adrenoceptors (Figure 7; Table 7).
Table 7.
Effect of subtype-selective antagonists on fluorescent ligand binding in mesenteric and carotid arteries
| Antagonist |
Average pixel intensity |
|||
|---|---|---|---|---|
|
WT |
α1B/D-KO |
|||
| Mesenteric arteries | Carotid arteries | Mesenteric arteries | Carotid arteries | |
| QAPB control | 79.9 ± 4.1 | 41.2 ± 9.2 | 24.3 ± 2.3 | 20.6 ± 2.9 |
| Prazosin | 13.3 ± 7.7*** | 5.9 ± 1.4*** | 7.4 ± 1.2*** | 4.7 ± 1.9** |
| Rauwolscine | 74.4 ± 10.7 | 55.3 ± 9.3 | 23.1 ± 1.3 | 17.3 ± 2.8 |
| BMY 7378 | 27.7 ± 3.1*** | 10.0 ± 0.6** | 24.6 ± 1.9 | 16.8 ± 2.6 |
| RS100-329 | 24.4 ± 1.2*** | 13.3 ± 2.9** | 9.1 ± 1.3*** | 5.5 ± 0.6*** |
Data are expressed as mean ± SEM; mesenteric arteries imaged at low power; carotid arteries imaged at high power (zoom 8).
α1B/D-KO, α1B/D-adrenoceptor knockout; BMY 7378, 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione; QAPB, BODIPY FL-Prazosin; RS100-329, N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride; WT, wild-type.
P < 0.01;
P < 0.001 compared with QAPB control (one-way anova, Bonferroni's post test).
BMY 7378 was employed at a concentration that should selectively compete with QAPB for α1D-adrenoceptor binding sites in the WT and to test its ability to compete with QAPB at the α1A-adrenoceptor in the α1B/D-KO. In the presence of BMY 7378 QAPB fluorescent binding was reduced in the WT but not in the α1B/D-KO (Figure 7; Table 7).
The selective α1A-adrenoceptor antagonist RS100-329 was used to compete with QAPB at a concentration that should be effective at the α1A-adrenoceptor but not at other potential binding sites. In the presence of RS100-329, fluorescent ligand binding was reduced in WT and abolished in α1B/D-KO (Figure 7; Table 7).
Carotid artery
Smooth muscle cells were clearly defined by QAPB binding throughout the media of the WT artery (see Figure S4) but fluorescence varied between individual cells (Figure 8). Quantitative analysis revealed that intensity of fluorescence of the 36 × 36 µm image was significantly lower in the α1B/D-KO, compared with the WT artery (Table 6). QAPB binding was more heterogeneous in the α1B/D-KO, with patches of cells virtually devoid of ligand binding. This is reflected by quantitative analysis, which showed that in smooth muscle cells showing fluorescent binding in the α1B/D-KO, the intensity of the fluorescence was not significantly different to that in the WT artery (Table 6). This suggests that the intensity of fluorescence measured for the 36 × 36 µm image reflects a reduction in the number of smooth muscle cells showing binding in the α1B/D-KO samples. In both strains, QAPB bound to both the cell surface and intracellular compartments with evidence of both perinuclear binding and punctate binding being observed intracellularly.
Figure 8.
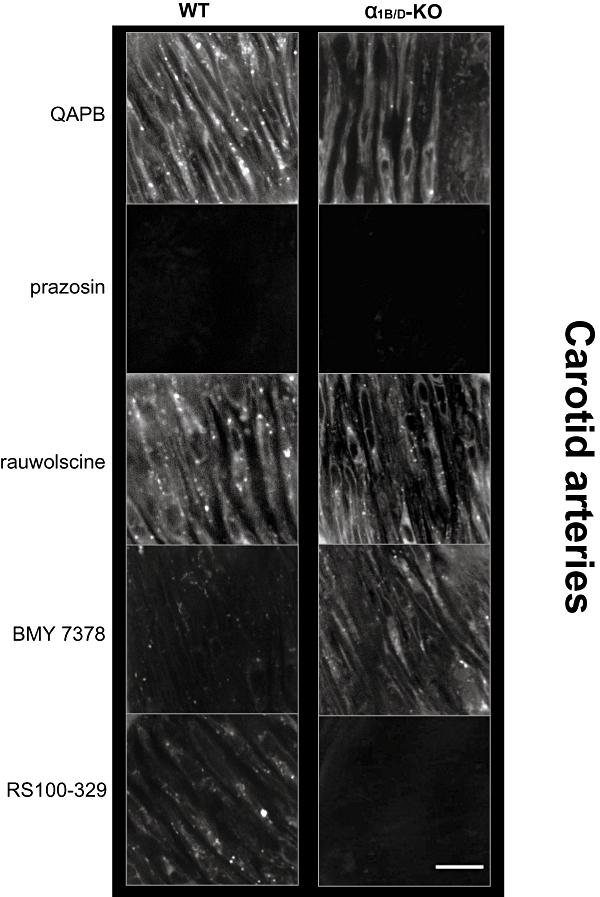
Comparison of QAPB fluorescent ligand binding in carotid arteries of WT and α1B/D-KO mice. Non-fluorescent antagonists (100 nM) were used to compete with QAPB (100 nM) after QAPB equilibrium was established. All images are collected under identical conditions of laser intensity, photomultiplier gain (contrast) and offset (brightness). Imaging was performed at high power (zoom 8) to include only smooth muscle cell and avoid elastic lamina in the media. An image size of 512 × 512 pixels produced a field size of 36 × 36 µm. Calibration bar indicates 10 µm. Each image is representative of those generated from four mice. α1B/D-KO, α1B/D-adrenoceptor knockout; BMY 7378, 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione; QAPB, BODIPY FL-Prazosin; RS100-329, N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride; WT, wild-type.
The use of prazosin and rauwolscine in carotid arteries confirmed that the fluorescent ligand was selective for α1-adrenoceptors and was not binding α2-adrenoceptors (Figure 8; Table 7).
In the WT the selective α1D-adrenoceptor antagonist BMY 7378 markedly reduced QAPB binding (Figure 8; Table 7), demonstrating that the α1D-adrenoceptor is present in the WT. In the α1B/D-KO, the lack of effect of BMY 7378 on fluorescence showed that BMY 7378 did not compete with QAPB for α1A-adrenoceptor binding sites (Figure 8; Table 7).
Using the selective α1A-adrenoceptor antagonist, RS100-329, to compete with QAPB showed reduced binding in the WT indicating that the α1A-adrenoceptor is present in this strain (Figure 8; Table 7). In the α1B/D-KO, QAPB binding was quantitatively reduced and could not be detected by visual inspection in the presence of RS100-329 (Figure 8; Table 7).
Discussion
The α1B/D-KO strain shows ‘pure’α1A-adrenoceptor pharmacology and elucidates the complex pharmacology of the WT. Potencies of agonists and antagonists in the WT did not correlate with any single subtype as shown in pure assay systems. By working back from the α1B/D-KO, the subtypes in the WT tissues could be identified. α1A-adrenoceptor-mediated contraction was identified in both arteries but was greater in the mesenteric artery. Both arteries showed α1D-adrenoceptor-mediated contraction but this was greater in the carotid. There was indirect evidence for contraction by α1B-adrenoceptors only in the carotid, but, in both arteries, removal of α1B-adrenoceptors from the WT enhanced α1D- or α1A-adrenoceptor-mediated contractions.
The tissues from the α1B/D-KO provide the first genuine example of α1A-adrenoceptor pharmacology in native tissues. In particular, the α1B/D-KO mesenteric artery represents a more accurate assay for functional pharmacology of the α1A-adrenoceptor than is otherwise available and can be used to interpret data from other species in which KO are impractical. Deviations from radioligand binding estimates are now shown to be consistent with ‘contamination’ of the assay vessels with other α1-adrenoceptor subtypes. Similarly, supposed ‘species’ or ‘tissue-specific’ differences may be due to varying proportions of different subtypes.
The only evidence for genetic compensation was enhancement of α1A-adrenoceptor-mediated contraction in the mesenteric artery: the maximum response of A-61603 increased when moving from the WT to α1B/D-KO. This must mean some increase in the efficiency of α1A-adrenoceptors, whether by up-regulation of receptor number or signalling system. This would have been missed by studying only phenylephrine, because loss of its potent α1D-adrenoceptor response negates the gain in α1A-adrenoceptor response. Imaging data did not provide evidence for an up-regulation of α1A-adrenoceptors in the α1B/D-KO mesenteric artery. This suggests that ‘compensation’ lies in the signalling process. This is specific to the α1A-adrenoceptor as the 5-hydroxytryptamine response was unaffected by the loss of the α1B- and α1D-adrenoceptors.
Pharmacology of α1A-adrenoceptors
In the α1B/D-KO, pharmacological analysis provided a clear profile for the α1A-adrenoceptor. In both arteries, A-61603 had greater efficacy and potency than phenylephrine: the potency ratio was similar (40-fold). This differential is less than that reported by Pediani et al. (2000) for recombinant bovine α1A-adrenoceptors (70-fold) or for WT tissues by Knepper et al. (1995) (rat vas deferens, 300-fold; canine prostate, 165-fold) but was closer to radioligand binding at bovine α1A-adrenoceptors [60-fold; (Pediani et al., 2000)]. Both agonists had lower potency in the carotid (approximately 50-fold) indicating a smaller receptor reserve for α1A-adrenoceptors.
In the carotid, the data from A-61603 clarify the phenylephrine data. Because responses to A-61603 are similar in the α1D-KO and α1B/D-KO this confirms that the reduction in the phenylephrine response is caused by the loss of an α1B-adrenoceptor-mediated contraction to phenylephrine, which A-61603 does not activate. Conversely, the similarity of the increases in response to A-61603 and phenylephrine in the α1B-KO suggests that this change is not due to the loss of an inhibitory effect of the agonists via α1B-adrenoceptors (as A-61603 is not a good agonist at α1B-adrenoceptors) but rather is a compensatory effect on the signalling process or receptor number for α1D- or α1A-adrenoceptors.
The functional pharmacology was straightforward in mesenteric arteries where α1A-adrenoceptor is dominant in the WT strain; the major gain in accuracy was for the α1D-adrenoceptor antagonist BMY 7378, whose estimated affinity fell to its α1A-adrenoceptor value in the α1B/D-KO. In contrast, the selective α1A-adrenoceptor antagonists, RS100-329 and 5-methylurapidil, retained high potency, as expected in the α1B/D-KO mice. A complication in the analysis of the α1B/D-KO mesenteric arteries was that prazosin was less potent in the WT than in the α1B/D-KO tissues. This was unexpected and, although this cannot be fully explained as yet, it reflects the complexity of analysis in the WT. It is interesting that the ‘α1L-adrenoceptor’ phenomenon, that is, low affinity for prazosin (Flavahan and Vanhoutte, 1986; Muramatsu et al., 1990) has been reported to be lost in the α1A-KO (Muramatsu et al., 2008), yet isolation of the α1A-adrenoceptor (in the present study) does not reveal it. Thus the ‘α1L-adrenoceptor’ phenomenon may require the presence of both the α1A-adrenoceptor and another subtype.
In carotid arteries, where the residual α1A-adrenoceptor response to agonists is small, the response was not sufficiently robust to sustain a comprehensive analysis. Threshold sensitivities to antagonists were as expected but were difficult to quantify in competitive terms. In addition, RS100-329 produced non-competitive blockade. Non-competitive blockade of recombinant α1A-adrenoceptors has been shown for prazosin and WB4101 and was attributed to the non-equilibrium nature of post-receptor events (Pediani et al., 2000). Similar factors may apply here, where the maximum response achievable is small, relative to the capability of the artery, as shown by 5-hydroxytryptamine.
Agonist activity at α1D-adrenoceptors by A-61603 was revealed in the KO. In the carotid, this occurred at high agonist concentrations as introducing the α1D-KO reduced the responses to A-61603. Furthermore, the A-61603-induced response has an α1D-adrenoceptor component in the WT, as there was a biphasic response to A-61603 in the presence of BMY 7378 10 nM and the potency estimate for BMY 7378 (100 nM) was comparable to the α1D-adrenoceptor values (Table 1). This did not occur in the mesenteric artery where the receptor reserve for α1D-adrenoceptor is smaller, consistent with A-61603 being a partial agonist at α1D-adrenoceptor. This detailed pharmacological information refines the ability to assess the presence of the subtypes in non-genetically modified species.
Location of α1A-adrenoceptors
When α1A-adrenoceptors were isolated in the α1B/D-KO they showed, in general, a more heterogeneous distribution than when the whole family of subtypes was present in the WT. This was most apparent in the carotid, where the α1A-adrenoceptor is secondary to the α1D-adrenoceptor. This suggests a general phenomenon of different phenotypes of vascular smooth muscle in respect of α1A-adrenoceptors. This may be relevant to function, for example, of pacemaker cells (Peng et al., 2001) and partly explains the reduced maximum response in the α1B/D-KO carotid, in which there is less likely to be a functional syncytium due to the separation of smooth muscle cells by collagen layers: consequently, the maximum response becomes dependent on the number of cells expressing the receptors, in contrast to a functional syncytium such as in the mesenteric artery where depolarization can spread through the tissue despite a limited number of cells expressing receptors.
In both arteries, receptors were found both on the cell surface and in intracellular compartments, reflecting findings in cell cultures with recombinant α1D- (Daly et al., 1998) and α1A-adrenoceptors (Pediani et al., 2005). This seems to be a genuine phenotype and not an artefact. Vascular fibroblasts in intact arteries also have a substantial population of intracellular α1-adrenoceptors (McGrath et al., 2005), so the phenomenon is a general one for native and recombinant α1-adrenoceptors, expressed in different cell types. In addition, fixed and permeabilized tissues also show these different cellular phenotypes (Miquel et al., 2005) eliminating differential ligand uptake as an explanation. There is no simple explanation based on different proportions of the subtypes in these different phenotypes or chaperoning by heterodimers. Nevertheless, increased heterogeneity in both types of artery, once the other subtypes were eliminated, shows a strong tendency for the α1A-adrenoceptor subtype to be heterogeneously distributed.
Potential interaction of α1-adrenoceptor subtypes
This is the first example of a native tissue in which it has been possible to observe the consequences, for receptor distribution, of moving from the WT strain with multiple subtypes of α1-adrenoceptor to a double KO expressing only one subtype. There was, however, no evidence that removing the α1B-adrenoceptor had any influence on the balance of surface to intracellular distribution of the remaining subtypes, as had been inferred from artificial systems (Stanasila et al., 2003; Hague et al., 2004b).
Physiological and therapeutic relevance
Our demonstration of the likely presence of all three subtypes in native vascular smooth muscle cells is extremely interesting for physiological function. The relative preponderance of the subtypes, as shown by fluorescent ligand binding, was similar between the two arteries despite the quantitatively different contribution of the subtypes to contraction. Thus, measuring the presence of the receptor or its RNA will not give a useful guide to function and, perhaps, the relative contribution of each subtype is more effectively regulated by other factors than by the amount of receptor present. This correlates also with functional compensation of the α1A-adrenoceptor for the loss of the other two subtypes, which showed as an increase in response without evidence for an up-regulation of the receptors present.
As binding and function both show multiple subtypes in these two arteries, it is not beneficial to assign one α1-adrenoceptor subtype to a specific vessel or to a class of vessel that performs a particular physiological function. α1-Adrenoceptor heterogeneity is an important consideration when assessing the selectivity of α1-adrenoceptor compounds. Blockade of either α1A-adrenoceptor or α1D-adrenoceptor can affect the response and, thus, can be expected to block adrenergic control of peripheral resistance, as has been demonstrated in vivo (Villalobos-Molina and Ibarra, 1996; Zhou and Vargas, 1996) and in vitro (Castillo et al., 1998; Zacharia et al., 2004; 2005;). This indicates that no one subtype plays a part in maintaining and regulating blood pressure as suggested by the mild hypotension in each single KO model (Rokosh and Simpson, 2002; Tanoue et al., 2002a).
Acknowledgments
This paper is dedicated to the memory of the late Ann McGee, preliminary studies carried out by Ann provided the impetus for this work. The authors thank Mrs Joyce Macmillan for her expert technical assistance. M McBride and L Methven received generous support from the British Heart Foundation (PG/05/140/20094 and FS/04/035) and the Ann B McNaught Bequest.
Glossary
Abbreviations:
- A-61603
N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulphonamide
- α1B/D-KO
α1B/D-adrenoceptor knockout
- BMY 7378
8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione
- KO
Knockouts
- QAPB
BODIPY FL-Prazosin
- RS100-329
N-[(2-trifluoroethoxy)phenyl],N′-(3-thyminylpropyl) piperazine hydrochloride
- SMC
smooth muscle cells
- WT
wildtype
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Example of abolishment of BODIPY FL-Prazosin (QAPB) binding by prazosin (left image) but presence of smooth muscle cells confirmed by the nuclear stain Syto 61 (right image) in carotid artery of wild-type (WT). Imaging was performed at high power (zoom 8) to include only smooth muscle cells and avoid elastic lamina in the media. An image size of 512 × 512 pixels produced a field size of 36 μm × 36 μm. Calibration bar indicates 5 μm.
Figure S2 Illustration of measurement of average pixel intensity from individual smooth muscle cells from wild-type (WT) and α1B/D-adrenoceptor knockout (α1B/D-KO) mesenteric and carotid arteries. Smooth muscle cells were outlined and measured as Regions of Interest. Imaging was performed at low power (zoom 3) for mesenteric arteries and high power (zoom 8) for carotid arteries. An image size of 512 × 512 pixels produced a field size of 96 × 96 μm for mesenteric arteries and 36 μm × 36 μm for carotid arteries.
Figure S3 Effect of 5-methylurapidil (5MeU) on contractile response to phenylephrine in mouse carotid arteries of α1B/D-adrenoceptor knockout (n = 6). Data points are expressed as percentage of control curve maximum as mean ± S.E.M.
Figure S4 Example images of BODIPY FL-Prazosin (QAPB) fluorescent binding on a plane imaged from the endothelial side (left image; average pixel intensity 78.3) and a plane imaged from the adventitial side (right image; average pixel intensity 77.7) from the same z-stack of a mesenteric artery. An image size of 512 × 512 pixels produced a field size of 96 μm × 96 μm.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2008;153:S14. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner SA, Oheim KW, Morse PA, Knepper SM, Hancock AA. Adrenoceptor-induced contractility in rat aorta is mediated by the alpha (1D) subtype. Eur J Pharmacol. 1996;297:241–248. doi: 10.1016/0014-2999(95)00755-5. [DOI] [PubMed] [Google Scholar]
- Castillo EF, López RM, Rodríguez-Silverio J, Bobadilla RA, Castillo C. Alpha 1D-adrenoceptors contribute to the neurogenic vasopressor response in pithed rats. Fundam Clin Pharmacol. 1998;12:584–589. doi: 10.1111/j.1472-8206.1998.tb00990.x. [DOI] [PubMed] [Google Scholar]
- Cavalli A, Lattion AL, Hummler E, Nenniger M, Pedrazzini T, Aubert JF, et al. Decreased blood pressure response in mice deficient of the alpha 1b-adrenergicreceptor. Proc Natl Acad Sci. 1997;94:11589–11594. doi: 10.1073/pnas.94.21.11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly CJ, McGrath I. Fluorescent ligands, antibodies, and proteins for the study of receptors. Pharmacol Ther. 2003;100:101–118. doi: 10.1016/j.pharmthera.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Daly CJ, Milligan CM, Milligan G, Mackenzie JF, McGrath JC. Cellular localization and pharmacological characterization of functioning alpha-1 adrenoceptors by fluorescent ligand binding and image analysis reveals identical binding properties of clustered and diffuse populations. of receptors. J Pharmacol Exp Ther. 1998;286:984–990. [PubMed] [Google Scholar]
- Daly CJ, Deighan C, McGee A, Mennie D, Ali Z, McBride M, et al. A knockout approach indicates a minor vasoconstrictor role for vascular α1B-adrenoceptors in mouse. Physiol Genomics. 2002;9:85–91. doi: 10.1152/physiolgenomics.00065.2001. [DOI] [PubMed] [Google Scholar]
- Deighan C, Woollhead A, Colston J, McGrath JC. Hepatocytes from α1B-adrenoceptor knockout mice reveal compensatory adrenoceptor subtype substitution. Br J Pharmacol. 2004;142:1031–1037. doi: 10.1038/sj.bjp.0705872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deighan C, Methven L, Naghadeh MM, Wokoma A, MacMillan J, Daly CJ, et al. Insights into the functional roles of α1-adrenoceptor subtypes in mouse carotid arteries using knockout mice. Br J Pharmacol. 2005;144:558–565. doi: 10.1038/sj.bjp.0706089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan NA, Vanhoutte PM. [alpha]1-adrenoceptor subclassification in vascular smooth muscle. Trends Pharmacol Sci. 1986;7:347–349. [Google Scholar]
- Garcia-Sainz JA, Romero-Avila M, Torres-Marquez ME. Characterization of the human liver alpha 1-adrenoceptors: predominance of the alpha 1A subtype. Eur J Pharmacol. 1995;289:81–86. doi: 10.1016/0922-4106(95)90171-x. [DOI] [PubMed] [Google Scholar]
- Goetz AS, Lutz MW, Rimele TJ, Saussy DLJ. Characterisation of alpha-1-adrenoceptor subtypes in human and canine prostate membrane. J Pharmacol Exp Ther. 1994;271:1228–1233. [PubMed] [Google Scholar]
- Goetz AS, King HK, Ward SDC, True TA, Rimele TJ, Saussy J. BMY 7378 is a selective antagonist of the D subtype of [alpha]1-adrenoceptors. Eur J Pharmacol. 1995;272:R5–R6. doi: 10.1016/0014-2999(94)00751-r. [DOI] [PubMed] [Google Scholar]
- Gross G, Hanft G, Rugevics C. 5-Methyl-urapidil discriminates between subtypes of the [alpha]1-adrenoceptor. Eur J Pharmacol. 1988;151:333–335. doi: 10.1016/0014-2999(88)90819-9. [DOI] [PubMed] [Google Scholar]
- Guimaraes S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53:319–356. [PubMed] [Google Scholar]
- Hague C, Chen Z, Pupo AS, Schulte NA, Toews ML, Minneman KP. The N terminus of the human alpha1D-adrenergic receptor prevents cell surface expression. J Pharmacol Exp Ther. 2004a;309:388–397. doi: 10.1124/jpet.103.060509. [DOI] [PubMed] [Google Scholar]
- Hague C, Uberti M, Chen Z, Hall R, Minneman KP. Cell surface expressin of alpha 1D-adrenergic receptors is controlled by heterodimerization with alpha 1B-adrenergic receptors. J Biol Chem. 2004b;279(15):15541–15549. doi: 10.1074/jbc.M314014200. [DOI] [PubMed] [Google Scholar]
- Hieble JP, Bylund DB, Clarke DE, Eikenburg DC, Langer SZ, Lefkowitz RJ, et al. International Union of Pharmacology. X. Recommendation for nomenclature of alpha 1-adrenoceptors: consensus update. Pharmacol Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- Hirasawa A, Sugawara T, Awaji T, Tsumaya K, Ito H, Tsujimoto G. Subtype-specific differences in subcellular localization of alpha 1-adrenoceptors: chlorethylclonidine preferentially alkylates the accessible cell surface alpha 1-adrenoceptors irrespective of the subtype. Mol Pharmacol. 1997;52:764–770. doi: 10.1124/mol.52.5.764. [DOI] [PubMed] [Google Scholar]
- Hosoda C, Koshimizu TA, Tanoue A, Nasa Y, Oikawa R, Tomabechi T, et al. Two alpha1-adrenergic receptor subtypes regulating the vasopressor response have differential roles in blood pressure regulation. Mol Pharmacol. 2005;67:912–922. doi: 10.1124/mol.104.007500. [DOI] [PubMed] [Google Scholar]
- Hrometz SL, Edelmann SE, McCune DF, Olges JR, Hadley RW, Perez DM, et al. Expression of multiple alpha 1-adrenoceptors on vascular smooth muscle: correlation with the regulation of Contraction. J Pharmacol Exp Ther. 1999;290:452–463. [PubMed] [Google Scholar]
- Kenny BA, Chalmers DH, Philpott PC, Naylor AM. Characterization of an α1D-adrenoceptor mediating the contractile response of rat aorta to noradrenaline. Br J Pharmacol. 1995;115:981–986. doi: 10.1111/j.1476-5381.1995.tb15907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potent alpha 1-adrenergic receptor agonist, selective for the alpha 1A receptor subtype. J Pharmacol Exp Ther. 1995;274:97–103. [PubMed] [Google Scholar]
- Leonardi A, Heible JP, Guarneri L, Naselsky DP, Poggesi E, Sironi G, et al. Pharmacological characterization of the uroselective alpha-1 antagonist Rec 15/2739 (SB 216469): role of the alpha-1L adrenoceptor in tissue selectivity, part I. J Pharmacol Exp Ther. 1997;281:1272–1283. [PubMed] [Google Scholar]
- McCune DF, Edelmann SE, Olges JR, Post GR, Waldrop BA, Waugh DJJ, et al. Regulation of the cellular localization and signaling properties of the alpha 1B- and alpha 1D-adrenoceptors by agonists and inverse agonists. Mol Pharmacol. 2000;57:659–666. doi: 10.1124/mol.57.4.659. [DOI] [PubMed] [Google Scholar]
- McGrath I, Deighan C, Briones A, Malekzadeh-Shafaroudi M, McBride M, Adler J, et al. New aspects of vascular remodelling: the involvement of all vascular cell types. Exp Physiol. 2005;90:469–475. doi: 10.1113/expphysiol.2005.030130. [DOI] [PubMed] [Google Scholar]
- Mackenzie JF, Daly CJ, Pediani JD, McGrath JC. Quantitative imaging in live human cells reveals intracellular alpha 1-adrenoceptor ligand-binding sites. J Pharmacol Exp Ther. 2000;294:434–443. [PubMed] [Google Scholar]
- Martinez-Salas SG, Campos-Peralta JM, Pares-Hipolito J, Gallardo-Ortiz IA, Ibarra M, Villalobos-Molina R. Alpha-1A adrenoceptors predominate in the control of blood pressure in mouse mesenteric vascular bed. Auton Autacoid Pharmacol. 2007;27:137–142. doi: 10.1111/j.1474-8673.2007.00403.x. [DOI] [PubMed] [Google Scholar]
- Meyer MD, Altenbach RJ, Hancock AA, Buckner SA, Knepper SM, Kerwin JF. Synthesis and in vitro characterization of N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulfonamide and its enantiomers: a novel selective alpha-1a receptor agonist. J Med Chem. 1996;39:4116–4119. doi: 10.1021/jm960354u. [DOI] [PubMed] [Google Scholar]
- Michel MC, Goepel M. Differential alpha-1 adrenoceptor labeling by [3H]-prazosin and [3H]-tamsulosin. Eur J Pharmacol. 1998;342:85–92. doi: 10.1016/s0014-2999(97)01419-2. [DOI] [PubMed] [Google Scholar]
- Miquel RM, Segura V, Ali Z, D'Ocon P, McGrath JC, Daly CJ. 3-D image analysis of fluorescent drug binding. Mol Imaging. 2005;4:1–13. doi: 10.1162/15353500200504172. [DOI] [PubMed] [Google Scholar]
- Morris DP, Price RR, Smith MP, Lei B, Schwinn DA. Cellular trafficking of human {alpha}1a-adrenergic receptors is continuous and primarily agonist-independent. Mol Pharmacol. 2004;66:843–854. doi: 10.1124/mol.104.000430. [DOI] [PubMed] [Google Scholar]
- Muramatsu I, Ohmura T, Kigoshi S, Hashimoto S, Oshita M. Pharmacological subclassification of α1-adrenoceptors in vascular smooth muscle. Br J Pharmacol. 1990;99:197–201. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu I, Morishima S, Suzuki F, Yoshiki H, Anisuzzaman ASM, Tanaka T, et al. Idenitifcation of α1L-adrenoceptor in mice and its abolition by α1A-adrenoceptor gene knockout. Br J Pharmacol. 2008;155:1224–1234. doi: 10.1038/bjp.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pediani J, Mackenzie JF, Heeley RP, Daly CJ, McGrath JC. Single-cell recombinant pharmacology: bovine alpha-1a adrenoceptors in rat-1 fibroblasts release intracellular calcium, display subtype-characteristic agonism and antagonism and exhibit an antagonist-reversible inverse concentration-response phase. J Pharmacol Exp Ther. 2000;293:895. [PubMed] [Google Scholar]
- Pediani JD, Colston JF, Caldwell D, Milligan G, Daly CJ, McGrath JC. Beta-arrestin-dependent spontaneous alpha-1a-adrenoceptor endocytosis causes intracellular transportation of alpha-blockers via recycling compartments. Mol Pharmacol. 2005;67:1004. doi: 10.1124/mol.104.008417. [DOI] [PubMed] [Google Scholar]
- Peng H, Matchkov V, Ivarsen A, Aalkjar C, Nilsson H. Hypothesis for the initiation of vasomotion. Circ Res. 2001;88:810–815. doi: 10.1161/hh0801.089603. [DOI] [PubMed] [Google Scholar]
- Piascik MT, Hrometz SL, Edelmann SE, Guarino RD, Hadley RW, Brown RD. Immunocytochemical localization of the alpha-1B adrenergic receptor and the contribution of this and the other subtypes to vascular smooth muscle contraction: analysis with selective ligands and antisense oligonucleotides. J Pharmacol Exp Ther. 1997;283:854–868. [PubMed] [Google Scholar]
- Rokosh DG, Simpson PC. Knockout of the alpha 1A/C-adrenergic receptor subtype: the alpha 1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci. 2002;99:9474–9479. doi: 10.1073/pnas.132552699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saussy DJ, Goetz AS, Queen KL, King HK, Lutz MW, Rimele TJ. Structure activity relationships of a series of buspirone analogs at alpha-1 adrenoceptors: further evidence that rat aorta alpha-1 adrenoceptors are of the alpha-1D-subtype. J Pharmacol Exp Ther. 1996;278:136–144. [PubMed] [Google Scholar]
- Stanasila L, Perez JB, Vogel H, Cotecchia S. Oligomerization of the {alpha}1a- and {alpha}1b-adrenergic receptor subtypes: potential implications in receptor internalization. J Biol Chem. 2003;278:40239–40251. doi: 10.1074/jbc.M306085200. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Hirasawa A, Hashimoto K, Tsujimoto G. Differences in the subcellular localization of α1-adrenoceptor subtypes can affect the subtype selectivity of drugs in a study with the fluorescent ligand BODIPY FL-prazosin. Life Sci. 2002;70:2113–2124. doi: 10.1016/s0024-3205(01)01533-8. [DOI] [PubMed] [Google Scholar]
- Tanoue A, Koba M, Miyawaki S, Koshimizu TA, Hosoda C, Oshikawa S, et al. Role of the {alpha}1D-adrenegric receptor in the development of salt-induced hypertension. Hypertension. 2002a;40:101–106. doi: 10.1161/01.hyp.0000022062.70639.1c. [DOI] [PubMed] [Google Scholar]
- Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, et al. The {alpha}1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest. 2002b;109:765–775. doi: 10.1172/JCI14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa R, Guarneri L, Angelico P, Poggesi E, Taddei C, Sironi G, et al. Pharmacological characterization of the uroselective alpha-1 antagonist Rec 15/2739 (SB 216469): role of the alpha-1L adrenoceptor in tissue selectivity, part II. J Pharmacol Exp Ther. 1997;281:1284–1293. [PubMed] [Google Scholar]
- Turnbull L, McCloskey DT, O'Connell TD, Simpson PC, Baker AJ. Alpha 1-adrenergic receptor responses in alpha 1AB-AR knockout mouse hearts suggest the presence of alpha 1D-AR. Am J Physiol Heart Circ Physiol. 2003;284:H1104–H1109. doi: 10.1152/ajpheart.00441.2002. [DOI] [PubMed] [Google Scholar]
- Uberti M, Hall R, Minneman KP. Subtype-specific dimerization of alpha1-adrenoceptors: effects on receptor expression and pharmacological properties. Mol Pharmacol. 2003;64:1379–1390. doi: 10.1124/mol.64.6.1379. [DOI] [PubMed] [Google Scholar]
- Villalobos-Molina R, Ibarra M. [alpha]1-Adrenoceptors mediating contraction in arteries of normotensive and spontaneously hypertensive rats are of the [alpha]1D or [alpha]1A subtypes. Eur J Pharmacol. 1996;298:257–263. doi: 10.1016/0014-2999(95)00781-4. [DOI] [PubMed] [Google Scholar]
- Williams TJ, Blue DR, Daniels DV, Davis B, Elworthy T, Gever JR, et al. In vitro α1-adrenoceptor pharmacology of Ro 70-0004 and RS-100329, novel α1A-adrenoceptor selective antagonists. Br J Pharmacol. 1999;127:252–258. doi: 10.1038/sj.bjp.0702541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshio R, Taniguchi T, Itoh H, Muramatsu I. Affinity of serotonin receptor antagonists and agonists to recombinant and native alpha1-adrenoceptor subtypes. Jpn J Pharmacol. 2001;86:189–195. doi: 10.1254/jjp.86.189. [DOI] [PubMed] [Google Scholar]
- Zacharia J, Hillier C, MacDonald A. 1-Adrenoceptor subtypes involved in vasoconstrictor responses to exogenous and neurally released noradrenaline in rat femoral resistance arteries. Br J Pharmacol. 2004;141:915–924. doi: 10.1038/sj.bjp.0705690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharia J, Hillier C, Tanoue A, Tsujimoto G, Daly CJ, McGrath JC, et al. Evidence for involvement of α1D-adrenoceptors in contraction of femoral resistance arteries using knockout mice. Br J Pharmacol. 2005;146:942–951. doi: 10.1038/sj.bjp.0706395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou l, Vargas HM. Vascular alpha 1D-adrenoceptors have a role in the pressor response to phenylephrine in the pithed rat. Eur J Pharmacol. 1996;305:173–176. doi: 10.1016/0014-2999(96)00229-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.