

Abstract
In the last decade, two tools, one drawn from information theory and the other from artificial neural networks, have proven particularly useful in many different areas of sequence analysis. The work presented herein indicates that these two approaches can be joined in a general fashion to produce a very powerful search engine that is capable of locating members of a given nucleic acid sequence family in either local or global sequence searches. This program can, in turn, be queried for its definition of the motif under investigation, ranking each base in context for its contribution to membership in the motif family. In principle, the method used can be applied to any binding motif, including both DNA and RNA sequence families, given sufficient family size.
Gatlin (1) first recognized that the Shannon expression for string entropy might prove useful in sequence analysis. This function is a statistical average for the distribution of possible characters at a particular position in a message. [Although it shares the form of the Gibbs–Boltzman entropy function
 |
1 |
it is independently derived and nonisomorphic with that function (2, 3).] Gatlin insightfully proposed that this function, originally developed to assay the fidelity with which strings could be transmitted in noisy communication channels, was also appropriate for the analogous transmission of string information represented in the central dogma of genetics. Schneider et al. (4) subsequently developed a redundancy index (RI), based on this function, to profile a given family of DNA-binding-site sequences. This index measured the reduction in Shannon entropy relative to the background DNA, represented in the strings of the sequences belonging to a particular motif.
Suppose we consider a simple example, a motif 3 bases long with five known members of the family. These could be: ATG, CTG, GTC, ATA, and ACA. For the first position, the pi values are the observed base frequencies
 |
2 |
The Shannon entropy for this position would be:
 |
3 |
or 1.13 bits (a bit being the unit of information in a decision between two equiprobable alternatives). For genomic base ratios in Escherichia coli, A = C = G = T, the background value for any position would be
 |
4 |
or 2 bits. The entropy reduction, indicating the redundancy or conservation above background, would be the difference or 0.87 bits. For the second and third positions in the example, the corresponding difference values would be 1.28 and 1.03; and the sum across all three positions of the motif would be 3.18 bits. In information theory terms, this would be the loss of Shannon entropy in the fixation of the motif as represented in the current sample. The degree of conservation at a particular position within the motif is equated with the functional importance of that position; in our example the order of importance would be position 2, then position 3, and then position 1. Thus it is the motif that is characterized in this analysis, not particular members of the family.
One would also like to be able to rank individual members of the family with respect to functionality. Somewhat later, a ranking function (5), relying solely on sequence information for a functional ordering of members of a motif, was developed based on two factors, one to measure the importance within the motif of each position (the Schneider redundancy index above) and a second to measure how well that position was filled with respect to a particular sequence, best assessed at each position with the Berg–von Hippel (BvH) function (6). Factoring the two was suggested by the need to improve the performance of the Berg-von Hippel function on ‘spacer positions’ within a motif (5, 6). The BvH function is
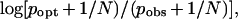 |
5 |
where popt is the frequency of the consensus base (e.g., 0.6 for position 1 in the example), pobs is the frequency found in the motif family for the first base in the sequence being examined (say cytidine or C, pobs = 0.2), N is the number of known family members, 5. Accordingly, the overall index for a C in the first position would then be 0.87 ∗ 0.30 or 0.26. This index would also be computed for the other positions and summed over the three positions; the higher the total index, the worse the fit of the query sequence to the family. For the five sequences of our example, the totals would be 0, 0.26, 0.26, 0.14, and 0.65, and the ranking would be 1 best, 4, 2 and 3, and 5 worst.
In parallel with these developments, neural networks, first perceptrons (7) and later back-propagation neural networks (8), were proving to be effective in searching for highly divergent binding sites. Artificial neural networks are computer programs that are designed to learn by example (an input-expected output pair) to fit a given computer input to a desired output by means of a number of weighted connections between the input and the output. During training, these weighted connections are continually adjusted until the program can give the correct output for most or all of the input examples, at which point the connection strengths are permanently fixed. If the training has been sufficiently broad, the program can then be used to evaluate additional new input data. The training input to these networks, in the case at hand, would most often be examples of the sequences of interest, i.e., sequences exemplifying a particular binding motif, contrasted with negative examples (nonmembers of the same size) drawn from random sequence. The output would be a yes or no decision on membership. In still other cases, the input might be only the most important parts of the sequence motif or possibly indices based on some significant aspect of the sequence.
The work presented below attempts to meld these two lines of research into a single general method. The method again uses back-propagation neural networks as the search engine, but in contrast to my earlier work (8, 9), uses the above-mentioned ranking function evaluation of the sequences as input in place of the sequences themselves. An additional ranking function, based solely on Shannon entropy evaluations, is also used as an alternative source of input. When earlier problems are revisited with this method, the false positive levels are reduced between 4- and 10-fold from what was then the state of the art. In practice, this means that in many searches the target sequence(s) can be located without false positives. With the use of a numerical differentiation of the weight matrix (see below), the neural network’s definition of the motif (i.e., the weight it places on any particular base at any position in making the correct classification) can be determined.
METHODS
The product of the RI and BvH, computed at each position of a sequence from a given motif, has been shown previously to provide a strong correlation with the functionality of that sequence as measured by DNA binding assays in vitro (5), being able to predict relative affinities of motif family members with correlations as high as 0.999. Both factors in this ranking function treat each position within the sequence as being independent of the other bases in the site, an assumption that seems fairly well met, on average, in the few cases studied to date. According to information theory (2), the greatest diversity of expression is made possible by an alphabet in which each letter is equiprobable and acts independently. Diversity provides the regulatory range in the nucleic acid binding sequences of regulatory proteins. (The assumption of positional independence would be less palatable in the case of peptide motifs where the joint occurrence of several amino acids may be correlated in the establishment of an active site and is, also, clearly abridged in certain specific cases of DNA binding sites.)
There are a few caveats associated with the use of this index. One is that it cannot give a quantitative assessment of rare mutant sequences. The base mutation would probably be represented in the prototype group (the current known sequence members of the motif that are to serve as the training definition of the motif) by a frequency of 0; another rare mutation at a different position in another sequence might also generate a frequency of 0. Both of these would get bad rankings, but in the absence of real frequency data for them [if added to the group, the assigned frequency would be 1/(N + 1), whereas in nature the real frequency might be 10−6 or 10−8], they cannot be quantitatively ranked. A second caveat has to do with symmetric or dyad repeat sequences. Investigators frequently turn to the half-sequence in analyzing such sites. While this is a legitimate manipulation for increasing the sample size in determining the half-sequence consensus, it results in an information loss in other applications, such as ranking any sequence that is not perfectly symmetric. This information loss due to averaging is often sufficient to destroy strong correlations that would be seen by using the entire sequences as opposed to disjoint half-sequences.
RI ∗ BvH, the ranking function computed at each position of a sequence of interest, is written
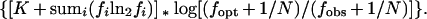 |
6 |
K in this function is the background entropy corrected for sample size, approximately 2 for E. coli (4), as was calculated in the example above. The frequencies, fi, are determined as in the example above from the aligned sequences of the prototype group for the given motif; the relative frequencies for the occurrence of each of the four bases are determined separately for each position of the motif. Fopt is the frequency found for the consensus base at a given position, and fobs is the frequency at which the base in the sequence of interest is found to occur in the same position of the prototype group.
A computer program was written that takes sequence as input, evaluates this function at each position within a window corresponding to the size of the motif of interest by using the prototype group’s frequencies, divides the range of resultant index values into six qualitative levels from “very good” to “very bad” (attempts with 10 levels yielded no better performance), and codes the result in binary form suitable as input for a neural network. (There has been some discussion whether fine or coarse coding of the neural network input is more effective. In the coarse-coded example, level 5 might be written “0 1 0 0 0 0,” whereas the fine-coded equivalent would be “1 0 1”; in binary, each position moving right to left represents a higher power of “2” beginning with 0 power on the right; 5 is a 2 squared plus a 2 to the 0 power. Fine coding is more compact, but it is also ambiguous, e.g., the rightmost position is “X X 1” when the level expressed is 1, 3, 5, or 7; this corresponds to a shared active input neuron among all of these inputs. Both forms of coding are used in examples below and both perform well.) The program then sums the function over all positions of the window, grades that result on four levels (attempts with seven levels did not improve performance), and coarsely codes that result. For example, a promoter sequence of 58 bases would be translated into 58 ∗ 6 + 4, or 352 characters, of which 59 would be ones, representing active input neurons in the case of coarse coding, the remainder being zeros. Let us consider our earlier example. Suppose we were to code our first sequence ATG for input to a network being trained for this motif. The range of index values could be: >2 = 1 0 0 0 0 0; >1 = 0 1 0 0 0 0; >0.5 = 0 0 1 0 0 0; >0.3 = 0 0 0 1 0 0; >0.15 = 0 0 0 0 1 0; ≤0.15 = 0 0 0 0 0 1. In the example, ATG has the consensus base for each position of the motif and thus scores 0 for each position; with six-level coding, this sequence would be represented as 0 0 0 0 0 1 0 0 0 0 0 1 0 0 0 0 0 1 in the input file to the network. In that it is the best possible sequence, its total value for the three positions would also be the best code 0 0 0 1; so the complete entry for this 3-base sequence would be 0 0 0 0 0 1 0 0 0 0 0 1 0 0 0 0 0 1 0 0 0 1, requiring 22 input neurons, and the network would be tasked to associate this input with an output of 1 0 for “yes”, confirming motif membership. A random sequence might read CGA with index values of 0.26, 0.90, and 0.14 and a coding from above of 0 0 0 1 0 0 0 1 0 0 0 0 0 0 0 0 0 1 0 1 0 0 (second worst total level) and this would be associated during training with an output of 0 1 for “no,” not a member of the motif family.
Note that all possible values for the ranking index can be determined and stored as soon as the prototype group is entered. There are only four possibilities for each position of the window. One of these is called each time a base and its position within the window are supplied until the window is exhausted. The program then offsets the window by one base and repeats the process until the sequence is exhausted. A similar program can convert discreet examples, one line at a time. (These programs and assistance in their use are available to investigators at moneill@umbc.edu). The output of these programs is used as the input file to train and test a back-propagation neural network. The network would have 6 ∗ (motif length) + 4 input neurons, 2 output neurons with 1 0 indicating a positive classification of the sequence and 0 1 indicating a negative classification.
Back-propagation networks, as opposed to earlier perceptrons, have an additional layer of neurons between the input and the output layers. These neurons sum and revalue the input from the first layer; the revaluation is nonlinear, using a sigmoid transfer function, output = 1/[1 + exp(−sum input)], and will output a value between zero and one to the connection to the output neuron(s). This middle layer makes it possible for these networks to fit nonlinear problems, which is to say that these networks can take context (cross-correlation in the input) into account in obtaining a solution. Various numbers of interneurons were tested for optimal performance, from 2 to 15, 8 being the final choice. Examples with fine coding, using 6 bits to generate 10 or 11 levels and producing a variable number of active input neurons per input base, are also included for comparison.
The Introduction mentions a new ranking function as a possible alternative to the RI ∗ BvH function. This second ranking function also calculates the RI− at each position for the prototype group as was done above. However, it then recalculates the same index after the sequence under evaluation has been added to the prototype group (RI+). As was the case above, it uses the redundancy index of the prototype group to assess the importance of that position to the motif and it uses the difference in the redundancy index, before and after the addition of the new sequence to the group, to evaluate how well that position has been filled in the new sequence. The function is therefore
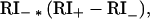 |
7 |
determined for each position and also summed over the entire sequence. This can again be fine or coarse coded for 352 input neurons in the subsequent neural network. Returning to our example, we have found RI− to be 0.87 for position 1, 1.28 for position 2, and 1.03 for position 3. Suppose the query sequence is CCG. Recalculating RI for each position with this sequence added to the original 5, we find RI+ is 0.55, 1.07, and 1.07 for position 1, 2, and 3, respectively, and the overall function values are 0.87 ∗ (−0.32), 1.28 ∗ (−0.21), and 1.03 ∗ (0.04) for a sum of −0.506. For this index, the higher the index the more likely that the query sequence is a member of the motif; negative values mean that the query sequence has fallen below the average for the members in the prototype group. The corresponding network input code might be 0 0 1 0 0 0 0 0 1 0 0 0 0 0 0 0 1 0 0 1 0 0, where first level on the right is best.
The promoter training and test files were drawn from the same DNA sequences used in earlier work (8, 9), 41 promoters of the 17-base-spacing class, 58 bases long, and 34 additional test promoters of the same class. That is, these 41 promoters constituted the prototype motif used in the evaluation and coding of all inputs to the networks. This list was deliberately not expanded to include hundreds of promoters to show what a prototype group in line with some of the other large families with relatively low RI values, such as the sites for E. coli catabolite receptor protein, can be expected to produce good results with this method. Investigators interested specifically in genomic searches might wish to use prototype groups of the maximum size possible. The training file itself consisted of the coded version of the training file used earlier (9) which included the 41 promoters permuted 1 base at a time in all nonessential positions as positives, the phage P22 ant double promoter-down mutants as negatives, and 10,833 prescreened random sequences as negatives. Prescreening, using the best earlier promoter-trained network, was necessary to remove the one or two bona fide promoter sequences, created at random, from the negative input set. Additional tests were run on plasmid pBR322, all 4,306 possible 58-base regions in each direction. Note that these networks only search for promoters of the 17-base-spacing class.
The operator training file was drawn from five of the six phage λ operator sites as positive examples (the same five were used to define the motif for coding purposes), reserving OL3 as a test site, plus 7,577 random sequences as negative examples; the test file in this case was drawn from the entire λ genomic sequence taken in both directions. Optimal training was generally achieved in approximately a single pass through the examples. Thus one could have a fully trained network within minutes of completing the construction of the training file. neuralware professional ii plus was used in this work, but the same result could be obtained with any standard back-propagation network program in the public domain.
In a trained network, the network’s definition of the problem is distributed in a nonlinear fashion over, possibly, thousands of weights. Generally, we cannot simply look at these weights and intuit the network’s operation except in a very qualitative sense. “Decompiling” a network describes the attempt to apportion the degree to which the correct output (classification) is dependent on each element of the input, on each base in our case. Numerical differentiation effects this decompiling by determining Δ output for a small Δ input change, one base at a time. Numerical differentiation of the weight matrix of a trained network was accomplished in each case in the context of a particular test sequence. These networks are not limited by an assumption that each base acts independently. The normal output value was obtained for the sequence. Since each base results in a single active input neuron, representing the index evaluation level in the case of coarse coding, each active input could be referred to a particular base in the sequence (with the exception of the final summation neuron). The weights (from the stored weight matrix of the trained network) between the single active input under consideration and the middle-layer neurons would be reduced by 5%, with all other weights unchanged and then the interneuron activations would be recalculated as would the sum and transform function at the output neuron. The new output value would be subtracted from the original output. (It is necessary to keep the precision of the output value in mind when performing these operations; the differences must be in significant figures.) This whole procedure would be repeated for each active input neuron in turn. A plot of the Δ value of the output versus the active input neurons (specific bases) shows the relative influence of each base on the reduction in the output. There is in this no assumption of linearity, nor are the transform functions limited to the linear regions of their ranges in making these calculations. Thus, if it happens that a trained network has the best operational definition of a particular motif, that definition can be quantitatively examined.
RESULTS
The first objective was to develop trained networks that would demonstrate an improved ability to classify DNA sequence with respect to the 17-base-spacing-class promoter motif, primarily with a goal of reducing the level of false positives. A number of neural networks were trained with a training file derived from the same group of sequences used in earlier work: included were 41 promoter sequences, singly permuted as described previously, as positive input; three sets of random sequences, prescreened to eliminate promoter-like sequences, as negative input; and the set of phage P22 ant promoters containing the complete set of double promoter-down mutations (10) described previously, as negative input. As described above, these sequences were evaluated at each position by a program; the coded evaluations contained the actual input of the training file in the place of the bases they represent. Typically, optimal training (average of 0.05% rms error between the actual network output and the desired output) occurred at approximately one pass through the complete set of training examples. Table 1 shows the results obtained with several distinct networks trained in this manner and tested on 34 previously unseen promoters and on sequences from pBR322 and λ.
Table 1.
Neural networks trained to recognize promoters of the 17-base-spacing class
| Network | Index | Index coding/levels | Prototype | No. test promoters >cutoff | Sequence† |
|---|---|---|---|---|---|
| NW1 (14.5) | RI ∗ BvH | Fine/11, 4 | 41 promoters 1× permuted (5,412 seq) | > 0.93 (28/34) | pBR >0.93 1, 4,135 pBR(c) >0.93 125, 1,2264,278 |
| NW2 (26) | RI ∗ BvH | Coarse/6, 4 | 41 promoters (41 seq) | >0.985 (28/34) | pBR >0.985 1, 4,135 pBR(c) >0.985 21, 125, 1226, 4278 |
| NW3 (21) | RI ∗ BvH | Coarse/6, 4 | 41 promoters (41 seq) | >0.96 (29/34) | pBR >0.96 1, 4135 pBR(c) >0.96 21, 125, 1226, 4278 |
| NW4 (192) | RI− ∗ (RI+ − RI−) | Fine/10, 0 | 41 promoters (41 seq) | >0.80 (29/34) | pBR >0.80 1, 4135 pBR(c) >0.80 125, 532, 1056, 1226, 4278 |
| NW5 (41.4) | RI− ∗ (RI+ − RI−) | Coarse/6,4 | 41 promoters (41 seq) | >0.77 (30/34) | pBR >0.77 1, 170, 3654, 4135 pBR(c) >0.77 125, 1056, 1226, 4278 |
| Average result | 29/34 | 1, 4135 | |||
| 85% TP | 125, 1226, 4278, 1FP | ||||
Numbers listed after the network in parentheses are the training iterations in thousands. NW3 had 15 middle-layer neurons; all others had 8. Learning rates and momentum values of the following networks: NW1, 0.5 and 0.2; NW2, 0.9 and 0.4; NW3, 0.6 and 0.4; NW4, 0.15 and 0.1; NW5, 0.6 and 0.4. Input neurons varied from 348 to 352. NW3 was tested on the first and last 16.4 kb of the λ r strand (the B region was omitted to avoid its many uncharted promoter sites) with performance virtually identical to that above for pBR322, 100% true positives (TP) and 0.024% false positives (FP). Underlined coordinants indicate known promoters.
pBR is the clockwise sequence of pBR322, beginning at position −5 to capture the tet promoter; pBR322(c) is the counterclockwise sequence beginning at the EcoRI site. The cutoff values for the minimum network output designating a promoter are listed for each network.
Table 1 shows five independently trained networks. In the first three networks, all sequence input was coded by the RI ∗ BvH function; in the last two networks, all sequence input was coded with the function RI− ∗ (RI+ − RI−). The coding was either fine, over 10 or 11 levels, with 0 or 4 levels for the sequence total (nets 1 and 4) or coarse, over 6 levels, with 4 levels for the total (nets 2, 3, and 5). The prototype group for all coding was 41 promoter sequences in nets 2, 3, 4, and 5; for net 1 the prototype group was collection of the same 41 promoters permuted at a single noncritical position each to produce 5,412 “promoters.” The cutoff values for the first output neuron on test sequences (above which membership in the motif was affirmed) varied with the degree of training, tending higher for longer training.
It can be seen that the recovery of true positives (known promoters) averages 85% or better and the level of false positives (sites not known to function as promoters but scoring above the cutoff value) is 0.025% or less. When polled (a technique that affords noise cancellation), these networks are in agreement in finding all of the promoters of the 17-base-spacing class in pBR322 and one false positive site, for an error rate of 0.01%. It should be noted that these false positive levels represent an upper limit in that an early transcript mapping study (11) indicated possible additional minor promoters in pBR322. The sequence of E. coli K12 (12) was also tested (data not shown), using NW3 in the forward direction and NW2 in the reverse direction, producing 2,746 and 2,640 hits, respectively, with the cutoffs of Table 1. Subject to assumptions about the relative frequency of 17-base-spacing-class promoters, these numbers may fit comfortably with those cited above for pBR322.
Although promoters constitute an important case, they are atypical with respect to the relatively large number of examples available even at the 41 sequence limit. The operators of phage λ may be more representative of the smaller sets that are represented in many binding-sequence motifs. Table 2 shows the results obtained in the first attempt with a network that was trained with a training file in which half the sequences were repeats of five of the operator sequences (missing OL3) and the other half was 7,577 random sequences as negative examples, all coarse-coded evaluations of the RI ∗ BvH function for each position of the sequence, graded in six levels and the total for the whole sequence, graded in four levels. Training to below the 0.05% rms error level was achieved in only 5,000 iterations. The λ genome was searched in both directions as the test of the network. All operator sites, including OL3, were correctly identified although the discrimination was high enough that each was found on only one strand, symmetry notwithstanding. There was a single other positive site at position 5,902 on the first strand (this site alone was identified on both strands). It should be noted that this additional positive site qualifies as a member of the operator motif. Using in vitro affinity projections for repressor (13), one finds the site at position 5,902 on a par with OR2; however, in the absence of cooperative effects, the intrinsic affinities of OR2 and OR3 are too low to be occupied by repressor in a mono-lysogen in vivo (14). Thus, though a legitimate member of the motif, the isolated site at position 5,902 is likely to be nonfunctional. Subject to this consideration, there were no false positives in a search of 97,000 bases.
Table 2.
Neural network trained to recognize λ operators
| Network | Index | Index coding/levels | Prototype | Test |
|---|---|---|---|---|
| NWOp (5) | RI ∗ (BvH) | Coarse/6, 4 | 5 operators (5 seq-OL3) >0.98 | λ l (48,485 seq) >0.98 5902, 35591, 35615, 35635, 37951, 37974 |
| λ r (48,485 seq) 37998 | ||||
| Result | 100% TP | |||
| 0% FP* |
See Discussion concerning sequence at position 5,902. Coordinates are given to the left edge of the site on the first strand. The underlined coordinates indicate known operator locations. NWOp had 106 input neurons, 8 middle-layer neurons, and 2 output neurons. The learning rate was 0.6 and the momentum was 0.4. The training file consisted of 2,000 copies of each of the five operator sequences plus 7,577 random sequences. OR2 and OR3 were flipped in orientation in the prototype group to maximize the redundancy index of that group; as a result, they appear to be located on the wrong strand. In all training cases, sequences were drawn from the training file in random order dictated by a random number generator. TP, true positives; FP, false positives.
Neural networks are often treated as “black boxes” that may produce useful answers but whose workings are veiled in that their solution is distributed over a large weight matrix (e.g., 5,328 weights), which, in turn, is used in a nonlinear fashion. To lift the veil, the first layer weights associated with a single active input neuron were slightly perturbed and the corresponding effect on the output was noted. In this way, one is effectively taking the partial derivative of the output with respect to one active input. When this is done successively for all active inputs dictated by a particular input sequence, one can determine the relative contributions of bases at given positions to the output classification. One can, of course, also compare different sequences. [It should be borne in mind that, while one can make up mutant sequences, if the prototype group does not contain solid frequency data on the mutation(s), the evaluation cannot be precise.]
Fig. 1 contains the results of such a decompiling of two networks (NW2 and NW3 from Table 1). The phage T7 A1 promoter was chosen as an example of a strong promoter and the bla promoter of plasmid pBR322 was chosen as a relatively weak promoter. When internally normalized to the maximum value within a plot, the qualitative profile between distinct networks (Fig. 1 A is T7A1 in NW2, B is T7A1 in NW3, C is bla in NW2, and D is bla in NW3) is quite similar for both the strong and the weak promoters (i.e., A is similar to B and C is similar to D, even though these networks were independently trained and had different numbers of middle layer neurons); however, the scale factors are quite different for the two promoters (e.g., 1.0 in B equals 0.000022, whereas 1.0 in D equals 0.00372). In the network’s view, the bla promoter is much more sensitive to change than the T7A1 promoter. Since the positive y axis represents the net decrease in network output with the reduction in weights on the input corresponding, individually, to each base shown on the x axis, the positive positions are those within the promoter that are most sensitive to degradative change. In Fig.1A, the largest values (most sensitive to change) are those for canonical bases in the −35 and −10 sequence regions that, if changed, would produce the most negative effect on promoter membership; conversely, the highly negative bases, if changed, could substantially improve membership status. Therefore, the higher the total, after common normalization say to the T7 scale factor, the worse the promoter; the total of all y values in Fig.1B is 4.7 for the T7A1 promoter and the total in Fig.1D, the same network and scale factor, for bla is 128.6. These networks can be drastically nonlinear in their operations. For example in NW3, changing the second base of the −10 sequence of the T7A1 promoter from “A” to “C” results in an output decrease of 0.003 to 0.993, whereas the same change in the bla promoter drops the output from 0.963 to 0.033, a nonpromoter classification.
Figure 1.

Promoter profiles generated by differentiating two promoter-trained networks. The y axis value represents the relative decrease in the network output value of the first output neuron for a 5% decrease in the input weight associated with that particular position (base) on the x axis; negative values would correspond to an increase in the network output value. All values have been normalized to the Δ maximum for each set of data, defined as 1.0. in A. For T7A1 in NW2, the output value was 0.992433; the Δ maximum for any position was 0.000121. (B) For T7A1 in NW3, the output value was 0.996465; the Δ maximum was 0.0000217. (C) For bla in NW2, the output value was 0.9850; the Δ maximum was 0.000338. (D) For bla in NW3, the output value was 0.963631; the Δ maximum was 0.00372.
DISCUSSION
Numerous ranking functions have been proposed over the period since DNA sequences became available for analysis (5, 6, 15–17). However, only the RI ∗ BvH function has been able to produce correlation coefficients in the 0.96–0.999 range for real data (5). A second closely related index does nearly as well (this work and unpublished results). This index, RI−(RI+ − RI−), again uses the redundancy index as an indicator of the position’s importance but uses the redundancy index difference, before and after adding the test sequence to the prototype group, as the measure of how well the position is filled by a particular base. Examples, NW4 and NW5 applying this index to promoters, are provided in Table 1. Using either index to preprocess sequence for neural network training and testing was found to significantly reduce the false-positive level without degrading the capture of true positives.
It is worth mentioning that there is some confusion about the terms true positives and false positives. A method that captures 100% of the true sequences and has 10% false positives might appear to be good if it meant that one would only get one wrong answer for every 10 correct answers. However, that is not what is meant. To evaluate capture of true positives, one must have a known proven collection; if there are 100 sequences in the collection and 90 are correctly classified, the yield is a 90% capture of true positives as all would expect. However, false positives are not determined in the same way. Since every base in DNA begins a sequence that is a potential site, false positive frequencies are reported on a per-base-searched basis. Therefore, in the example above, 100% true positives per 10% false positives, a search of pBR322 would find five promoters (one of which belongs to the 18-base-spacing class) plus about 860 false positives, obviously not a very useful method. Indeed, one can see that false-positive levels of even 1% are still unacceptably high. Apparently confusion over these terms has allowed a growing number of essentially useless methods to see publication and has caused at least one pair of reviewers to discard neural nets as incapable of low false-positive levels (18).
The ingredient still missing in analyses of this type is that we still do not know the explicit relationship between a motif’s redundancy index and the number and range of sequence examples likely to produce a successful classification network. Clearly, as the redundancy index goes up, the number of examples required goes down; the redundancy index of the λ operators is a healthy 16.75 bits even after reductions for small sample size. The development of a useful rule must await the study of many motifs by this method.
The differentiation of the trained networks gives one the network’s evaluation, in the specific context of a particular sequence example, of the sensitivity of each base within that sequence to change, given the network’s current image of the motif under study.
The methods developed in this report are general in their application to any binding-sequence family. They have the potential to work well even with small families if they are not too divergent as seen in the operator case. Furthermore, false-positive levels are brought down to a level at which genomic searches may be feasible for many motifs, requiring no other information than the motif family sequences.
Acknowledgments
I thank Dr. Peter H. von Hippel and Dr. Richard E. Wolf, Jr., for their extensive insightful comments that contributed substantially to the final form of this paper.
ABBREVIATIONS
- RI
redundancy index
- BvH
the Berg–von Hippel function
References
- 1.Gatlin L L. Information Theory and the Living System. New York: Columbia Univ. Press; 1972. [Google Scholar]
- 2.Fano R M. Transmission of Information. New York: MIT Press; 1961. [Google Scholar]
- 3.Yockey H P. Information Theory and Molecular Biology. Cambridge, U.K.: Cambridge Univ. Press; 1992. pp. 62–70. [Google Scholar]
- 4.Schneider T D, Stormo G D, Gold L, Ehrenfeucht A. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 5.O’Neill M C. J Mol Biol. 1989;207:301–310. doi: 10.1016/0022-2836(89)90256-8. [DOI] [PubMed] [Google Scholar]
- 6.Berg O G, von Hippel P H. J Mol Biol. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- 7.Stormo G D, Schneider T D, Gold L, Ehrenfeucht A. Nucleic Acids Res. 1982;10:2997–3010. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Neill M C. Nucleic Acids Res. 1991;19:313–318. doi: 10.1093/nar/19.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Neill M C. Nucleic Acids Res. 1992;20:3471–3477. doi: 10.1093/nar/20.13.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moyle H, Waldburg C, Suskind M M. J Bacteriol. 1991;173:1944–1950. doi: 10.1128/jb.173.6.1944-1950.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stuber D, Bujard H. Proc Natl Acad Sci USA. 1981;78:167–171. doi: 10.1073/pnas.78.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blattner F R, Plunkett G, III, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277(5331):1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 13.Sarai A, Takeda Y. Proc Natl Acad Sci USA. 1989;86:6513–6517. doi: 10.1073/pnas.86.17.6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson A D, Meyer B J, Ptashne M. Proc Natl Acad Sci USA. 1979;76:5061–5065. doi: 10.1073/pnas.76.10.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulligan M E, Hawley D K, Entriken R, McClure W R. Nucleic Acids Res. 1984;12:789–800. doi: 10.1093/nar/12.1part2.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucher P. J Mol Biol. 1990;212:563–578. doi: 10.1016/0022-2836(90)90223-9. [DOI] [PubMed] [Google Scholar]
- 17.Stormo G D. Methods Enzymol. 1991;208:458–468. doi: 10.1016/0076-6879(91)08024-c. [DOI] [PubMed] [Google Scholar]
- 18.Hertz G Z, Stormo G D. Methods Enzymol. 1996;273:30–42. doi: 10.1016/s0076-6879(96)73004-5. [DOI] [PubMed] [Google Scholar]