

Abstract
Motivation: Identification and characterization of protein–protein interactions (PPIs) is one of the key aims in biological research. While previous research in text mining has made substantial progress in automatic PPI detection from literature, the need to improve the precision and recall of the process remains. More accurate PPI detection will also improve the ability to extract experimental data related to PPIs and provide multiple evidence for each interaction.
Results: We developed an interaction detection method and explored the usefulness of various features in automatically identifying PPIs in text. The results show that our approach outperforms other systems using the AImed dataset. In the tests where our system achieves better precision with reduced recall, we discuss possible approaches for improvement. In addition to test datasets, we evaluated the performance on interactions from five human-curated databases—BIND, DIP, HPRD, IntAct and MINT—where our system consistently identified evidence for ∼60% of interactions when both proteins appear in at least one sentence in the PubMed abstract. We then applied the system to extract articles from PubMed to annotate known, high-throughput and interologous interactions in I2D.
Availability: The data and software are available at: http://www.cs.utoronto.ca/∼juris/data/BI09/.
Contact: yniu@uhnres.utoronto.ca; juris@ai.utoronto.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Identification and characterization of protein–protein interactions (PPIs) is one of the key aims in biological research. Although numerous PPIs have been manually curated into databases such as BIND (Bader et al., 2001), DIP (Xenarios et al., 2000), HPRD (Peri et al., 2003), IntAct (Kerrien et al., 2007) and MINT (Zanzoni et al., 2002), information about many PPIs is still only available through the PubMed database. The PubMed database contains over 17 million papers, making complete coverage by manual curation difficult. Several systems have been developed to make the curation process more accurate, faster and effective (Donaldson et al., 2003; Hoffmann and Valencia, 2004). Information extraction (IE) aims to identify prespecified types of relations from unstructured text, and thus can help in automated PPI detection (Bunescu and Mooney, 2005; Fundel et al., 2007; Romano et al., 2006; Temkin and Gilder, 2003; Yakushiji et al., 2005). An IE approach to PPI detection has two major and relatively independent subtasks: entity recognition and relation detection. First, protein names must be identified in text. Entity recognition is challenging due to non-standard name forms, multiple synonyms for most proteins and the ambiguity of protein names across organisms. Second, interaction identification, i.e. relation detection, identifies protein pairs in the sentence with additional support for their physical interaction. Since the two problems are usually addressed by different computational techniques, they are often investigated separately. In this article, we use entity recognition from (Otasek et al., 2006), and focus on relation detection. Our goal is to systematically evaluate multiple linguistic features, context features, keywords and patterns as cues in the automatic extraction of interaction relation to improve our understanding of how individual features or their combination affect system performance. We then applied the system to find evidence for PPIs in the I2D database. This annotation of PPIs resulted in: (i) identifying more evidence for curated PPIs and providing more biological context for them, and (ii) providing some level of validation for PPIs detected by high-throughput methods and predicted computationally (human interpretation and curation of extracted evidence is still required).
Most automatic relation detection approaches use pattern matching techniques (Bunescu et al., 2005; Hao et al., 2005; Jang et al., 2006; Plake et al., 2005; Romano et al., 2006; Yakushiji et al., 2005) in which a set of syntax patterns are usually developed to specify how an interaction is described in literature. A pattern, serving as a rule, is a sequence of words, or part-of-speech tags, describing an interaction. The locations of the two interaction partners are defined in each pattern. In the pattern matching process, a sentence matches the rule if it satisfies the word constraints in the given order. When a pattern is found in a sentence, the text at defined locations will be extracted as the two interaction partners.
The pattern-based method is intuitively straightforward and conceptually simple. However, manually constructing patterns is a time-consuming process, and is not feasible to cover all the possibilities or compute and evolve all the necessary rules. Thus, several alternative approaches have been proposed (Bunescu and Mooney, 2005; Fundel et al., 2007; LLL, 2005; Temkin and Gilder, 2003). Fundel et al. (2007) made use of rules based on dependency parse trees of sentences. Grammar-based approaches use a parser with grammar production rules for the detection of interactions (Temkin and Gilder, 2003). The rules were derived by manually analyzing sentences that contain interactions in a corpus. Bunescu and Mooney (2005) developed a machine learning approach that explores a sequence kernel measuring the similarity of two sentences by evaluating the common string sequences that they contain. Systems that participated in the 4th Learning Language in Logic Workshop (LLL, 2005) explored knowledge representation methods to detect PPIs. Despite the substantial success of automatic PPI detection approaches, there is still a need to improve the precision and recall of the process. In the BioCreAtIvE 2 interaction pairs extraction subtask, the best results of all the participating systems (measured by precision and recall) were about ∼0.30 (BioCreAtIvE, 2006).1
We aim to improve automatic PPI detection by combining multiple linguistic clues via machine learning. While systems using linguistic features achieve good performance in extracting social, part-of and locational relations between entities such as person, organization and location (Zhou et al., 2005), they have not been fully explored for identifying PPIs. Nielsen (2006) built a system to detect PPIs, using manually identified ‘interaction words’ and some lexical and context features. The system was evaluated on a small corpus containing 255 relations. Mitsumori et al. (2006) only applied context features. Haddow and Matthews (2007) explored n-grams and context features. In the BioCreAtIvE 2 interaction pairs extraction subtask, Erkan et al. (2006) used features from dependency trees, while Huang et al. (2007) used profile features.
In this article, we evaluate the contribution that context, lexical and syntactic information, as well as keywords and patterns have on PPI detection. In addition, we propose a new feature, ‘interaction sentence’, and evaluate its contribution to improving accuracy. Our goal is to identify the most important features for detecting interaction relations in sentences, and after validation, apply the system to validate curated, high-throughput and predicted PPIs. The evaluation results on the AImed dataset (Bunescu and Mooney, 2005) show that our approach outperforms other systems. Automatic interaction extraction may directly contribute to our knowledge of PPIs. For instance, it can be used to enhance human-curated databases (e.g. BIND, DIP, HPRD, IntAct and MINT), since in these databases an interaction is often supported by a few or only one PubMed references. Generally, an increased number of PPI sources results in higher confidence in the interaction as multiple literature links may provide alternative biological methods and biological context, and provide further evidence about its nature, dynamics and affinity. In addition, relation extraction can also be used to verify PPIs predicted computationally or detected by high-throughput experiments. We have built a system and applied it to find evidence for interactions in the Interologous Interaction Database (I2D, http://ophid.utoronto.ca/i2d; Brown and Jurisica, 2007).
2 METHOD
A sentence may contain multiple protein mentions. For each pair of protein mentions, referred to as a candidate hereafter, the task is to determine whether the text describes an interaction by observing the whole sentence. We treat this as a classification problem and use support vector machines (SVMs) as the classification model. SVMs have been effective in many classification problems. Their goal is to find an optimal hyperplane so that examples on the same side of the hyperplane share the same label. SVMs learn decision functions:
where 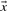 denotes data vector,
denotes data vector, 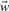 and b are parameters. Each function corresponds to a hyperplane in the feature space. The optimal hyperplane that SVMs chose is the one with the largest margin. The classification task is then to determine on which side of the hyperplane a data point lies. In the experiment, we use SVMlight implementation of SVMs (Joachims, 2005) with a linear kernel.
and b are parameters. Each function corresponds to a hyperplane in the feature space. The optimal hyperplane that SVMs chose is the one with the largest margin. The classification task is then to determine on which side of the hyperplane a data point lies. In the experiment, we use SVMlight implementation of SVMs (Joachims, 2005) with a linear kernel.
3 FEATURES
We assume that a candidate is represented by a vector of features, including context, lexical forms and positions within the sentence. M1 and M2 refer to the two target protein mentions of a candidate (M1 is to the left of M2 in the sentence). We investigate multiple factors that may contribute to identifying interactions, and evaluate the effectiveness of their use as features.
3.1 Position
Three types of features are used to describe positions of the two protein mentions in a sentence: indices of M1 and M2 in the sentence, the distance (in tokens) between M1 and M2 and the number of other proteins between M1 and M2.
3.2 Lexical forms and context
The lexical forms of proteins may provide clues about interactions. Neighbors of M1 and M2 in a sentence often provide context information about their relation, which may be important, and it comprises lexical forms of three parts: n tokens on the left of M1, n tokens on the right of M2 and all tokens between M1 and M2. Since multiple proteins often occur within a sentence (∼40% in the dataset), a small window of context would cause less ambiguity. We set n to 3 in our experiment, as it was shown to be the optimal number of tokens in context (Mitsumori et al., 2006).
3.3 Keywords
Some words are important in identifying interaction relations, and have been used in pattern-matching approaches as cues in rule construction, e.g. bind, activate. Our set of keyword features is developed based on the list in Plake et al. (2005).2 The final keyword collection comprises each word in the list together with all its morphological variations. Several features are generated:
The lexical forms of keywords occurring in a sentence.
Position of a keyword—to the left or to the right of the candidate, or between M1 and M2.
The distance (in tokens) between a keyword and the protein mention (M1 or M2) nearest to it.
3.4 Patterns
We incorporate patterns constructed in pattern-matching methods by generating a binary feature for each pattern. If M1 and M2 in a sentence match a specific pattern, then the corresponding feature will be turned on. We adopt patterns from Plake et al. (2005) in the feature set, and present some simplified examples below:
proteinA word* [form|forms] word* complex with word* proteinB;
proteinA word* [inhibitor|repressor] of word* proteinB;
interaction between word* proteinA word* and word* proteinB.
In these patterns, bold words have to be matched exactly. The symbol ‘|’ separates options. Tag word* indicates a word gap that will be filled with at most n tokens. Plake et al. (2005) developed an approach to optimize n. In our experiment described in Section 4, n is set to 5, as this achieves the best performance (Plake et al., 2005).
3.5 Phrase information
In order to characterize PPIs at the syntax level, we consider the phrases and word dependency in a sentence. Phrase information may be helpful in detecting relations such as part-of and location (Zhou et al., 2005). Word dependency may be informative since it examines which word a protein is related to. In our experiment, phrase information is derived from full parse trees of sentences obtained using the Collins' parser (Collins and Singer, 1999). An example of a sentence and its parse tree (in bracket format) is shown in Figure 1.
Fig. 1.

A sentence parsed by Collins' parser.
The parse tree shows that the sentence comprises a prepositional phrase ‘in the complex’, a noun phrase ‘one interferon gamma homo-dimer’ and a verb phrase ‘binds two receptor molecules’.
For easy processing, detailed phrase information is then extracted from the full parse tree using a Perl script (http://ilk.uvt.nl/∼sabine/chunklink/). The output of the script for the sentence in Figure 1 is shown in Table 1 (to reduce space requirement, some irrelevant columns are omitted in the table).
Table 1.
The output of the phrase extraction script
| 0 | C-PP | IN | In | PP | B-S/B-PP |
| 1 | B-NP | DT | the | NOFUNC | I-S/I-PP/B-NP |
| 2 | I-NP | NN | complex | NP | I-S/I-PP/I-NP |
| 3 | O | COMMA | COMMA | NOFUNC | I-S/E-PP/E-NP |
| 4 | B-NP | CD | one | NOFUNC | I-S/B-NP |
| 5 | I-NP | NN | interferon | NOFUNC | I-S/I-NP |
| 6 | I-NP | NN | gamma | NOFUNC | I-S/I-NP |
| 7 | E-NP | NN | homo-dimer | NP | I-S/E-NP |
| 8 | C-VP | VBZ | binds | VP/S | I-S/B-VP |
| 9 | B-NP | CD | two | NOFUNC | I-S/I-VP/B-NP |
| 10 | I-NP | NN | receptor | NOFUNC | I-S/I-VP/I-NP |
| 11 | I-NP | NNS | molecules | NP | I-S/I-VP/I-NP |
| 12 | O | . | . | NOFUNC | E-S/E-VP/E-NP |
Each token in the sentence is presented on a single line. Every column represents one type of information. The first column is the index of the current token within the sentence. The second column indicates whether this token is the beginning (B-), end (E-) or inside (I-) of a specific phrase. The fourth and third columns list the words and their part-of-speech tags. The function of a phrase is described in the fifth column on the line of the head word of this phrase. Other words in the same phrase are marked by NOFUNC in this column. For example, the head of the noun phrase one interferon gamma homo-dimer is homo-dimer. In the sixth column, the syntactic categories of all the constituents on the path from the root to the leaf node of the parse tree are presented. Given the index of M1 and M2, phrase features are extracted from these three columns, including:
First, last and other phrase heads between M1 and M2;
First and second phrase heads before M1;
First and second phrase heads after M2;
Path of phrase labels connecting M1 and M2;
Path of phrase labels connecting M1 and M2 augmented with head words, if at most two phrases are in between.
3.6 Dependency information
This feature considers words that M1 (M2) is dependent on, i.e. the word that M1 (M2) has a direct syntactic relation with. Dependency relations of words in a sentence are obtained using a dependency parser Minipar (Lin, 1994). The output of Minipar for the sentence in Figure 1 is shown in Table 2. The information for each token in the sentence is on a single line. Column 1 is the index of a token. Lexical form of the token is at column 2. Column 4 lists the index of its dependent word, and column 3 has its part-of-speech information. Column 5 specifies the type of syntactic relation. As shown in the table, molecules is the object of binds.
Table 2.
An output example from Minipar (Lin, 1994)
| E0 | (() | fin | C | * | ) |
| 1 | (In | Prep | E0 | mod | (gov fin)) |
| 2 | (the | Det | 3 | det | (gov complex)) |
| 3 | (complex | N | 1 | pcomp-n | (gov in)) |
| 4 | (, | U | E0 | punc | (gov fin)) |
| 5 | (one | N | 8 | nn | (gov homo-dimer)) |
| 6 | (interferon | N | 8 | nn | (gov homo-dimer)) |
| 7 | (gamma | N | 8 | nn | (gov homo-dimer)) |
| 8 | (homo-dimer | N | 9 | s | (gov bind)) |
| 9 | (binds | V | E0 | i | (gov fin)) |
| 10 | (two | N | 12 | nn | (gov molecule)) |
| 11 | (receptor | N | 12 | nn | (gov molecule)) |
| 12 | (molecules | N | 9 | obj | (gov bind)) |
| 13 | (. | U | * | punc | ) |
To construct dependency features, the dependent word and its part-of-speech tag are extracted for M1 and M2, respectively. A protein name often contains more than one token, and each token may have a dependent word. Thus, the dependent word of M1 (M2) is determined by selecting the word with the smallest index (indicates its location in the sentence), which is related to one of the tokens in M1 (M2) and is not part of M1 (M2).
3.7 Overlap
A recognizable protein name is sometimes embedded in another longer protein name. In such cases, it is unlikely that the embedded protein participates in an interaction. Although we do not address entity recognition in this paper, we still can measure the overlaps of the two target proteins that are known to us. The overlap feature takes this into account by evaluating whether M1 (M2) is embedded in M2 (M1), and whether M1 (M2) is embedded in another protein name. We also evaluate whether the two proteins are mentioned in the same noun phrase, prepositional phrase or verb phrase.
3.8 Interaction sentences
Identifying protein name co-occurrence ensures high recall but poor precision for the system. Determining that a sentence contains at least one interaction relation, i.e. it is an interaction sentence, may help to increase precision. For sentences with multiple protein mentions, this information may promote recovering interactions in the sentences. When multiple protein mentions are present in a sentence of no interaction, no protein pairs should be classified as interactions. To verify this hypothesis, we determine whether a sentence is an interaction sentence. We applied a mixture model combining a supervised model with a rule-based method to automatically identify interaction sentences and achieved an F-score of 63.3%. Details of this model are described in Niu et al. (2008).3 The feature value is set to 1 for a candidate protein pair if it appears in an interaction sentence; otherwise, the value is 0.
4 PERFORMANCE EVALUATION
Most of the systems have been evaluated on different datasets, specific to a given work (Zhou and He, 2008). In an attempt to standardize the validation of text mining algorithms for PPI detection, BioCreAtIvE workshops established a PPI corpus (BioCreAtIvE, 2004, 2006). BioCreAtIvE 2 evaluation considers the full complexity of PPI recognition, and does not separate the task of finding protein names, normalizing a protein name to its SwissProt IDs, and identifying the interaction relations. In this article, we describe a method that focuses only on the last task, and thus it is difficult to make a fair comparison with the participants of BioCreAtIvE 2. Nonetheless, we report the results of our system on the test set of the BioCreAtIvE 2—protein interaction pairs subtask. In the following experiments, we first evaluate our system on AImed corpus [developed at University of Texas at Austin (Bunescu and Mooney, 2005)], where interactors in a sentence are explicitly annotated. Since AImed has been used to evaluate several systems on a PPI detection task, we have used this dataset to directly compare performance of our approach to the other systems. We then summarize our results using the BioCreAtIvE 2 test set.
AImed contains about 200 PubMed abstracts (1944 sentences in total) from the Database of Interacting Proteins (DIP; Xenarios et al., 2000) with manually annotated interactions. Each abstract is in sentence-per-line format. Protein mentions and interactions are marked with specific XML tags. Multiple interactions in a sentence are numbered so they can be easily distinguished.
Since our work in this article focuses on relation detection, we assumed the knowledge of protein names in this dataset. A pair of proteins is a positive case if it is manually labeled as an interaction; otherwise, it is a negative case. We applied our approach for interaction relation detection to this data, and compared the labels predicted by the SVM classifier with this benchmark. All results reported are using 10-fold cross-validation.
4.1 Feature comparison
We first evaluate how individual features and their combination affect classification performance. Similar to other approaches, precision (TP/TP+FP), recall (TP/TP+FN) and F-score (2 × precision × recall/(precision+recall)) are used as metrics. In the evaluation, every pair of distinct proteins in a sentence is a candidate. This is a pair-level evaluation, i.e. the correctness of each candidate is measured. Results are summarized in Table 3.
Table 3.
Pair-level evaluation of individual features and their combination on data from AImed (Bunescu and Mooney, 2005), using 10-fold cross-validation and measuring precision, recall and F-score
| Features | Precision | Recall | F-score |
|---|---|---|---|
| (%) | (%) | (%) | |
| position+lexical forms+context (1) | 57.4 | 25.3 | 35.1 |
| (1)+keywords | 56.2 | 17.6 | 26.8 |
| (1)+patterns | 66.7 | 11.0 | 18.9 |
| (1)+phrase | 58.6 | 29.8 | 39.5 |
| (1)+dependency | 64.0 | 29.3 | 40.2 |
| (1)+phrase+dependency (2) | 61.6 | 31.6 | 41.7 |
| (2)+overlap (3) | 65.4 | 33.0 | 43.9 |
| (3)+interaction sentence | 65.0 | 35.9 | 46.2 |
As shown in the first row of Table 3, combining position, lexical forms and context has an F-score of 35.1%, considered as the baseline. Adding keywords does not improve the performance. Instead, recall drops by ∼8%, indicating that the keywords do not provide good clues in this setting. (We see a decline of recall in Table 3 because of the combination result of keywords and baseline features using SVM.) In fact, of the 1330 sentences that do not express any PPIs, we found 731 containing at least one keyword. However, among the 614 sentences that describe at least one PPI, 498 have a keyword. As expected, it shows that keywords alone provide high recall but low precision for PPIs. Thus, additional features will be necessary to improve precision.
Patterns are rules combining keywords with extra constraints. Compared with the baseline, adding the patterns feature achieves high precision but low recall. In the third row of Table 3, we can see that precision is improved by ∼9%, but recall decreases by ∼14%. Thus, patterns are more useful to systems in which precision is more important, such as for providing verification evidence for high-throughput or computationally predicted interactions, and for providing additional evidence for human-curated interactions.
As shown in Table 3, the two syntactic features are both helpful. While dependency improves precision by ∼7% and recall by ∼4%, incorporating the phrase feature improved recall and to a certain degree precision. F-score is 5% higher in comparison with the baseline. It shows that the dependent words of two proteins are strong cues indicating whether they interact.
After adding overlap to features, the performance is further improved. This simple feature increases precision by ∼4% compared with feature set (2). The highest F-score is achieved, which is significantly better than the baseline (P < 0.001). Both precision and recall are improved by ∼8%.
In general, good precision but low recall is achieved. After adding the automatically generated interaction sentence feature, recall is improved by 2.9%, while precision drops by 0.4%. In order to determine the upper bound of possible performance improvement by incorporating this feature, we tested the ideal case by manually labeling sentences as interaction or non-interaction. The results show an increase of ∼11% in recall at the same precision as feature set (3) in the table. This increase is larger than adding any other feature alone. Therefore, knowing that a sentence describes an interaction greatly increases the chance of recovering PPIs from that sentence. This suggests a possible direction for building a better PPI detection system. However, since an automatic approach for obtaining an interaction sentence still needs to be improved, this feature is not included in the following experiments.
In this article, we used a dataset with sentences containing both two proteins (∼60%) and three or more proteins (∼40%). Therefore, our evaluation is not strongly biased to either case. In order to provide some additional information, we further calculate precision and recall on three groups of sentences separately. Group 1 contains sentences with only two proteins; precision is 83.3%, and recall is 33.7%. Group 2 includes sentences with three proteins; precision is 67.8%, and recall is 36.3%. Group 3 contains sentences with more than three proteins; precision is 58.0%, and recall is 30.8%.
4.2 Comparison to other systems
AImed has been used to evaluate several systems (Bunescu et al., 2005; Bunescu and Mooney, 2005; Romano et al., 2006; Yakushiji et al., 2005); thus, to enable a direct comparison we adopt their evaluation scheme. Abstract-level evaluation is conducted by measuring correct identification of an interaction in an abstract. The interaction is considered correct if at least one of possible several descriptions is detected by the system. The feature set that has the best performance in the previous pair-level evaluation is used [without interaction sentence, feature set (3) in Table 3]. The results are shown in Table 4. The results of Bunescu et al. (2005) and Bunescu and Mooney (2005) are obtained by taking the points in the precision–recall curve that has approximately the same recall as in our system (43%). The results of Mitsumori et al. (2006) are obtained by using their features in the same experimental settings of SVM that is used to test our own features (linear model, default parameters). As shown in the table, with similar recall, our approach achieves the highest precision. The SD of the 10 runs in terms of F-score is 3.55. We also trained our system on a training set of 60% of the whole dataset (randomly selected), and tested on the rest 40% of the data. The precision is 67.1%, recall 46.8% and F-score 55.1%.
Table 4.
Abstract-level evaluation of different systems on AImed dataset.
| System | Recall | Precision | F-score |
|---|---|---|---|
| (%) | (%) | (%) | |
| Bunescu and Mooney (2005) | 43 | 60 | 50.1 |
| (10-fold cross-validation) | |||
| Bunescu et al. (2005) | 43 | 48 | 45.4 |
| (10-fold cross-validation) | |||
| Yakushiji et al. (2005) | 45.3 | 37.3 | 40.9 |
| (10-fold cross-validation) | |||
| Mitsumori et al. (2006) | 36.7 | 64.2 | 46.7 |
| (10-fold cross-validation) | |||
| Romano et al. (2006) (unsupervised) | 29 | 42 | 34.3 |
| (development: 60%, test: 40%) | |||
| Our approach | 43.2 | 70.2 | 53.5 |
| (10-fold cross-validation) |
The 10-fold splits follow (Bunescu et al., 2005).
4.3 Evaluation using BioCreAtIvE 2 test set
In order to evaluate our system on the BioCreAtIvE 2 protein-interaction pair subtask test set, we performed the three steps. First, the system identifies protein names in the text. We developed a machine learning approach using SVM to identify protein names in text. We use several features that have been shown effective in this task and are commonly used to detect protein or gene names (Fundel et al., 2005; Hakenberg et al., 2005). These features include unigrams, part-of-speech tags, orthography, prefix/suffix and the previous and following tokens. Second, our interaction detection system is applied to find interactions. Third, a protein name is normalized to its UniProtKB ID (http://www.uniprot.org/) using the dictionary in our database. Since one protein name may be mapped to several UniProtKB IDs because of different organisms, we attempted to constrain the mapping using the Medical Subject Heading (MeSH) terms. Unfortunately, sometime the MeSH terms are not always available. For some articles, there are more than one MeSH terms. In such cases, we did not apply further techniques to resolve the ambiguity. Since our database does not contain CHEBI IDs (http://www.ebi.ac.uk/chebi/), we evaluate our system on the SwissProt-only article set, i.e. ‘the set of articles that exclusively mention interaction pairs that can be normalized to SwissProt’ (Krallinger et al., 2007). This set comprises 228 articles. After automatically identifying protein names in the test articles, our interaction detection system identified 677 positive pairs in 166 (of the 228) articles (over 85% of interaction relations). However, using the normalization method described above, we uniquely identified UniProtKB IDs for both interactors for only 14 pairs in 11 articles. There are two major reasons for this loss. One is that the UniProtKB ID could not be found for at least one of the interactors, which happened to 527 pairs. The second reason is that zero or multiple MeSH terms were found, which occurs in 231 pairs. The average results on the 11 articles and the whole test set are shown in Table 5. The micro-averaged performance is evaluated by weighing equally every pair in the test set. To get the macro-averaged scores, each document is evaluated and then the result is averaged on the whole set (Krallinger et al., 2008).
Table 5.
Results obtained for the SwissProt-only test set
| Precision | Recall | F-score | |
|---|---|---|---|
| Average (11 articles) | 0.5454 | 0.3918 | 0.4560 |
| Macro-average | 0.0246 | 0.0176 | 0.0205 |
| Micro-average | 0.5714 | 0.0094 | 0.0186 |
The micro-average precision is higher compared with systems at BioCreAtIvE 2 (although evaluated on a small subset of articles, caused by the issues described above). As no sophisticated normalization method was explored, most detected interactors cannot be mapped to unique UniProtKB IDs. Therefore, the macro- and micro-average recall are very low. However, when averaged on the articles with at least one prediction (the 11 articles), the precision is similar to the micro-score, whereas the recall is substantially higher. Note that we evaluated all pairs with unique UniProtKB IDs for both interactors. While the 14 pairs is a small sample set, it represents a selection of the positive pairs detected by our system.
5 VERIFYING PROTEIN–PROTEIN INTERACTIONS
5.1 System architecture
The automatic detection of PPIs can be applied to find supporting evidence for predicted and high-throughput interactions, and additional evidence for curated PPIs. The I2D (http://ophid.utoronto.ca/i2d) includes interactions from 6 species (Homo sapiens, Rattus norvegicus, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae), and I2D version 1.71 integrates 444 277 PPIs (Brown and Jurisica, 2007). These interactions fall into three broad groups: (i) known interactions from PubMed; (ii) experimental interactions from high-throughput screens; and (iii) computationally predicted interactions inferred between species using homology (Brown and Jurisica, 2005).
For human, I2D comprises PPIs from curated databases (Bader et al., 2001; Kerrien et al., 2007; Peri et al., 2003; Xenarios et al., 2000; Zanzoni et al., 2002), combined with human PPIs from high-throughput experiments (e.g. Barrios-Rodiles et al., 2005; Ingham et al., 2005; Jones et al., 2006; Rual et al., 2005; Stelzl et al., 2005) and predicted from model organism PPI databases (Bader et al., 2001; Gavin et al., 2002; Giot et al., 2003; Ho et al., 2002; Ito et al., 2000; Li et al., 2004; Mewes, 2002; Mering et al., 2002), resulting in 100 083 unique human PPIs (I2D ver. 1.71). The architecture of the verification system is shown in Figure 2.
Fig. 2.

System architecture for PPI verification. To speed up analysis, we use a copy of NLM Medline/PubMed stored in an IBM DB2 ver. 9.2 database on IBM p595 server. Individual abstracts are indexed using the relevant MeSH terms and protein name synonyms found in each abstract.
In I2D, interactions are represented by a pair of UniProKB IDs. For automated PPI verification using text mining, IDs are first mapped to multiple corresponding protein names using a synonym dictionary. The dictionary includes synonyms for all proteins listed in UniProtKB. Since, unfortunately, gene names are often used in the protein interaction literature instead of protein names, all gene names from the available data in Entrez Gene are also added to the dictionary. Then, the NLM Medline/PubMed abstract database is searched to find the co-occurrence of the two proteins. The lack of standards in research writing allows referencing a single protein in multiple ways. For example, cyclophilin 3, cyclophilin III, cyp3, Cyclophilin F, PPIF, PPIase, Rotamese and Peptidyl-prolyl cis-trans isomerase are all valid references to the same protein (P30405). This presents great challenges in protein name identification (Tsuruoka and Tsujii, 2004). In order to increase coverage in co-occurrence finding, both synonyms and abstracts were normalized before searching as follows: (i) removing dashes, spaces, and commas; (ii) Converting letters from upper case to lower case; and (iii) converting Roman digits to Arabic digits.
Evidence of PPIs is presented at three levels. First, abstracts with two proteins co-occurring are listed (target abstracts). Second, these abstracts are searched for sentences that contain both proteins (target sentences). Third, interactions in the sentences are selected by the feature-based classifier. Our confidence in an interaction increases as more refined filter is applied. Interactions identified by the classifier provide direct evidence, and target abstracts and target sentences can provide complementary information to aid interaction confirmation. Moreover, even if target abstracts and sentences do not describe an interaction relation, they may suggest useful relations between the two proteins. This could be particularly valuable in investigating previously unknown PPIs.
5.2 PPI verification on curated databases
Here, we describe verification of PPIs from five human-curated sources in I2D: BIND (Bader et al., 2001), DIP (Xenarios et al., 2000), HPRD (Peri et al., 2003), IntAct (Kerrien et al., 2007), and MINT (Zanzoni et al., 2002). Each source specifies a set of PPIs and one or more PubMed abstracts that support these interactions. Each source has a different focus and distribution of incorporated organisms, and thus overlap among them is small (Ramani et al., 2005). (With the effort of IMEx, http://imex.sourceforge.net, the data combining multiple sources is also available.) In order to understand how this difference will impact PPI detection, we search for PPI evidence in each of the sources separately (note that our experiments are not organism specific).
We first collected the UniProtKB IDs of all PPIs from the five curated sources in I2D. The number of PPIs from each source is shown in the first row in Table 6. For evaluation purpose, it is sufficient to search for evidence in the supporting abstracts specified in each source instead of all of PubMed. All the results reported in Sections 5.2.1–5.2.3 are based on these abstracts.
Table 6.
Number of distinct interactions in individual human-curated databases
| HPRD | BIND | DIP | MINT | IntAct | |
|---|---|---|---|---|---|
| No. of PPIs in a database | 34 177 | 44 391 | 4443 | 58 733 | 55 182 |
| No. of PPI partners in abstracts | 11 182 | 3410 | 473 | 1620 | 1892 |
Please note that human curation uses full papers, not just abstracts.
5.2.1 Co-occurrence in abstracts
The number of distinct interactions for the co-occurrence of the two interaction partners in at least one abstract is shown by the second row of Table 6. HPRD has the highest number of abstract-level co-occurrence. Target abstracts are found for 32.7% of PPIs in HPRD. About 10% of the interactions in BIND and DIP are found to co-occur in at least one abstract, and occur at a rate of ∼3% for MINT and IntAct. Multiple factors may account for this. First, some PPIs are described only in the full article rather than in the abstract. It has been noted that only approximately half of the PPIs in HPRD can be found in PubMed abstracts (Fundel et al., 2007). Second, individual curator teams use different strategies to extract PPIs, and focus either on broader PubMed coverage or on topical subset of journals (as described at http://imex.sourceforge.net). Third, an interaction in a database is often supported by only one or two abstracts. Fourth, protein name identification may improve with enhanced synonym coverage. As discussed in Section 5.1, the dictionary used by our system comprises synonyms from two major databases of protein and gene names, UniProtKB and Entrez Gene. As more sources are incorporated, higher coverage will be achieved. The downside is that more false positives may be generated, and the searching time may increase. Fifth, spelling variations in protein synonyms cannot be solved perfectly using current approaches. Also, while other databases provide evidence for all PPIs, 2530 interactions in BIND do not have the supporting abstracts available. As shown in the table, compared with other databases, more abstracts were found for PPIs in HPRD, which focuses on human interactions. This may be due to the synonym dictionary having a better coverage for human proteins, especially disease-related proteins.
5.2.2 Co-occurrence in sentences
For further analysis, we randomly selected 300 distinct interactions from each database in Table 6 as examples to demonstrate our three-level system (ensuring each had at least one target abstract). All corresponding target abstracts (1634 in total) are presented in Supplementary Table 1.
When two proteins co-occur in a single sentence, they are more likely to interact. In this step, sentences containing both proteins of an interaction were retrieved. The number of distinct pairs that co-occur in at least one sentence is shown Table 7 (2nd row). About 80% of proteins co-occurring in abstracts also co-occur in sentences, and this number is consistent for all five sources.
Table 7.
Number of distinct interactions, considering simple co-occurrence and SVM classification
| HPRD | BIND | DIP | MINT | IntAct | |
|---|---|---|---|---|---|
| Co-occur in abstracts | 300 | 300 | 300 | 300 | 300 |
| Co-occur in sentences | 247 | 258 | 234 | 266 | 241 |
| Detected by the classifier | 149 | 158 | 128 | 168 | 126 |
Number of target abstracts in HPRD, BIND, DIP, MINT, IntAct: 357, 315, 315, 331, 326, respectively.
5.2.3 Automatically detecting interactions
As a final filter, we applied our classifier, trained on AImed, using the best feature set [feature set (3) in Table 3] to determine whether two proteins mentioned in a sentence interact. The last row in Table 7 shows the number of distinct interactions detected by the classifier. For each database, evidence was found for ∼60% of interactions where both proteins appear in at least one sentence. This indicates that interaction descriptions are consistent despite the great variations in the content of the five databases.
5.2.4 Identifying new evidence
It is worth noting that results in the previous three sections are obtained by searching for only one or two supporting abstracts for each interaction. We further compared this with the result of looking for evidence in the entire PubMed collection (PubMed07). As shown in Table 7, a smaller number of distinct interactions were found co-occurring in sentences for the 300 interactions from Bind or DIP compared with those from the other three databases. Since fewest distinct interactions were detected by the classifier for DIP, we used DIP as an example and searched PubMed for evidence supporting the 300 randomly selected PPIs. The results are summarized in Table 8.
Table 8.
Results of identifying new evidence for 300 pairs in DIP (showing numbers of distinct pairs)
| ABS | SEN | SVM > 0 |
SVM > 0.5 |
|||
|---|---|---|---|---|---|---|
| SIN | MUL | SIN | MUL | |||
| Search provided abstracts | 300 | 234 | 123 | 5 | 84 | 1 |
| Search an entire PubMed | 300 | 295 | 26 | 243 | 69 | 128 |
ABS, co-occur in abstracts; SEN, co-occur in sentences; SIN, PPIs with single abstract as evidence; MUL, PPIs with multiple abstracts as evidence.
At the abstract level, co-occurrence was found for all interactions. In total, 131 522 abstracts were detected by searching the entire PubMed. Therefore, on average, each pair has 438 co-occurrence abstracts. These abstracts include 309 out of the 315 abstracts provided in DIP. Among the six abstracts that were missing, only one contained both of the two target proteins. Two hundred and ninety-five pairs were found co-occurring in at least one sentence in PubMed. In comparison, only 234 pairs were found in abstracts provided in DIP. The SVM classifier produces a score for each protein pair candidate, and this score implies the confidence of a candidate being an interaction. Applying different threshold to the scores, we can adjust precision and recall of the classification results. Using the default threshold, we take all candidates with scores above zero as positive interactions. In this case, the classifier identified evidence for 269 interactions. Among them, evidence was found in multiple abstracts for 243 pairs, as shown in Table 8. For 95 out of the 243 pairs, each was detected in more than 10 abstracts. By searching abstracts provided in DIP, evidence was found for only 128 pairs. As mentioned in Section 5.2.2, only one or two manually annotated abstracts are shown in DIP as an evidence of an interaction. In the second case, we set the threshold to 0.5, the average score of the provided evidence (manually curated evidence in DIP). With this higher threshold (the distribution of the scores of all the positive data points can be found in Supplementary Figure 1), evidence was found for 197 pairs, and multiple abstracts were indicated as supporting evidence for 128 of them (52% is overlapped with the 128 pairs in Table 7). Compared with manual curation in DIP, the automatic approach collected much more evidence for these interactions, and thus provided additional information on the method and system used for PPI detection.
Our interaction detection system has been used to identify evidence for human PPIs in I2D (Brown and Jurisica, 2007), which contains 148 580 distinct pairs of proteins (self-interactions excluded). By searching PubMed with our system, we found 58 489 pairs with the two interaction partners co-occurring in at least one abstract. On average, the number of co-occurrence abstracts for one pair is 78.11. The total number of pairs with SVM score >0.5 is 14 054, and each pair has 1.8 abstracts in average. We have also explored proteins of degree zero (no PPIs available) in I2D. The system assessed 23 831 pairs, and identified PubMed evidence for 184 pairs of them with SVM score >0.5. Highlight of the network related to this new evidence is presented in Supplementary Figures 2–3. First, we show 110 PPIs with novel evidence are identified by the system (Supplementary Fig. 2). Second, we extend this network to include all related PPIs from I2D (Supplementary Fig. 3). Third, we focus on a subgraph in this network, and highlight PPIs with new evidence for human-curated, high-throughput and predicted PPIs, as well as novel evidence (Fig. 3).
Fig. 3.

PPIs with new evidence from automatic text mining. Expanding the original set of 1433 proteins and 845 interactions by including PPIs from I2D results in a network with 8250 proteins and 21 652 interactions. Subgraph from Supplementary Figure 3, showing only the largest connected component and highlighting new evidence for human-curated PPIs, predicted PPIs and PPIs from high-throughput experiments. Combined network comprises 392 interactions on 322 proteins. Node color is according to Gene Ontology, as shown in the legend. Node size is based on node degree in the network. For simplicity, only protein names of high degree nodes are shown. Edge color is according to the PPI source, as described in the legend. The figure was generated in NAViGaTOR 2.1.12 (http://ophid.utoronto.ca/navigator), (Brown et al., 2009).
6 CONCLUSION
In this article, we described a system for automatic PPI detection in the text. Diverse features were studied and their effectiveness, individually or in combination, were evaluated. We tested system performance on the AImed and BioCreAtIvE 2 datasets. Compared with other systems using AImed, our approach achieves the highest precision at approximately the same recall. On the BioCreAtIvE 2 test set, we get high micro-average precision, although as noted in Section 4.3, while the system identified 677 positive pairs in 166 articles, the final comparison reported in Table 5 was done only on the 11 articles. The results in Table 3 show that using sentence-level interaction information as a feature can improve recall of PPI detection. Improving the performance of this additional filter may lead to a more accurate text mining system for PPI detection and characterization. Analyzing the performance of individual steps is useful to identify bottlenecks in the pipeline. However, combining them to achieve the best performance may require additional analysis, since it is possible that combining the best algorithms for individual steps in the pipeline for PPI recognition may not result in the most optimal system (Ponzielli et al., 2008). In the future, we also plan to integrate the analysis with BioCreative Meta Server (BCMS; Leitner et al., 2008, http://bcms.bioinfo.cnio.es/), to identify known PPIs from PubMed.
We applied our system for PPI detection to two tasks: (i) to find more evidence for PPIs from five human-curated databases, BIND, DIP, HPRD, IntAct and MINT and (ii) to validate high-throughput and predicted interactions in I2D database. Although individual human-curated PPI databases are often used to evaluate automatic PPI identification systems, to the best of our knowledge, no comparison study of multiple databases has been reported.
Implementing the IE as a pipeline system enables us to identify bottlenecks with high precision or recall impact. Since the process of finding co-occurrence in abstracts incurs the largest loss, it suggests that the spelling variation of protein names is still a challenge in automatic PPI detection, although multiple factors may account for this. Nevertheless, when only a few supporting abstracts are available, evidence can be found for ∼60% of the interactions with the two proteins co-occurring in at least one sentence using our automatic PPI detection approach. Furthermore, this coverage is fairly consistent across the five databases that have different distributions of incorporated organisms. The entire PubMed collection was searched for evidence of 300 interactions in DIP. With high confidence (SVM scores >0.5), evidence was found for about 2/3 of them, and new evidence was found for ∼40% of the pairs. Searching the whole PubMed increases average number of supporting abstracts from 1.05 to 9.5.
ACKNOWLEDGEMENTS
We would like to acknowledge Richard Lu and Frederic Breard for supporting the I2D database, Max Kotlyar for providing the predicted interactions and Kevin R. Brown for being involved with I2D development and many stimulating discussions. We gratefully acknowledge access to the U.S. National Library of Medicine (NLM) MEDLINE/PubMed database through the NLM Data Distribution Program, using the license code FAZ.
Finding: Genome Canada via the Ontario Genomics Institute; the Canada Foundation for Innovation (grants #12301 and #203383); the Canada Research Chair Program; an Ontario Research Fund Research Excellence grant; IBM Canada.
Conflict of Interest: none declared.
Footnotes
1Note that this is a comprehensive evaluation in which proteins must be disambiguated with respect to the organism source.
2 Plake et al. (2005) describes a pattern-based PPI detection approach and provides complete information on the list of cues and the set of patterns used.
3Detecting interaction sentences cannot provide information on the specific proteins involved in an interaction. Therefore, the method and evaluation has no overlap with this paper.
REFERENCES
- Bader GD, et al. BIND – the biomolecular interaction network database. Nucleic Acids Res. 2001;29:242–245. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrios-Rodiles M, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- BioCreAtIve. Critical assessment for information extraction in biology. 2004 Available at http://biocreative.sourceforge.net/index.html. [Google Scholar]
- BioCreAtIvE. Critical assessment for information extraction in biology. 2006 Available at http://biocreative.sourceforge.net/index.html (last accessed date January 2009) [Google Scholar]
- Brown KR, Jurisica I. Unequal evolutionary conservation of human protein interactions in interologous networks. Genome Biol. 2007;8:R95. doi: 10.1186/gb-2007-8-5-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KR, Jurisica I. Online Predicted Human Interaction Database OPHID. Bioinformatics. 2005;21:2076–2082. doi: 10.1093/bioinformatics/bti273. [DOI] [PubMed] [Google Scholar]
- Brown KR, et al. NAViGaTOR: Network analysis, visualization & graphing Toronto. Bioinformatics. 2009 doi: 10.1093/bioinformatics/btp595. [Epub ahead of print, doi: 10.1093/bioinformatics/btp595] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunescu RC, et al. Comparative experiments on learning information extractors for proteins and their interactions. Artif. Intell. Med. 2005;33:139–155. doi: 10.1016/j.artmed.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Bunescu RC, Mooney RJ. Proceedings of the 19th Annual Conference on Neural Information Processing Systems. Cambridge, MA, USA: MIT Press; 2005. Subsequence kernels for relation extraction; pp. 171–178. [Google Scholar]
- Collins M, Singer Y. Proceedings of the Joint SIGDAT Conference on Empirical Methods in Natural Language Processing and Very Large Corpora. MD, USA: Association for Computational Linguistics; 1999. Unsupervised models for named entity classification; pp. 100–110. [Google Scholar]
- Donaldson I, et al. PreBIND and Textomy - mining the biomedical literature for protein-protein interactions using a support vector machine. BMC Bioinformatics. 2003;4:11–23. doi: 10.1186/1471-2105-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkan G, et al. Proceedings of the 2nd BioCreAtivE Challenge Evaluation Workshop. Madrid, Spain: Fundación CNIO Carlos III; 2006. Extracting interacting protein pairs and evidence sentences by using dependency parsing and machine learning techniques; pp. 287–292. [Google Scholar]
- Fundel K, et al. A simple approach for protein name identification: prospects and limits. BMC Bioinformatics. 2005;6(Suppl. 1):s15. doi: 10.1186/1471-2105-6-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fundel K, et al. RelEx – relation extraction using dependency parse trees. Bioinformatics. 2007;23:365–371. doi: 10.1093/bioinformatics/btl616. [DOI] [PubMed] [Google Scholar]
- Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Giot L, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Haddow B, Matthews M. The extraction of enriched protein-protein interactions from biomedical text. Proceedings of the BioNLP Workshop at ACL. 2007:145–152. [Google Scholar]
- Hakenberg J, et al. Systematic feature evaluation for gene name recognition. BMC Bioinformatics. 2005;6(Suppl. 1):s9. doi: 10.1186/1471-2105-6-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, et al. Discovering patterns to extract protein-protein interactions from the literature: Part II. Bioinformatics. 2005;21:3294–3300. doi: 10.1093/bioinformatics/bti493. [DOI] [PubMed] [Google Scholar]
- Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Valencia A. A gene network for navigating the literature. Nat. Genet. 2004;36:664. doi: 10.1038/ng0704-664. [DOI] [PubMed] [Google Scholar]
- Huang M, et al. Proceedings of the 2nd BioCreAtivE Challenge Evaluation Workshop. Madrid, Spain: Fundación CNIO Carlos III; 2007. Mining physical protein-protein interactions by exploiting abundant features; pp. 237–245. [Google Scholar]
- Ingham RJ, et al. WW domains provide a platform for the assembly of multi-protein networks. Mol. Cell Biol. 2005;25:7092–7106. doi: 10.1128/MCB.25.16.7092-7106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, et al. Toward a protein-protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl Acad. Sci. USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, et al. Finding the evidence for protein-protein interactions from PubMed abstracts. Bioinformatics. 2006;22:e220–e226. doi: 10.1093/bioinformatics/btl203. [DOI] [PubMed] [Google Scholar]
- Joachims T. SVMlightSupport Vector Machine. 2002 Available at http://svmlight.joachims.org/ (last accessed date April 2007) [Google Scholar]
- Jones RB, et al. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- Kerrien S, et al. IntAct – open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:d561–d565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krallinger M, et al. Proceedings of the 2nd BioCreative Challenge Evaluation Workshop. Madrid, Spain: Fundación CNIO Carlos III; 2007. Assessment of the second BioCreative PPI task: automatic extraction of protein-protein interactions; pp. 41–54. [Google Scholar]
- Krallinger M, et al. Overview of the protein-protein interaction annotation extraction task of BioCreative II. Genome Biol. 2008;9(Suppl. 2):S4. doi: 10.1186/gb-2008-9-s2-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner F, et al. Introducing meta-services for biomedical information extraction. Genome Biol. 2008;9(Suppl. 2):S6. doi: 10.1186/gb-2008-9-s2-s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D. Proceedings of the 15th International Conference on Computational Linguistics. NJ, USA: Association for Computational Linguistics Morristown; 1994. Principar – an efficient, broad-coverage, principle-based parser; pp. 482–488. [Google Scholar]
- LLL. Proceedings of the 4th Learning Language in Logic Workshop. 2005 Available at http://www.cs.york.ac.uk/aig/lll/lll05/ [Google Scholar]
- Mewes HW. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mering CV, et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002;417:399–403. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- Mitsumori T, et al. Extracting protein-protein interaction information from biomedical text with SVM. IEICE Trans. Inf. Syst. 2006;E89-D:2464–2466. [Google Scholar]
- Nielsen L. Proceedings of the BioNLP Workshop at HLT/NAACL. NJ, USA: Association for Computational Linguistics Morristown; 2006. Extracting protein-protein interactions using simple contextual features; pp. 120–121. [Google Scholar]
- Niu Y, et al. Detecting protein-protein interaction sentences using a mixture model. Proceedings of NLDB08, Lecture Notes in Computer Science. 2008;5039:352–354. [Google Scholar]
- Otasek D, et al. Proceedings of SIAM Conference on Text Mining. Bethesda, Maryland: 2006. Confirming protein-protein interactions by text mining. April 20-22, 2006. [Google Scholar]
- Peri S, et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plake C, et al. Proceedings of the ACM Symposium on Applied Computing. New York, USA: ACM Press; 2005. Optimizing syntax patterns for discovering protein-protein interactions; pp. 195–201. [Google Scholar]
- Ponzielli R, et al. Optimization of experimental design parameters for high-throughput chromatin immunoprecipitation studies. Nucleic Acids Res. 2008;36:e144. doi: 10.1093/nar/gkn735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani A, et al. ACL-ISMB Workshop on Linking Biological Literature, Ontologies and Databases: Minging Biological Semantics. NJ, USA: Association for Computational Linguistics Morristown; 2005. Using biomedical literature mining to consolidate the set of known human protein-protein interactions; pp. 46–53. [Google Scholar]
- Romano L, et al. Proceedings of the 11th Conference of the European Chapter of the Association for Computational Linguistics. NJ, USA: Association for Computational Linguistics Morristown; 2006. Investigating a generic paraphrase-based approach for relation extraction; pp. 409–416. [Google Scholar]
- Rual JF, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Stelzl U, et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Temkin JM, Gilder MR. Extraction of protein interaction information from unstructured text using a context-free grammar. Bioinformatics. 2003;19:2046–2053. doi: 10.1093/bioinformatics/btg279. [DOI] [PubMed] [Google Scholar]
- Tsuruoka Y, Tsujii J. Improving the performance of dictionary-based approaches in protein name recognition. J. Biomed. Inform. 2004;37:461–470. doi: 10.1016/j.jbi.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Xenarios I, et al. DIP: the database of interacting proteins. Nucleic Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakushiji A, et al. Proceedings of the 1st International Symposium on Semantic Mining in Biomedicine. Cambridgeshire, UK: CEUR-WS.org; 2005. Biomedical information extraction with predicate-argument structure patterns; pp. 60–69. [Google Scholar]
- Zanzoni A, et al. MINT: A Molecular INTeraction database. FEBS Lett. 2002;513:135–140. doi: 10.1016/s0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- Zhou D, He Y. Extracting interactions between proteins from the literature. J. Biomed. Inform. 2008;41:393–407. doi: 10.1016/j.jbi.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Zhou G, et al. Proceedings of the 43rd Annual Meeting of ACL. NJ, USA: Association for Computational Linguistics Morristown; 2005. Exploring various knowledge in relation extraction; pp. 427–434. [Google Scholar]