

Here, the crystal structure of adenylosuccinate lyase from Escherichia coli was determined to 1.9 Å resolution.
Keywords: adenylosuccinate lyase, ASL, PurB, purine biosynthesis
Abstract
Adenylosuccinate lyase (ASL) is an enzyme from the purine-biosynthetic pathway that catalyzes the cleavage of 5-aminoimidazole-4-(N-succinylcarboxamide) ribonucleotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and fumarate. ASL is also responsible for the conversion of succinyladenosine monophosphate (SAMP) to adenosine monophosphate (AMP) and fumarate. Here, the crystal structure of adenylosuccinate lyase from Escherichia coli was determined to 1.9 Å resolution. The enzyme adopts a substrate-bound conformation as a result of the presence of two phosphate ions bound in the active site. Comparison with previously solved structures of the apoenzyme and an SAMP-bound H171A mutant reveals a conformational change at His171 associated with substrate binding and confirms the role of this residue as a catalytic acid.
1. Introduction
Adenylosuccinate lyase (ASL) is an enzyme that catalyzes two distinct steps in de novo purine biosynthesis. One involves the cleavage of 5-aminoimidazole-4-(N-succinylcarboxamide) ribonucleotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and fumarate. ASL also carries out the chemically similar cleavage of succinyladenosine monophosphate (SAMP) to adenosine monophosphate (AMP) and fumarate in the final step of de novo AMP synthesis (Ratner, 1972 ▶). Together with the reactions catalyzed by adenylosuccinate synthetase and AMP deaminase, the latter reaction is a part of the purine pathway that regulates the levels of free AMP in the cell. The importance of ASL is emphasized by its conservation across all kingdoms of life and the existence of ASL-associated diseases in humans. ASL deficiencies arising from point mutations in the ASL gene result in autism, muscle wasting and epilepsy (Stone et al., 1992 ▶; Verginelli et al., 1998 ▶; Maaswinkel-Mooij et al., 1997 ▶).
ASL is a member of the fumarase/aspartase superfamily of enzymes, which also includes argininosuccinate lyase, aspartase and class II fumarase. Despite having low sequence similarity, the proteins have similar three-dimensional structures, catalyze β-elimination reactions and yield fumarate as one of the products (Simpson et al., 1994 ▶; Weaver et al., 1995 ▶; Turner et al., 1997 ▶; Shi et al., 1997 ▶; Toth & Yeates, 2000 ▶; Yang et al., 2004 ▶). Structurally, ASL forms a homotetramer of approximately 200 kDa in which each of the four equivalent catalytic sites in the protein is formed by a combination of conserved sequences from three different subunits. The catalytic reaction proceeds via a general acid–base mechanism in which the Cβ proton of the substrate is abstracted by the general base, yielding a carbanion intermediate (Porter et al., 1983 ▶; Bulusu et al., 2009 ▶). This step is followed by proton donation by a catalytic acid, resulting in C—N bond cleavage and product release (Hanson & Havir, 1972 ▶). Recent studies on Plasmodium falciparum ASL showed that the C—N bond cleavage is the rate-limiting step (Bulusu et al., 2009 ▶). Earlier studies of Bacillus subtilis ASL implicated the conserved residue His68 (His91 in Escherichia coli ASL) and residue His141 (His171 in E. coli ASL) as the catalytic acid and base, respectively (Lee et al., 1997 ▶, 1998 ▶). Recent structures of the E. coli ASL H171A and H171N mutants in complex with substrate (SAMP) and reaction products (AMP and fumarate), respectively, provided a significant advance in the understanding of the catalytic mechanism (Tsai et al., 2007 ▶). These structures showed that His91 is too far from the proton to be abstracted and seems to be involved in succinyl-group binding rather than catalytic activity. Instead, the invariantly conserved Ser295 appears to function as a catalytic base owing to its proximity to Cβ of the SAMP succinyl group. His171 is likely to be a catalytic acid, which is in agreement with results showing a severe or complete loss of catalytic activity upon mutagenesis of this residue in the E. coli (Tsai et al., 2007 ▶) and B. subtilis enzymes (Lee et al., 1999 ▶; Brosius & Colman, 2002 ▶). The position of the His171 side chain in the substrate- and product-bound proteins cannot be observed in the structures of the mutant enzymes, which may also undergo local conformational distortions. In the structure of the E. coli apoenzyme, this residue adopts a catalytically inactive position and would require a conformational rearrangement upon substrate binding in order to act as a catalytic acid (Tsai et al., 2007 ▶).
Here, we present the crystal structure of ASL from E. coli at 1.9 Å resolution. The catalytic loop, comprising residues 287–303, is ordered as a result of the binding of two phosphate ions in the active site. The position of the side chain of His171 reveals that it functions as a catalytic acid via a charge-relay interaction with a glutamate residue, Glu308, that is similarly conserved across the ASL superfamily.
2. Materials and methods
2.1. Cloning, protein expression and purification
ASL from E. coli (residues 2–456) was cloned into a derivative of pET15b vector (Amersham-Pharmacia) modified by introducing the TEV protease-cleavage site and expressed in E. coli BL21 (DE3) in rich (LB) medium fused with an N-terminal His tag (MGSSHHHHHHHHDYDIPTTENLYFQGS). The fusion protein was purified by anion-exchange chromatography using a Q-Sepharose column followed by size-exclusion chromatography.
2.2. Crystallization, data collection and processing
Crystallization conditions were identified utilizing hanging-drop vapour diffusion with the Classics II crystallization suite (Qiagen). The best crystals were obtained by equilibrating a 1.0 µl drop of ASL (10 mg ml−1) in 10 mM HEPES pH 7.0, 50 mM NaCl mixed with 1.0 µl reservoir solution containing 1.4 M sodium/potassium phosphate pH 7.5 and 4%(v/v) glycerol. Crystals grew in 3–10 d at 295 K. For data collection, the crystals were picked up in a nylon loop and flash-cooled in an N2 cold stream. The solution for cryoprotection contained the reservoir solution with the addition of 15%(v/v) glycerol. Diffraction data from a single crystal of ASL were collected using an ADSC Quantum-210 CCD detector (Area Detector Systems Corp.) on beamline A1 at the Cornell High-Energy Synchrotron Source (CHESS; Table 1 ▶). The crystals contained one ASL molecule in the asymmetric unit, corresponding to a V M of 2.66 Å3 Da−1 and a solvent content of 53.8% (Matthews, 1968 ▶). Data processing and scaling were performed with HKL-2000 (Otwinowski & Minor, 1997 ▶).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray wavelength (Å) | 0.9700 |
| Space group | I222 |
| Unit-cell parameters (Å) | a = 77.32, b = 100.26, c = 150.10 |
| Resolution range (Å) | 50.0–1.90 (1.97–1.90) |
| Unique reflections | 40348 |
| Completeness (%) | 91.9 (84.7) |
| I/σ(I) | 16.0 (4.5) |
| Rmerge† | 0.070 (0.227) |
| Model refinement | |
| Rwork/Rfree‡ | 0.170/0.214 |
| No. of non-H atoms/average B factor (Å2) | |
| Protein | 3745/27.61 |
| Waters | 317/38.92 |
| Phosphate ions | 10/36.11 |
| Sodium ions | 2/16.61 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths (Å) | 0.014 |
| Bond angles (°) | 1.47 |
| Ramachandran plot | |
| Most favourable (%) | 91.5 |
| Allowed (%) | 8.1 |
| Generously allowed (%) | 0.5 |
| Disallowed (%) | 0.0 |
R
merge = 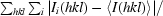
 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl,  is the sum over all reflections and
is the sum over all reflections and 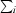 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
R = 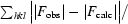
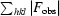 , where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
, where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
2.3. Structure determination and refinement
The structure was determined by molecular replacement with Phaser (Read, 2001 ▶) using the coordinates of the ASL apoenzyme from E. coli (PDB entry 2pts). The initial model obtained from Phaser was completed and adjusted with the program Coot (Emsley & Cowtan, 2004 ▶) and was improved by several cycles of refinement using the program REFMAC5.2 (Murshudov et al., 1999 ▶) and model refitting. The refinement statistics are given in Table 1 ▶. The final model has good stereochemistry according to the program PROCHECK (Laskowski et al., 1993 ▶). Only two residues (Ala120 and Trp333) were in the generously allowed region of the Ramachandran plot, but the excellent quality of the electron-density map allowed reliable modelling of these residues. The coordinates and structure factors have been deposited in the RCSB Protein Data Bank (PDB code 3gzh).
3. Results and discussion
ASL from E. coli was crystallized in space group I222 and its structure was determined at 1.9 Å resolution by molecular replacement using PDB entry 2pts. There is a single molecule in the asymmetric unit, which generates the homotetramer via crystallographic symmetry (Fig. 1 ▶). The structure of the individual subunit contained three distinct α-helical domains. The N- and C-terminal domains are relatively compact, while the middle domain consists of a bundle of five long α-helices which account for the majority of the intermolecular contacts. Of the 482 residues in the His-tag fusion protein, only 13 N-terminal residues from the cloning linker were missing from the electron-density map. 14 of the His-tag residues are ordered, with some of them participating in crystal contacts between tetramers. Unexpectedly, the electron-density map also revealed the long catalytic loop (residues 287–303), which was not observed in the previous apoenzyme structure. Analysis of the electron-density maps also identified two metal ions, which were both coordinated to negatively charged groups with square-pyramidal geometry. Based on the crystallization-solution composition and their B factors after refinement, they were modelled as sodium ions. One of them caps a helix in the N-terminal domain and is coordinated by the carbonyls of Leu47, Ala48, His50, Ile53 and Val56. The other sodium ion binds at the interface between the ASL subunits and is coordinated by the side chains of Asp270 and Asp274 of one subunit and the carbonyls of Ser331 and Trp333 of an adjacent subunit. The fifth coordination site is an ordered water molecule. Both sodium ions are relatively far from the ASL catalytic site and are unlikely to influence catalysis.
Figure 1.

(a) Crystal structure of a single subunit of adenylosuccinate lyase (ASL) from E. coli. The protein is coloured from the N-terminus (blue) to the C-terminus (red). The N- and C-termini and the catalytic loop are labelled. The phosphate (magenta) and sodium (grey) ions are shown as space-filling representations. (b) Structure of the tetramer of E. coli ASL. Two orthogonal views are shown. Figures were produced with PyMOL (http://pymol.sourceforge.net/).
Superposition of our structure with the previously determined apoenzyme structure (PDB entry 2pts) results in a relatively high root-mean-square deviation (r.m.s.d.) of 0.89 Å and in particular shows significant differences in the active site (Fig. 2 ▶ a). The structural rearrangements are caused by two bound phosphate ions. One of them occupies a genuine phosphate-binding site that overlays perfectly with the phosphate group of SAMP bound to ASL (Tsai et al., 2007 ▶). The second phosphate ion has two O atoms that mimic one of the carboxylates of SAMP. The latter phosphate is responsible for structuring the catalytic loop and forms hydrogen bonds to the side chains of Ser295 and Ser296.
Figure 2.

E. coli ASL adopts a substrate-bound conformation with bound phosphates. (a) Overlay of the phosphate-bound enzyme (green) with the unliganded enzyme (PDB code 2pts; yellow). Hydrogen bonds between the phosphate ion and the side chains of Ser295 and Ser296 contribute to the stability of the catalytic loop and are shown as dotted lines. Most of the loop is absent in the apoenzyme structure. In the phosphate-bound enzyme structure the additional catalytically important residues, His171 and Glu308 are labelled. The side chain of Glu308 is hidden from view. (b) Overlay of the phosphate-bound enzyme (green) with the substrate-bound H171A mutant (PDB code 2ptr; magenta). Phosphate ions (purple) and SAMP (light grey) are shown in stick representation. Ser295, Ser296 and SAMP are labelled.
The overlay of the phosphate-bound and SAMP-bound structures shows their striking similarity, with an overall r.m.s.d. of 0.42 Å using Cα atoms including the catalytic loop (Fig. 2 ▶ b). Interestingly, the structures of duck delta 1 crystallin (Sampaleanu et al., 2001 ▶) and argininosuccinate lyase (Bhaumik et al., 2004 ▶), two other members of the aspartase/fumarase superfamily, also showed folding of their catalytic loops upon binding sulfate and phosphate ions. Despite the very limited number of contacts between the catalytic loop and the bound phosphate ion, the loop adopts a nearly identical conformation throughout its length compared with the substrate- and product-bound ASL structures. Unsurprisingly, the sequence of the loop is highly conserved in the ASL family. Notably, the active site of our ASL structure is in a catalytically competent state, with the proper orientation of Ser295, which has been proposed to function as a catalytic base (Tsai et al., 2007 ▶). Mutagenesis of corresponding serines in the B. subtilis and human enzymes has recently shown that this residue is essential for catalysis (Sivendran & Colman, 2008 ▶). Interestingly, the side chain of His171 is perfectly positioned to act as a catalytic acid by donating a proton to the N6 atom of the targeted C—N bond (Fig. 3 ▶ a). Previously, the unliganded ASL structure showed this residue in an inactive conformation requiring a conformational change for its activation (Tsai et al., 2007 ▶). The conformation of the His171 side chain in the phosphate-bound ASL is fixed by a salt bridge between the side chain of the invariant Glu308 and Nδ1 of His171. The role of this glutamate is to prime His171 for function as a catalytic acid; this charge-relay interaction is conserved in other superfamily members (Weaver et al., 1995 ▶; Turner et al., 1997 ▶; Chakraborty et al., 1999 ▶; Brosius & Colman, 2002 ▶).
Figure 3.

Involvement of His171 in catalysis. (a) Overlay of the active sites of phosphate-bound (green), unliganded (yellow) and SAMP-bound H171A mutant (magenta) E. coli ASL. His171 of the phosphate-bound structure is perfectly positioned to donate a proton to the N6 atom of SAMP, supported by a charge-relay interaction with the side chain of Glu308. The side chains of His171 and Glu308 are shown and labelled. (b) Overlay of the phosphate-bound (green) and product-bound H171N mutant (PDB code 2ptq; cyan). The side chain of His171 in the phosphate-bound structure prevents the binding of AMP owing to a steric clash (1.9 Å) between the N6 atom of AMP and the histidine ring.
Comparison of substrate- and product-bound structures shows that the nucleotide rotates slightly following cleavage in order to fit into the active site together with fumarate (Tsai et al., 2007 ▶). Overlay of the product-bound and phosphate-bound ASL structures reveals a potential steric clash between the side chain of His171 and AMP (Fig. 3 ▶ b). AMP and fumarate have been observed in complex with the H171N mutant of E. coli ASL (Tsai et al., 2007 ▶). The shorter side chain of asparagine allows the accommodation of both products in the active site, whereas a histidine in this position requires a minor structural rearrangement for simultaneous binding of AMP and fumarate.
A major conformational change resulting in active-site opening would be the translational movement of the N-terminal domain away from AMP and opening of the catalytic loop to release fumarate. The former would be likely to require higher energy, favouring the initial release of fumarate, which is in agreement with earlier studies that point to a two-step product release in which fumarate is followed by AMP (Bridger & Cohen, 1968 ▶).
Supplementary Material
PDB reference: PurB, 3gzh, r3gzhsf
Acknowledgments
This work was funded by a Canadian Institutes of Health Research (CIHR) Genomics grant GSP-48370 for the Montreal-Kingston Bacterial Structural Genomics Initiative (M. Cygler). KG is a Chercheur National of the Fonds de la Recherche en Santé du Québec (FRSQ). Data acquisition at the Macromolecular Diffraction (MacCHESS) facility at the Cornell High Energy Synchrotron Source (CHESS) was supported by National Science Foundation award DMR 0225180 and National Institutes of Health award RR-01646.
References
- Bhaumik, P., Koski, M. K., Bergmann, U. & Wierenga, R. K. (2004). Acta Cryst. D60, 1964–1970. [DOI] [PubMed]
- Bridger, W. A. & Cohen, L. H. (1968). J. Biol. Chem.243, 644–650. [PubMed]
- Brosius, J. L. & Colman, R. F. (2002). Biochemistry, 41, 2217–2226. [DOI] [PubMed]
- Bulusu, V., Srinivasan, B., Bopanna, M. P. & Balaram, H. (2009). Biochim. Biophys. Acta, 1794, 642–654. [DOI] [PubMed]
- Chakraborty, A. R., Davidson, A. & Howell, P. L. (1999). Biochemistry, 38, 2435–2443. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Hanson, K. R. & Havir, E. A. (1972). The Enzymes, edited by P. D. Boyer, Vol. 7, pp. 75–166. New York: Academic Press.
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lee, T. T., Worby, C., Bao, Z. Q., Dixon, J. E. & Colman, R. F. (1998). Biochemistry, 37, 8481–8489. [DOI] [PubMed]
- Lee, T. T., Worby, C., Bao, Z. Q., Dixon, J. E. & Colman, R. F. (1999). Biochemistry, 38, 22–32. [DOI] [PubMed]
- Lee, T. T., Worby, C., Dixon, J. E. & Colman, R. F. (1997). J. Biol. Chem.272, 458–465. [PubMed]
- Maaswinkel-Mooij, P. D., Laan, L. A., Onkenhout, W., Brouwer, O. F., Jaeken, J. & Poorthuis, B. J. (1997). J. Inherit. Metab. Dis.20, 606–607. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Porter, D. J., Rudie, N. G. & Bright, H. J. (1983). Arch. Biochem. Biophys.225, 157–163. [DOI] [PubMed]
- Ratner, S. (1972). The Enzymes, edited by P. D. Boyer, Vol. 7, pp. 167–197. New York: Academic Press.
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Sampaleanu, L. M., Vallée, F., Slingsby, C. & Howell, P. L. (2001). Biochemistry, 40, 2732–2742. [DOI] [PubMed]
- Shi, W., Dunbar, J., Jayasekera, M. M., Viola, R. E. & Farber, G. K. (1997). Biochemistry, 36, 9136–9144. [DOI] [PubMed]
- Simpson, A., Bateman, O., Driessen, H., Lindley, P., Moss, D., Mylvaganam, S., Narebor, E. & Slingsby, C. (1994). Nature Struct. Biol.1, 724–734. [DOI] [PubMed]
- Sivendran, S. & Colman, R. F. (2008). Protein Sci.17, 1162–1174. [DOI] [PMC free article] [PubMed]
- Stone, R. L., Aimi, J., Barshop, B. A., Jaeken, J., Van den Berghe, G., Zalkin, H. & Dixon, J. E. (1992). Nature Genet.1, 59–63. [DOI] [PubMed]
- Toth, E. A. & Yeates, T. O. (2000). Structure, 8, 163–174. [DOI] [PubMed]
- Tsai, M., Koo, J., Yip, P., Colman, R. F., Segall, M. L. & Howell, P. L. (2007). J. Mol. Biol.370, 541–554. [DOI] [PMC free article] [PubMed]
- Turner, M. A., Simpson, A., McInnes, R. R. & Howell, P. L. (1997). Proc. Natl Acad. Sci. USA, 94, 9063–9068. [DOI] [PMC free article] [PubMed]
- Verginelli, D., Luckow, B., Crifo, C., Salerno, C. & Gross, M. (1998). Biochim. Biophys. Acta, 1406, 81–84. [DOI] [PubMed]
- Weaver, T. M., Levitt, D. G., Donnelly, M. I., Stevens, P. P. & Banaszak, L. J. (1995). Nature Struct. Biol.2, 654–662. [DOI] [PubMed]
- Yang, J., Wang, Y., Woolridge, E. M., Arora, V., Petsko, G. A., Kozarich, J. W. & Ringe, D. (2004). Biochemistry, 43, 10424–10434. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: PurB, 3gzh, r3gzhsf
PDB reference: PurB, 3gzh, r3gzhsf