

Summary
Insulin drives the global anabolic response to nutrient ingestion, regulating both carbohydrate and lipid metabolism. Previous studies have demonstrated that Akt2/protein kinase B is critical to insulin’s control of glucose metabolism, but its role in lipid metabolism has remained controversial. Here we show that Akt2 is required for hepatic lipid accumulation in obese, insulin-resistant states induced by either leptin-deficiency or high fat diet feeding. Lepob/ob mice lacking hepatic Akt2 failed to amass triglycerides in their livers, associated with and most likely due to a decrease in lipogenic gene expression and de novo lipogenesis. However, Akt2 is also required for steatotic pathways unrelated to fatty acid synthesis, as mice fed high fat diet had reduced liver triglycerides in the absence of hepatic Akt2 but did not exhibit changes in lipogenesis. These data demonstrate that Akt2 is a requisite component of the insulin-dependent regulation of lipid metabolism during insulin resistance.
Introduction
Among its numerous functions, the mammalian liver serves both as a repository for stored nutrients and an organ that senses, integrates and controls the metabolic state of the organism. As a metabolic tissue, the liver responds to multiple inputs, including hormones, neuronal impulses and nutrients. Following the ingestion of food, the dominant signals to the liver are insulin and absorbed nutrients, particularly glucose, which serves as a key substrate and a first messenger providing information to the liver (Postic et al., 2007). Understanding how the liver processes these diverse inputs becomes even more challenging when one considers pathological states associated with over-nutrition, the incidence of which are reaching epidemic proportions throughout the world (Doria et al., 2008). Here it becomes difficult to distinguish protective from maladaptive processes, though there is little doubt that the accumulation in the liver of large quantities of macromolecules, such as neutral lipid, has unmistakable potential for serious toxicity (Browning and Horton, 2004; Savage et al., 2007). Thus the study of hepatic metabolism has justifiably attracted considerable attention in recent years.
During the transition from fasting to the fed state, insulin stimulates hepatic glycogen synthesis and suppresses hepatic gluconeogenesis, resulting in suppression of glucose production by the liver (Petersen et al., 1998; Saltiel, 2001). Loss of function experiments have shown that the protein kinase Akt (also known as protein kinase B) is central to the hepatic actions of insulin on glucose output, though a recent report has questioned the role of Akt as a cell-autonomous intermediate in insulin signaling (Chen et al., 2009; Cho et al., 2001a; Garofalo et al., 2003; Gross et al., 2008). Nonetheless, the prevalent model of hepatic insulin signaling is that activated Akt phosphorylates and inhibits the transcription factor FoxO, peroxisome proliferator-activated receptor-coactivator-1α (PGC-1α) and others, thereby terminating expression of the rate-controlling enzymes of gluconeogenesis (Gross et al., 2008; Li et al., 2007). During insulin resistance, this process is blunted and the persistence of hepatic glucose output following a meal compounds diminished glucose uptake into muscle, resulting in postprandial hyperglycemia (Saltiel, 2001). However, in a seminal essay, McGarry argued persuasively that the view of type 2 diabetes mellitus (T2DM) as a disease of glucose metabolism is purely historical, and that the lipid abnormalities are equally intrinsic to the pathophysiology (McGarry, 1992). Certainly, T2DM is associated with a characteristic dyslipidemia, and the vast majority of obese T2DM individuals with insulin resistance have abnormal accumulation of triglyceride in their livers, so-called non-alcoholic fatty liver disease (NAFLD) (Petersen et al., 2005; Postic and Girard, 2008a). Moreover, the high serum triglycerides and low HDL cholesterol of insulin resistance predispose to cardiovascular disease, the predominant morbidity associated with T2DM, and NAFLD itself can progress to hepatitis (Browning and Horton, 2004; Williams, 2008). However, our understanding of the mechanistic processes governing hepatic lipid metabolism is still rudimentary.
During times of nutritional abundance, the liver converts substrate into triglyceride for local storage as well as export in the form of very low-density lipoprotein (VLDL). This process is controlled at multiple steps by regulation of gene expression, posttranslational modification and substrate supply (Postic and Girard, 2008b). Most recent experiments have been directed towards investigating the transcriptional control of de novo lipogenesis, for which insulin and glucose activate overlapping sets of genes via the sterol regulatory element-binding protein-1c (SREBP1c) and the carbohydrate response element-binding protein (ChREBP), respectively (Koo et al., 2001; Postic et al., 2007; Postic and Girard, 2008a). Though each is required for the maximal accumulation of hepatic triglyceride during insulin resistance in mice, the relative roles of these two transcription factors in the development of NAFLD and under more physiological conditions remains unclear (Dentin et al., 2006; Postic and Girard, 2008a; Yahagi et al., 2002). Moreover, the relative contribution of de novo lipogenesis to hepatic triglyceride accumulation has been controversial, though recent data suggest that it might account for as much as 25–30% of the triglyceride in livers of patients with NAFLD (Donnelly et al., 2005). In the studies described below, we consider the downstream signaling pathway that insulin utilizes to promote hepatic lipid accumulation and, in particular, the induction of SREBP1c, a question for which a consensus answer has yet to emerge. For example, hepatic overexpression of constitutively active Akt increases lipid synthesis, resulting in NAFLD and hypertriglyceridemia, as does hepatic deficiency of PTEN, a negative regulator of PI3K-dependent protein kinases including Akt (Horie et al., 2004; Ono et al., 2003; Stiles et al., 2004). Akt mediates insulin’s effect on SREBP1c expression in cell culture, but studies in vivo have argued that atypical protein kinase C (aPKC) is the responsible intermediary signaling protein (Fleischmann and Iynedjian, 2000; Hegarty et al., 2005; Matsumoto et al., 2003; Porstmann et al., 2005; Taniguchi et al., 2006). Here we present an in vivo model that is inherently defective in insulin signaling due to loss of hepatic Akt2, the major isoform expressed in liver, and utilize these mice to evaluate the role of Akt2 in lipid synthesis and accumulation.
Results
Germline Akt2 Deficiency Prevents NAFLD in Lepob/ob mice
The serine-threonine kinase Akt exists as three highly related isoforms, Akt1-3, each encoded by a distinct gene. Based on studies of mice with interruptions in each locus, Akt2 is the major paralog mediating insulin’s effects on glucose metabolism, whereas Akt1 and Akt3 are more important to growth (Chen et al., 2001; Cho et al., 2001a; Cho et al., 2001b; Easton et al., 2005; Garofalo et al., 2003). Akt2 is the predominant isoform expressed in insulin target tissues, such as liver, muscle and adipose tissue, and Akt2−/− mice exhibit glucose intolerance and a mild diabetic phenotype (Cho et al., 2001a; Garofalo et al., 2003). While the role of Akt2 in glucose homeostasis has been established, its influence on lipid metabolism in vivo remains controversial. To resolve this, we crossed Akt2−/− mice with leptin-deficient Lepob/ob mice. The Lepob/ob Akt2+/+ mice were substantially heavier than Lep+/+ Akt2+/+ littermates at 12 weeks of age and had large, lipid-laden livers. However, deletion of both alleles of Akt2 led to a dramatic reduction of both body and liver weight in the Lepob/ob mouse, with loss of one allele of Akt2 resulting in an intermediate decrease (Figure 1A and 1B). Triglyceride accumulation in the livers of Lepob/ob mice under both fed and fasted conditions was completely prevented by loss of both alleles of Akt2, and, as above, mice heterozygous for Akt2 demonstrated intermediate protection from fatty liver (Figure 1C and 1D). Deletion of Akt2 exacerbated the impairment of glucose homeostasis in Lepob/ob mice, as indicated by an elevation in fasted glucose and insulin levels compared with Lepob/ob Akt2+/+ mice (Figure 1E and 1F). Interestingly, hyperglycemia was not worsened in the Lepob/ob Akt2+/− mice, suggesting that redistribution of hepatic lipid to other organs was not the cause of the aggravated insulin resistance.
Figure 1. Deletion of Akt2 in Lepob/ob mice results in decreased hepatic triglycerides and de novo lipogenesis.
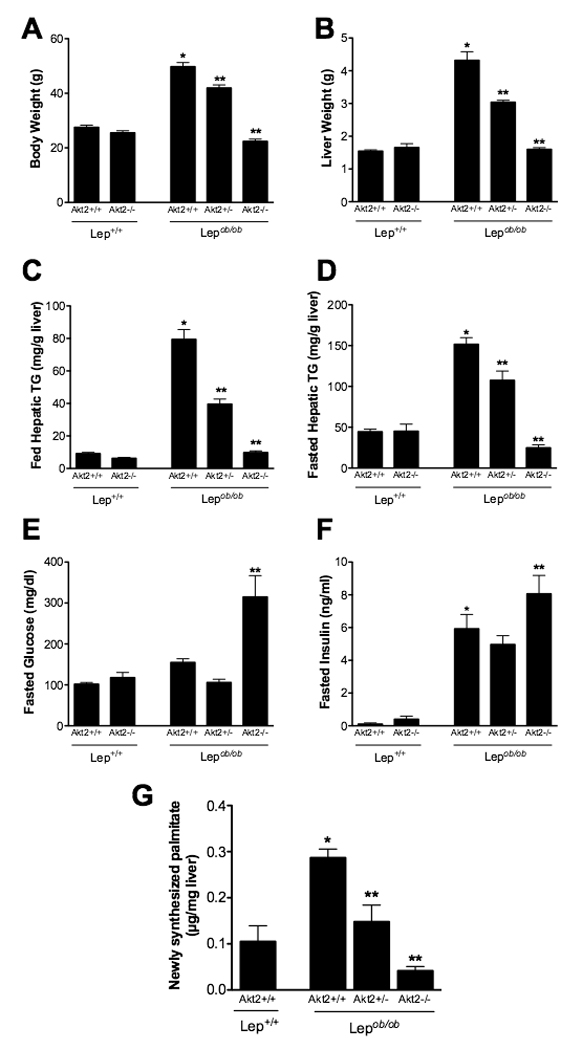
A,B. Body weight (A) or liver weight (B) of 12-week old fed male mice. C,D. Hepatic triglyceride levels of 12-week old fed (C) or overnight fasted (D) male mice. E,F. Blood glucose (E) and insulin (F) levels in 8-week old overnight fasted male mice. G. De novo lipogenesis: 12 week-old male mice were injected with D2O after a 5 hour fast, sacrificed after 3 hours, and liver was removed and analyzed for palmitate by GC/MS. All values are expressed as mean ± SEM. n=5–6; *p<0.05 vs Lep+/+;Akt2+/+ and **p<0.05 vs Lepob/ob;Akt2+/+ by one-way ANOVA using ewman-Keuls post-test.
Accumulation of hepatic triglyceride in Lepob/ob mice is associated with an increase in de novo lipogenesis (Bray and York, 1979; Shimomura et al., 1999). Therefore, we measured hepatic fatty acid synthesis and incorporation into triglycerides using deuterated water as a biosynthetic tracer. Lepob/ob Akt2+/+ mice demonstrated greater de novo lipogenesis compared to Lep+/+ Akt2+/+ mice, but this was reduced in both Lepob/ob Akt2+/− and Lepob/ob Akt2−/− mice (Figure 1G). Non-esterified free fatty acids (NEFA) were unchanged or increased in Lepob/ob Akt2−/− compared to Lepob/ob Akt2+/+ mice, and there was an increase in serum triglyceride levels (Suppl. Table 1). In addition, loss of Akt2 resulted in a modest decrease in serum cholesterol but no change in serum ketone bodies in Lepob/ob mice (Suppl. Table 1).
Generation of Lepob/ob Liver-Specific Akt2 Null Mice
In the experiments described above, the non-cell autonomous effects of germline deletion of Akt2 confound the interpretation of changes in hepatic lipid content and metabolism. To clarify this, we generated mice with liver-specific deletion of Akt2. Mice containing an allele of Akt2 in which exons 3 and 4 are flanked by loxP sires (Akt2lox/lox) were crossed with mice expressing Cre recombinase under the control of the albumin promoter with an alpha-fetoprotein enhancer (AFP). Progeny were crossed with Lepob/ob mice, and offspring were assessed for deletion of Akt2 selectively in the liver. As shown in Suppl. Figure 1, AFP;Akt2lox/lox and Lepob/ob AFP;Akt2lox/lox mice lacked hepatic Akt2 protein, while showing no compensatory changes in the levels of Akt1 or decrease in Akt2 protein in other tissues. While AFP;Akt2lox/lox mice exhibited no difference in body weight compared to Akt2lox/lox mice or AFP;Akt2+/+ mice, Lepob/ob AFP;Akt2lox/lox mice weighed less than both Lepob/ob Akt2lox/lox and Lepob/ob AFP;Akt2+/+ mice (Figure 2A and not shown). This decrease in body weight was due to proportional reductions in both lean and fat mass, such that relative body composition remained unchanged (Suppl. Figure 2A). The decreased body weight could not be attributed to changes in food intake or energy expenditure, which were indistinguishable in Lepob/ob mice with or without hepatic Akt2 (Suppl. Figure 2B and 2C). AFP;Akt2lox/lox mice had mildly elevated fasting glucose levels compared with Lepob/ob Akt2lox/lox mice but did not exhibit any change in insulin levels (Figure 2C and 2D).
Figure 2. Lepob/ob AFP;Akt2lox/lox mice have decreased hepatic triglyceride levels, which correlate with a decrease in de novo lipogenesis.
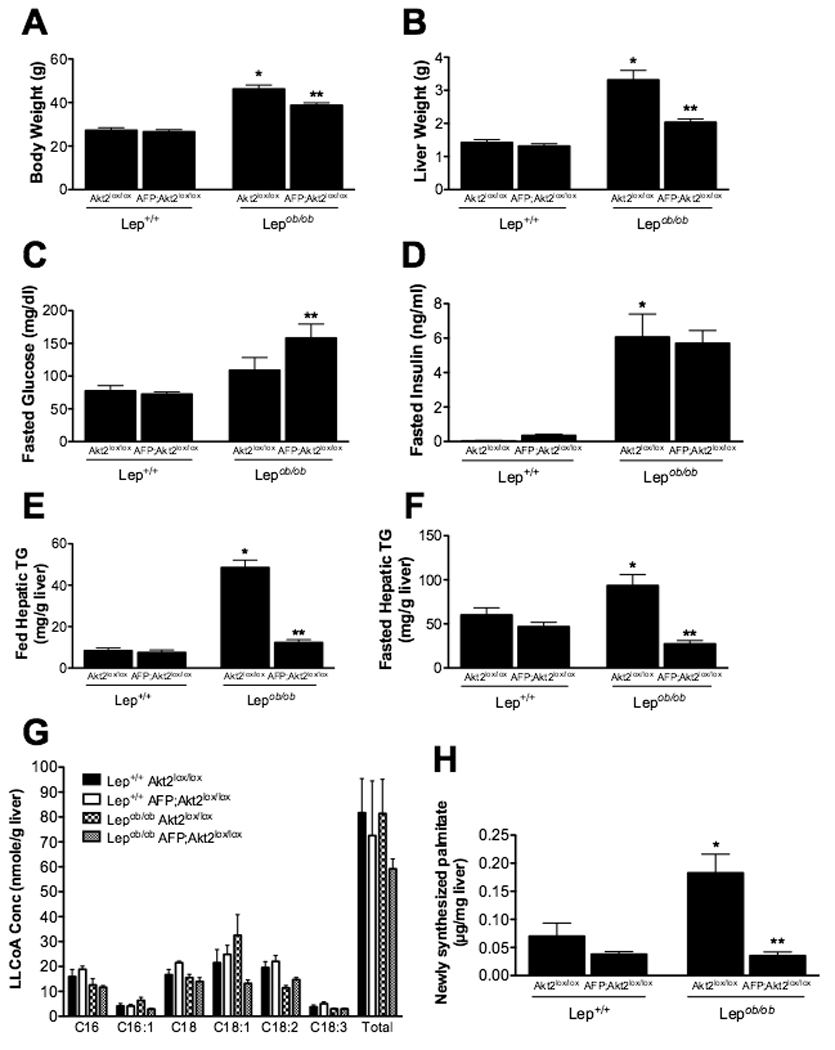
A,B. Body weight (A) or liver weight (B) of 12-week old fed male mice. n=6–8. C,D. Blood glucose (C) and insulin (D) levels in 8-week old overnight fasted male mice. n=6–9. E,F. Hepatic triglyceride levels of 12-week old fed (E) or overnight fasted (F) male mice. n=6–9. G. Hepatic long-chain fatty acid CoA (LLCoA) concentrations from 12 week-old overnight fasted male mice. n=3–4. H. De novo lipogenesis measured as in Fig. 1. n=5–6. All values are expressed as mean ± SEM. *p<0.05 vs Lep+/+;Akt2lox/lox and **p<0.05 vs Lepob/ob;Akt2lox/lox by one-way ANOVA using Newman-Keuls post-test.
Loss of hepatic Akt2 reduces hepatic triglyceride levels, de novo lipogenesis and lipogenic gene expression in the Lepob/ob mouse
Hepatic deletion of Akt2 resulted in decreased liver weight in the Lepob/ob mouse, due at least in part to a reduction in triglyceride content under both fed and fasted conditions (Figure 2B, 2E and 2F). This decrement does not solely reflect the decreased body weight in Lepob/ob AFP;Akt2lox/lox mice, as even compared to weight-matched controls, Lepob/ob AFP;Akt2lox/lox mice exhibited reduced hepatic triglyceride content under fed conditions (Lepob/ob Akt2lox/lox: 56.7 ± 3.34 mg/g TG, 34.9 ± 1.6 g body weight;, Lepob/ob AFP;Akt2lox/lox:12.3 ± 1.43 mg/g TG, 38.7 ± 1.1 g body weight). Fasted serum triglyceride levels were decreased in Lepob/ob AFP;Akt2lox/lox: mice compared with Lepob/ob Akt2lox/lox mice, while fed values were unchanged (Suppl. Table 2). Serum cholesterol was decreased in Lepob/ob mice upon liver-specific deletion of Akt2, but other serum markers of lipid metabolism were largely unaffected (Suppl. Table 2). The content and distribution of long-chain fatty acyl-CoA were not significantly different among any of the genotypes, though there was a trend towards a decrease in the two monounsaturated fatty acids palmitoleate (C16:1) and oleate (C18:1) in Lepob/ob AFP;Akt2lox/lox compared with Lepob/ob Akt2lox/lox livers (Figure 2G). De novo lipogenesis was elevated in the Lepob/ob Akt2lox/lox mice, and loss of hepatic Akt2 reversed this effect, as was previously observed with whole-body loss of Akt2 (Figure 2H). These results demonstrate that Akt2 is required in the liver for the increases in both hepatic de novo lipogenesis and triglyceride accumulation in Lepob/ob mice.
The augmented de novo lipogenesis in the Lepob/ob mouse is associated with an increase in lipogenic gene expression, in particular SREBP-1c and its transcriptional targets (Shimomura et al., 1999). In the present studies, we confirmed the predicted increases in the mRNAs for SREBP-1c, steroyl CoA desaturase (SCD1), fatty acid synthase (FAS), acetyl COA carboxylase (ACC), ATP citrate lyase (ACL), glycerol phosphate acyltransferase (GPAT) and glucokinase (GCK) in Lepob/ob Akt2lox/lox livers, and moreover found that all of these increases were prevented by loss of hepatic Akt2 (Figure 3). Expression of ChREBP was unchanged, but its target pyruvate kinase (L-PK) was increased in Lepob/ob Akt2lox/lox mice and normalized in livers from Lepob/ob AFP;Akt2lox/lox mice. Peroxisome proliferator-activated receptor-γ (PPARγ) and Cidec/Fsp27, gene products required for development of hepatic steatosis in Lepob/ob mice, were induced in Lepob/ob Akt2lox/lox livers and this was prevented by loss of hepatic Akt2 (Figure 3) (Matsusue et al., 2008). Lepob/ob Akt2lox/lox mice exhibited a slight increase in the expression of PGC-1α, which was further enhanced in Lepob/ob AFP;Akt2lox/lox mice (Figure 3). However, expression of medium-chain acetyl-CoA dehydrogenase (MCAD), a transcriptional target of PGC-1α and rate-determining enzyme in fatty acid oxidation, was not altered in Lepob/ob AFP;Akt2lox/lox compared with Lepob/ob Akt2lox/lox livers (Schreiber et al., 2003). Additionally, the expression of carnitine palmitoyltransferase I (CPT1), another PGC-1α target, was actually decreased in Lepob/ob AFP;Akt2lox/lox livers, while the expression of the mitochondrial gene cytochrome c oxidase subunit 7a (Cox7a) was not different between Lepob/ob Akt2lox/lox and Lepob/ob AFP;Akt2lox/lox mice (Song et al., 2004).
Figure 3. Hepatic gene expression in Lepob/ob AFP;Akt2lox/lox mice.
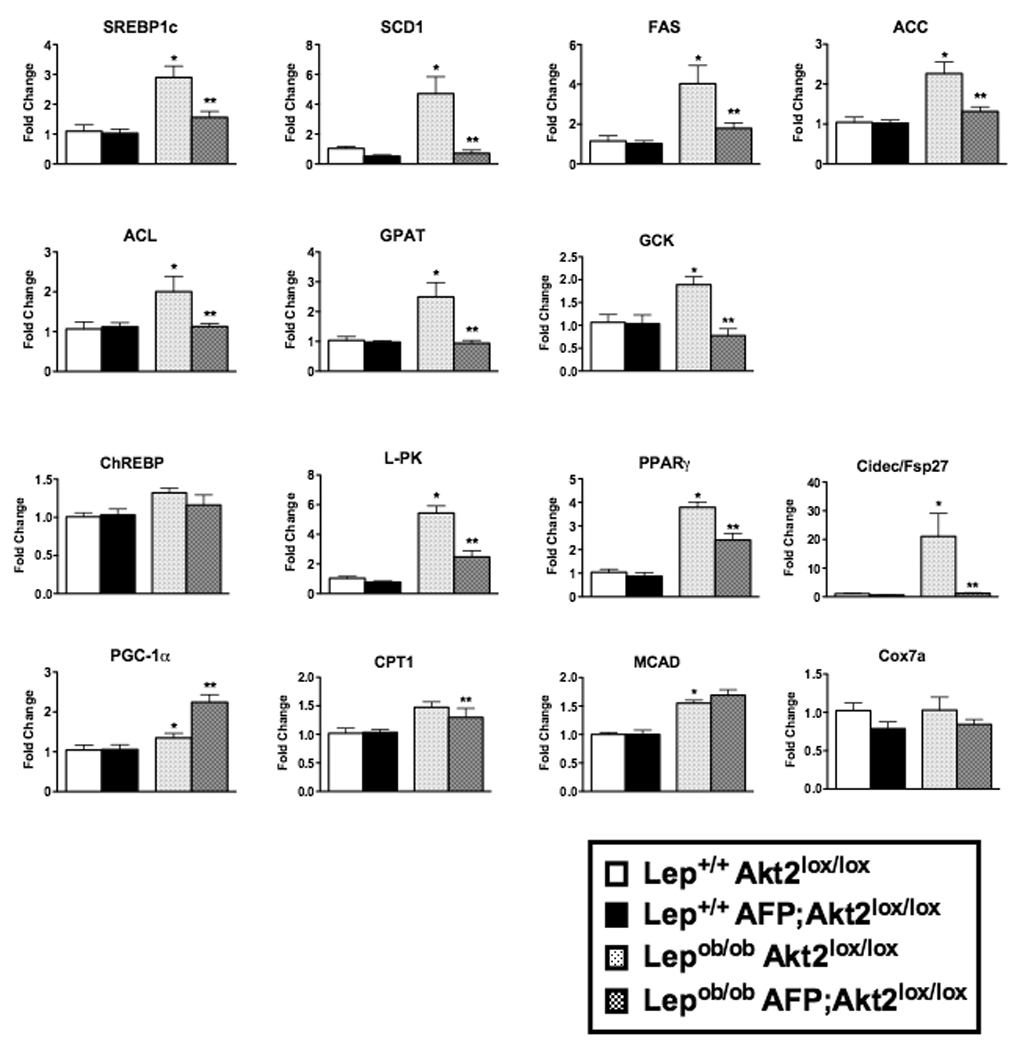
Hepatic gene expression as measured by rtPCR of 12 week-old fed male mice. Data are presented as mRNA expression relative to that of TATA binding-protein (TBP) and normalized to expression in Akt2lox/lox, which is set to 1.0 using the ddCT method. All values are expressed as mean ± SEM. n=6; *p<0.05 vs Lep+/+;Akt2lox/lox and **p<0.05 vs Lepob/ob;Akt2lox/lox by one-way ANOVA using Newman-Keuls post-test.
Germline Akt2 deletion decreases hepatic lipid accumulation resulting from Surwit high fat diet (HFD)
Though the Lepob/ob mouse is a robust model of obesity and NAFLD, these mice also lack leptin, which itself plays a role in metabolism. To complement our studies in Lepob/ob mice, we fed Akt2−/− mice a Surwit diet, which induces obesity and insulin resistance in mice through dietary enrichment of fat (58% kcal from fat) and simple carbohydrates (25.5% kcal from carbohydrates, approximately half from sucrose) (Surwit et al., 1995; Surwit et al., 1988). Male mice were given a Surwit HFD at approximately 5 weeks of age, and maintained on the diet for one or 4 months; chow-fed age-matched male mice served as controls. After 4 months on Surwit diet, Akt2+/+ mice gained more weight than their chow-fed counterparts, an effect that was attenuated by whole-body loss of Akt2 (Figure 4A). Though liver weight was not significantly different between any of the groups (Figure 4B), the increase in fed hepatic triglycerides observed in Akt2+/+ mice after 4 months on the diet was abrogated with loss of Akt2 (Figures 4C and 4D). Germline deletion of Akt2 did not change serum triglyceride levels on Surwit HFD, but did decrease fasted serum free fatty acids and serum ketone bodies (Suppl. Table 3). Interestingly, while Akt2−/− mice developed severe hyperglycemia on a Lepob/ob background, Akt2−/− mice on a Surwit HFD for 4 months were able to maintain fasting euglycemia by increasing insulin levels (Figure 1E, Figure 4E and 4F).
Figure 4. Akt2−/− mice have decreased hepatic triglyceride levels after 4 months on Surwit HFD.
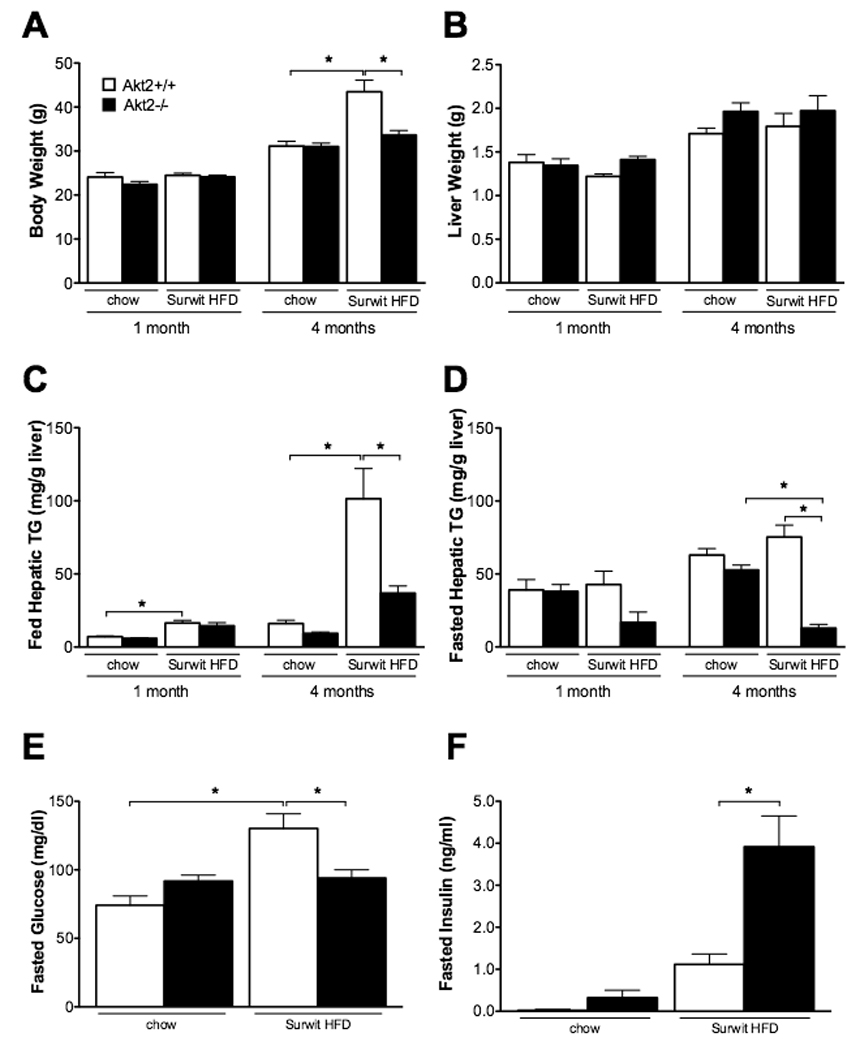
Male mice were started on Surwit HFD at approximately 5 weeks of age and sacrificed after either 1 or 4 months along with age-matched chow controls. Body weight (A), liver weight (B); hepatic triglyceride levels of fed (C) or overnight fasted (D); blood glucose (E) and insulin (F) levels from overnight fasted mice after 4 months on HFD. Values are expressed as mean ± SEM. n=5-9; *p<0.05 by one-way ANOVA using Newman-Keuls post-test.
AFP;Akt2lox/lox mice display decreased hepatic triglycerides on Surwit HFD without significant changes in de novo lipogenesis or lipogenic gene expression
As in the previous experiments, we wanted to determine if the requirement for Akt2 in hepatic triglyceride accumulation resulting from Surwit HFD was cell autonomous. Starting at approximately 5 weeks of age, we fed male AFP;Akt2lox/lox mice Surwit HFD for one or 4 months and used chow-fed age-matched male mice as controls. During this period, there were no differences in the weight gained on Surwit HFD between Akt2lox/lox mice and AFP;Akt2lox/lox mice or in liver weight between any of the groups (Figure 5A, 5B and Suppl. Figure 3A). AFP;Akt2lox/lox mice did not differ from Akt2lox/lox mice with regards to fasting glucose or insulin levels, food intake, body composition, or energy expenditure (Figures 5C and 5D and Suppl. Figure 3B–D). After one month on Surwit HFD, Akt2lox/lox mice had significantly increased fed hepatic triglyceride levels compared to their chow-fed counterparts, an effect that was partially ameliorated by loss of hepatic Akt2 (Figure 5E). The same held true after 4 months on Surwit HFD, as the 4-fold increase in fed hepatic triglycerides in Akt2lox/lox mice was reduced by approximately 25% with loss of hepatic Akt2. Total long chain fatty acyl-CoA and linoleoyl-CoA (C18:2) were decreased in livers from mice on Surwit HFD, as the latter is an essential fatty acid and its abundance is relatively low in coconut oil, the major source of fat in the Surwit diet. However, there were no differences between mice having or lacking hepatic Akt2 (Figure 5F). Fed serum triglycerides were increased in mice after 4 months on the diet with or without Akt2 in the liver (Suppl. Table 4).
Figure 5. AFP;Akt2lox/lox mice on Surwit HFD have decreased hepatic triglyceride levels, but do not exhibit changes in de novo lipogenesis.
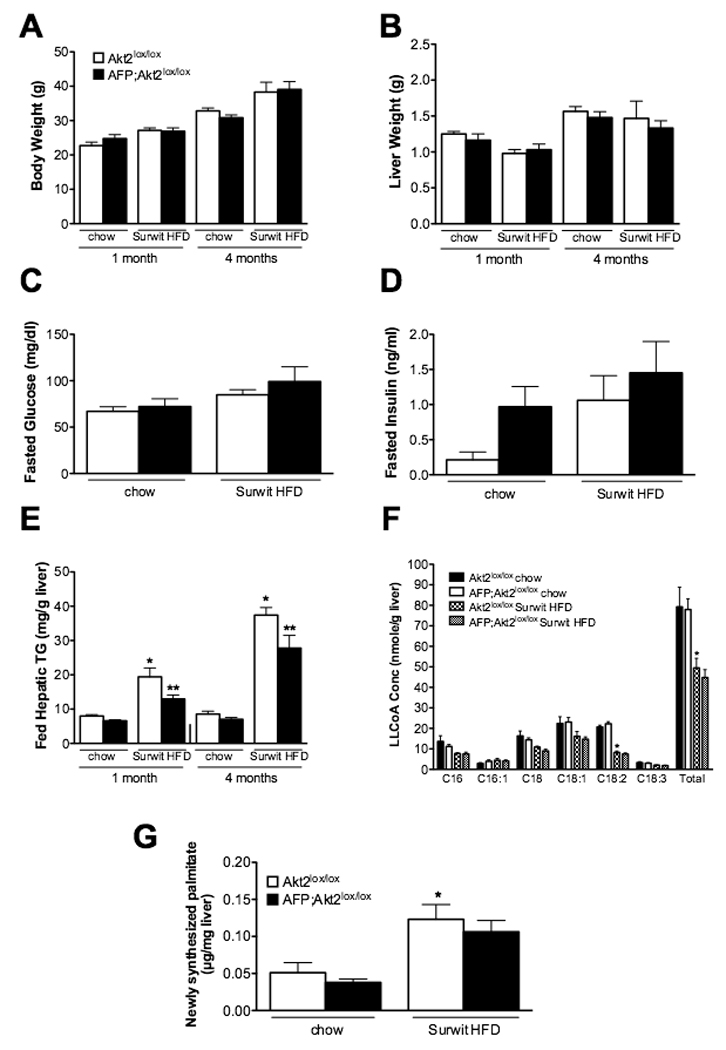
Male mice were started on Surwit HFD at approximately 5 weeks of age and sacrificed after either 1 or 4 months along with age-matched chow controls. Body weight (A), liver weight (B); blood glucose (C) and insulin (D) levels from overnight fasted mice after 4 months on HFD; hepatic triglyceride levels of fed mice (E). n=5–9; *p<0.05 vs. Akt2lox/lox chow and **p<0.05 vs Akt2lox/lox HFD by one-way ANOVA using Newman-Keuls post-test. F. Hepatic long-chain fatty acyl CoA (LLCoA) concentrations from overnight fasted male mice after 1 month on Surwit HFD. n=4. *p<0.05 vs Akt2lox/lox chow by two-way ANOVA using Bonferroni post-test. G. De novo lipogenesis: Lipogenesis was assayed in male mice after 4 months on Surwit HFD as in Fig. 1. n=5; *p<0.05 vs Akt2lox/lox chow by one-way ANOVA using Newman-Keuls post-test. All values are expressed as mean ± SEM.
While there was a significant increase in de novo lipogenesis in livers from mice on a Surwit HFD, this was not prevented by deletion of Akt2 (Figure 5G). Lipogenic gene expression paralleled this finding, as there was no decrease in hepatic expression of SREBP-1c or its targets in AFP;Akt2lox/lox fed Surwit HFD for one month; in fact there was a slight increase in SREBP1c, FAS, ACC and L-PK expression (Figure 6). However, only SCD1 showed a significant increase in livers from Akt2lox/lox mice on Surwit HFD compared chow-fed animals. Additionally, there were not changes in the expression of PGC-1α, CPT-1, MCAD, or Cox7a in the AFP:Akt2lox/lox livers, and fasted or fed serum ketone levels were similar in AFP;Akt2lox/lox mice on Surwit HFD, suggesting that β-oxidation was not altered in these animals (Figure 6, Suppl. Table 4).
Figure 6. Hepatic gene expression in AFP;Akt2lox/lox mice on Surwit HFD.
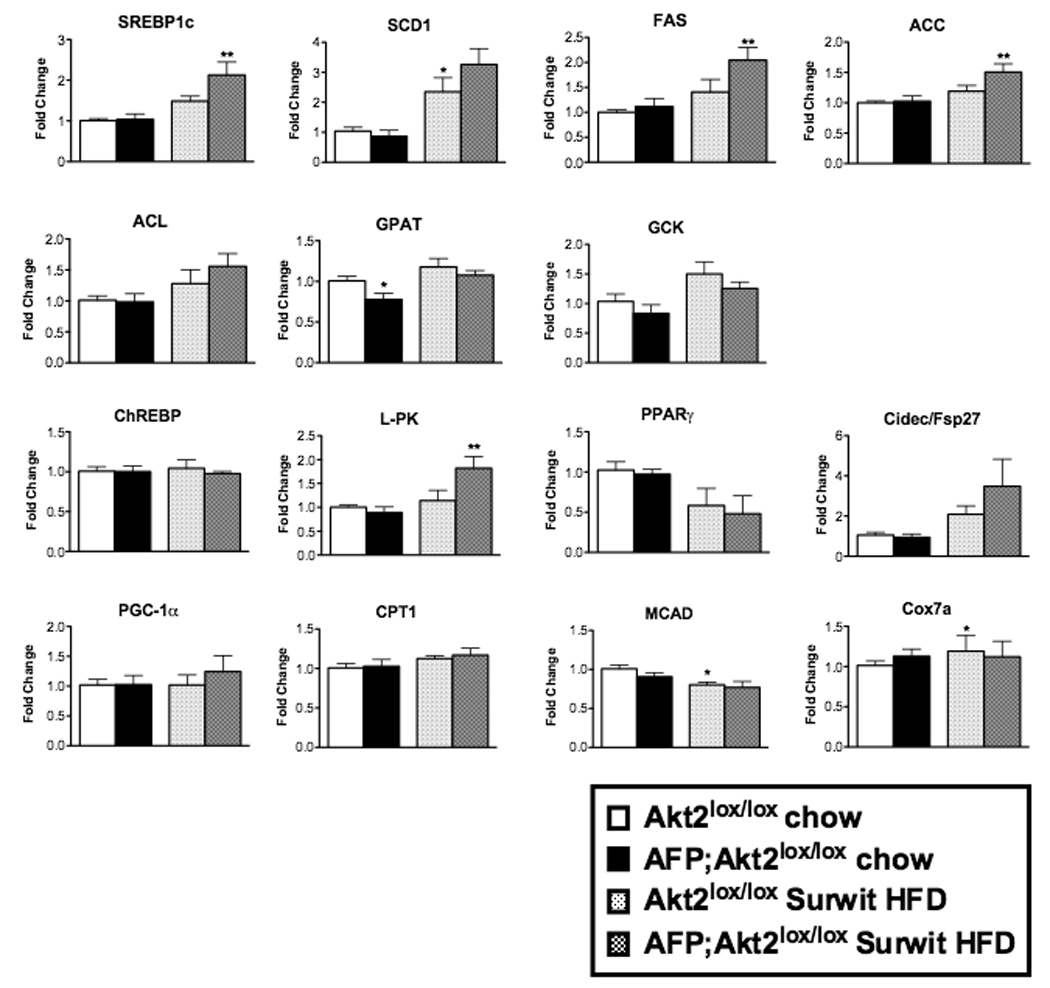
Hepatic gene expression as measured by rtPCR of male mice after 1 month on Surwit HFD or age-matched chow-fed controls sacrificed under fed conditions. Data are presented as mRNA expression relative to that of TBP and normalized to expression in Akt2lox/lox chow-fed, which is set to 1.0 using the ddCT method. All values are expressed as mean ± SEM. n=6; *p<0.05 vs Akt2lox/lox chow and **p<0.05 vs Akt2lox/lox HFD by one-way ANOVA using Newman-Keuls post-test.
AFP;Akt2lox/lox mice display decreased hepatic triglycerides upon being fed a lard HFD
Given the differences in phenotype between mice deficient for leptin and those fed a Surwit HFD, we tested the generality of the requirement for Akt2 in hepatic steatosis by feeding male AFP;Akt2lox/lox mice another commonly used HFD, one enriched in fat from lard (60% kcal from fat) with a mixture of both starch and sucrose (20% kcal from carbohydrates). After one month on the lard HFD, there was an increase in hepatic triglycerides in Akt2lox/lox mice, which was ameliorated by loss of hepatic Akt2 (Figure 7A). However, unlike the results with the Surwit HFD, there was a significant increase in SREBP-1c expression in the livers of Akt2lox/lox mice after one month on a lard HFD, an increase that was significantly reduced in the AFP;Akt2lox/lox mice (Figure 7B). Other lipogenic genes were not increased on the lard HFD, though there was a trend for reduced expression in the absence of Akt2. Nonetheless, these data again show clearly that hepatic Akt2 is required for hepatic lipid accumulation ensuing from HFD feeding.
Figure 7. AFP;Akt2lox/lox mice on lard HFD have decreased hepatic triglyceride levels and SREBP-1c expression.
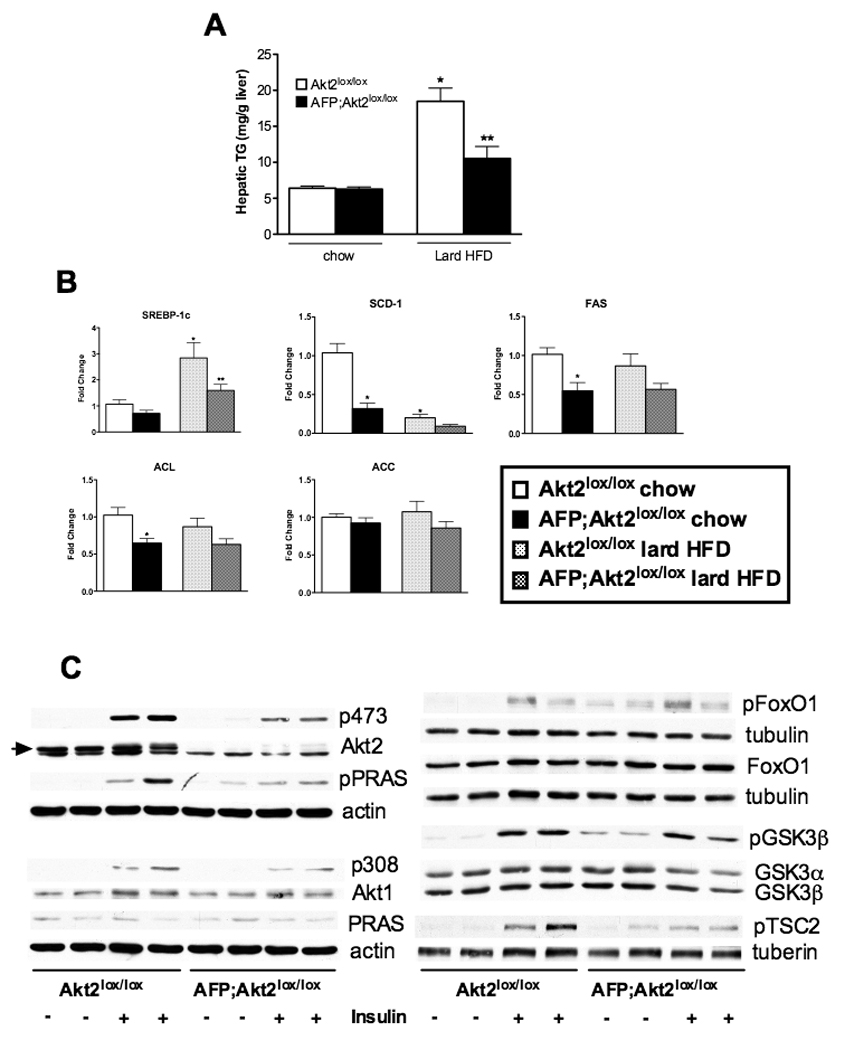
Male mice were started on lard HFD at approximately 6 weeks of age and sacrificed after 1 month on diet under fed conditions along with age-matched chow-fed controls. A. Hepatic triglycerides. B. Hepatic gene expression as measured by rtPCR. Data are presented as mRNA expression relative to that of TBP and normalized to expression in Akt2lox/lox chow-fed, which is set to 1.0 using the ddCT method. All values are expressed as mean ± SEM. n=5-6; *p<0.05 vs Akt2lox/lox chow and **p<0.05 vs Akt2lox/lox HFD by one-way ANOVA using Newman-Keuls post-test. C. Western blot of phospho-Akt (p473 and p308) and downstream signaling targets in hepatic lysates from Akt2lox/lox and AFP;Akt2lox/lox mice. 8-week old male mice were fasted overnight and IP injected with either saline or 1U/kg insulin, then sacrificed after 20 minutes. Each lane represents an individual mouse. Loading controls are included when blots from different gels were used for phospho-protein and total protein, though the same protein extracts were used. The arrow indicates Akt2 as there is a slightly more mobile non-specific band.
Loss of hepatic Akt2 decreases insulin signaling in lean mice, but does not change downstream protein phosphorylation in insulin-resistant livers
In order to gain insight into the pathways that could be mediating Akt2’s effect on hepatic lipid metabolism, we performed Western blots on liver extracts from Akt2lox/lox and AFP;Akt2lox/lox mice following intraperitoneal (IP) injection of saline or insulin (Figure 7C). Phosphorylation of Akt at residues S473 and T308 was increased in Akt2lox/lox livers injected with insulin, and this increase was blunted in the absence of Akt2. The same pattern held true for the phosphorylation of Proline-Rich Akt Substrate of 40kDa (PRAS), glycogen synthase kinase 3-β (GSK3β) and phospho-tuberous sclerosis complex-2 (TSC2), while phosphorylation of FoxO1 did not appear to be decreased in the absence of hepatic Akt2. Interestingly, phosphorylation of these Akt targets was increased under basal fasted conditions in AFP;Akt2lox/loxmice, suggesting relief of a negative feedback loop in the absence of Akt2 (Figure 7C). We also determined if loss of Akt2 had significant effects on hepatic insulin signaling in Lepob/ob and HFD-fed mice as Akt2 mediates different lipid metabolic pathways in these two models (Suppl. Figure 4). In order to assess insulin signaling in both experimental groups in the presence of comparable levels of insulin and glucose, Lepob/ob Akt2lox/lox and Lepob/ob AFP;Akt2loxlox mice were submitted to a hyperinsulinemic euglycemic clamp and liver extracts were prepared; glucose and insulin levels did not differ between Akt2lox/lox and AFP;Akt2lox/lox on a Surwit HFD, so liver extracts were made from these animals after one month on the diet under fed conditions. Insulin signaling was significantly impaired in both models of insulin resistance, such that phosphorylation of Akt and its targets was significantly below stimulated conditions in Akt2lox/lox animals; moreover, loss of hepatic Akt2 in either model did not reduce protein phosphorylation further (Suppl. Figure 4).
Discussion
Though insulin is well known to stimulate de novo lipogenesis in liver, the precise mechanism by which this is accomplished is not well understood. SREBP1c is clearly an important intermediate in the transcriptional control of lipogenesis, but the pathway by which insulin activates both processing and expression of SREBP1c remains uncertain (Raghow et al., 2008). To some extent, the difficulty in investigating this problem relates to the low rates of de novo lipogenesis in the normal postabsorptive liver, which have been estimated to contribute as little as 5% of total triglyceride fatty acids in humans (Diraison et al., 2003). For this reason, it has proven instructive to manipulate the metabolic state of experimental organisms in order to increase fat accumulation in the liver. In the present study, we have done so employing both genetic and dietary strategies and found a strong dependency on the presence of hepatic Akt2 for the development of steatosis and an absolute requirement for the increase in lipogenesis. Though these data are derived from mouse models of human disease, there is much to support the notion that the obligatory role for Akt2 in lipid synthesis is not unique to the insulin-resistant, steatotic condition. We often see a trend towards lower hepatic triglycerides, fatty acid synthesis and lipogenic gene expression in livers from normal mice on a chow diet and in several cohorts these changes have reached statistical significance (Figure 6, 7). Thus, we can conclude that Akt2 is required definitively for full accretion of hepatic triglyceride during pathological states and it is likely that this is an extension of a requirement for the kinase in normal anabolic lipid metabolism.
One surprising result of these studies is that even though the necessity of Akt2 for steatosis applies to multiple models, in at least one, i.e. obesity induced by high fat and sucrose feeding, hepatic triglyceride content is reduced but lipogenesis and lipogenic gene expression are unchanged (Figure 5). This contrasts with development of NAFLD in the Lepob/ob mouse, in which the protection from steatosis is mediated, at least in part, through preventing the stimulation of de novo lipogenesis (Figure 2). Thus, Akt2 likely mediates insulin’s induction of triglyceride accumulation by stimulating fatty acid synthesis as well as processes other than de novo lipogenesis (Figure 5). One obvious candidate mechanism it that loss of Akt2 abrogates the normal suppression of β-oxidation produced by insulin. For example, insulin suppresses the PGC-1α-dependent stimulation of fatty acid oxidation, both by reducing hepatocyte cyclic AMP and by promoting the Akt-dependent inhibition of PGC-1α activity (Li et al., 2007). However, by several criteria, an increase in β-oxidation is unlikely to explain the protection from steatosis in our studies. First, though PGC-1α mRNA increased in Lepob/ob AFP;Akt2lox/lox livers, expression of the critical targets MCAD and CPT1 were not elevated in parallel (Figure 3) (Schreiber et al., 2003; Song et al., 2004). Second, there were no differences in expression of hepatic oxidative genes comparing Akt2lox/lox to AFP;Akt2lox/lox mice placed on a Surwit HFD (Figure 6). Lastly, RER and serum ketones bodies were largely indistinguishable in wildtype versus AFP;Akt2lox/lox mice on either an Lepob/ob background or Surwit HFD (Suppl. Figures 2 and 3, Suppl. Tables 2 and 4). Another possible mechanism through which Akt2 could be decreasing hepatic lipid accumulation is increased VLDL export, as serum triglycerides were elevated in Lepob/ob Akt2−/− mice (Suppl. Table 1). However, neither Lepob/ob AFP;Akt2lox/lox nor AFP;Akt2lox/lox mice on Surwit HFD exhibited increased serum triglyceride levels under fasted or fed conditions and direct measurement of triglyceride export failed to reveal a requirement for Akt2 (Suppl. Tables 2 and 4 and not shown). Thus, the necessity for Akt2 in the development of hepatic steatosis relates to its role in mediating induction of SREBP1c and de novo lipogenesis as well as other presently unidentified anabolic processes.
The role of Akt as the primary mediator of insulin’s action to increase SREBP1c and promote lipogenesis has been a point of some controversy. Hepatic overexpression of constitutively active Akt increases hepatic neutral lipid dramatically by a pathway only partially dependent on SREBP1c (Ono et al., 2003). Similarly, forced activation of endogenous Akt by liver-specific deletion of the lipid phosphatase Pten produces substantial accumulation of hepatic triglyceride and increased lipogenic gene expression, though this model is complicated by the concomitant activation of other PI3K-dependent kinases (Stiles et al., 2004). On the other hand, a dominant inhibitory Akt does not block insulin’s induction of SREBP1c in tissue culture cells and Lepob/ob mice have markedly increased SREBP1c mRNA in spite of significantly reduced levels of phospho-Akt (Matsumoto et al., 2002; Shimomura et al., 2000). Atypical PKC (PKCλ/ζ) proteins have received considerable attention as obligate mediators of the effects of insulin and PI3K on anabolic lipid metabolism, and have specifically been advanced as an alternative to Akt (Matsumoto et al., 2003; Taniguchi et al., 2006). Matsumoto et al. showed that mice with liver-specific deletion of PKCλ have decreased SREBP1c expression and triglyceride content, though reduced serum insulin levels complicated the interpretation of the in vivo findings in the study (Matsumoto et al., 2003). Kahn and colleagues undertook a different approach, eliminating both aPKC and Akt activity by ablation of all PI3K in the liver, and then selectively introducing constitutively activate versions of the two kinases by adenovirus-mediated delivery (Taniguchi et al., 2006). They found that aPKC, but not Akt, restored SREBP1c mRNA, but the effects on hepatic lipids were not reported. The current studies do not address a potential role for aPKC and thus are compatible with a requirement for this kinase. However, in contrast to Taniguchi et al., they strongly support a critical role for Akt2. Importantly, Lepob/ob mice heterozygous for Akt2 displayed a reduction in liver triglyceride content and de novo lipogenesis intermediate between that of mice wildtype and null for Akt2 (Figures 1C, 1D and 1G). This indicates that Akt2 is not only permissive for anabolic lipid metabolism, but is actually rate-determining. Moreover, Lepob/ob Akt2+/− mice did not display the increase in serum glucose compared to Lepob/ob mice evident in the Akt2 null mice, so the protection in hepatic steatosis cannot be attributed to a worsening of the diabetes. Thus, these data support a obligate role for Akt2 in the development of the steatosis of obesity and insulin resistance, most likely reflecting the function of Akt2 in normal insulin signaling to lipid metabolism.
As shown in Figure 1E, deletion of both Akt2 and leptin resulted in fasting hyperglycemia considerably more severe than that in mice lacking either of the two proteins alone. This is associated with glycosuria, which likely contributes to caloric loss and reduction in body size in these mice (Figure 1A). In a recent paper, Chen and colleagues attribute the severe diabetes observed in compound Akt1+/− Akt2−/− mice to leptin deficiency secondary to lipodystrophy (Chen et al., 2009). Our findings cannot be explained by this model as Akt2 deficiency enhances the diabetes and presumably insulin resistance in spite of the complete absence of leptin in either mouse line. Rather, these data suggest that Akt and leptin control glucose metabolism in parallel pathways.
A recent study examining lipid abnormalities in humans with genetic syndromes of insulin resistance reported increased liver fat content, lipogenesis and serum triglycerides in several individuals with a dominant-negative mutation in Akt2 (Semple et al., 2009). However, as pointed out by Semple et al., the one patient studied in most detail might have had other, confounding metabolic abnormalities. Of note, like patients with an Akt2 mutation, Lepob/ob mice null for Akt2 had elevated serum triglyceride levels, whereas Lepob/ob mice with liver-specific deletion of Akt2 exhibit normal or reduced circulating triglyceride (Suppl. Tables 1 and 2). This emphasizes the role of non-hepatic tissues in determining lipid levels and the difficulties in interpreting metabolic data. Nonetheless, additional translational experiments will be required to establish whether the requirement for Akt in NAFLD is unique to rodents or does indeed recapitulate the pathophysiology in humans.
A longstanding paradox has been that people with T2DM and the metabolic syndrome or rodents with equivalent metabolic disorders have systemic insulin resistance in the face of increased hepatic lipogenesis, a classical insulin response (Petersen et al., 2007). Though a number of models could explain this, the concept of selective or partial insulin resistance has received increasing recent attention (Brown and Goldstein, 2008). Both humans with insulin resistance due to inherited mutations in the insulin receptor and mice with liver-specific deletion of the insulin receptor exhibit hyperglycemia and hyperinsulinemia but are protected against steatosis and hypertriglyceridemia (Biddinger et al., 2008; Semple et al., 2009). This finding is consistent with the idea that in classical “insulin-resistant” states, not all signaling is blunted, but rather some is preserved, in particular that to lipid synthesis. While it is likely that the pathways regulating glucose and lipid metabolism diverge somewhere downstream of the IRS proteins but upstream of FoxO1 and SREBP1c, respectively, the precise biochemical site is unknown (Dong et al., 2008; Kubota et al., 2008; Matsumoto et al., 2007). In a recent consideration of selective insulin resistance, Brown and Goldstein wrote that the “Identification of the branch point is a central question for future research” (Brown and Goldstein, 2008). In the studies presented in this paper, we have demonstrated that the point of selective insulin resistance lies downstream of Akt. A major unresolved question is the nature of those pathways responsible for divergent signaling to glucose output and lipogenesis.
Experimental Procedures
Metabolic measurements and analytical procedures
Overnight fasted mice were used for measurements of blood glucose using a glucometer (OneTouch Ultra, Lifescan). Insulin assays were conducted on blood collected from mice after an overnight fast by tail bleed into heparinized tubes using an ELISA kit (Ultra Sensitive Rat Insulin ELISA kit, Crystal Chem, Inc.). Hepatic triglycerides were measured from animals sacrificed by CO2 inhalation, snap-frozen in liquid nitrogen, and stored at −80°C until processed. Frozen livers were weighted and homogenized in lysis buffer (140mM NaCl, 50mM Tris, 0.1% Triton-X) using a Tissuelyser (Qiagen). Liver homogenates were then incubated at 37°C with 1% deoxycholate, and triglycerides measured colorimetrically using Infinity Triglyceride Reagent (ThermoDMA, Inc). Serum triglycerides, cholesterol, free fatty acids (NEFA), and ketone bodies were analyzed from blood collected after sacrifice by cardiac puncture using colorimetric assay kits (Infinity TG and CH reagents, ThermoDMA, Inc; NEFA-HR kit, Wako; β-hydroxybutyrate LiquiColor kit, Stanbio Laboratories, respectively). Hepatic long-chain fatty acyl-CoAs were isolated as previously described and measured by using an API 4000 tandem mass spectrometer (Applied Biosystems) in conjunction with 2 PerkinElmer 200 Series micro pumps and a 200 Series autosampler (PerkinElmer) (Neschen et al., 2005).
De novo lipogenesis assay
Male mice were fasted for 5 hours (8am to 1pm), injected with D2O (400µl per 20g body wt), and sacrificed 3 hours later. Blood was collected by pipette after cutting the aorta/IVC using the diaphragm as a barrier to the peritoneal cavity. Liver was removed and snap-frozen in liquid nitrogen. Palmitate was analyzed as its trimethylsilyl derivative using gas chromatography-electron impact ionization mass spectrometry. The oven temperature was initially held for 1 min at 150° C, then increased by 20° C per min to 310° C and maintained for 8 min. The split ratio was 20:1 with helium flow 1 ml per min. The inlet temperature was set at 270° C and MS transfer line was set at 310° C. Under these conditions, palmitate elutes at ~ 5.7 min. The 2H-enrichment was determined by using selective ion monitoring under electron impact ionization of m/z 313 and 314 (M+0 and M+1), 10 ms dwell time per ion. The concentration of palmitate was determined by comparing the corrected abundance of m/z 313 to 314 to that of heptadecanoate (17:0, m/z 327). To account for possible differences in the ionization efficiency of each fatty acid, the profile was compared against standards prepared by mixing known quantities of each fatty acid. Rate of lipid synthesis was determined as the percent contribution of newly made using the equation: % newly made palmitate = [total 2H-labeling palmitate / (2H-labeling body water × n)] × 100 where n is the number of exchangeable hydrogens, assumed to equal 22. The absolute amount of newly made palmitate was determined by multiplying the % newly made palmitate by the concentration of palmitate (Brunengraber et al., 2003; Diraison et al., 1997; Lee et al., 1994a; Lee et al., 1994b).
RNA isolation and gene expression studies
Total RNA was prepared from liver using Trizol reagent (Invitrogen), followed by chloroform extraction and DNAse treatment (DNA-free kit, Ambion). cDNA was synthesized with random decamers using the RetroScript Kit (Ambion), and mixed with Brilliant SYBR Green QPCR Master Mix (Stratagene) and primers as noted in Supplementary Table 5. Reactions were performed on an M×3000P Quantitative PCR System (Stratagene). The relative amounts of specific transcripts were calculated using TATA binding protein (TBP) mRNA as an invariant control by the comparative CT (ddCT) method, with the control genotype set to 1.0 (either Lep+/+ Akt2lox/lox or Akt2lox/lox chow-fed).
Statistics
Data are presented as mean ± SEM. Data were analyzed using one-way ANOVA using Newman-Keuls post-test, two-way ANOVA using Bonferroni post-test, or Student’s t-test assuming unequal variance with 2-tailed analysis as described in the figure legends. Values of p < 0.05 were defined as statistically significant.
Supplementary Material
Acknowledgements
This work was supported by NIH grants RO1 DK56886 (MJB), T32 GM07229 (KFL), and 1F30 DK081283 (KFL). The Transgenic/Knockout, Mouse Phenotyping, and Biomarker Cores of the University of Pennsylvania Diabetes and Endocrinology Research Center (NIH grant P30 DK19525) and the NIH-supported Mouse Metabolic Phenotyping Center (Case Western Reserve University) were instrumental in this work. We thank Dr. Klaus Kaestner (University of Pennsylvania) for the kind gift of the AFP Cre mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Aleman JO, Suzuki R, Scapa EF, Agarwal C, Carey MC, Stephanopoulos G, Cohen DE, King GL, Ginsberg HN, Kahn CR. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev. 1979;59:719–809. doi: 10.1152/physrev.1979.59.3.719. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunengraber DZ, McCabe BJ, Kasumov T, Alexander JC, Chandramouli V, Previs SF. Influence of diet on the modeling of adipose tissue triglycerides during growth. Am J Physiol Endocrinol Metab. 2003;285:E917–E925. doi: 10.1152/ajpendo.00128.2003. [DOI] [PubMed] [Google Scholar]
- Chen WS, Peng XD, Wang Y, Xu PZ, Chen ML, Luo Y, Jeon SM, Coleman K, Haschek WM, Bass J, Philipson LH, Hay N. Leptin deficiency and beta-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol Cell Biol. 2009 doi: 10.1128/MCB.01792-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 2001a;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001b;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, Girard J, Postic C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003;29:478–485. doi: 10.1016/s1262-3636(07)70061-7. [DOI] [PubMed] [Google Scholar]
- Diraison F, Pachiaudi C, Beylot M. Measuring lipogenesis and cholesterol synthesis in humans with deuterated water: use of simple gas chromatographic/mass spectrometric techniques. J Mass Spectrom. 1997;32:81–86. doi: 10.1002/(SICI)1096-9888(199701)32:1<81::AID-JMS454>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metab. 2008;8:186–200. doi: 10.1016/j.cmet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann M, Iynedjian PB. Regulation of sterol regulatory-element binding protein 1 gene expression in liver: role of insulin and protein kinase B/cAkt. Biochem J. 2000;349:13–17. doi: 10.1042/0264-6021:3490013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- Hegarty BD, Bobard A, Hainault I, Ferre P, Bossard P, Foufelle F. Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc Natl Acad Sci U S A. 2005;102:791–796. doi: 10.1073/pnas.0405067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, Naito M, Enomoto K, Watanabe S, Mak TW, Nakano T. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Dutcher AK, Towle HC. Glucose and insulin function through two distinct transcription factors to stimulate expression of lipogenic enzyme genes in liver. J Biol Chem. 2001;276:9437–9445. doi: 10.1074/jbc.M010029200. [DOI] [PubMed] [Google Scholar]
- Kubota N, Kubota T, Itoh S, Kumagai H, Kozono H, Takamoto I, Mineyama T, Ogata H, Tokuyama K, Ohsugi M, Sasako T, Moroi M, Sugi K, Kakuta S, Iwakura Y, Noda T, Ohnishi S, Nagai R, Tobe K, Terauchi Y, Ueki K, Kadowaki T. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 2008;8:49–64. doi: 10.1016/j.cmet.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Lee WN, Bassilian S, Ajie HO, Schoeller DA, Edmond J, Bergner EA, Byerley LO. In vivo measurement of fatty acids and cholesterol synthesis using D2O and mass isotopomer analysis. Am J Physiol. 1994a;266:E699–E708. doi: 10.1152/ajpendo.1994.266.5.E699. [DOI] [PubMed] [Google Scholar]
- Lee WN, Bassilian S, Guo Z, Schoeller D, Edmond J, Bergner EA, Byerley LO. Measurement of fractional lipid synthesis using deuterated water (2H2O) and mass isotopomer analysis. Am J Physiol. 1994b;266:E372–E383. doi: 10.1152/ajpendo.1994.266.3.E372. [DOI] [PubMed] [Google Scholar]
- Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Ogawa W, Akimoto K, Inoue H, Miyake K, Furukawa K, Hayashi Y, Iguchi H, Matsuki Y, Hiramatsu R, Shimano H, Yamada N, Ohno S, Kasuga M, Noda T. PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J Clin Invest. 2003;112:935–944. doi: 10.1172/JCI18816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Ogawa W, Teshigawara K, Inoue H, Miyake K, Sakaue H, Kasuga M. Role of the insulin receptor substrate 1 and phosphatidylinositol 3-kinase signaling pathway in insulin-induced expression of sterol regulatory element binding protein 1c and glucokinase genes in rat hepatocytes. Diabetes. 2002;51:1672–1680. doi: 10.2337/diabetes.51.6.1672. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, Gonzalez FJ. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab. 2008;7:302–311. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992;258:766–770. doi: 10.1126/science.1439783. [DOI] [PubMed] [Google Scholar]
- Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, Romanelli AJ, Cline GW, Pongratz RL, Zhang XM, Choi CS, Coleman RA, Shulman GI. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Ono H, Shimano H, Katagiri H, Yahagi N, Sakoda H, Onishi Y, Anai M, Ogihara T, Fujishiro M, Viana AY, Fukushima Y, Abe M, Shojima N, Kikuchi M, Yamada N, Oka Y, Asano T. Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes. 2003;52:2905–2913. doi: 10.2337/diabetes.52.12.2905. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, Papademetris X, Rothman DL, Shulman GI. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104:12587–12594. doi: 10.1073/pnas.0705408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Laurent D, Rothman DL, Cline GW, Shulman GI. Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J Clin Invest. 1998;101:1203–1209. doi: 10.1172/JCI579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–6481. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008a;118:829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Girard J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008b;34:643–648. doi: 10.1016/S1262-3636(08)74599-3. [DOI] [PubMed] [Google Scholar]
- Raghow R, Yellaturu C, Deng X, Park EA, Elam MB. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab. 2008;19:65–73. doi: 10.1016/j.tem.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Saltiel AR. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 2001;104:517–529. doi: 10.1016/s0092-8674(01)00239-2. [DOI] [PubMed] [Google Scholar]
- Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha) J Biol Chem. 2003;278:9013–9018. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, Vottero A, Kanabar D, Charlton-Menys V, Durrington P, Soos MA, Carpenter TA, Lomas DJ, Cochran EK, Gorden P, O'Rahilly S, Savage DB. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. 1999;274:30028–30032. doi: 10.1074/jbc.274.42.30028. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- Song S, Zhang Y, Ma K, Jackson-Hayes L, Lavrentyev EN, Cook GA, Elam MB, Park EA. Peroxisomal proliferator activated receptor gamma coactivator (PGC-1alpha) stimulates carnitine palmitoyltransferase I (CPT-Ialpha) through the first intron. Biochim Biophys Acta. 2004;1679:164–173. doi: 10.1016/j.bbaexp.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, Sherwin R, Devaskar S, Lesche R, Magnuson MA, Wu H. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci U S A. 2004;101:2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, Kuhn CM, Rebuffe-Scrive M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism. 1995;44:645–651. doi: 10.1016/0026-0495(95)90123-x. [DOI] [PubMed] [Google Scholar]
- Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Kondo T, Sajan M, Luo J, Bronson R, Asano T, Farese R, Cantley LC, Kahn CR. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006;3:343–353. doi: 10.1016/j.cmet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Williams KJ. Molecular processes that handle -- and mishandle -- dietary lipids. J Clin Invest. 2008;118:3247–3259. doi: 10.1172/JCI35206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.