

Abstract

This manuscript describes the development and scope of the asymmetric rhodium-catalyzed [2+2+2] cycloaddition of terminal alkynes and alkenyl isocyanates leading to the formation of indolizidine and quinolizidine scaffolds. The use of phosphoramidite ligands proved crucial for avoiding competitive terminal alkyne dimerization. Both aliphatic and aromatic terminal alkynes participate well, with product selectivity a function of both the steric and electronic character of the alkyne. Manipulation of the phosphoramidite ligand leads to tuning of enantio- and product selectivity, with a complete turnover in product selectivity seen with aliphatic alkynes when moving from Taddol-based to biphenol-based phosphoramidites. Terminal and 1,1-disubstituted olefins are tolerated with nearly equal efficacy. Examination of a series of competition experiments in combination with analysis of reaction outcome shed considerable light on the operative catalytic cycle. Through a detailed study of a series of X-ray structures of rhodium(cod)chloride/phosphoramidite complexes, we have formulated a mechanistic hypothesis that rationalizes the observed product selectivity.
Keywords: Asymmetric Catalysis, [2+2+2] cycloaddition, Rhodium, Isocyanate, Alkyne, Alkene, Phosphoramidite
Introduction
Indolizidines and quinolizidines are common motifs found in many biologically active natural products isolated from arthropod, amphibian, plant, and marine sources (Figure 1).1 Interest in the synthesis of these compounds is threefold: scarcity of natural sources, pharmacological activity, and unique structural features. The assembly of these nitrogen heterobicycles is a proving ground for synthetic methodology.2 Classic syntheses rely on SN2 cyclization of amines, lactamization, and ring closing metathesis.3 New methodology showcased in the construction of these natural products depend on chiral pool substrates or require lengthy starting material syntheses.4,5,6,7,8,9 Therefore, the development of new, rapid, and enantioselective methods to generate nitrogen-containing bicycles would be a useful contribution to the synthetic community.
Figure 1.

Indolizidine and quinolizidine natural products.
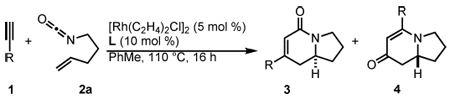
We envisioned the coupling of three separate π-components: C=N, C=C, and C≡C in a metal-catalyzed, [2+2+2] cycloaddition10 to construct these bicycles (Scheme 1). Due to their modest basicity, isocyanates11 have been shown to be competent partners in transition metal-catalyzed reactions.12 Yamazaki and Hoberg demonstrated that cobalt and nickel catalyze the [2+2+2] cycloaddition of an isocyanate and two equivalents of an alkyne to form 2-pyridone (eq 1, 2).13 Vollhardt later found that cobalt can couple an alkynyl isocyanate with an exogenous alkyne to form bicyclic pyridones (eq 3) and applied this methodology to the synthesis of camptothecin.14 More recently, rhodium, cobalt, nickel, and ruthenium have been shown to catalyze [2+2+2] cycloadditions of alkynes and isocyanates, forming pyridones.15 Although useful for the synthesis of heterocycles, these methods use two alkynes to form achiral cycloadducts. A notable exception is Tanaka’s use of a chiral rhodium complex to access pyridone atropisomers.16 It would be a clear benefit if an alkene could be incorporated in place of one of the alkynes to form an sp3 stereocenter in a reaction that could be rendered asymmetric.
Scheme 1.

[2+2+2] cycloadditions with alkynes and isocyanates.
 |
(1) |
 |
(2) |
 |
(3) |
 |
(4) |
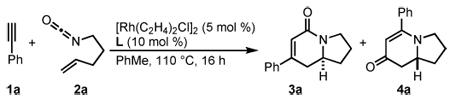
Prior to our work, three component [2+2+2] cycloadditions between an isocyanate, alkene, and alkyne were unknown.17 In 2006, we demonstrated that a rhodium(I)/tris(para-methoxyphenyl)phosphine complex catalyzes the [2+2+2] cycloaddition of 4-pentenyl isocyanate with symmetrical internal alkynes (eq 4).17a Our initial studies revealed that dialkyl alkynes provide lactam 3 while diaryl alkynes favor vinylogous amide 4, which arises from fragmentation of the isocyanate moiety (vide infra). To increase the utility of the reaction, we expanded the substrate scope to readily available terminal alkynes and rendered the transformation asymmetric with chiral phosphoramidite ligands.17b A variety of structurally and electronically different terminal alkynes and isocyanates are tolerated, enabling the synthesis of a wide range of indolizidines and quinolizidines. Herein, we disclose a full description of the development of this reaction, the effects of steric and electronic changes of the phosphoramidite ligand, single X-ray crystal analysis of six previously unpublished rhodium(cod)chloride/phosphoramidite complexes, and mechanistic insight into the rhodium-catalyzed [2+2+2] cycloaddition of terminal alkynes and alkenyl isocyanates.
Initial ligand screen
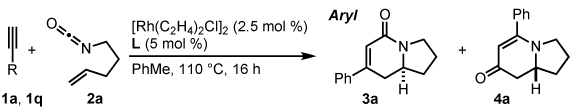
Our initial efforts to incorporate terminal alkynes began with an examination of the conditions that were effective for internal alkynes.17a We found that the ligand tris(para-methoxyphenyl)phosphine provides less than 20% of 3a and 4a in a 1:1 ratio (Table 1, entry 1); the low yield is due to the known dimerization of terminal alkynes (eq 5).18 BINAP gives no desired product while dppb affords only trace amounts of 3a, suggesting that a monodentate ligand is required (Table 1, entries 2, 3). MonoPhos (B1) affords a 32% yield and induces a modest 55% ee (entry 4). In addition to higher yields, phosphoramidites do not promote dimerization of terminal alkynes ((eq 6). Binol-based phosphoramidite B2 provides a slightly higher yield of vinylogous amide but poor enantioselectivity, while phosphite B3 shows lower yields than B1 and B2 (Table 2, entries 5, 6). Finally, we were delighted to find that Taddol-based phosphoramidite T1 increases the yield (80%), product selectivity toward vinylogous amide (1:7.0), and enantioselectivity (94%, entry 7). After modifying the amine, we found T2 was the optimal ligand for this transformation (entry 8).
Table 1.
Initial ligand screen.a
 | |||||
|---|---|---|---|---|---|
| entry | L | 3a:4ab | yield (%)c | ee (%)d3a | ee (%)d4a |
| 1 | P(4-MeO-C6H4)3 | 1:1 | <20 | - | - |
| 2 | BINAP | - | NR | - | - |
| 3 | dppb | >20:1 | <5 | - | - |
| 4 | B1 | 1:2.2 | 32 | 5e | 55e |
| 5 | B2 | 1:4.5 | 50 | 45 | 8 |
| 6 | B3 | 1:2.7 | 26 | 5e | 45e |
| 7 | T1 | 1:7.0 | 80 | 83 | 94 |
| 8 | T2 | 1:7.3 | 87 | 89 | 94 |
| 9 | T3 | 1:3.3 | 76 | 90 | 81 |
Reaction conditions: 1 (2 equiv), 2, [Rh(C2H4)2Cl]2 5 mol %, L 10 mol % in PhMe at 110 °C for 16 h.
Ratio determined by 1H NMR of crude reaction.
Combined isolated yield.
Determined by HPLC analysis on a chiral stationary phase.
Opposite enantiomer.
Table 2.
Terminal aryl alkyne scope.a
 | |||||
|---|---|---|---|---|---|
| entry | Ar | 3:4b | yield (%)c | ee (%) 3d | ee (%) 4d |
| 1 |  |
<1:20 | 72 | - | 94 |
| 2 | <1:20 | 70 | - | 90 | |
| 3 |  |
<1:20 | 64 | - | 94 |
| 4 | <1:20 | 78 | - | 87 | |
| 5 | 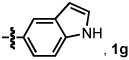 |
<1:20 | 65 | - | 90 |
| 6 | 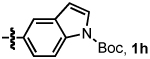 |
<1:20 | 85 | - | 91 |
| 7e | 1:9.0 | 64 | - | 86 | |
| 8 | 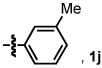 |
1:8.3 | 65 | - | 94 |
| 9 | 1:7.3 | 86 | 89 | 94 | |
| 10 | 1:3.8 | 65 | 93 | 91 | |
| 11 | 1:3.2 | 72 | 90 | 89 | |
| 12 | 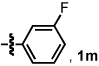 |
1:1.8 | 68 | 94 | 94 |
| 13 | 1:1.5 | 65 | 94 | 81 | |
| 14 | 2.5:1 | 50 | 94 | - | |
| 15 | <1:20 | 96 | - | 92 | |
See Table 1.
T3 used as ligand.

 |
(5) |
 |
(6) |
Scope of Terminal Alkynes
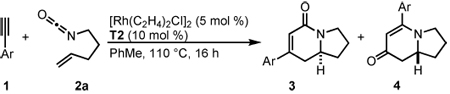
We investigated the scope of this reaction with terminal aryl alkynes (Table 2) and found electron-rich aryl alkynes provide exclusively vinylogous amide 4 in good yield and high enantioselectivity (entries 1–4).19 In addition, sterically bulky o-substituted aryl alkynes are tolerated (entry 3). Alkynes bearing a variety of heterocycles, including protected and unprotected indoles, readily participate, providing vinylogous amide with high enantioselectivity (entries 5–7). As the alkyne is made more electron-deficient, increasing amounts of lactam 3 are seen (entries 10–14). Vinylogous amide product formation is not limited to aryl acetylenes; 1-ethynylcyclohexene 1p gives 4p exclusively in a 96% yield and 92% ee (entry 15). The reaction clearly shows that substrate electronics can be used to tune product selectivity. A plot of electron-rich and deficient aryl alkynes versus Hammett σm/p values20 indicates a clear linear free energy relationship, demonstrating that the electronics of the alkyne can be used to tune product selectivity (Figure 2). In general, the electronics of the aryl alkyne bias product selectivity such that electron-rich favor vinylogous amide while strongly electron-deficient generate lactam.
Figure 2.

Linear free energy relationship of aryl acetylenes.

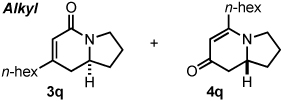
Alkyl alkynes shift product selectivity toward the lactam adduct with moderate to good yields (Table 3). An initial ligand screen using Taddols T1-T3 revealed piperidine-substituted T3 to be optimal (for a complete comparison, see Table 4). Enantioselectivities with alkyl alkynes and T3 are moderate (76–87%, entries 1–5, 7, 8). Biphenol based phosphoramidite A1 leads to an inversion in product selectivity with aliphatic alkynes, preferentially forming vinylogous amide adducts 4 with excellent enantioselectivities (88–94% ee, entries 1–4, 6, 8–13). Esters, amides, and silyl ethers are tolerated with moderate yields and enantioselectivities (entries 2, 5, 6 and 13). Interestingly, bulky cyclohexylacetylene 1w leads to an increased amount of vinylogous amide with T1 relative to other aliphatic alkynes (1.2:1), while ligand A1 generates the vinylogous amide with high product selectivity and excellent ee (14:1, 91% ee, entry 8). Noteworthy is the successful incorporation of 1,7-octadiyne and 1,8-nonadiyne, with ligand A1 affording vinylogous amides 4y and 4z in good yields and high ee’s (entries 10–11). On the other hand, 1,6-heptadiyne exclusively furnishes the substituted benzene arising from intramolecular cyclization of the diyne followed by incorporation of a second equivalent of alkyne. These results suggest that, except for the case of a 3-carbon tether linking the diyne, an isocyanate is incorporated in the initial oxidative cycloaddition in preference to an intramolecular coordination and activation of a second terminal alkyne.21
Table 3.
Scope of alkyl alkynes.a
 | |||||||
|---|---|---|---|---|---|---|---|
| T3 |
A1f |
||||||
| entry | R | 3:4b | yield (%)c | ee (%) 3/4d | 3:4b | yield (%)c | ee (%) 4d |
| 1 | n-hex, 1q | 5.0:1 | 78 | 80/33 | 1:6.0 | 66 | 91e |
| 2 | (CH2)4CO2Me, 1r | 5.8:1 | 65 | 80/- | 1:5.0 | 66 | 90e |
| 3 | (CH2)2Ph, 1b | >20:1 | 47 | 84/- | 1:5.0 | 56 | 91e |
| 4 | Bn, 1s | >20:1 | 50 | 84/- | 1:3.0 | 52 | 90e |
| 5 | (CH2)2OTBS, 1t | >20:1 | 65 | 87/- | |||
| 6 | (CH2)4OTBS, 1u | 1:5.0 | 62 | 90e | |||
| 7 | CH2OMe, 1v | >20:1 | 46 | 76/- | |||
| 8 | Cy, 1w | 1.2:1g | 82g | 77/95g | 1:14 | 86 | 91e |
| 9 | (CH2)4CI, 1x | 1:5.0 | 57 | 94e | |||
| 10 | (CH2)4CCH, 1y | 1:7.2 | 60 | 90e | |||
| 11 | (CH2)5CCH, 1z | 1:5.0 | 55 | 91e | |||
| 12 | t-Bu, 1aa | 1:10 | 67 | 79e | |||
| 13 | 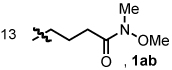 |
1:5 | 54 | 90e | |||
See Table 1.
Reaction conditions: 1 (2 equiv), 2a, [Rh(C2H4)2CI]2 2.5 mol %, L 5 mol % in PhMe at 110 °C for 16h.
T1 used as ligand.
Table 4.
Electronic and steric investigation of phosphoramidites with alkyl and aryl acetylenes.a
 |
 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | ligand substituent | 3a:4ab | yield (%)c | ee (%) 3ad | ee (%) 4ad | 3q:4qb | yield (%)c | ee (%) 3qd | ee (%) 4qd | ||
| L | R1 | R2 | |||||||||
| 1 | T1 | Ph | 1:7.0 | 80f | 83 | 94 | 3.2:1 | 81f | 81 | 73 | |
| 2 | T2 | Ph | 1:7.3 | 87f | 89 | 94 | 2.4:1 | 80f | 83 | 70 | |
| 3 | T3 | Ph | 1:3.3 | 76f | 90 | 81 | 5.0:1 | 78f | 80 | 33 | |
| 4 | T4 | Ph | 1:2.0 | 14 | - | - | >20:1 | 5 | - | - | |
| 5 | T5 | 1:6.5 | 84 | 91 | 95 | 2.5:1 | 55 | 87 | 76 | ||
| 6 | T6 | 1:2.8 | 68 | 87 | 85 | 8.3:1 | 68 | 83 | 46 | ||
| 7 | T7 | 1:>20 | 67 | 69 | 84 | 1:1 | 31 | 79 | 74 | ||
| 8 | T8 |  |
1:5.6 | 84 | 96 | 97 | 4.0:1 | 77 | 90 | 74 | |
| 9 | T9 | 1:1.6 | 75 | 94 | 94 | 12.5:1 | 72 | 82 | 51 | ||
| 10 | B1 | H | 1:2.2 | 32f | 5 | 55e | 1:1.9 | 19 | 14e | 67e | |
| 11 | B2e | Me | 1:4.5 | 50f | 45e | 8 | 1:4.2 | 34 | 18 | 59 | |
| 12 | B4 | TMS | 1:7.1 | 74 | 36 | 45e | 1:3.6 | 53 | 32e | 95e | |
| 13 | B5 | TMS | 1:12.5 | 77 | 37 | 55e | 1:3.6 | 51 | 50e | 95e | |
| 14 | B6 | TMS | 1:12 | 82 | 43 | 55e | 1:3.2 | 63 | 47e | 92e | |
| 15 | A1 | 1:>20 | 77 | 41 | 3e | 1:6.2 | 75 | 8 | 91e | ||
Reaction conditions: 1 (2 equiv), 2a, [Rh(C2H4)2CI]2 2.5 mol %, L 5 mol % in PhMe at 110 °C for 16 h.
See Table 1.
Reaction conditions: 1 (2 equiv), 2a, [Rh(C2H4)2CI]2 5 mol %, L 10 mol % in PhMe at 110 °C for 16 h.
Ligand Exploration
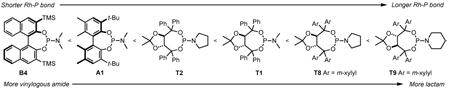
An examination of the structure and electronics of the phosphoramidite revealed that the reaction is sensitive to both (Table 4).22 In general, Taddol-based phosphoramidites (T1-T9) give more lactam product with aliphatic alkynes, while Binol and biaryl phosphoramidites (B1-B6, A1) favor vinylogous amide. Interestingly when the amine of Taddol ligands is changed from pyrrolidine T2 to piperidine T3, product selectivity shifts in favor of lactam 3 from 1:7.3 to 1:3.3 with phenyl acetylene and 2.4:1 to 5:1 with 1-octyne (entries 2, 3), and this trend can be extended to all of the Taddol ligands (entries 1–9). We suspect that it is the size of the amine that is affecting product selectivity, as the basicities of pyrrolidine (11.27 pKa) and piperidine (11.22 pKa) are very similar.23 Furthermore, very large amines, such as dicyclohexyl amine, completely favor the lactam product, albeit in low yield (entry 4). Interestingly, the size of the amine has less effect on product selectivity with Binol/biaryl phosphoramidites (entries 12–14). These results show that the sterics of the phosphoramidite play a major role in determining product selectivity.
To investigate the effect of ligand electronics, we synthesized a ligand (T7) that differs from T6 only in the aryl substituents: p-MeC6H4 vs p-CF3C6H4 (entry 7). This more electron-deficient ligand increases product selectivity for vinylogous amide with both aryl (1:>20 from 1:2.8) and alkyl alkynes (1:1 from 8.3:1), demonstrating that the ligand electronics can enhance product selectivity. Also, electron-rich m-xylyl Taddol T9 gives a relative increase in the amount of lactam product formed with aryl (1:1.6) and alkyl acetylenes (12.5:1, entry 9).
Taddol ligands provide the best enantioselectivities for aryl alkynes (81–97%, entries 1–9) with T8 being optimal, while Binol and biaryl ligands give higher enantioselectivity for alkyl alkynes (59–95%, entries 10–14) with B4 and B5 being the highest. Substitution at the 3,3’-positions of the Binol/biaryl proves to be essential to improving the enantioselectivity with alkyl acetylenes. Biaryl A1 provides the highest product ratio (1:6.2) and excellent enantioselectivity (91%) for vinylogous amide with alkyl acetylenes (entry 15). Finally, A1 made possible the construction of indolizidine (−)-209D24 in a 5-step synthesis from commercially available starting materials, using 4q as the key intermediate.17e

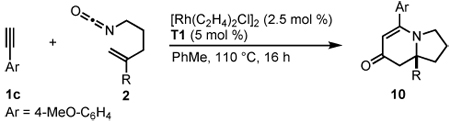
Alkenyl Isocyanate Scope
The successful incorporation of homologous alkenyl isocyanates in this transformation would allow access to quinolizidine natural products. Indeed a longer tether length is tolerated, but in lengthening the tether, we observe a significant amount of 2-pyridone 6 side product (eq 7). A further increase in tether length to heptenyl isocyanate does not provide the desired 1-azabicyclo[5.4.0]undecane, forming only 2-pyridone 7 (eq 8). The synthetic utility of this approach to quinolizidine motifs was demonstrated in the 4-step synthesis of (+)-lasubine II.17b
 |
(7) |
 |
(8) |
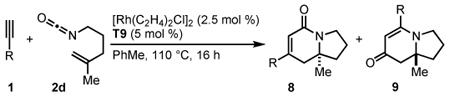
The cycloaddition of 1,1-disubstituted alkenyl isocyanate 2d with a variety of conjugated aryl and alkenyl alkynes provides indolizinones bearing a tetrasubstituted stereocenter (Table 5). Electron-rich alkyne 1d gives exclusively vinylogous amide 9d in 80% yield with 91% ee (entry 1). Heterocyclic and conjugated terminal alkynes react smoothly to afford 9 in good yields and enantioselectivity (entries 2–6). We see a general trend that as the aryl alkyne becomes more electron-deficient, increasing amounts of the lactam 8 are generated (entry 7). This is consistent with the results seen with 4-pentenyl isocyanate 2a (vide supra).
Table 5.
Conjugated alkynes with 1,1-disubstituted alkenyl isocyanates.a
Alkyl alkynes and 1,1-disubstituted alkenyl isocyanates provide mostly lactam 8 with enantioselectivities ranging from 91% to 95% ee (Table 6). Functional groups including protected alcohols, halogens, and internal alkynes are tolerated (entries 5–8). Silyl alkynes fail to participate in this reaction; this finding allowed the chemoselective incorporation of mono-protected 1,6-diynes (entry 8).
Table 6.
Alkyl alkynes with 1,1-disubstituted alkenyl isocyanates.a
Variations of the 1,1-disubstituted alkene furnish tetrasubstituted indolizinones with a range of substitution at the 9 position (Table 7). Substrates bearing primary alkyl groups on the olefin react smoothly with high enantioselectivity (entries 1–5). However, the reaction is sluggish with secondary alkyl substituents and increasing amounts of pyridone are seen (entries 6, 7). Protected alcohols, halogens, and alkenes are tolerated in the reaction, providing handles for further chemical manipulation (entries 9–11).
Table 7.
Scope of 1,1-disubstituted alkenyl isocyanates.
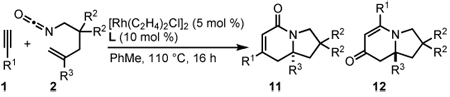
Substitution on the alkenyl isocyanate tether is tolerated in the cycloaddition (Table 8). Diethyl malonate derived alkenyl isocyanate 2p provides yields and enantioselectivities comparable to 2a (entries 1, 2). Geminal dimethyl substituents on the tether react smoothly providing good yields and selectivities with T1 (entries 3, 4). This backbone substitution is also tolerated with 1,1-disubstituted alkenes, affording optimal selectivities when ligand A1 is used (entry 6). Oxygen substitution in the tether provides 13 in moderate yields and excellent enantioselectivities up to 96% ee (eq 9).
Table 8.
Substitution on the alkyl tether.a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | L | R1 | R2 | R3 | 11:12b | yield (%)c | ee (%) 11/12d |
| 1 | T1 | 4-MeO-C6H4 1d | CO2Et | H, 2p | 1:15 | 88 | 74/91 |
| 2 | B4 | 4-MeO-C6H4 1d | CO2Et | H, 2p | 1:1 | 69 | <5/16e |
| 3 | T1 | 4-MeO-C6H4 1d | Me | H, 2q | 1:>20 | 72 | -/83 |
| 4 | B4 | 4-MeO-C6H4 1d | Me | H, 2q | 1:5.0 | 62 | -/30e |
| 5 | B4 | PMB, 1af | Me | Me, 2r | 1:2.2 | 65 | 52/94e |
| 6 | A1 | PMB, 1af | Me | Me, 2r | 1:4.0 | 72 | 56/82e |
See Table 1.
Reaction conditions: 1 (2 eq), 2, [Rh(C2H4)2Cl]2 2.5 mol %, L 5 mol % in PhMe at 110 °C for 16 h.
 |
(9) |
Mechanistic Investigation
The proposed catalytic cycle for the [2+2+2] cycloaddition of alkenyl isocyanates with exogenous alkynes to form lactam, vinylogous amide, 2-pyridone, and 4-pyridone products is described in Scheme 2. All products may be accessed from a common coordination of the alkyne and isocyanate to form the Rh(I)/phosphoramidite complex (I). From this complex, oxidative cyclization and C–C bond formation provides IIa.25 Migratory alkene insertion into IIa generates seven-membered rhodacycle IIIa, which reductively eliminates to generate lactam 3. Alternatively, the rhodium(I) coordination complex (I) can oxidatively cyclize to form IIb, where C–N bond formation occurs first. However, the alkene is unable to insert into the resultant rhodium(III) species IIb presumably due to a strained, bridged geometry in the transition state. A CO migration via IIIb then takes place to form IVb.26 At this point, migratory alkene insertion gives rhodacycle Vb, and reductive elimination provides vinylogous amide 4. Additionally, formation of 2-pyridone can be generated from an exogenous alkyne intercepting either IIa or IIb.17c 4-pyridone can be formed from alkyne interception of rhodacycle IVb, and its isolation from a similar catalytic system is further evidence for the formation of IVb.27
Scheme 2.

Proposed catalytic cycle.
Another catalytic cycle can be imagined in which the alkene of the alkenyl isocyanate coordinates to the rhodium (VI) in lieu of an alkyne (Scheme 3). Such coordination would lead to oxidative cyclization, C–N bond formation, and set the stereochemistry for both lactam and vinylogous amide products in the formation of the bicyclic rhodacycle VII. Migratory alkyne insertion would give IIIa and subsequent reductive elimination would generate lactam 3. Alternatively, a CO migration via VIII could take place prior to alkyne insertion making bicycle IX, which would then undergo alkyne insertion (X) and reductive elimination to form vinylogous amide 4.
Scheme 3.

Alternative catalytic cycle
Several observations suggest that this alternative mechanism is not the operative catalytic cycle. First, two competition experiments were conducted between mono (2a) and 1,1-disubstituted (2d) alkenyl isocyanates (eqs 10, 11) in the presence of either aryl (1d) or alkyl (1s) acetylenes. These yield a 1:1 ratio of vinylogous amide or lactam products respectively. We would predict that isocyanate 2a should react at a different rate than 2d leading to an unequal product mixture.28 Because olefin substitution has no effect on the ratio of products obtained, we propose that the olefin is not involved in the turnover-limiting step. Second, higher reaction concentrations (0.12 M vs 0.04 M) (eq 12) or bulky alkene substituents increase pyridone formation.29 Higher concentrations favor intermolecular interception of metallacycle IIa/b by a second equivalent of alkyne over intramolecular migratory insertion of the tethered alkene. Furthermore, bulky substituents inhibit migratory insertion of the alkene favoring intermolecular insertion of a second alkyne to form pyridone. These results suggest that the alkyne and isocyanate are the first components to oxidatively cyclize. This is further supported by the observation that 1,7-octadiyne 1y and 1,8-nonadiyne 1z furnish vinylogous amides 4y and 4z showing an apparent kinetic preference for intermolecular coordination and activation of the isocyanate in spite of the entropically favored intramolecular coordination of the second terminal alkyne (Table 3, entries 10, 11). While individually the preceding pieces of evidence do not eliminate the alternate catalytic cycle shown in Scheme 3, their aggregate suggests them to be unlikely. Finally, the vinylogous amide and lactam products obtained with the same Taddol phosphoramidite are opposite major enantiomers. This suggests that both products are not formed from rhodacycle VII as stereoinduction would occur prior to CO migration. Considering these observations, we propose that the alkene is the last π-component to be incorporated and the operative catalytic cycle is likely that shown in Scheme 2.30
 |
(10) |
 |
(11) |
 |
(12) |
We see remarkable regioselectivity in this rhodium-catalyzed cycloaddition, where the vinyl hydrogen is α to the carbonyl in all terminal alkyne products. As part of our ongoing efforts to explain the regio- and product selectivity of the reaction, single crystal X-ray analyses31 of rhodium(I)(cod)chloride/phosphoramidite complexes32 were undertaken. In each of these structures, rhodium is in a square planar geometry and a strong phosphoramidite trans influence is displayed. Generally, Taddol-based phosphoramidites have a weaker trans influence than Binol/biaryl phosphoramidite as seen by the Rh–alkene distance (Table 9).
Table 9.
Selected bond lengths of rhodium(I)(cod)chloride/phosphoramidite complexes.
 | ||||||
|---|---|---|---|---|---|---|
| Trans to P | Trans to Cl | |||||
| Ligand | Rh–P | Rh–Cl | Rh–C=C* | C=C | Rh-C=C* | C=C |
| T9 | 2.2703(4) | 2.3654(4) | 2.132 | 1.409(2) | 1.994 | 1.413(3) |
| T8 | 2.2688(6) | 2.3765(6) | 2.131 | 1.377(4) | 1.992 | 1.395(4) |
| T1 | 2.2648(6) | 2.3777(6) | 2.140 | 1.376(4) | 1.997 | 1.417(3) |
| T2 | 2.2547(3) | 2.3842(3) | 2.152 | 1.3781(18) | 1.995 | 1.4131(16) |
| A1 | 2.2451(4) | 2.3485(4) | 2.162 | 1.369(3) | 2.013 | 1.409(2) |
| B4 | 2.2388(9) | 2.3455(8) | 2.151 | 1.365(5) | 2.003 | 1.396(5) |
Bond length from Rh to centroid of alkene
After examining the X-ray crystal structures, we hypothesize that the steric environment created by monodentate, C2-symmetric phosphoramidite ligands explains the exceptional regioselectivity in the cycloaddition. As we have depicted in Figure 3, the phosphoramidite ligands sterically hinder one face of the square planar rhodium(I) complex. One of the m-xylyl groups sits above the Rh square plane in the structure of T8•Rh(cod)Cl, with the opposite side of the complex much more exposed. A naphthyl ring plays this role in the B4•Rh(cod)Cl complex, reinforced by the TMS group. These effects are also evident in other Rh•phosphoramidite crystal structures.32 We propose that this hindrance biases the coordination of the alkyne and isocyanate such that the sterically smaller substituents are in the same hemisphere of the square plane as shown in I (Fig 2). We suggest that the alkynes displace the ethylene prior to the association of the isocyanate, and as the phosphoramidite has the greater trans influence than chloride, we predict that the isocyanate coordinates trans to the phosphoramidite. From this single coordination, oxidative cyclization accounts for the single regioisomer of the lactam and vinylogous amide. Thus, we propose that regioselectivity is controlled predominately by the sterics of the phosphoramidite ligand.
Figure 3.

X-ray crystal structures of Rh(cod)Cl/phosphoramidite complexes
It may be argued that the alkyne and isocyanate coordinate to the metal parallel to the square plane.33 However, Wakatsuki and Yamazaki’s calculations of cobaltacyclopentadienes34 and ground state X-ray crystal analysis of other d8 complexes35 suggest that an orthogonal coordination of π-components is operative because steric repulsion between π-components is minimized and better back donative stabilization is possible when orthogonal. Wakatsuki-Yamazaki’s regioselectivity model is based on observations and calculations of cobaltacyclopentadiene formation (Scheme 4). The major product results from a head to tail orientation of the large alkyne substituents to form cobaltacyclopentadiene 14. We do not observe either lactam or vinylogous amide products derived from such an orientation. Wakatsuki and Yamazaki claim the minor product 15 is kinetically favored over 16 due to steric interactions between large alkyne substituents as shown in TSIII (Scheme 4). When the alkynes are not head to tail, they suggest that the large substituents prefer to be α to the metal. This model accounts for the regioselectivity seen with rhodacycle IIa, which generates lactam (Scheme 2). Using this argument, one would predict that larger alkyne substituents would favor lactam; however, more vinylogous amide is seen (Table 3, entry 1 vs 8). Finally, this model does not account for vinylogous amide formation, since the larger groups are β to the metal in rhodacycle IIb (Scheme 2). As product selectivity could not be completely explained by substrate steric control alone, we sought another model to rationalize selectivity based on stereoelectronic effects.
Scheme 4.

Regioselectivity models for metallacycle formation.
Stockis and Hoffman in their theoretical treatise on metallacyclopentane formation discuss the effects that polarized π components have on regioselectivity in the absence of steric contributions.36 As a result of their calculations and subsequent correlation with experimental data, they propose a model based on stereoelectronic effects to explain regioselectivity. They hypothesize that polarized π components oxidatively cyclize so that the greatest LUMO coefficient is β to the metal, due to enhanced π* mixing with filled dxy orbitals in the transition state (Scheme 4). In our system, we observe a propensity for lactam formation when the sterics of the phosphoramidite and alkyne substrate decrease. Presumably, this is due to the large LUMO coefficient of the isocyanate at the β position, lowering the activation energy for TSIV en route to metallacycle IIa (Scheme 5). However, it does not explain the regioselectivity of the isocyanate in TSV for vinylogous amide formation or lactam products derived from electron-rich aryl acetylenes or alkyl acetylenes. In each of the cases discussed, the largest LUMO of one of the π-components is α to the metal in the resultant metallacycle contrary to Stockis-Hoffman.
Scheme 5.

Model to rationalize product selectivity.
As neither the Wakatsuki-Yamazaki steric nor the Stockis-Hoffman stereoelectronic model adequately explain the product selectivity seen in the cycloaddition, we hypothesize that the reaction is controlled primarily by sterics and enhanced or diminished by electronic factors (Scheme 5). Single regioisomeric products are explained by alkyne and isocyanate coordination dictated by phosphoramidite sterics, placing the small substituent of each π-component in a cis orientation. The two π-components are coordinated orthogonal to the square plane, as proposed by Wakatsuki-Yamazaki and corroborated by crystal structures.37 To generate rhodacycle IIa en route to lactam from the orthogonal coordination seen in Ia, the CO of the isocyanate and the terminal C–H of the alkyne must bend away from the Rh center and toward each other passing through TSIV forming the C–C bond. Similarly, the N–R2 group of the isocyanate and the C–R1 bond of the terminal alkyne bend away from the Rh center, forming the C–N bond via TSV, en route to rhodacycle IIb in the vinylogous amide pathway.
In terms of product selectivity, the LUMO of the isocyanate favors formation of lactam, but is overridden by sterics of the substrate and ligand. Smaller alkynes (1-octyne, Table 3, entry 1) generate lactam, while larger alkynes (cyclohexylacetylene, Table 3, entry 7) produce vinylogous amide due to ligand-alkyne interactions in the transition state (Scheme 5, TSIV). Alkyne electronics modify product selectivity as seen in Figure 2. Electron-deficient aryl alkynes override steric control to favor lactam, as the alkyne and isocyanate LUMOs are β to the metal in TSIVb. Electron-rich aryl alkynes generate vinylogous amide due to unfavorable steric interactions between the alkyne and ligand (TSIV), and this selectivity is reinforced by the alkyne LUMO because it is β to the metal in TSVa.
These unfavorable alkyne-phosphoramidite interactions are augmented by changes in the Rh–P bond length, where a shorter bond favors vinylogous amide while a longer bond favors lactam (Table 9). Electron-deficient phosphoramidites have shorter Rh–P bond lengths, which exacerbate the ligand alkyne interaction in the transition state leading to more vinylogous amide. On the other hand, electron-rich phosphoramidites have longer Rh–P bonds, which alleviate the steric interactions between the alkyne and ligand to generate more lactam. Additionally, we have seen that in the Taddol series large amines favor lactam product presumably by increasing the Rh–P bond length.
Stereochemical Model
A proposed model that rationalizes the observed enantioselectivity is depicted in Scheme 6. In metallacycle IIa leading to lactam, we postulate that there are two factors controlling enantioinduction: facial selectivity of the alkene dictated by the geometry of the tether and facial selectivity at rhodium as influenced by the phosphoramidite ligand. Each of the rhodium(cod)chloride•phosphoramidite crystal structures show that the ligand hinders one of the open faces on rhodium and we suggest this facial hindrance is present in the rhodium(III) intermediates. For migratory insertion to occur the alkene must be syn-coplanar with the Rh—N bond, and as a result, only four alkene coordination scenarios can be envisioned. In the first two scenarios, IIaA and IIaB, the alkene has to coordinate to the hindered rhodium face, which is disfavored. Additionally, IIaA requires the alkene tether to be in an undesirable twist-boat conformation further disfavoring it; in IIaB the tether can adopt a chair conformation and 1,2-migratory insertion leads to the minor enantiomer. In the second two scenarios, IIaC and IIaD, we propose that the alkene coordinates to the less hindered face of rhodium. Alkene coordination is disfavored in IIaC because the tether would be in a strained twist-boat conformation. On the other hand, the tether shown in metallacycle IIaD is in a favorable chair conformation and migratory insertion provides the major lactam enantiomer. This model for lactam enantioselectivity rationalizes the correct absolute stereochemistry observed; however, it assumes no rearrangement of the unobservable rhodium(III) intermediates.

Rationalizing enantioselectivity for vinylogous amide is more difficult because enantioinduction presumably occurs after several ligand rearrangements on rhodium(III) species. CO migration from IIb probably occurs from amide bond cleavage and amine migration to the unhindered open coordination site, resulting in metallacycle IIIbA,B. We speculate that the alkenyl carbon migrates to the CO ligand as seen in IIIbA leading to metallacycle IVbA. However, coordination of the alkene and migratory insertion of this metallacycle leads to the minor enantiomer observed for vinylogous amide. The major enantiomer could be formed by CO migration onto the alkenyl carbon (IIIbB), but such a migration is unprecedented.26 Rapid rearrangement of IVbA to IVbB and subsequent alkene insertion would provide the major observed enantiomer. This rearrangement is potentially influenced by steric interactions between the alkenyl tether and ligand disfavoring IVbA.38 As these models are for an unobservable rhodium(III) species, we can only speculate as to the actual ligand rearrangements. These models are tentative and we are currently undertaking computational studies to rationalize enantioinduction.
Conclusion
In conclusion, we have developed a rapid, efficient, and highly enantioselective cycloaddition of alkenyl isocyanates and exogenous alkynes to access indolizidine and quinolizidine cores found in biologically relevant natural products. We have found that single regioisomeric products are obtained in good yields and excellent enantioselectivities. X-ray crystal analysis of rhodium(cod)chloride/phosphoramidite complexes has led us to a mechanistic hypothesis that accounts for the single regioisomeric products obtained. In conjunction with competition experiments and pyridone formation, we have suggested a catalytic cycle that accounts for both the lactam and vinylogous amide products. We conclude that the regio- and product selectivity are controlled primarily by the sterics of the phosphoramidite ligand and substrate, while the electronics of both either enhance or diminish product selectivity.
Supplementary Material
Acknowledgements
We thank NIGMS (GM80442) for support. DMD thanks NSF-LSAMP Bridge to the Doctorate Program for support. SP thanks the FQRNT for a postdoctoral fellowship. RTY thanks Lilly for a graduate fellowship. KMO and DMD thank Oren Anderson and Susie Miller (CSU) for support and guidance. TR thanks the Monfort Family Foundation for a Monfort Professorship. We thank Johnson Matthey for a loan of rhodium salts.
Footnotes
Supporting information available. Experimental procedures and spectral data for all new compounds (PDF), as well as cif files for all new crystal structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Daly JW. J. Med. Chem. 2003;46:445–452. doi: 10.1021/jm0204845. [DOI] [PubMed] [Google Scholar]; (b) Daly JW, Spande TF, Garraffo HM. J. Nat. Prod. 2005;68:1556–1575. doi: 10.1021/np0580560. [DOI] [PubMed] [Google Scholar]
- 2.For reviews of recent syntheses, see: Michael JP. Nat. Prod. Rep. 2000;17:579–602. doi: 10.1039/a904849i. Michael JP. Nat. Prod. Rep. 2002;20:458–475. doi: 10.1039/b208137g. Michael JP. Nat. Prod. Rep. 2005;22:603–626. doi: 10.1039/b413748p. Michael JP. Nat. Prod. Rep. 2007;24:191–222. doi: 10.1039/b509525p. Michael JP. Nat. Prod. Rep. 2008;25:139–165. doi: 10.1039/b612166g.
- 3.For selected syntheses: Kim G, Jung S-d, Kim W-j. Org. Lett. 2001;3:2985–2987. doi: 10.1021/ol0163171. Toyooka N, Fukutome A, Nemoto H, Daly JW, Spande TF, Garraffo HM, Kaneko T. Org. Lett. 2002;4:1715–1717. doi: 10.1021/ol025775m. Hermet J-PR, McGrath MJ, O’Brien P, Porter DW, Gilday J. Chem. Commun. 2004:1830–1831. doi: 10.1039/b406632d. Zaja M, Blechert S. Tetrahedron. 2004;60:9629–9634. Toyooka N, Kawasaki M, Nemoto H, Awale S, Tezuka Y, Kadota S. Synlett. 2005:3109–3110. Toyooka N, Dejun Z, Nemoto H, Garraffo HM, Spande TF, Daly JW. Tetrahedron Lett. 2006;47:577–580.
- 4.For tandem conjugate additions, see :Back TG, Nakajima K. J. Org. Chem. 1998;63:6566–6571. Ma D, Zhu W. Org. Lett. 2001;3:3927–3929. doi: 10.1021/ol016802w. Back TG, Hamilton MD, Lim VJJ, Parvez M. J. Org. Chem. 2005;70:967–972. doi: 10.1021/jo048284j. Cai G, Zhu W, Ma D. Tetrahedron. 2006;62:5697–5708.
- 5.For cascade reactions, see:Amorde SM, Judd AS, Martin SF. Org. Lett. 2005;7:2031–2033. doi: 10.1021/ol050544b. Padwa A, Bur SK. Tetrahedron. 2007;63:5341–5378. doi: 10.1016/j.tet.2007.03.158.
- 6.For the use of dihydro-4-pyridones as synthons, see: Joseph S, Comins DL. Curr. Opin. Drug Discovery Dev. 2002;5:870. Young DW, Comins DL. Org. Lett. 2005;7:5661–5664. doi: 10.1021/ol052313a.
- 7.For intramolecular Schmidt reaction, see: Aubé J, Milligan GL. J. Am. Chem. Soc. 1991;113:8965–8966. Gracis V, Zeng Y, Desai P, Aubé J. Org. Lett. 2003;5:4999–5001. doi: 10.1021/ol035965c.
- 8.For a three component coupling using silyl dithianes, see:Smith AB, III, Kim D-S. Org. Lett. 2004;6:1493–1495. doi: 10.1021/ol049601b. Smith AB, III, Kim D-S. J. Org. Chem. 2006;71:2547–2557. doi: 10.1021/jo052314g.
- 9.For a catalytic, asymmetric aza-Diels-Alder reaction, see:García-Mancheño O, Gómez-Arrayás R, Carretero JC. J. Am. Chem. Soc. 2004;126:456–457. doi: 10.1021/ja038494y. García-Mancheño O, Gómez-Arrayás R, Adrio J, Carretero JC. J. Org. Chem. 2007;72:10294–10297. doi: 10.1021/jo702076j.
- 10.For reviews on [2+2+2] cycloadditions of alkynes see:Saito S, Yamamoto Y. Chem. Rev. 2000;100:2901–2915. doi: 10.1021/cr990281x. Agenet N, Busine O, Slowinski F, Gandon V, Aubert C, Malacria M. Org. React. 2007;68:1–302. Galan BR, Rovis T. Angew. Chem. Int. Ed. 2009;48:2830–2834. doi: 10.1002/anie.200804651. For reviews on [2+2+2] cycloadditions of alkynes and nitrogen moieties see:Varela JA, Saá C. Chem. Rev. 2003;103:3787–3801. doi: 10.1021/cr030677f. Heller B, Hapke M. Chem. Soc. Rev. 2007;36:1085–1094. doi: 10.1039/b607877j. Chopade PR, Louie J. Adv. Synth. Catal. 2006;348:2307–2327.
- 11.Ozaki S. Chem. Rev. 1972;72:457–496. [Google Scholar]
- 12.Braunstein P, Nobel D. Chem. Rev. 1989;89:1927–1945. [Google Scholar]
- 13.(a) Hong P, Yamazaki H. Synthesis. 1977;1:50–52. [Google Scholar]; (b) Hong H, Yamazaki H. Tetrahedron Lett. 1977;15:1333–1336. [Google Scholar]; (c) Hoberg H, Oster BW. Synthesis. 1982:324–325. [Google Scholar]; (d) Hoberg H, Oster BW. J. Organomet. Chem. 1983;234:C35–C38. [Google Scholar]; (e) Hoberg H, Oster BW. J. Organomet. Chem. 1983;252:359–364. [Google Scholar]
- 14.(a) Earl RA, Vollhardt KPC. J. Am. Chem. Soc. 1983;105:6991–6993. [Google Scholar]; (b) Earl RA, Vollhardt KPC. J. Org. Chem. 1984;49:4786–4800. [Google Scholar]
- 15.Cobalt: Diversi P, Ingrosso G, Lucherini A, Malquori S. J. Mol. Catal. 1987;40:267–280. Bonaga LVR, Zhang H-C, Moretto AF, Ye H, Gauthier DA, Li J, Leo GC, Maryanoff BE. J. Am. Chem. Soc. 2005;127:3473–3485. doi: 10.1021/ja045001w. Nickel: Takahashi T, Tsai F, Li Y, Wang H, Kondo Y, Yamanaka M, Nakajima K, Kotora M. J. Am. Chem. Soc. 2002;124:5059–5067. doi: 10.1021/ja017507+. Duong HA, Cross MJ, Louie J. J. Am. Chem. Soc. 2004;126:11438–11439. doi: 10.1021/ja046477i. Duong HA, Louie J. J. Organomet. Chem. 2005;690:5098–5104. Duong HA, Louie J. Tetrahedron. 2006;62:7552–7559. Ruthenium: Yamamoto Y, Takagishi H, Itoh K. Org. Lett. 2001;3:2117–2119. doi: 10.1021/ol016082t. Yamamoto Y, Kinpara K, Saigoku T, Takagishi H, Okuda S, Nishiyama H, Itoh K. J. Am. Chem. Soc. 2005;127:605–613. doi: 10.1021/ja045694g. Rhodium:Flynn ST, Hasso-Henderson SE, Parkins AW. J. Mol. Catal. 1985;32:101–105. Kondo T, Nomura M, Ura Y, Wada K, Mitsudo T. Tetrahedron Lett. 2006;47:7107–7111.
- 16.(a) Tanaka K, Wada A, Noguchi K. Org. Lett. 2005;7:4737–4739. doi: 10.1021/ol052041b. [DOI] [PubMed] [Google Scholar]; (b) Tanaka K. Synlett. 2007:1977–1993. [Google Scholar]; (c) Tanaka K, Takahashi Y, Suda T, Hirano M. Synlett. 2008:1724–1728. [Google Scholar]
- 17.(a) Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:2782–2783. doi: 10.1021/ja057803c. [DOI] [PubMed] [Google Scholar]; (b) Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:12370–12371. doi: 10.1021/ja064868m. [DOI] [PubMed] [Google Scholar]; (c) Lee EE, Rovis T. Org. Lett. 2008;10(6):1231–1234. doi: 10.1021/ol800086s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yu RT, Rovis T. J. Am. Chem. Soc. 2008;130:3262–3263. doi: 10.1021/ja710065h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yu RT, Lee EE, Malik G, Rovis T. Angew. Chem. Int. Ed. 2009;48:2379–2382. doi: 10.1002/anie.200805455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Keller Friedman R, Rovis T. J. Am. Chem. Soc. 2009;131:10775–10782. doi: 10.1021/ja903899c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Albano P, Aresta M. J. Organomet. Chem. 1980;190:243–246. [Google Scholar]; (b) Schäfer H, Marcy R, Rüping T, Singer H. J. Organomet. Chem. 1982;240:17–25. [Google Scholar]; (c) Ohshita J, Furumori K, Matsuguchi A, Ishikawa M. J. Org. Chem. 1990;55:3277–3280. [Google Scholar]
- 19.In our experience, this chemistry has proven to be very robust. Roughly 25% of all reactions described herein have been conducted multiple times often by different coworkers. We have found that yields vary slightly between runs (~5%) while product selectivities (± 0.2, e.g. 7.0:1 to 7.2:1) and enantioselectivities (± 0.5%) change little. Product selectivity is determined by analysis of 1H NMR spectra of unpurified reaction mixtures, and can be complicated by unreacted starting material and symmetrical ureas arising from partial hydrolysis of the isocyanate.
- 20.Hansch C, Leo A, Taft RW. Chem. Rev. 1991;91:165. [Google Scholar]
- 21.TMS-acetylene does not participate under these reaction conditions.
- 22.During this study, we found that catalyst loading could be reduced from 10 mol % to 5 mol % without sacrificing yield or enantioselectivity.
- 23.Hall HK. J. Am. Chem. Soc. 1957;79:5441. [Google Scholar]
- 24.(−) Indolizidine-209D is a non-natural indolizidine derived from the misassignment of pyrrolizidine 209-K; see ref 1b.
- 25.For selected publications with similar metallacycles involving an isocyanate and alkyne see:Hoberg H. J. Organomet. Chem. 1988;358(1–3):507–517. Duong HA, Louie J. J. Organomet. Chem. 2005;690(23):5098–5104. Louie J. Curr. Org. Chem. 2005;9(7):605–623. Yamamoto Y, Kinpara K, Saigoku T, Takagishi H, Okuda S, Nishiyama H, Itoh K. J. Am. Chem. Soc. 2005;127:605–613. doi: 10.1021/ja045694g. Kondo T, Nomura M, Ura Y, Wada K, Mitsudo T. Tetrahedron Lett. 2006;47(39):7107–7111. Duong HA, Louie J. Tetrahedron. 2006;62(32):7552–7559. Kondo T, Nomura M, Ura Y, Wada K, Mitsudo T-a. J. Am. Chem. Soc. 2006;128:14816–14817. doi: 10.1021/ja066305g.
- 26.For selected CO migration references on rhodium(III) species see: Cavallo L, Sola M. J. Am. Chem. Soc. 2001;123:12294–12302. doi: 10.1021/ja016468z. Gonsalvi L, Adams H, Sunley GJ, Ditzel E, Haynes A. J. Am. Chem. Soc. 2002;124:13597–13612. doi: 10.1021/ja0176191. Gonsalvi L. Organometallics. 2003;22(5):1047–1054.
- 27.Oberg KM, Lee EE, Rovis T. Tetrahedron. 2009;65:5056–5061. doi: 10.1016/j.tet.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.For a study of the effect of olefin substitution on the thermodynamics of alkene-nickel coordination, see: Tolman CA. J. Am. Chem. Soc. 1974;96:2780–2789.
- 29.If pyridone were to be formed by oxidative cycloaddition of two equivalents of alkyne prior to isocyanate incorporation, a different regioisomer would be predicted. Additionally, different alkynes should give different regioisomers as seen by multiple authors (see references 15 and 16). Using Rh(I)•A1, we have noted the formation of pyridones as single regioisomers when the reaction is conducted using isocyanates lacking the tethered alkene; see reference 27.
- 30.A mechanism where the alkyne and alkene oxidatively cyclize to make a rhodacyclopentene followed by isocyanate insertion can be proposed, but this is unlikely because there is no plausible pathway to vinylogous amide and 4-pyridone. For computations proposing alkyne-alkyne cyclization followed by C=N insertion, see:Schmid R, Kirchner K. J. Org. Chem. 2003;68:8339–8344. doi: 10.1021/jo034985p. Dazinger G, Schmid R, Kirchner K. New J. Chem. 2004;28:153–155. Dazinger G, Torres-Rodrigues M, Kirchner K, Calhorda MJ, Costa PJ. J. Organomet. Chem. 2006;691:4434–4445.
- 31.For complete details of the rhodium/phosphoramidite X-ray crystal analysis, see the supplementary information.
- 32.For publications of rhodium and iridium phosphoramidite crystal structures see: Bartels B, García-Yebra C, Rominger F, Helmchen G. Eur. J. Inorg. Chem. 2002:2569–2586. Leitner A, Shekhar S, Pouy MJ, Hartwig JF. J. Am. Chem. Soc. 2005;127:15506–15514. doi: 10.1021/ja054331t. Faller JW, Milheiro SC, Parr J. J. Organomet. Chem. 2006;691:4945–4955. Giacomina F, Meetsma A, Panella L, Lefort L, de Vries AHM, de Vries JG. Angew. Chem. Int. Ed. 2007;46:1497–1500. doi: 10.1002/anie.200603930. Mikhel IS, Ruegger H, Butti P, Camponovo F, Huber D, Mezzetti A. Organometallics. 2008;27:2937–2948. Filipuzzi S, Maennel E, Pregosin PS, Albinati A, Rizzato S, Veiros LF. Organometallics. 2008;27:4580–4588.
- 33.Imabayashi T, Fujiwara Y, Nakao Y, Sato H, Sakaki S. Organometallics. 2005;24:2129–2140. [Google Scholar]
- 34.Wakatsuki Y, Nomura O, Kitaura K, Morokuma K, Yamazaki H. J. Am. Chem. Soc. 1983;105(7):1907–1912. [Google Scholar]
- 35.For crystal structures, see:Davies GR, Hewerston WH, Maise RHB, Owston PG, Patel CG. J. Chem. Soc. 1970:1873. Beauchamp AL, Rochon FR, Theophanides T. Can. J. Chem. 1973;51:126. For computational analysis of alkyne coordination to Pd, see: de Vaal P, Dedieu A. J. Organomet. Chem. 1994;478:121–1129.
- 36.Stockis A, Hoffmann R. J. Am. Chem. Soc. 1980;102:2952–2962. [Google Scholar]
- 37.Kemmitt and coworkers report the crystal structures of Rh(acac)(C2H4)(CF3CCCF3) and the cyclooctene analog. Both structures show coordination of the olefin and alkyne orthogonal to the square plane. Intriguingly, both complexes show unsymmetrical coordination of the two p-components, with the a atoms having shorter Rh–C distances than the b atoms, even with a ligand as sterically undemanding as acac; see:Barlow JH, Clark GR, Curl MG, Howden ME, Kemmitt RDW, Russell DR. J. Organomet. Chem. 1978;144:C47–C51. Barlow JH, Curl MG, Russell DR, Clark GR. J. Organomet. Chem. 1982;235:231–241.
- 38.It is also possible that the trans influence of the phosphoramidite and chloride dictate the relative stability of IVbA and IVbB (or the corresponding transition states). For a review on computational approaches to studying olefin insertion into metal hydrides, see: Niu S, Hall MB. Chem. Rev. 2000;100:363–405. doi: 10.1021/cr980404y. and references therein
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.