

Abstract
Within the archaea, the thermoacidophilic crenarchaeote Sulfolobus solfataricus has become an important model organism for physiology and biochemistry, comparative and functional genomics, as well as, more recently also for systems biology approaches. Within the Sulfolobus Systems Biology (“SulfoSYS”)-project the effect of changing growth temperatures on a metabolic network is investigated at the systems level by integrating genomic, transcriptomic, proteomic, metabolomic and enzymatic information for production of a silicon cell-model. The network under investigation is the central carbohydrate metabolism. The generation of high-quality quantitative data, which is critical for the investigation of biological systems and the successful integration of the different datasets, derived for example from high-throughput approaches (e.g., transcriptome or proteome analyses), requires the application and compliance of uniform standard protocols, e.g., for growth and handling of the organism as well as the “–omics” approaches. Here, we report on the establishment and implementation of standard operating procedures for the different wet-lab and in silico techniques that are applied within the SulfoSYS-project and that we believe can be useful for future projects on Sulfolobus or (hyper)thermophiles in general. Beside established techniques, it includes new methodologies like strain surveillance, the improved identification of membrane proteins and the application of crenarchaeal metabolomics.
Electronic supplementary material
The online version of this article (doi:10.1007/s00792-009-0280-0) contains supplementary material, which is available to authorized users.
Keywords: Crenarchaeon, Standard operating procedures, Genomics, Transcriptomics, Proteomics, Metabolomics, Biochemistry, Systems biology
Introduction
The thermoacidophilic archaeon Sulfolobus solfataricus represents one of the best studied members of the (hyper)thermophilic organisms within the phylum crenarchaeota, and thus represents a most suitable archaeal representative for “Hot Systems Biology”.
Systems Biology represents a relatively young scientific area that is applied at various levels of living systems, i.e., a metabolic network, cells or interacting organisms. Systems Biology aims to systematically decipher the communication between parts and modules or complex biological systems and how these lead to functioning of these systems (Snoep and Westerhoff 2005). Furthermore, Systems Biology enables the potential to realize a quantitative view on, for instance, metabolic processes of an organism including the regulatory mechanisms.
S. solfataricus optimally grows at 80°C (60–92°C) and pH 2–4. The S. solfataricus strain P2 (DSM 1617) was originally isolated from Pisciarelli, Italy (Zillig et al. 1980), but closely related strains reside in high numbers in virtually all acidic hot springs around the globe. The organism is a strict aerobe and grows heterotrophically on a variety of organic compounds as carbon and energy source such as sugars (e.g., glucose, galactose, arabinose, sucrose), amino acids or peptides (Grogan 1989), thus, S. solfataricus can be easily maintained in the laboratory with relatively little special equipment (Grogan 1989). The complete genome sequence is available (She et al. 2001) and functional genomics approaches have been applied to study this organism, including transcriptomics, proteomics and comparative genomics (e.g., Verhees et al. 2003; Snijders et al. 2006). Furthermore, several in vitro assay systems to analyse aspects of information processing in (hyper-)thermophiles, such as replication, transcription or translation, have been established for S. solfataricus (Ruggero et al. 1993; Bell and Jackson 2001; Kelman and White 2005; Barry and Bell 2006) and many of its proteins have been crystallized. The development of genetic tools for S. solfataricus has been a major breakthrough that allows for the study of gene functions and the potential to perturb the system (Jonuscheit et al. 2003; Worthington et al. 2003; Albers et al. 2006; Albers and Driessen 2008; Wagner et al. 2009).
The Sulfolobus systems biology (“SulfoSYS”)-project (Albers et al. 2009) represented the first (hyper-)thermophilic Systems Biology project, funded within the European trans-national research initiative “Systems Biology of Microorganisms” (SysMO; http://www.sysmo.net/). Within the SulfoSYS-project, focus lies on studying the effect of temperature variation on the central carbohydrate metabolism (CCM) of S. solfataricus (Albers et al. 2009) that is characterized by the branched Entner–Doudoroff (ED)-like pathway for sugar (glucose, galactose) degradation (Ahmed et al. 2005; Lamble et al. 2003, 2005; Kim and Lee 2005, 2006) and the Embden–Meyerhof–Parnas (EMP)-like pathway, which is employed during gluconeogenesis (Snijders et al. 2006; for review see Van der Oost and Siebers 2007; Zaparty et al. 2008).
The effect of temperature changes on the CCM network of S. solfataricus is analyzed by the tight integration of bioinformatics, genome, transcriptome, proteome, metabolome, and enzymatic data, with all –omic and biochemical data being produced from identical batches of biomass. Beside providing experimental data, one main part of this highly integrative project is the in silico analysis of the CCM network, including the design of a sufficiently precise model according to the silicon cell type model (http://www.siliconcell.net, Olivier and Snoep 2004). This model will allow for the computation of the S. solfataricus CCM, and in particular to investigate its robustness to changes in temperature at the system level.
Prerequisites for reproducibility and reliability of the produced datasets and the successful integration of the different data are the establishment and application of uniform standards, e.g., for the handling of the organism as well as the realization of the coordinated experiments. A basic necessity for the project was the evaluation of a suitable S. solfataricus strain and control of its genomic stability, followed by the optimization and standardization of growth conditions, handling of glycerol stocks and biomass production. First pilot experiments have been performed with S. solfataricus grown at 80°C (optimal growth temperature) compared to 70°C in order to improve and implement the SOPs, as well as establish the new methodologies applied to S. solfataricus.
Here, we report on the establishment and application of standard operating procedures (SOPs) regarding genomic, transcriptomic, proteomic, metabolomic as well as biochemical techniques applied for a comprehensive analysis of the CCM of the thermoacidophile S. solfataricus in the course of the SulfoSYS-project. Within the scientific archaeal community, this project represents the first effort to prepare common standards. Furthermore, new methodologies like the iTRAQ method for membrane proteome analysis have been established and applied successfully. Moreover, to our knowledge, this is the first report on metabolome analyses performed with a crenarchaeon.
In general, working with (hyper)thermophilic organisms (Bacteria or Archaea) or (hyper)thermophilic enzymes, is not always favorable due to the sometimes substantial technical challenges. However, it also harbors several experimental advantages, for example recombinant (hyper)thermophilic proteins can be easily purified from mesophilic hosts via heat precipitation, and because of their high rigidity they tend to crystallize easier. With our work we want to further contribute to establish S. solfataricus and also other (hyper)thermophiles as model organisms.
The S. solfataricus “Hot standards” will be updated on a regular basis and will be available, together with additional information (e.g., workflows), at the SulfoSYS homepage http://www.sulfosys.com/.
Strain evaluation and test for genomic stability of S. solfataricus strains P1 and P2
A special feature of the S. solfataricus genome is the presence of about 20 different types of mobile transposable elements (IS-elements) that occur at 10–25 copies each in the genome and that have been demonstrated to actively move or multiply (Schleper et al. 1994; Martusewitsch et al. 2000; She et al. 2001; Redder et al. 2001). Therefore, a particularly strict control of the genomic integrity of the organism is required over the course of the experiments. To avoid accumulation of mutations, it is common practice in most laboratories working with Sulfolobus, to prepare a large number of stocks from a primary culture obtained from DSMZ, from which experiments are started freshly, but the effectiveness of this procedure has not been examined.
In order to evaluate this maintenance procedure and to select a suitable strain for a Systems Biology project, seven different stocks of the S. solfataricus strains P1 and P2 (DSM 1616 and 1617) were compared. They were collected from the partners within the consortium as well as from the German Collection of Microorganisms and Cell Cultures (DSMZ), where stocks had been deposited about 15 years ago.
Cells from each stock were grown in parallel under identical conditions and chromosomal DNA was prepared (SOP_SSO_080901). Probes targeting four different IS elements (ISC1058, ISC1217, ISC1439 and ISC1359), were used in Southern hybridizations to produce characteristic footprints of the genomic DNA (Fig. 1). Three out of three tested S. solfataricus P1 stocks showed highly similar patterns in these hybridizations, as did four out of five different stocks from S. solfataricus P2. Only one stock that had been subcultured for several months in the laboratory showed major changes in the chromosomal footprints with all four probes tested (two of these are shown in Fig. 1, stock 2). All other stocks stemmed from laboratories in which cultures were routinely discarded after three to four passages in order to avoid the accumulation of spontaneous mutations. This analysis showed for the first time, that the maintenance of the strains as performed in most laboratories is indeed quite effective. The stock of S. solfataricus P2 (DSM1617) deposited at DSMZ was selected to be used in the SulfoSYS-project, in order to allow comparability to studies from other laboratories and because the complete genome of this strain is available (She et al. 2001). The strain has not undergone major genomic rearrangements during its maintenance at the DSMZ, since its chromosomal patterns were mostly identical to the four other stable stocks, including one that stems from the W. Zillig’s laboratory and has not been touched over the last 15 years (lane 2, Fig. 1).
Fig. 1.

Southern hybridization of AflIII-cut chromosomal DNAs hybridized with DIG-DNA probes of IS-element ISC1439 (a) and ISC1058 (b), respectively. Lanes 1–3 Strain S. solfataricus P1 (DSM 1616), lanes 4–8 strain P2 (DSM1617), lane 9 strain PBL2025 (used for constructions of knockout mutants (Worthington et al. 2003). DSMZ stock obtained freshly from DSMZ, stock 1–3 obtained from three different laboratories of this consortium, in which S. solfataricus is regularly grown. Stocks 3/1999 and 3/2004 were kept in the same laboratory, but were obtained in two different years
A detailed SOP procedure has been established for the production of glycerol stocks (SOP_SSO_080906a, b; for details see supplement S1) and for the evaluation of genomic integrity of the strain after fermentations in the SulfoSYS project (SOP_SSO_080901). For each fermentation, cells were grown from stock cultures to avoid the accumulation of mutations. In addition, Southern hybridizations are used to make sure that the stocks have not been contaminated by the virus SSV1 or its derivatives that are routinely used in the laboratories for genetic manipulations (SOP_SSO_080901).
Procedures
Test for genomic stability (SOP_SSO_080901)
The different S. solfataricus strains are grown at 78°C and pH 3 in Brock’s basal salt medium supplemented with 0.2% d-arabinose and 0.1% tryptone. Pyrimidine-auxotrophic mutants (PH1-16) are grown in media supplemented with 10 μg/ml uracil. For the isolation of chromosomal DNA 10 ml of an exponentially grown liquid culture (A 600nm = 0.25–0.4) are precooled on ice and centrifuged for 10 min at 4,000 rpm and 4°C. The cells are resuspended in 500 μl TEN solution (20 mM Tris/HCl, 1 mM EDTA, 100 mM NaCl) and 500 μl TEN solution supplemented with 1.6% N-laurylsarcosine and 0.12% Triton X-100. After an incubation of 30 min at room temperature, the chromosomal DNA is extracted with phenol:chloroform:isoamylalcohol (25:24:1) twice and two times with chloroform, finally the DNA is precipitated with ethanol. For southern hybridizations, 3 μg of chromosomal DNA are incubated with AflIII and separated on a 0.7% agarose gel. The DNA is blotted on nylon membranes and hybridized with digoxigenin-labeled double stranded DNA probes (approx. 1,000 bp) specific for each of the four IS-elements used in the analysis or the virus SSV1, respectively.
Standardized fermentation of S. solfataricus P2
S. solfataricus is an obligate aerobe and a chemoorganoheterotroph, growing on various carbon sources, such as yeast extract, tryptone or various sugars, amino acids and peptides (Grogan 1989). The thermoacidophilic organism optimally grows at 80°C (60–92°C) and pH 2–4. Cultivation of the organism under well-defined conditions represents one of the most important prerequisites for reproducibility and reliability of the produced data derived from the different technologies as well as subsequent data integration. Determination of the optimal growth conditions and the fermenter set-up, have been performed at the optimal growth temperature of 80°C (Fig. 2; SOP_SSO_080903).
Fig. 2.
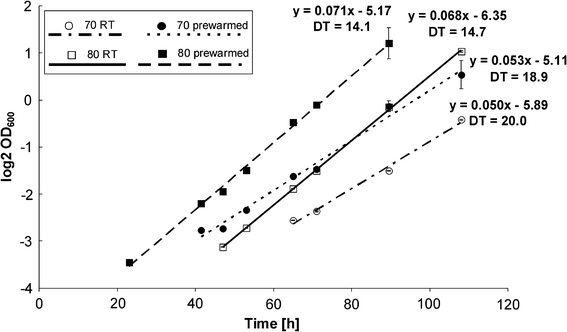
Log phase of S. solfataricus growth at 70 and 80°C (log2 scale). Inoculation of the medium preheated to desired temperature (filled circle, filled square), inoculation at room temperature (RT) and subsequently heated to desired temperature (open circle, open square). Growth at 70°C (filled circle, open circle) and growth at 80°C (filled square, open square) is shown. Lines represent trend lines for given conditions with equation and doubling time (DT) (h), R 2 values are in all cases >0.988
Procedures
Minimal medium (SOP_SSO_080902)
The minimal medium according to Brock et al. (1972, modified) contains (amount per litre): 1.3 g (NH4)2SO4, 0.28 g KH2PO4, 0.25 g MgCl2 × 7H2O, 0.07 g CaCl2 × 2H2O, 0.02 g FeCl2 × 4H2O, 1.8 mg MnCl2 × 4H2O, 4.5 mg Na2B4O7 × 10H2O, 0.22 mg ZnSO4 × 7H2O, 0.06 mg CuCl2 × 2H2O, 0.03 mg Na2MoO4 × 2H2O, 0.03 mg VOSO4 × 2H2O and 0.01 mg CoCl2 × 6H2O. Demineralized water with a value of resistivity not lower than 18.2 MΩ cm at 25°C is used for all solutions. Thus, the medium is uniform, independent from geography or used demineralization technique. Prior to autoclaving, the pH of the medium is set to 3.5 using H2SO4 The sterile filtered iron solution is kept in the dark at RT and added to the medium just before inoculation. The filter sterilized carbon sources such as glucose (30%) are added just before inoculation to reach a final of concentration of 0.3%.
Batch fermentation in flasks (SOP_SSO_080903)
The aerobic cultivation of S. solfataricus is carried out in 25–100 ml batch cultures in long-neck Erlenmeyer flasks (50–500 ml) at 70 and 80°C in minimal medium containing 0.3% glucose as carbon source (for exometabolome analysis only 0.15% glucose are used, SOP_SSO_080912) according to SOP_SSO_080902. An optimal oxygen supply is given by shaking (160 rpm) using a Thermotron shaker. Prewarmed medium (70 or 80°C, respectively) is inoculated with 200 μl glycerol stock (working stock; SOP_SSO_080906b, supplement S1) and growth is monitored spectrophotometrically at 600 nm. Afterwards, cells are chilled on ice and harvested by centrifugation (6,000×g, 15 min, 4°C) in the exponential growth phase (OD600 = 0.8–1) approximately after 96 h of growth and either directly used for analysis or stored at −80°C. For subsequent metabolome analysis cells are harvested by centrifugation (4,629×g, 5 min, 25°C), cell pellet is resuspended in 20 ml 0.9% NaCl (w/v) at RT and washed twice (4,629×g, 3 min, 25°C; 5810 R) (SOP_SSO_080912a).
Fermenter set-up and fermentation (SOP_SSO_080904)
Fermentation of S. solfataricus is performed in a 1.5 l fermenter (Applikon) with controlled temperature and pH settings. Also, oxygen dissolution (dO2 [%]) is algorithm controlled. Cells are aerated using air.
The organism is grown at respective temperatures and a pH of 3.5 in the minimal medium according to Brock et al. (1972; SOP_SSO_080902). The temperature of the medium (without glucose and the iron solution) is pre-set 1 day before fermentation start. Calibration of the pH and dO2 is completed, when the temperature in the fermenter is stable for 16 h.
The buffers used to calibrate the pH electrode for the fermenter (pH 7.0: 0.12 g NaH2PO4 in 90 ml H2O, set pH to 7.15, adjust to 100 ml; pH 3.0: 0.156 g NaH2PO4 in 90 ml H2O, adjust pH to 2.85, adjust volume to 100 ml) are pre-warmed to the respective growth temperature. The oxygen electrode is pre-calibrated prior to fermentation at the respective temperature. At 80°C experimentally determined dO2 = 80% is the optimal value for S. solfataricus for the used setup. As it relates to 3.5 mg/l of dissolved oxygen, this value is used for lower temperatures. The algorithm used to grow S. solfataricus P2 cells (for details see supplement S2) is designed to keep the dissolved oxygen at a level as close as possible to 80%. It is based on regulating stirrer speed and aeration intensity, and taking the growth phase estimate into account (for details see supplement S2).
For the SulfoSYS-experiments cells have been grown on 0.3% glucose as carbon source. Optical densities of liquid cultures are monitored at 600 nm (OD600). The fermenter is inoculated with 0.05 l of a pre-culture OD600 = 1.0 (±0.2). Pre-cultures are prepared using −80°C glycerol stocks to inoculate pre-heated medium (respective growth temperature) as it is shown in Fig. 2 to significantly reduce the lag phase of growth.
Cell harvest (SOP_SSO_080905)
When the culture reaches an OD600 = 0.85 (±0.15), the cells are sampled in aliquots of 20 ml (for transcriptomics and proteomics), 50 ml (for enzyme assays) or custom amounts dependant on OD600 (for the metabolomics). Further samples are taken for strain integrity evaluation. Cells are quickly cooled down to 4°C by dipping the collected cells in centrifugation tubes in liquid nitrogen for 30 s and finishing the cooling down in iced water to prevent sample freezing. Subsequently, cells are collected by centrifugation (3,500×g, 12 min, 4°C), catalogued and stored at −80°C in cell samples stock.
Preparation S. solfataricus glycerol stocks (SOP_SSO_080906a,b)
Beside the development of standard fermentation procedure, uniform handling has been established to prepare S. solfataricus glycerol stock solutions. The S. solfataricus strain 1617 has been acquired from the DMSZ and a master stock has been prepared (SOP_SSO_080906a, for details see supplement S1). Based on this master stock, the working stocks are prepared (SOP_SSO_080906b; for details see supplement S1), which are used for inoculation of fermentations.
The master stock is obtained after limited amount of transfers from the DMSZ stock, thus, guaranteeing genetic stability. Part of the master stock has been re-inoculated to create a bulk quantity of working stock used in the experiments. In case of the working stock running out, it can be recreated using the master stock (for details see supplement S1).
Glucose uptake measurements in S. solfataricus
The genome of S. solfataricus harbors several primary and secondary transporters (She et al. 2001), but as in all Archaea with only a few exceptions (e.g., Thermofilum pendens, Anderson et al. 2008) the organism lacks the phosphoenolpyruvate-dependent phosphotransferase system (PTS). Some of the primary active transporters represent sugar binding-protein-dependent ATP-binding cassette (ABC) transporters, and systems have been identified for the uptake of glucose, arabinose, trehalose, cellobiose, maltose and maltotriose (Albers et al. 1999, 2000; Elferink et al. 2001; Albers et al. 2001, 2004). Recently, the pH-dependent uptake of glucose via a high affinity ABC transporter has been characterized (Albers et al. 1999; Elferink et al. 2001). Compared to other sugars, such as galactose, glucose has been shown to be most effectively transported.
Procedures
Preparation of cells (SOP_SSO_080907a)
S. solfataricus P2 cells are grown in 50 ml of Brock medium according to the SOP (SOP_SSO_080902) except containing 0.4% glucose at 80°C until an OD600 of 0.3–0.4. Cells are collected by centrifugation (3,000×g, 15 min, 4°C) and resuspended in 50 ml of minimal Brock medium (SOP_SSO_080903). This procedure is repeated three times, and cells are finally resuspended to 1/10 of the starting volume at a protein concentration of about 10 mg/ml. Protein concentrations are determined by the BioRad Protein Assay (Bradford 1976, modified) with BSA as the standard.
Glucose uptake measurements (SOP_SS_080907b)
Uptake measurements using (14C-) labeled glucose (291 mCi/mmole, GE Healthcare) are performed at 60, 65 and 70°C (Table 1) using a previously described filter based assay (Albers et al. 1999). The concentrated cell suspension (10 μl) is added to 90 μl of minimal Brock medium and the solution is pre-warmed for 2 min at 60°C. Next 1 μl of the labelled glucose solution that is diluted with unlabeled glucose to the desired concentration is added yielding a final glucose concentration of 0.1–20 μM. After 10 s, the reaction is stopped by the addition of 2 ml of ice-cold 0.1 M LiCl and the mixture is rapidly filtered through a nitrocellulose filter (0.45 μm pore size, BA 85 nitrocellulose, Schleicher & Schuell). Filters are washed with 2 ml of 0.1 M LiCl and dissolved in 2 ml of scintillation fluid (Emulsifier Scientillator Plus, Perkin Elmer) and counted with a liquid scintillation analyzer 1600CA (Perkin Elmer).
Table 1.
Results for glucose uptake in S. solfataricus cells grown at 65 and 70°C
| Growth temperature (°C) | Uptake temperature(°C) | OD600 | Protein concentration (mg/ml) | K m (μM) | V max (nmol min−1 (mg protein)−1) |
|---|---|---|---|---|---|
| 65 | 65 | 0.368 | 15.43 | 0.44 | 0.45 |
| 65 | 70 | 0.368 | 15.43 | 0.56 | 0.62 |
| 70 | 65 | 0.298 | 6.29 | 0.12 | 0.61 |
| 70 | 70 | 0.298 | 6.29 | 0.23 | 0.85 |
Results
The in vitro uptake assay system for glucose has previously been established (Albers et al. 1999; Fig. S1 in the supplemental material) and the apparent K m for glucose uptake at 60°C and a pH 3.5 has been determined to be 1.9 μM with a V max value of 0.9 nmol min−1 (mg protein)−1. The assay has been established and performed at 65 and 70°C (Table 1). The assay is currently optimized for use at higher temperatures around 80°C, at which metabolism occurs so fast that label is evaporating as CO2 very rapidly. The measurements will be tried with only 5 and 2.5 s incubation time.
Genomics
Reconstruction of the central carbohydrate metabolism (CCM) network by comparative genomics
On the basis of the genome sequence information (She et al. 2001) and previous bioinformatic and experimental studies (Verhees et al. 2003; Ahmed et al. 2005; Snijders et al. 2006; van der Oost and Siebers 2007) the respective pathways of the CCM of S. solfataricus have been reconstructed (Albers et al. 2009). CCM reconstruction revealed the presence of: (i) The branched Entner–Doudoroff (ED) pathway that is promiscuous for glucose and galactose degradation (Ahmed et al. 2005, Lamble et al. 2003, 2005; Kim and Lee 2005, 2006). The pathway is characterized by two different branches, a non- and a semiphosphorylative branch. (ii) The Embden-Meyerhof-Parnas (EMP) pathway that is employed during gluconeogenesis. (iii) An oxidative TCA cycle (including glyoxylate shunt), which is responsible for the complete oxidation of glucose to carbon dioxide by using oxygen as terminal electron acceptor. (iv) The reverse ribulose-monophosphate (RuMP) pathway, which is utilized in pentose phosphate metabolism. (v) Finally, pathways for the synthesis and degradation of the storage compound glycogen (Skorko et al. 1989) as well as the disaccharide trehalose, which is known as compatible solute involved in stress response, are present.
Procedures
Reconstruction of the CCM network (SOP_SSO_080908)
The genome sequence information of S. solfataricus and other organisms as well as additional bioinformatic data have been derived from the UCSC Archaeal Genome Browser (http://archaea.ucsc.edu/). Blast search analyses are performed by using the nucleotide and protein blast tools (e.g., blastn, blastp, psi-blast) from the National Center for Biotechnology Information (NCBI; http://blast.ncbi.nlm.nih.gov/Blast.cgi). For genomic context analyses the STRING database (http://string.embl.de/) and for comparative genomics the respective tools from IMG (http://img.jgi.doe.gov/cgi-bin/pub/main.cgi?page=home) and from the LBMGE Genomics ToolBox (http://www-archbac.u-psud.fr/genomics/GenomicsToolBox.html) are applied. For pathway reconstruction the KEGG PATHWAY tool from the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) and for gaining detailed enzymatic information (e.g., enzyme reactions, specificities or enzymatic parameters) the BRENDA database (http://www.brenda-enzymes.org/) is used. The network reconstruction and annotations are regularly updated by using the above described methods and tools.
Results
A total of 97 genes have been identified that encode homologs with either a confirmed or a predicted function in the CCM network of S. solfataricus (Fig. 3; Albers et al. 2009). For several of these identified candidate genes, different functions are predicted, thus, their physiological function needs to be verified. To confirm the gene assignments the enzymatic activities of the recombinant gene products are analyzed (see SOPs_SSO_080913).
Fig. 3.
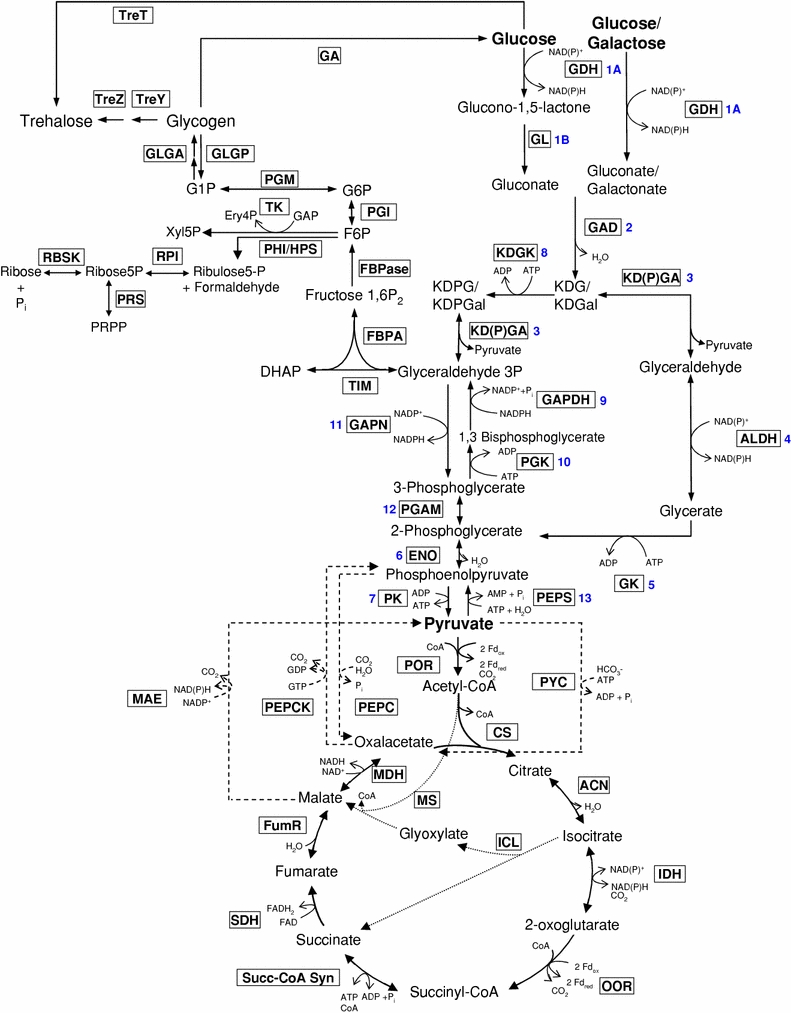
Reconstructed CCM of S. solfataricus. Identified CCM reactions (enzyme abbreviations boxed) involved in the branched ED and the EMP pathway [reactions numbered, corresponding to Table 3)], the citric acid cycle including the glyoxylate shunt (dotted arrow) the reversed ribulose monophosphate pathway, C3/C4 conversions (dashed arrow) as well as glycogen and trehalose metabolism. Intermediates: DHAP dihydroxy acetonephosphate, Ery4P erythrose 4-phosphate, F6P fructose 6-phosphate, fructose 1,6P2, fructose 1,6-bisphosphate, GAP glyceraldehyde 3-phosphate, G6P glucose 6-phosphate, KD(P)G 2-Keto-3-deoxy-6-(phospho)gluconate, KD(P)Gal 2-Keto-3-deoxy-6-(phospho)galactonate. Enzymes (including EC number): ACN aconitase (EC 4.2.1.3), CS citrate synthase (EC 2.3.3.1), ENO enolase (6; EC 4.2.1.11), FBPA fructose-1,6-bisphosphate aldolase (EC 4.1.2.13), FBPase fructose-1,6-bisphosphatase (EC 3.1.3.11), FumR fumarate hydratase (EC 4.2.1.2), GA glucan-1,4-α-glucosidase (EC 3.2.1.3), GAD gluconate dehydratase (2; EC 4.2.1.39), GADH glyceraldehyde dehydrogenase (4; EC 1.2.1.3), GAPDH glyceraldehyde-3-phosphate dehydrogenase (9; EC 1.2.1.12/13), GAPN non-phosphorylating GAP dehydrogenase (11; EC 1.2.1.9), GDH glucose dehydrogenase (1A; EC 1.1.47), GK glycerate kinase (5; EC 2.7.1-), GL gluconolactonase (1B; EC 3.1.17), GLGA glycogen synthase (EC 2.4.1.11), GLGP glycogen phosphorylase (EC 2.4.1.1), ICL isocitrate lyase (EC 4.1.3.1), IDH isocitrate dehydrogenase (EC 1.1.1.41), KD(P)GA KD(P)G aldolase (3; active on KDG as well as KDPG; EC 4.1.2.-), KDGK KDG kinase (8; EC 2.7.1.45), MAE malic enzyme (EC 1.1.1.38), MDH malate dehydrogenase (EC 1.1.1.37), MS malate synthase (EC 2.3.3.9), OOR α-oxoglutarate ferredoxin oxidoreductase (EC 1.2.7.3), PEPC PEP carboxylase (EC 4.1.1.31), PEPCK PEP carboxykinase (EC 4.1.1.32), PEPS phosphoenolpyruvate synthetase (13; EC 2.7.9.2), PGAM phosphoglycerate mutase (12; EC 5.4.2.1), PGI glucose-6-phosphate isomerase (EC 5.3.1.9), PGK phosphoglycerate kinase (10; EC 2.7.2.3), PGM phosphoglucomutase (EC 5.4.2.2), PHI/HPS 3-hexulose-6-phosphate isomerase/3-hexulose-6-phosphate synthase (EC 5.-.-.-/4.1.2.-), PK pyruvate kinase (7; EC 2.7.1.40), POR pyruvate synthase (EC 1.2.7.1), PRS ribose phosphate pyrophosphokinase (EC 2.7.6.1), PYC pyruvate carboxylase (EC 6.4.1.1), RBSK ribokinase (EC 2.7.1.15), RPI ribose-5-phosphate isomerase (EC 5.3.1.6), SDH succinate dehydrogenase (EC1.3.99.1), Succ-CoA Syn succinyl-cenzymA synthetase (EC 6.2.1.5), TIM triosephosphate isomerase (EC 5.3.1.1), TK transketolase (EC 2.2.1.1), TreT trehalose glycosyltransferring synthase (2.4.1.B2), TreY maltooligosyltrehalose synthase (EC 5.4.99.15), TreZ trehalose hydrolase (EC 3.2.1.141)
Comparative genomics
A comparative genomics approach is used to identify potential transcription factors (TFs) involved in the regulation of the CCM of S. solfataricus P2. This analysis basically followed a two-step strategy: first, all putative TFs in the genome of S. solfataricus P2 were identified globally. Subsequently, potential CCM regulators were selected by a genomic context scan.
Procedures and results
Global identification of putative TFs (SOP_SSO_080909a)
The global identification of putative TFs included different approaches. One source of information was the genome annotation, which was accessed via IMG (Markowitz et al. 2008; http://img.jgi.doe.gov/) and revealed a total of 51 predicted TFs in the genome of S. solfataricus P2. In addition to the annotation, two online databases ArchaeaTF (Wu et al. 2008; http://bioinformatics.zj.cn/archaeatf/) and DBD (Wilson et al. 2008; www.transcriptionfactor.org/), which both are specialized for the prediction of TFs, were analyzed to receive a more reliable and comprehensive set of predicted TFs. Following this SOP (additional information available at http://www.sulfosys.com), the predicted TFs of the three online databases IMG, ArchaeaTF and DBD were compared and united to a total set of 138 (Fig. 4).
Fig. 4.
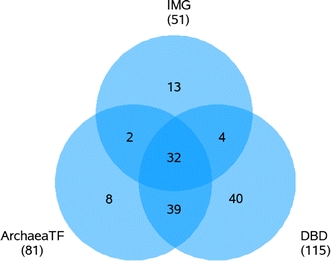
Venn diagram depicting the overlaps between the predicted sets of TFs in the genome of S. solfataricus P2, according to three different online databases. The numbers of predicted TFs in IMG, ArchaeaTF and DBD are 51, 81 and 115, respectively. The total amount of all three databases results in 138 different putative TFs
Identification of putative TFs by psi-BLAST-based approach (SOP_SSO_080909b)
Like in all other prokaryotes with sequenced genomes, not all protein functions of S. solfataricus P2 are known. Within the total of 3,048 protein-coding genes, 1,487 (i.e., 49%) are without or with uncertain function prediction, according to the annotation of IMG. In order to identify putative TFs in this fraction of genes, a psi-BLAST-based (Altschul et al. 1997) approach was performed. Following this procedure (SOP_SSO_080909b; details available at http://www.sulfosys.com), weak sequence similarities between proteins of unknown function and proteins of reported function in transcriptional regulation could be detected very sensitively.
Context-based approach for identifying putative TFs of the CCM (SOP_SSO_080909c)
The resulting set of 696 psiBLAST predicted TF candidates was examined by a genomic context scan, together with the total of 138 additional TFs which were predicted following SOP_SSO_080909a (see above and supplemental material S4). Here, the genomic neighborhoods of 57 of the identified CCM genes (see SOP_SSO_080908) were searched for the presence of the predicted TF candidates. The results were then manually examined, to determine if the corresponding pair of CCM-gene and TF candidate is likely to be co-transcribed in an operon or co-regulated bidirectionally. This resulted in a set of 81 candidate transcriptional regulators of the CCM, 34 of those are considered to be „strong candidates” for one of the following reasons: (1) the e value of a hit between candidate TF and a known transcription factor in the psi-BLAST-report is smaller than 1e-15, or (2) the candidate TF was predicted by (at least) one of the online databases IMG, ArchaeaTF or DBD.
The psi-BLAST approach detected four genes as candidate TFs, which also belong to the reported CCM-genes: SSO0286, SSO2281, SSO3041 and SSO3226; the latter three are considered to be strong candidates for TFs. These genes possibly have both functions (moonlighting), CCM-gene and TF. One of these four moonlighting candidates, SSO2281 is a glucose-6-phosphate-isomerase and another one SSO3226 is a fructose-1,6-bisphosphate aldolase. For these proteins, moonlighting functions have been reported in Eukaryotes (Jeffery et al. 2000; Sherawat et al. 2008). Although these two proteins are likely to have multiple functions, a role as TF has not been described so far, nor has a DNA-binding property been reported. Experimental verification and available corresponding protein structures, structural comparisons with transcription factors or DNA-binding proteins might give further insight. The other two moonlighting candidates are SSO0286, a fructose-1,6-bisphosphate phosphatase, and SSO3041, a putative gluconolactonase. For these proteins, no further evidence for moonlighting functions was found in the present literature.
Functional genomics
Transcriptome analyses
In order to investigate temperature adaptation strategies on the transcriptional level, different methods, i.e., DNA microarray analyses and real-time reverse transcription qPCR are used. The qPCR experiments mainly serve to verify the results obtained from the microarray analyses and a protocol will be available for download from the SulfoSYS homepage (http://www.sulfosys.com).
Microarray analyses
The 70-mer oligonucleotide DNA microarray has been designed and constructed in the group of John van der Oost (Wageningen University, NL, USA) by using the OligoWiz 2.0 (Wernersson and Nielsen 2005) software for oligonucleotide prediction. The array harbors a total of 8,860 spots, including probes for roughly 3,500 S. solfatricus genes, which are spotted in duplicate on the array, as well as those of viruses and plasmids of Sulfolobus. As negative controls 32 human sequences and 268 targets from Arabidopsis thaliana are comprised on the microarray in duplicate. In former studies, the RNA and cDNA preparation techniques had been optimized (Snijders et al. 2006; Fröls et al. 2007) revealing good and reproducible results with this oligoarray.
Procedures
Preparation of mRNA from S. solfataricus cells (SOP_SSO_080910a)
Total RNA is extracted from S. solfatricus cells that have been rapidly frozen in liquid nitrogen as described in fermentation protocols (SOP_SSO_080902-5).
For the isolation of S. solfataricus mRNA, the MirVana miRNA Isolation Kit (AMBION) according to the instructions of the manufacturer with slight modifications of the protocol is used. Cell pellets harvested from 20 ml of culture at OD600 = 0.85(±0.15) are taken from the sample stock. For optimal results all reagents in the initial steps of the protocol are used in double amounts. The samples are separated in two tubes during the acid phenol:chloroform:IAA (125:24:1, Ambion) extraction and proceeded according to manufacturers protocol. Finally, bound RNA is eluted by using 50 μl of pre-heated (95°C) H2O instead of 100 μl as recommended by the manufacturer [detailed protocol in supplementary materials (S3)]. RNA concentration is determined by using a Nanodrop RNA protocol (Thermo). The concentration of the prepared mRNA should be at least 1.3 μg/μl.
cDNA synthesis and labeling by reverse transcription (SOP_SSO_080910b)
Reverse transcription has been performed using a mix of standard nucleotides, with a 1:4 mixture of dTTP and aminoallyl dUTP (Ambion). The 50x aadUTP + dNTP mixture is prepared by dissolving 10 μl each of 100 mM dATP, dGTP, dCTP, 16 μl 50 mM aminoallyl-dUTP (AMBION–AM8439) and 2 μl 100 mM dTTP in 0.1 M KPO4 (pH 8.0). Single stranded cDNA is generated out of 20 μg total RNA by using a standard protocol for Superscript III (Invitrogen). The reaction is stopped with 4.5 μl 0.1 M EDTA pH 8.0. By the addition of 3 μl 1 M NaOH, followed by further incubation at 70°C for 15 min, the RNA template is degraded. The sample is neutralized by adding 3 μl of 1 M HCl.
The samples are purified by using the Cleanup–MinElute Kit (Qiagen) according to the manufacturer’s instructions, except slight modifications: 80% ethanol is used for the wash steps and elution is performed by the addition of NaHCO3 pH 8.6.
For the following labeling reaction using the Alexa dyes 647 and 555 (Invitrogen), cDNA concentration should be at least 80 ng/μl. Quantification is performed using a Nanodrop. For the labeling, add 18.4 μl of the cDNA sample to 3 μl of appropriate dye dissolved in DMSO and incubate for 1.5 h at RT in darkness.
For purification using the Cleanup–MinElute Kit (Qiagen), combine samples to be co-hybridized. All subsequent steps are performed according to the manufacturer’s instructions. The concentration of the pooled and labeled cDNA should be at least 120 ng/μl, as verified by Nanodrop and microarray measurements. In both cases the dye concentrations should be >0.7 pmol/μl.
Hybridization (SOP_SSO_080910c)
Prior to hybridization of the labeled cDNA to the microarrays, the slides are pre-hybridized in pre-warmed 5 × SSC containing 0.1% SDS and 10 μg/ml BSA, at 42°C for 40 min. Afterwards, the slides are washed thoroughly (30 s steps) in three Coplin jars with A.bidest. followed by briefly dipping them in isopropanol. Finally, the slides are dried in Microarray High-Speed Centrifuge (MHC, Arrayit; 2,000×g, 30 s, RT) and used for hybridization within 1 h.
For hybridization, 17.4 μl of the labeled cDNA is mixed with 1 μl tRNA (10 μg/μl), 1 μl herring sperm DNA (10 μg/μl) and 42.6 μl hybridization mixture containing 27 μl deionized formamide, 15 μl 20 ×SSC and 0.75 μl SDS (10%). The sample is incubated for 2 min at 95°C and subsequently cooled on ice for 1 min.
After quick-spin (10,000×g, 10 s, RT) the sample is applied on a slide (under a lifterslip). A.bidest (15 μl) is added to appropriate wells in the hybridization chamber to prevent evaporation. The slides are sealed for incubation at 42°C in darkness for 16–20 h. Afterwards, the slides are incubated in 2 ×SSC, 0.1% SDS for 5 min and in 0.1 ×SSC, 0.1% SDS for 20 min (both steps performed in the dark at 42°C). Later slides are washed 5× in Coplin jars containing 0.1 ×SSC and finally dried by centrifugation in MHC (2,000×g, 30 s, RT).
Scanning, extraction features, normalization and data analyses (SOP_SSO_080910d)
Each hybridization experiment using the 70-mer oligonucleotide DNA array has been performed as a dye swap, which provides a mean to exclude spots, where hybridization errors occur. Scans are performed with the GenePix Pro 4000B scanner (Axon). In a first scan of each array, 60% of laser intensity and in a second scan only 10% of laser intensity have been used, in order to be able to determine the proper ratios in spots saturated at 60%.
Features are extracted with GenePixPro 6.0 software (Axon) and flagged bad if intensities are below 3 times of the background in case of both dyes.
A feature is also excluded from further analysis, if the R 2 of the spot is <0.6, which indicates lack of homogeneity of the spot. Results acquired in the form of *.gpr file are converted to *.mev and normalized using Midas software (TIGR). The main normalization tool is Lowess (Quackenbush 2002; Yang et al. 2002) and log mean centering. By this means, extracted and normalized data can be transferred to Microsoft Excel sheets that allow for quick analysis and annotation of the data. Since the main interest is in up- and down-regulated genes, which corresponds to log2 ratio values >1 and <−1, respectively, the initial confirmation of statistical soundness of the data can be performed using Z test, testing if population of results with a given standard deviation is higher or lower than input value. By setting the input values at 1 and −1 we can statistically assess significance of the up-regulation of a given gene (for value >1, z value ≤ 0.05; for value <1, z value ≥ 0.95). Further analysis can be performed using SAM analysis in MeV program (Tusher et al. 2001).
Results
The pilot experiment involving transcriptomics has been performed by comparing cells grown in batch fermenter cultures at 80 and 70°C. Two biological samples have been used and a total of four microarrays have been hybridized. It has been assumed that log2 ratios higher than 1 and lower than −1 indicate significant fluctuation of the gene expression of the gene. Upregulation has been assessed using the Z test with 95% confidence level. Apart from the set of regulated genes, all genes involved in CCM have been compared.
In total, 24 genes are significantly up-regulated at 80°C and 43 genes are down-regulated. The up-regulated genes include a superoxide dismutase, indicating higher presence of reactive oxygen intermediates at higher temperature. Furthermore, nadA gene was overexpressed, suggesting higher rate of NAD synthesis. Other annotated genes include those coding for a large subunit of the replication factor C (RFC), a transcription activator in the thiamine synthesis pathway (tenA-2) and a small heat shock protein from hsp20 family. Four genes up-regulated are involved in amino acid synthesis, transport and proteolysis, suggesting scavenging of the dead cell material from the culture.
Surprisingly, the biggest group of down-regulated genes at 80°C consists of small and large subunit ribosomal genes (Table 2). A total of ten ribosome-related genes are down-regulated. This may indicate that in suboptimal conditions protein synthesis is one of the limiting factors for the population growth. It has to be noted here that nine of them are found in a large operon, which tend to have lower stability. It has been shown (Andersson et al. 2006) that all of these transcripts have a half life of no longer than 3 min. Another interesting finding is the down-regulation of the γ subunit of the thermosome (Table 2), which is consistent with findings of Kagawa et al. (2003). Other genes include two subunits of the cytochrome c complex, two putative RNA helicases related to deaD family (Table 2) There are also six genes coding for putative ABC transporter binding proteins, which are downregulated at 80°C (Table 2). This might indicate scavenging debris from cells that die due to cold shock, as two of the transporters are binding sugars not present in the medium, in which cells have been grown (arabinose and maltose) and other two bind dipeptides. The remaining two transporters have not yet been assigned a function, but based on sequence similarity they might play a role in oligosaccharide uptake. Other candidates have no assigned function or are distantly related to proteins from other species.
Table 2.
Significantly regulated genes comparing growth at 80 versus 70°C revealed from transcriptomic analysis
| Gene ID | Annotation | 80 versus 70°C log2 ratio (±SD) |
|---|---|---|
| SSO0068 | SSU ribosomal protein S9AB (rps9AB) | −1.29 (±0.38) |
| SSO0489 | Phosphate binding periplasmic protein precursor (pstS) | −1.91 (±0.25) |
| SSO0697 | LSU ribosomal protein L30AB (rpl30AB) | −1.85 (±0.84) |
| SSO0698 | SSU ribosomal protein S5AB (rps5AB) | −2.07 (±0.70) |
| SSO0700 | LSU ribosomal protein L19E (rpl19E) | −1.73 (±0.67) |
| SSO0704 | LSU ribosomal protein L5AB (rpl5AB) | −1.44 (±0.35) |
| SSO0707 | LSU ribosomal protein L24AB (rpl24AB) | −1.60 (±0.60) |
| SSO0716 | LSU ribosomal protein L2AB (rpl2AB) | −1.73 (±0.72) |
| SSO0718 | LSU ribosomal protein L4AE (rpl4AE) | −1.25 (±0.29) |
| SSO1274 | Oligo/dipeptide transport, permease protein (dppB-1) | −1.80 (±0.74) |
| SSO1275 | Oligo/dipeptide transport, permease protein (dppC-1) | −1.19 (±0.27) |
| SSO1889 | ATP-dependent RNA helicase | −1.74 (±0.73) |
| SSO2036 | ATP-dependent RNA helicase | −1.26 (±0.24) |
| SSO3000 | Thermosome gamma subunit | −2.11 (±0.60) |
| SSO3043 | ABC transporter, binding protein | −2.05 (±0.99) |
| SSO3047 | ABC transporter, permease | −1.37 (±0.55) |
| SSO3053 | Maltose ABC transporter, maltose binding protein | −2.29 (±0.85) |
| SSO3066 | Arabinose ABC transporter, arabinose binding protein | −1.51 (±0.61) |
| SSO3120 | Metabolite transport protein, putative | −1.69 (±0.94) |
| SSO3198 | Muconate cycloisomerase related protein | −1.28 (±0.49) |
| SSO6391 | SSU ribosomal protein S14AB (rps14AB) | −1.44 (±0.53) |
| SSO6401 | LSU ribosomal protein L23AB (rpl23AB) | −1.85 (±0.64) |
| SSO2088 | Peptidase, putative | 1.12 (±0.12) |
| SSO0316 | Superoxide dismutase [Fe] (sod) | 1.17 (±0.20) |
| SSO2603 | Small heat shock protein hsp20 family | 1.33 (±0.52) |
| SSO2598 | Transcriptional activator (tenA-2) | 1.35 (±0.52) |
| SSO0998 | Quinolinate synthetase (nadA) | 1.99 (±0.27) |
| SSO2549 | Amino acid transporter, putative | 2.27 (±0.45) |
| SSO0769 | Activator 1, replication factor C (RFC) large subunit (rfcL) | 2.56 (±0.89) |
A log2 ratio >1 indicates up-regulation at 80°C, log2 < −1 indicates down-regulation at 80°C. For all genes Z test reaveld values ≤0.05
SD standard deviation
Of the 97 genes hypothesized to be involved in the CCM network, 91 have been found using the transcriptome analysis. Most genes do not show statistically significant differential expression. The genes of the branched ED pathway (Fig. 3) also do not show differential expression between the two conditions with the exception of SSO3198 coding for gluconate dehydratase and SSO3194 encoding the non-phosphorylating glyceraldehyde 3-phosphate dehydrogenase (GAPN) (Table 3). The encoding genes are twofold down-regulated at 80°C. They are located in the ED operon (SSO3198-3197-3195-3194; Ahmed et al. 2005), and the other genes from the same cluster indicate a similar regulation (with the exception of SSO3195 KDG kinase; Table 3). Also the proteomic data (SOPs_SSO_080911) show no significant differences except for the GAPN, which is in accordance to the transcriptomic data, downregulated at 80°C at the proteomic level (Table 3). These first results suggest that the regulation of the CCM in S. solfataricus is placed on different regulatory levels.
Table 3.
Results of the initial transcriptomic and proteomic analyses of the glycolytic, branched ED pathway of S. solfataricus in response to growth at 80 versus 70°C
| Gene ID | Reaction no. (Fig. 3) | Gene product | EC no. | Transcriptomics 80 versus 70°C log2 ratio (±SD) | Proteomics 80 versus 70°C log2 ratio (±SD) |
|---|---|---|---|---|---|
| SSO3003 | 1A | Glucose-1-dehydrogenase (GDH)a | 1.1.1.47 | −0.34 (±0.11) | NF |
| SSO2705 | 1B | Gluconolactonase (GL) | 3.1.1.17 | −0.16 (±0.20) | 0.34 (±0.06) |
| SSO3041 | 1B | Gluconolactonase (GL) | 3.1.1.17 | −0.42 (±0.32) | NF |
| SSO3198 | 2 | Gluconate dehydratase (GAD)b | 4.2.1.39 | −1.28 (±0.49) | −0.44 (±0.06) |
| SSO3197 | 3 | 2-keto-3-deoxy-(6-phospho)-gluconate/galactonate aldolase (KD(P)GA)b | 4.1.2.- | −0.78 (±0.15) | −0.27 (±0.60) |
| SSO2636 | 4 | Aldehyde ferredoxin oxidoreductase, β-subunit (AOR) | 1.2.7.- | −0.54 (±0.23) | 0.29 (±0.04) |
| SSO2637 | 4 | Aldehyde ferredoxin oxidoreductase, γ-subunit (AOR) | 1.2.7.- | −1.12 (±0.53) | 0.36 (±0.17) |
| SSO2639 | 4 | Aldehyde ferredoxin oxidoreductase, α-subunit (AOR) | 1.2.7.- | −1.28 (±0.88) | −0.05 (±0.10) |
| SSO0666 | 5 | Glycerate kinase (GK) | 2.7.1.- | −0.45 (±0.21) | −0.40 (±0.14) |
| SSO0913 | 6 | Enolase (ENO) | 4.2.1.11 | 0.02 (±0.09) | −0.25 (±0.21) |
| SSO0981 | 7 | Pyruvate kinase (PK) | 2.7.1.40 | 0.63 (±0.43) | 0.07 (±0.13) |
| SSO3195 | 8 | 2-keto-3-deoxy-gluconate/galactonate kinase (KDGK)b | 2.7.1.45 | −0.09 (±0.21) | NFb |
| SSO0528 | 9 | Glyceraldehyde-3-phosphate (GAP) dehydrogenase (GAPDH) | 1.2.1.12/13 | −0.12 (±0.32) | 0.62 (±0.13) |
| SSO0527 | 10 | Phosphoglycerate kinase (PGK) | 2.7.2.3 | −0.50 (±0.44) | 0.45 (±0.16) |
| SSO3194 | 11 | Non-phosphorylating GAP dehydrogenase (GAPN)c | 1.2.1.9 | −1.18 (±0.44) | −1.47 (±0.65) |
| SSO0417 | 12 | Phosphoglycerate mutase (PGMA) | 5.4.2.1 | −0.51 (±0.36) | −1.36 (±0.47) |
| SSO0883 | 13 | Phosphoenolpyruvate synthetase (PEPS) | 2.7.9.2 | −0.65 (±0.37) | −0.40 (±0.20) |
Proteome analyses
In course of the SulfoSYS-project one goal is to quantitatively measure and understand protein expression changes, protein interaction networks, non-covalent interactions and post-translational modifications of the CCM proteins of S. solfataricus in response to temperature changes.
Different approaches for protein quantitation for membrane proteomes are applied within this project, since membrane proteins play most important roles during cell life. The iTRAQ method is used for global expression profiling, to compare up to eight fully adapted cell states.
Procedures
Cellular extraction (SOP_0809011a)
Frozen cells are firstly washed twice with ice-cold water, then they are centrifuged at 6,000×g before being resuspended in 1 mL of extraction buffer, which contains 43 mM NaCl, 81 mM MgSO4 and 27 mM KCl (Bisle et al. 2006). Protein extraction is carried out using an ultra sonicator (Sonifier 450, Branson) 4 times (alternatively 1 min of sonication and 1 min on ice) at 70% duty cycle. Samples are then centrifuged at 3,000×g for 5 min at ×4°C to discard unbroken cells and debris, the supernatant is collected before centrifugation again at 100,000×g for 90 min 4°C using a sucrose gradient detailed as elsewhere (Bisle et al. 2006). The pellets are collected as enriched membrane fractions. These membrane fractions are then delipidated using chloroform/methanol as detailed by Wessel and Flugge (1984) with some modifications. Briefly, the membrane is resuspended in 400 μl of methanol, vortexed at 1,500 rpm for 30 s and centrifuged at 9,000×g for 20 s at room temperature. The pellet is collected by discarding the supernatant, then resuspended in 100 μl of chloroform and 1,500 rpm for 30 s, and centrifuged at 9,000×g for 20 s room temperature. The recovery of membrane is performed using phase separation, where 300 μl of water is added to the sample, followed by 1,500 rpm for 30 s and centrifugation at 9,000×g for 90 s. While the upper phase is discarded carefully, 300 μl of methanol are added to the interphase (containing precipitated proteins) and lower phase. This sample is mixed by vortexing at 1,500 rpm for 1 min, followed by centrifugation at 9,000×g for 2 min to pellet membrane proteins. The pellet is collected by discarding the supernatant and then drying in a vacuum concentrator before being resuspended in 100 μl of 0.5 M TEAB pH 8.5 buffer containing 0.095% SDS. The sample is dissolved totally by sonicating for 5 min before the total protein concentration is determined using the RC-DC Protein Quantification Assay (Bio-Rad, UK). This sample is then ready for the iTRAQ labeling step. For soluble protein analysis, cells are resuspended in 0.5 M TEAB pH 8.5 before being extracted as detailed above.
iTRAQ labeling (SOP_0809011b)
A total of 100 μg protein of each phenotype is used for iTRAQ analysis. Protein samples are reduced, alkylated, digested and labeled with iTRAQ reagents according to the manufacturer’s protocol (Applied Biosystems, USA). Briefly, samples are reduced by adding 2 μl of 50 mM tris-(2-carboxyethyl) phosphine (TCEP) and incubating at 60°C for 1 h; then cysteines are alkylated with 1 μl of 200 mM methyl methanethiosulfonate (MMTS) for 10 min at room temperature. The digestion step at 37°C overnight is carried out using trypsin MS grade (Promega, UK) with the ratio of trypsin:proteins 1:20. Then these samples were labeled with iTRAQ reagents in isopropanol (or ethanol). After incubation at room temperature for 4 h, labeled samples were combined before being dried in a vacuum concentrator.
In the case of the combination of both, trypsin and chymotrypsin, for the digestion step, samples are firstly digested with trypsin on the first day (at a ratio of 1:40) and then a mixture of chymotrypsin and trypsin (ratio enzyme: protein = 1:40 for each) on the second day. After digestion by trypsin, the partially digested sample is centrifuged at 13,000×g for 1 h at room temperature to pellet undigested proteins, then, while supernatant was collected and transferred to a new tube, the pellet is resuspended again in methanol before a mixture of trypsin and chymotrypsin is added (refer to Fischer et al. 2006 for chymotrypsin digestion details). The sample is then incubated overnight at 37°C. After digestion, this sample is centrifuged again at 13,000×g to pellet undigested proteins, the supernatant is collected and mixed with the previous trypsin digested supernatant. The mixture of digested peptides is then dried in a vacuum concentrator before being resuspended in 30 μl of 0.5 M TEAB pH8.5 for the iTRAQ labeling step. To enhance the protein digestion step for the membrane fractions, the use of sodium deoxycholate (SDC) with a final concentration of 0.007% has also been applied (see Masuda et al. 2008) for more detail).
Strong cation exchange (SCX; SOP_0809011c)
The dried iTRAQ samples are resuspended in buffer A (details below) and then fractionated using a SCX technique on a BioLC HPLC system (Dionex, UK) to clean the sample, as well as reduce its complexity. The SCX fractionation is carried out using a PolySulfoethyl A column (PolyLC, USA) 5 μm particle size in a length of 20 cm × 2.1 mm in diameter, 200 Å pore size. The system is operated at a flow rate of 0.2 ml/min, and with an injection volume of 120 μl. The mobile phase is used consisting of buffers A and B. While buffer A contains 10 mM KH2PO4, 25% acetonitrile, pH3, buffer B consists of 10 mM KH2PO4, 25% acetonitrile and 500 mM KCl, pH3. A gradient of 60 min is used, 5 min at 100% buffer A, followed by ramping from 5 to 30% buffer B for 40 min, 30–100% B over 5 min and finally 100% A for 5 min. A UV detector UVD170U and Chromeleon Software (Dionex, The Netherlands) are used to record the chromatogram. Labeled peptide fractions are collected every minute, subsequently each fraction is dried in a vacuum concentrator.
Mass spectrometry analysis (SOP_0809011d)
Selected dried labeled peptides samples are redissolved in 50 μl of buffer A consisting of 0.1% formic acid and 3% acetonitrile, and then MS analysis is performed on a QStar XL Hybrid ESI Quadrupole time-of-flight tandem mass spectrometer, ESI-qQ-TOF–MS/MS (Applied Biosystems, Canada), coupled with a nano-LC system comprising a combination of a LC Packings Ultimate 3000 (Dionex, UK). An injection of 15 μl of sample is submitted to the nano-LC–MS/MS system. The LC gradient is operated at a flow rate of 300 μl/min, consisting of 5% buffer B (0.1% formic acid and 97% acetonitrile) to 30% buffer B over 85 min, followed by a 5 min ramp to 95% buffer B, and then 10 min at 5% buffer B. The ESI–MS detector mass range is set at 350–1800 m/z. The MS data acquisition is performed in the positive ion mode. During the scan, peptides with a +2, +3, or +4 charge state are selected for fragmentation, and the time for summation of MS/MS events is set up at 3 s.
Data searching (SOP_0809011e)
MS/MS data are analyzed using Phenyx software v.2.6 (Geneva Bioinformatics, Switzerland) with the S. solfataricus P2 protein database (2977 ORFs) downloaded June 2007 from NCBI (http://www.ncbi.nlm.nih.gov/). The search parameters for peptides and MS/MS tolerance are as follows: 0.2 Da peptide tolerance, default parent charge were +2, +3 and +4 with trust parent charge: yes. Acceptance parameters are set as following: minimum peptide length, peptides z score, maximum P value and AC score were 5, 5, 10−5 and 5, respectively. Fixed modifications of MMTS, cys_CAM, iTRAQ_K, iTRAQ_Ntermi are used, and enzymes used for searching are trypsin alone or a combination of trypsin and chymotrypsin (in Experiment 3) with one missed cleavage for both. The results are exported to Excel (Microsoft 2008, USA) for further analyses. Although Phenyx software is used for searching and exporting data, the data analysis is carried out as suggested by the Protein Pilot v2.0 software documentation (Applied Biosystems, USA), since Phenyx does not automatically calculate iTRAQ quantitation. All peptides are converted to log10 space before the calculation of the protein ratio is applied, as per the equation adapted from the Protein Pilot software documentation. Subsequently, the correcting of the bias median ratio of each protein is also applied. Moreover, the estimation of false determination rate is also carried using spectra derived from a decoy databases (generated from S. solfataricus reversed sequences) as described by Elias and Gygi (2007). We adjusted parameters for MS/MS searching to get the false determination rate (for each experiment) less than 0.2%.
Results
Protein identification for quantitative membrane proteomic analysis of S. solfataricus
In this investigation, three different iTRAQ-8plex experiments have been analyzed for enriched membrane fractions, including one experiment carried out as suggested by the original protocol (Experiment 1), and two experiments for modified protocols (Experiment 2 for trypsin and chymotrypsin, Experiment 3 trypsin and chymotrypsin with the presence of SDC). Cells grown at 80°C have been used as the controls and labeled with iTRAQ reagents 118, 119 and 121 (119 and 121 used as an independent biological replicate whilst 118 and 119 used as technical replicate), and samples at 70°C were labeled with reagents 115, 116 and 117 (115 and 116 used as an independent biological replicate, 116 and 117 used as a technical replicate).
As a result, the numbers of proteins detected for three different iTRAQ experiments are shown in Fig. 5. It is clear that more proteins were detected for Experiments 2 and 3 as a result, more membrane proteins and trans-membrane proteins were also detected for Experiments 2 and 3 compared to Experiment 1 (for more details see Fig. 5). These data agree with a previous study, since more membrane proteins were found with the presence of SDC (Masuda et al. 2008). There also seems to be more membrane and transmembrane proteins being found in Experiment 3 compared to Experiment 2 (for more details see Fig. 6). Moreover, in term of cell localization, the highest number of integral membrane proteins was identified for Experiment 3.
Fig. 5.
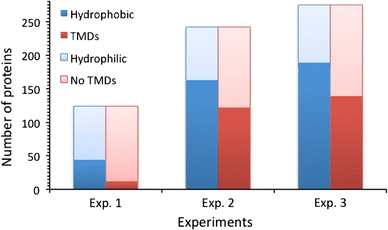
Number of proteins detected in the three different iTRAQ experiments. The identification of these proteins’ membrane properties based on hydrophobic (dark blue) and transmembrane domains (TMDs, dark red) found, are shown
Fig. 6.
Total numbers of proteins detected for enriched membrane fractions from three different iTRAQ experiments. Peptide detection
Therefore, we can assert that the combination of both SDC and chymotrypsin for trypsin digestion is suitable for S. solfataricus integral membrane proteins. A slightly increased total number of detected proteins are also found in Experiment 3, because more peptides are released during the digestion step, when using a combination of trypsin and chymotrypsin with a presence of SDC.
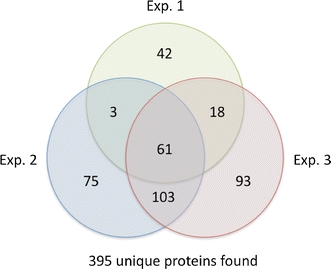
By combining proteins detected in all three different iTRAQ experiments for enriched membrane fractions 395 proteins were found as shown in Fig. 6.
For bottom-up proteomic analysis, the identification and quantitation of protein are based on peptide-level assignments; therefore, it is necessary to discuss this issue here. The numbers of distinct peptides detected for each experiment are 749, 1374 and 1635 for Experiments 1, 2 and 3, respectively.
Since SDS and SDC are applied in this study, and these compounds are known to be unfriendly compounds for mass spectrometry, and excess amounts of these compounds affect the labeling step. Therefore, we evaluated the affect of these chemicals to the iTRAQ labeling step, as well as nano-LC MS/MS operation via the efficiency of iTRAQ labeling, where the evaluation was calculated based on the percentage of labeled peptides compared to the total number of detected peptides (labeled and unlabeled peptides). However, we could not detect any difference within these experiments, since there were a small percentage of unlabeled peptides being detected; actually only two unlabeled peptides were solely identified in Experiment 3. Therefore, we can conclude that the SDC concentration used in this study was acceptable for the iTRAQ labelling step.
Membrane proteins
As discussed above, more peptides than proteins are detected for enriched membrane fractions in Experiments 2 and 3. To ensure that all proteins detected here contained membrane properties, these proteins were examined based on membrane properties including hydrophobic (Gravy score), TMDs found (TMHMM, http://www.cbs.dtu.dk/services/TMHMM/) and cell localization (http://www-archbac.u-psud.fr/projects/sulfolobus/). As a result, of 395 merged proteins (from all 3 experiments), 373 proteins were found to be membrane proteins, where 233 were proteins observed with more than two different membrane properties.
In summary, we have applied successfully iTRAQ for S. solfataricus (P2) quantitative membrane proteomic analysis (Fig. 7), since of 284 proteins detected, 246 proteins were found as membrane proteins. A merged data from all different iTRAQ data led to 395 unique proteins were detected, in which 373 were found as membrane proteins. All merged proteins from iTRAQ experiments and more details about membrane proteins’ regulations can be found in “Quantitative Proteomic Analysis of Sulfolobus solfataricus Membrane Proteins” (Pham et al. 2009).
Fig. 7.
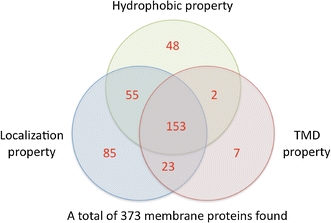
Classification of merged proteins base on membrane properties
Metabolome analyses
The metabolic composition reflects the set of metabolites within a cell at a certain timepoint. Metabolites take part in regulatory mechanisms, directly in allosteric regulation of enzyme activities but also indirectly by influencing transcriptional and translational control. Therefore, the integration of metabolome data (relative metabolite concentrations) can (i) highlight regulatory mechanisms taking place due to the temperature change, (ii) help to complete functional gene annotations by identification of missing enzymatic activities, (iii) being used in order to identify and analyze specific metabolic pathways and, (iv) provide data for the computational cell simulations.
First quantitative analysis of changes of metabolite concentrations due to temperature changes comparing 80 versus 70°C have been performed with cell mass derived from batch flask fermentation (SOP_SSO080903; Tables 4 and 5). In addition, exometabolome analyses have been performed, comprehending all metabolites that are excreted into the growth medium and therefore depict a picture of the metabolome during a period of metabolic and biological activity prior to sampling.
Table 4.
Ratios of detected metabolites in samples derived from cells grown at 80 versus 70°C
| Metabolites | Ratio | |
|---|---|---|
| CCM metabolism | ||
| KDG/KDGal | 0.11 | |
| Glyceraldehyde | 0.58 | |
| Citrate | 3.13 | |
| 3-Phosphoglycerate | 2.86 | |
| Succinate | 1.75 | |
| Glycerate | 1.56 | |
| Glucose 6-phosphate | 1.51 | |
| Trehalose | 1.45 | |
| Glucose | 1.33 | |
| Fructose 6-phosphate | 1.25 | |
| Malate | 1.18 | |
| Fumarate | 1.11 | |
| Galactose | 0.09 | |
| Pyruvate | NF | |
| 2-Oxoglutarate | NF | |
| Glucono-1,5-lactone | NF | |
| Glucose-1-phosphate | NF | |
| Dihydroxyacetonphosphate | NF | |
| 2-Phosphoglycerate | NF | |
| Phosphoenolpyruvate | NF | |
| Fructose 1,6-bisphosphate | NF | |
| 1,3 Bisphosphoglycerate | NF | |
| Glyceraldehyde 3-phosphate | NF | |
| Isocitrate | NF | |
| Oxaloacetate | NF | |
| KDPG/KDPGal | Not available | |
CCM compounds and metabolites of amino acid and nucleic acid metabolism as well as of glycosylated protein and lipid biosynthesis. Higher metabolite concentrations at 70°C are indicated in bold fonts and lower concentrations at 70°C are itaclicized. Others represent no significant changes
NF not found (below observation limit)
Table 5.
Ratios of detected metabolites in samples derived from cells grown at 80 versus 70°C
| Metabolites | Pathway | Ratio |
|---|---|---|
| Other metabolites | ||
| Valine | Amino acid metabolism | 0.12 |
| Isoleucine | Amino acid metabolism | 0.1 |
| Glucosamine | Precursor of glycosylated proteins and lipids | 0.16 |
| Leucine | Amino acid metabolism | 0.19 |
| Spermidine | Nucleic acid and protein synthesis | 0.21 |
| Alanine | Amino acid metabolism | 0.31 |
| Thymine | Pyrimidine metabolism | 0.35 |
| Putrescine | Amino acid metabolism | 0.39 |
| Glutamic acid | Amino acid metabolism | 0.4 |
| Lysine | Amino acid metabolism | 0.42 |
| Threonine | Amino acid metabolism | 0.57 |
| Aspartic acid | Amino acid metabolism | 0.62 |
| Beta-Alanine | Amino acid metabolism | 2.5 |
| Glycine | Amino acid metabolism | 1.61 |
| Serine | Amino acid metabolism | 2.32 |
| Phenylalanine | Amino acid metabolism | 3.7 |
As one important prerequisite for the set-up of the protocols for S. solfataricus metabolome analysis, cell growth and handling of the organism have been performed according to the developed SOPs (SOP_SSO080902-4). However, a special protocol for cell treatment directly after harvest by centrifugation had to be established (SOP_SSO_080912a).
Procedures
Sample preparation (SOP_SSO_080912a)
Cell mass is obtained from batch fermentation (SOP_SSO_080903). 20 mg cell dry weight (that is equivalent to 38/OD600 nm = x ml S. solfataricus culture) is harvested by centrifugation (4,629×g, 5 min, 25°C; 5810 R, Eppendorf). After harvesting, the cell pellet is resuspended (by shaking) in 20 ml 0.9% NaCl (w/v) at RT and washed twice (4,629×g, 3 min, 25°C; 5810 R, Eppendorf).
Subsequently, cells are resuspended in 1.5 ml methanol (containing 60 μl ribitol (c = 0.2 g l−1) and lyzed in an ultrasonic bath for 15 min at 70°C. Afterwards, the sample is incubated on ice for 2 min, 1.5 ml of deionized water is added and the sample is vortexed. For extraction of metabolites 1 ml chloroform is added and the sample is mixed by vortexing. After centrifugation (4,629×g, 5 min, 4°C; 5810 R, Eppendorf) the upper, polar phase is transferred into a fresh tube (2 ml) and dried in a vacuum concentrator (SpeedVac, Eppendorf) for 1 h with rotation and overnight without rotation. Final step is the derivatization of the metabolites for subsequent GC–MS analysis: Hereunto, 20 μl pyridine, containing 20 mg ml−1 methoxyamine hydrochloride are added to the dried sample (vortex for 1 min). After incubation in a thermomixer (600 rpm, 90 min, 30°C; Thermomixer comfort, Eppendorf) 32 μl N-methyl-N-trimethylsilyltrifluoroacetamide (MSTFA) is added (vortex for 1 min). Samples are incubated again for 30 min at 37°C (shaking speed 600 rpm) followed by 120 min at 25°C (shaking speed 600 rpm). After subsequent centrifugation (18,400×g, 5 min, RT; 5424, Eppendorf) 50 μl of the sample are transferred in a glass vial containing a micro cartridge for GC–MS analysis.
For exometabolome analysis cells of a S. solfataricus batch culture are grown on 0.15% glucose (instead of 0.3%) and harvested in the exponential growth phase by centrifugation (4,629 × g, 5 min, 25°C, 5810 R, Eppendorf). The supernatant is collected and 40 μl ribitol (c = 0.2 g l−1) as internal standard are added to 500 μl of culture supernatant. Subsequently, the sample is transferred in a 2 ml eppendorf tube and dried in a vacuum centrifuge (SpeedVac, Eppendorf) for 1 h with rotation and overnight without rotation. Afterwards metabolites are derivatized for GC/MS analysis (SOP_SSO_080912a) that is performed following SOP_SSO_080912b.
GC–MS analysis (SOP_SSO_080912b)
The system consists of a TRACE mass spectrometer coupled to a TRACE gas chromatograph with an AS 3000 autosampler (all devices from Thermo Finnigan GmbH, Egelsbach, Germany). The system operates under the Xcalibur software (version 1.2, Thermo Finnigan GmbH, Egelsbach, Germany). Positive electron ionization (EI +) mode at 70 eV is used for ionization. Tuning is done according to the operating manual using perfluorotri-N-butylamine (Fluorochem Ltd., Derbys, UK) as reference gas. Full scan mass spectra are acquired from 40 to 800 m/z with a scan rate of 2/s and a solvent delay time of 6 min. The chromatography was performed using a 30 m, 0.25 mm, 0.25 μm film thickness, DB-5MS column (J&W Scientific, Folsom, USA) with a helium flow of 1 ml min−1. For measurements a derivatized sample volume of 2 μl was injected in split mode (25:1) at 70°C and the solvent was evaporated in 0.2 min. Injections were made using a programmed temperature vaporizer (PTV) injector supplied with a 12 × 2 mm glass liner manually filled with glass wool (Restek GmbH, Bad Homburg, Germany). For sample transfer the temperature was increased to 280°C at a rate of 14°C s−1 followed by an additional constant temperature period at 280°C for 2 min. The oven temperature is increased at 1°C min−1 to 76°C and then with 6°C min−1 to 325°C, after 10 min isothermal cool-down to 70°C.
Results
A total of 70 metabolites from widely different metabolic pathways can be detected in the exponential growth phase for S. solfataricus (Table S1, supplemental material). Derived data have been compared to available bacterial metabolome data. The most obvious difference is that S. solfataricus shows a much smaller number of metabolites compared to Bacteria, such as Corynebacterium glutamicum (Strelkov et al. 2004) or Pseudomonas aeruginosa (Frimmersdorf et al., unpublished). These data are of special interest, because to our knowledge this is the first metabolome analysis for a thermoacidophilic organism.
Some of the detected metabolites in samples derived from cells grown at 80°C (optimal growth temperature) and 70°C show differences in relative concentrations (Tables 4 and 5). Especially some amino acids have considerably increased concentrations at the lower growth temperature (70°C). Valine, leucine, isoleucine, alanine, aspartic acid, lysine, threonine and glutamic acid have been detected in higher concentrations at 70°C. In accordance with this finding, an up-regulation of genes and proteins involved in amino acid biosynthesis at lower cultivation temperatures than 80°C has been observed by the transcriptomic and proteomic analyses (70°C) and has been reported previously for the hyperthermophilic euryarchaeon Pyrococcus furiosus (Weinberg et al. 2005).
Interestingly, the polyamines putrescine and spermidine are detected in high concentrations in S. solfataricus and it has previously been shown that polyamines play an important role in stabilizing DNA and RNA at high temperatures in the hyperthermophilic bacterium Thermus thermophilus (Cava et al. 2009). However, from the comparison of S. solfataricus cells grown at 80 versus 70°C putrescine and spermidine are detected in higher amounts in cells grown at 70°C.
In contrast, the CCM metabolism shows only small differences in metabolite concentrations comparing growth at 80 versus 70°C. Citrate and 3-phosphoglycerate are present in lower concentrations, whereas glyceraldehyde and 2-keto-3-deoxy gluconate (KDG) are detected in higher concentrations at 70°C.
The exometabolome analysis revealed only a small number of detectable compounds (only a few peaks identified in the GC–MS analysis). The identified metabolites are glucose, glycerol, erythritol and inositol. The detected glycerol probably comes from the glycerolstock that has been used for inoculation and glucose has been used as carbon source (0.15%). The sugar alcohols erythritol and inositol are found in high concentrations in the supernatant as well as in the cell. The accumulation of these known compatible solutes is discussed as a thermoprotective trait in the extremely hyperthermophilic Pyrolobus fumarii (Goncalves et al. 2008) and therefore, a role as compatible solutes can also be assumed for S. solfataricus.
Biochemistry of the CCM enzymes
Goals of the biochemical analyses are to identify and confirm the key players of the CCM network of S. solfataricus suggested from the genomic reconstruction (SOP_080908; Fig. 3) and particularly, to provide detailed enzymatic and biochemical information of the recombinant CCM enzymes in order to study the behavior and regulation of the network under temperature change. Focus lies on providing detailed information on substrate specificity, kinetic information (V max-, K m-, K cat-values) as well as regulatory properties of key enzymes predicted by modeling.
A prerequisite for the biochemical and enzymatic analyses is the availability of recombinant proteins. Therefore, the respective CCM candidate genes are cloned and heterologously expressed in Escherichia coli, which is performed according to standard protocols (SOP_SSO_080913a). However, if the recombinant expression in E. coli fails, i.e., expression in an insoluble form (inclusion bodies formation) or no expression at all, the respective candidates are expressed in S. solfataricus by using the recently developed virus vector based expression system in S. solfataricus (SOP_SSO_080913b; Albers et al. 2006). Moreover, homologous expression is used to identify post-translational modifications or to unravel protein–protein interactions, which have not been identified yet. In addition, the constructed over-expression strains (perturbation experiments) will be further analyzed to challenge and improve the established models via transcriptome, proteome as well as the metabolome analyses.
The obtained recombinant proteins from E. coli or S. solfataricus, respectively, are purified to homogeneity by standard purification methods, like heat precipitation, ion exchange or hydrophobic interaction chromatography, gelfiltration, and subsequently characterized according to their biochemical, kinetic and regulatory properties (for examples see SOP_SSO_080913c and SOP_SSO_080913d).
The effect of temperature variation at the enzyme level is also studied by determining enzyme activities in crude extracts of S. solfataricus grown at different temperatures (SOP_0809012e). Assays for the respective enzymes involved in the branched ED pathway, which is the initial focus of the project (Albers et al. 2009), have been established at high temperature. The cell mass of S. solfataricus grown at the optimal growth temperature of 80°C has been obtained from the central fermentation unit. The derived data (V max values) play an important role for the parameterization of the constructed models of the CCM network (Drengstig et al. 2008; Ni et al. 2009; Ni et al. in preparation).
Procedures
Cloning and heterologous expression in E. coli (SOP_SSO_080913a)
In order to prove the gene assignments of the identified CCM candidates, the respective genes are cloned into the vector pBlueScript (Novagen) via PCR mutagenesis. The E. coli strain K12 DH5α (Hanahan 1983) is used for cloning, storage and preparation of the recombinant plasmid-DNA. For heterologous expression of recombinant S. solfataricus proteins the genes are cloned via PCR-mutagenesis (oligonucleotide primers are purchased from Invitrogen) into the pET vector system (Novagen; Table 6) and the strains E. coli BL21(DE3), BL21(DE3) pLysS (Studier and Moffat 1986), BL21-CodonPlus(DE3)-RIL (Stratagene; Carstens and Waesche 1999) and Rosetta (DE3) pRIL (Novagen) are used for the production of the recombinant proteins. The BL21-CodonPlus(DE3)-pRIL and the Rosetta (DE3) pRIL strains contain plasmids encoding (argU, ileY, leuW and argU, argW, glyT, IleX, leuW, proL, respectively) and therefore, these hosts allow for the expression of genes encoding tRNAs for the rare argenine (AGA, AGG, CGA), glycine (GGA), isoleucine (AUA), leucine (CUA), and proline (CCC) codons.
Table 6.
Plasmids and their application
| Vector | Resistance | Application | Source of supply, reference |
|---|---|---|---|
| pET15b & pET11c | Ampr | Heterologous expression of S. solfataricus proteins in E. coli | Novagen, Merck Biosciences |
| pET24a & pET24d | Kanr | Heterologous expression of S. solfataricus proteins in E. coli | Novagen, Merck Biosciences |
| pMZ1 | Ampr | Cloning of S. solfataricus genes for homologous expression contains C-terminal tandem (strep-his)-tag | Zolghadr et al. (2007) |
| SSV1 | S. solfataricus shuttle vector | Jonuscheit et al. (2003) and Albers et al. (2006) | |
| pLysS | Camr | Heterologous expression of T7 lysozyme in E. coli | Novagen, Merck Biosciences |
| pRIL | Camr | Expression of rare tRNA genes (argU, ileY, leuW) | Stratagene, La Jolla (USA) |
The aerobic cultivation of the different E. coli strain is carried out in 3–400 ml batch cultures in test glasses or Erlenmeyer flasks at 37°C in Luria–Bertani (LB) medium (1% tryptone, 0.5% yeast extract, 0.5% NaCl (w/v), pH 7) or on solid medium plates (LB medium containing 1.5% (w/v) agar–agar). An optimal oxygen supply of the smaller liquid cultures (3–400 ml) is given by vigorously shaking (220 rpm; Thermotron). Mass cultures of the expression strains are grown at 37°C in a 4 l fermenter [Minifors, Infors AG Bottmingen (CH)] in LB medium. Antibiotics are added according to the plasmid-encoded antibiotic resistance in the following concentrations: ampicillin 100 μg/ml, kanamycin 50 μg/ml and chloramphenicol 34 μg/ml. Liquid LB medium containing the appropriate antibiotic is inoculated with a preculture (1% (v/v)) and growth is monitored spectrophotometrically at 578 nm. Recombinant protein expression is induced at an OD578 of 0.6–0.8 by the addition of 1 mM isopropyl-β-d-thiogalactopyranosid (IPTG) and cultivation is continued for 3–4 h. Afterwards, cells are chilled on ice, harvested by centrifugation (6,000×g, 15 min, 4°C) and stored at −80°C.
Cloning and homologous expression in S. solfataricus (SOP_SSO_080913b)
This virus vector based expression system relies on the complementation of uracil auxotrophic mutants of the S. solfataricus strain PH1-16 with the selectable marker genes pyrEF (Jonuscheit et al. 2003; Albers et al. 2006). Many efforts failed to heterologously express, for example gluconate dehydratase (GAD, SSO3198) in an active, soluble form in E. coli. Therefore, SSO3198 was one of the first candidates cloned into the entry vector pMZ1 (via NcoI/BamHI), which contains a C-terminal tandem-tag (Strep-His-tag) and the araS promoter (arabinose inducible promoter).
After the transfer of the expression cassette containing the SSO3198 gene into the virus shuttle vector pMJ05 (via BlnI/EagI; Jonuscheit et al. 2003; Albers et al. 2006), the resulting plasmid (pSVA124) was used to transform the S. solfataricus expression strain PH1-16 via electroporation (25 μF, 2.5 kV, 400 Ω; time constant should be between 4–5.2 ms) as described previously (Schleper et al. 1992). Positive transformants have been selected, growth has been performed in Brock medium (SOP_SSO_080902, lacking uracil) containing 0.1% NZ-amine at 80°C and expression is induced by the addition of 0.2% d-arabinose at OD600 of ~0.3. Cultivation is continued until an OD600 of 0.8–0.9. Afterwards, cells are chilled, harvested by centrifugation (7,000×g, 15 min, 4°C) and stored at −80°C. For enzyme preparation a 40 l fermenter has been performed.
Preparation of recombinant enzymes (SOP_SSO_080913c)
Recombinant E. coli cells are resuspended (1:3) in chilled lysis buffer: 0.1 M HEPES/KOH buffer, pH 7 at room temperature. Recombinant S. solfataricus cells are resuspended (1:3) in chilled 50 mM HEPES/KOH, pH 8.5, 100 mM KCl, containing 250 μl complete Protease Inhibitor (7x, Roche). Cell disruption is carried out by sonication (4 times: 2 min pulse/1 min cooling). After centrifugation (45 min, 16,000×g, 4°C) the supernatant is decanted and for determination of protein concentration the BioRad Protein Assay based on the Bradford protein quantitation method (Bradford 1976, modified) is used.
Preparation of S. solfataricus crude extracts (SOP_SSO_080913d)
Resuspension of 0.5 g (wet weight) cells in 1.5 ml 0.1 M HEPES/KOH buffer, pH 7 at room temperature, containing 5 mM DTT and 250 μl complete Protease Inhibitor (7×, Roche). Cell disruption is carried out by sonication (4×, 2 min pulse/1 min cooling). After centrifugation (45 min, 16,000×g, 4°C) the supernatant is dialyzed overnight against 0.1 M HEPES/KOH pH 7 at room temperature. For determination of protein concentration the BioRad Protein Assay based on the Bradford protein quantitation method (Bradford 1976, modified) is used. Between 0.25–1 mg total protein is used for the different enzyme assays using crude extracts.
Non-phosphorylating glyceraldehyde-3-phosphate (GAP) dehydrogenase (GAPN; E.C. 1.2.1.9) and gluconate dehydratase (GAD; EC 4.2.1.39) activity in cell-free extracts (Table 7; SOP_SSO_080913e, f)
Table 7.
Enzymatic activities of GAPN (SSO3194) and GAD (SSO3198) assayed at 80 and 70°C in cell-free extracts of S. solfataricus grown at 80 and 70°C
| Growth temperature: | 80°C | 70°C | ||
|---|---|---|---|---|
| Assay temperature: | 80°C | 70°C | 80°C | 70°C |
|
E: GAD (U/mg) S: gluconate (U/mg) |
0.167 ±0.0108 |
0.127 ±0.0001 |
0.114 ±0.012 |
0.092 ±0.0047 |
|
E: GAD (U/mg) S: galactonate (U/mg) |
0.077 ± 0.0005 |
0.052 ±0.0024 |
0.043 ±0.0029 |
0.029 ±0.0024 |
|
E: GAPN (U/mg) S: GAP (U/mg) |
0.036 ±0.0014 |
0.021 ±0.0003 |
0.054 ±0.004 |
0.021 ±0.0014 |
GAPN activity is determined in a continuous enzyme assay at 70°C and 80°C (Table 7). The assay is performed in 0.1 M HEPES/KOH (pH 6.5 is set at 80°C assay temperature) containing 5 mM NADP+ and 300 μg of crude extract in a total volume of 0.5 ml. Reactions are started by the addition of GAP (final concentration 10 mM). Enzymatic activity is measured by monitoring the formation of NADPH and the increase of absorbance at 340 nm by using a specord 210 photometer (Analytik Jena). For each assay three independent measurements are performed.
GAD activity in crude extracts (350 μg crude extract) is measured in a discontinuous enzyme assay at 70 and 80°C (Table 7). The assay is performed in 0.1 M HEPES/KOH (pH 6.5 at the respective assay temperature (70 or 80°C) containing 10 mM MgCl2 and 10 mM galactonate or 15 mM gluconate, respectively. Reactions are started by the addition of substrate. The sample is incubated in a thermoblock, after 0, 2.5, 5, 7.5 and 10 min of incubation, 25 μl sample is withdrawn on ice and the reaction is stopped by the addition of 2.5 μl of 12% (w/v) trichloroacetic acid.
Enzymatic activity is determined using the TBA assay (modified, Buchanan et al. 1999): Precipitated proteins are removed by centrifugation (16,000×g, 15 min at 4°C) and 20 μl of the supernatants are oxidized by the addition of 125 μl of 25 mM periodic acid/0.25 M H2SO4 and incubated at RT for 20 min. Oxidation is terminated by the addition of 250 μl of 2% (w/v) sodium arsenite in 0.5 M HCl. 1 ml of 0.3% (w/v). Subsequently, TBA is added and the chromophore is developed by heating at 100°C for 10 min. Subsequently, a sample (0.5 ml) of the solution is then removed and the color is intensified by adding to an equal volume of DMSO. The change in absorbance is followed at 549 nm (εchromophore = 67.8 × 103 M−1 cm−1). For each assay three independent measurements are performed.
Western blotting and detection of the recombinant S. solfataricus proteins (SOP_SSO_080913g)
Electrophoretically separated tagged proteins are transferred from the PAA gel to a hydrophobic membrane (PVDF-(ProBlott) or Nylon-membrane (Roth)) by wet electroblotting.
The transfer is carried out using a tank blot system (Biometra). Therefore, after the electrophoresis run, the gel and two Whatman paper (Schleicher & Schuell) are equilibrated in transfer buffer (50 mM Tris, 380 mM Glycin, 0.1% SDS, 20% methanol) for 15 min. The membrane is briefly moistened with 100% (v/v) methanol and afterwards also equilibrated in transfer buffer. The blot assembly is performed as recommended by the blot system manufacturer (Biometra). The transfer is carried out with 12 V over night (~20 h) at 4°C and after blotting the membrane is air dried. Blotting efficiency is controlled by the transfer of the applied pre-stained protein marker (PageRuler, Fermentas) on the PAA gel.
For immunodetection the membrane is incubated for 5 min in 100% (v/v) methanol, washed three times for 5 min with PBST-buffer (1× PBS (63.2 mM Na2HPO4, 11.7 mM KH2PO4, 68 mM NaCl pH ~7.3) + 0.3% Tween-20) at RT on a rotary shaker, blocked for 1 h at RT by either using PBST-buffer containing 5% skim milk (his-Tag detection) or PBST-buffer containing 0.2% I-Block (Applied Biosystems;StrepII-tag detection). After three times washing for 5 min using PBST-buffer either containing 2.5% skim milk or 0.1% I-Block, 1:2,000 Anti-His antibody AP conjugate (rabbit; Abcam) or 1:4,000 Strep-Tactin AP conjugate (IBA BioTAGnology) are added to the respective PBST-buffer. Incubation is carried out for at least 1 h 30 min at RT on a rotary shaker. Afterwards, the membrane is washed six times for 5 min at RT using PBST-buffer either containing 2.5% skim milk or 0.1% I-Block. Finally, the membrane is washed two times for 10 min in A.bidest. and incubated for 15 min at 37°C in 9 ml pre-warmed A.bidest., containing 1 ml CDP-Star (Invitrogen). Chemiluminescence is detected by using the VersaDoc System (BioRad).
Results
Purification of obtained recombinant GAPN (SSO3194; Fig. 8) and the GAD (SSO3198; Fig. 9) (SOP_SSO_0809013c, d)
Fig. 8.
Purification of the heterologously expressed GAPN from S. solfataricus by using the E. coli pET expression system. HP Heat precipitation at 70°C, IEC ion exchange chromatography, GF gelfiltration, M protein ladder (Page ruler™, fermentas)
Fig. 9.
SDS PAGE gel (a) and western blot (b) showing homologous expression and purification of the S. solfataricus GAD (SSO3198). a Coomassie stained 12.5% PAA gel of His tag-specific affinity chromatography fractions. b Detection of the blotted S. solfataricus GAD using Strep-Tactin, revealing a protein of about 49 kDa (including tandem tag). M Protein standard, CE crude extract, FT flow through, W1-3 washing fractions, E1-3 elution fractions
For enrichment of the recombinant GAPN, the resulting E. coli crude extract is diluted 1:1 with 0.1 M HEPES/KOH buffer, pH 7 at RT and subjected to a heat precipitation for 20 min at 70°C. After heat precipitation, the samples are cleared by centrifugation (16,000×g for 30 min at 4°C). The supernatant is dialyzed overnight against 20 mM HEPES/KOH (pH 6.5, 70°C), containing 5 mM dithiothreitol, subjected to ion exchange chromatography on UNO Q-12 (Bio-Rad Laboratories) pre-equilibrated by using the respective buffer, and eluted with a salt gradient from 0 to 1 M NaCl. Fractions containing the GAPN (checked by SDS–PAGE) are pooled and concentrated via centrifugal concentrators (Vivaspin6, Sartorius Stedim Biotech). Afterwards, the sample is dialyzed overnight against 50 mM HEPES/KOH (pH 6.5, 70°C), containing 5 mM dithiothreitol, 300 mM NaCl, and subjected to gelfiltration on HiLoad 26/60 Superdex 200 prep grade (Amersham Biosciences) preequilibrated in the respective buffer (Fig. 8).
The homologously expressed recombinant GAD from S. solfataricus is isolated via the attached His-tag by Immobilized Metal Affinity Chromatography (IMAC) using a His-Select column (Qiagen, Hilden) and HIS-Select® Nickel Affinity Gel (Sigma). Hereunto, the resulting S. solfataricus crude extract is applied onto nickel-nitrilotriacetic acid (Ni–NTA) affinity columns (5 ml volume, Qiagen) equilibrated with 50 mM HEPES/KOH, pH 8.5 containing 100 mM KCl (buffer 1). The column is washed three times with 2× column volume buffer 1 containing 25 mM imidazole. Bound GAD is eluted in three steps with buffer 1 containing 250 mM imidazole. After monitoring purification by SDS–PAGE, the protein has been blotted and stained with Strep-Tactin (streptavidine analogue; IBA; Fig. 9).
Activity of the recombinant GAPN (EC 1.2.1.9; SOP_SSO_0809013e)
GAPN activity is determined in a continuous enzyme assay at 80 and 70°C (Table 8). The standard assay is performed in 0.1 M HEPES/KOH (pH 6.5 is set at the respective assay temperature (70 or 80°C) containing 2 mM NADP+ and 5 μg of purified protein in a total volume of 0.5 ml. Reactions are started by the addition of 3 mM D,L-GAP. Enzymatic activity is measured by monitoring the change in absorbance due to the increase of NADPH at 340 nm (εNADPH, 70°C = 5.71 mM−1(cm−1). For each assay three independent measurements are performed.
Table 8.
Kinetic parameters of the GAPN (SSO3194) assayed at 80 and 70°C
| D,L-GAP (mM) | NADP (mM) | Assay temp. (°C) | V max (U/mg) | K m (mM) | K cat (min−1) (s−1) | K cat/K m (mM−1 s−1] |
|---|---|---|---|---|---|---|
| 3 | 2 | 80 | 10.58 | 0.95 | 544.97 9.08 | 9.51 |
| 3 | 2 | 70 | 7.46 | 1.51 | 384.17 6.40 | 4.25 |
The kinetic parameters (V max and K m) are calculated by iterative curve-fitting (Hanes) using the program Origin (Microcal Software, Northampton, MA, USA).
Activity of the recombinant GAD (EC 4.2.1.39; SOP_SSO_0809013f)
Recombinant GAD activity has been confirmed via the modified thiobarbituric acid (TBA)-assay (Buchanan et al. 1999) by using 7.5 μg of the purified protein (enriched elution fraction). Activity is determined in a discontinous enzyme assay at 80°C. The assay is performed in 0.1 M HEPES/KOH (pH 6.5 is set at the respective assay temperature 80°C) containing 10 mM MgCl2 and 10 mM gluconate or 10 mM galactonate, respectively. Reactions are started by the addition of substrate.
For initial enzymatic analysis the sample is incubated at 80°C and after 0 and 10 min, 25 μl of the sample is transferred on ice. The reaction is stopped by the addition of 2.5 μl of 12% (w/v) trichloroacetic acid. Precipitated protein is removed by centrifugation (16,000×g, 15 min, 4°C). Enzymatic activity is determined by using a modified thiobarbituric acid (TBA)-assay (Buchanan et al. 1999; see above).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors thank the Federal Ministry of Education and Resarch (BMBF), Germany, the Netherlands Organization for Scientific Research (NWO), the Research Council of Norway (RCN), and the Biotechnology, Biological Research Council (BBSRC), United Kingdom, as well as the partner universities (University of Bergen (Norway), University of Duisburg-Essen (Germany), Wageningen University and University of Groningen (The Netherlands), University of Sheffield and the University of Manchester (The United Kingdom), Free University Amsterdam (The Netherlands) for financial support of the SulfoSYS-project (SysMo P–N-01-09-23).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- CCM
Central carbohydrate metabolism
- ED
Entner–Doudoroff
- EMP
Embden–Meyerhof–Parnas
- SOP
Standard operating procedure
- SulfoSYS
Sulfolobus Systems Biology
Footnotes
M. Zaparty, D. Esser, S. Gertig, P. Haferkamp, T. Kouril, T. K. Pham, P. Sierocinski, M. von Jan and P. Wieloch contributed equally to this project.
References
- Ahmed H, Ettema TJ, Tjaden B, Geerling AC, Van der Oost J, Siebers B. The semi-phosphorylative Entner–Doudoroff pathway in hyperthermophilic archaea—a re-evaluation. Biochem J. 2005;390:529–540. doi: 10.1042/BJ20041711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers SV, Driessen AJM. Conditions for gene disruption by homologous recombination of exogenous DNA into the Sulfolobus solfataricus genome. Archaea. 2008;2:145–149. doi: 10.1155/2008/948014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers SV, Elferink MGL, Charlebois RL, Sensen CW, Driessen AJM, Konings WN. Glucose transport in the extremely thermoacidophilic Sulfolobus solfataricus involves a high affinity membrane-integrated binding protein. J Bacteriol. 1999;181:4258–4291. doi: 10.1128/jb.181.14.4285-4291.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers SV, van de Vossenberg JLCM, Driessen AJM, Konings WN. Adaptations of the archaeal cell membrane to heat stress. Front Biosci. 2000;5:813–820. doi: 10.2741/albers. [DOI] [PubMed] [Google Scholar]
- Albers SV, van de Vossenberg JLCM, Driessen AJM, Konings WN. Bioenergetics and solute uptake under extreme conditions. Extremophiles. 2001;5:285–294. doi: 10.1007/s007920100214. [DOI] [PubMed] [Google Scholar]
- Albers SV, Koning SM, Konings WN, Driessen AJM. Insights into ABC transport in Archaea. J Bioenerg Biomembr. 2004;36:5–15. doi: 10.1023/B:JOBB.0000019593.84933.e6. [DOI] [PubMed] [Google Scholar]
- Albers SV, Jonuscheit M, Dinkelaker S, Urich T, Kletzin A Tampe R, Driessen AJM, Schleper C (2006) Production of recombinant and tagged proteins in the hyperthermophilic archaeon Sulfolobus solfataricus. Appl Environ Microbiol 72(1):102–111 [DOI] [PMC free article] [PubMed]
- Albers SV, Birkeland NK, Driessen AJM, Gertig S, Haferkamp P, Klenk HP, Kouril T, Manica A, Pham TK, Ruoff P, Schleper C, Schomburg D, Sharkey KJ, Siebers B, Sierocinski P, Steuer R, Van der Oost J, Westerhoff HV, Wieloch P, Wright PC, Zaparty M. SulfoSYS—Sulfolobus Systems Biology: towards a Silicon Cell Model for the central carbohydrate metabolism of the archaeon Sulfolobus solfataricus under temperature variation. Biochem Soc Trans. 2009;37:58–64. doi: 10.1042/BST0370058. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson I, Rodriguez J, Susanti D, Porat I, Reich C, Ulrich LE, Elkins JE, Mavromatis K, Lykidis A, Kim E, Thompson LS, Nolan M, Land M, Copeland A, Lapidus A, Lucas S, Detter C, Zhulin IB, Olsen GJ, Whitman W, Mukhopadhyay B, Bristow J, Kyrpides N. Genome sequence of Thermofilum pendens reveals an exceptional loss of biosynthetic pathways without genome reduction. J Bacteriol. 2008;190(8):2957–2965. doi: 10.1128/JB.01949-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson AF, Lundgren M, Eriksson S, Rosenlund M, Bernander R, Nilsson P. Global analysis of mRNA stability in the archaeon Sulfolobus. Genome Biol. 2006;7:R99. doi: 10.1186/gb-2006-7-10-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry ER, Bell SD. DNA replication in the archaea. Microbiol Mol Biol Rev. 2006;70:876–887. doi: 10.1128/MMBR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SD, Jackson SP. Mechanism and regulation of transcription in archaea. Curr Opin Microbiol. 2001;4(2):208–213. doi: 10.1016/S1369-5274(00)00190-9. [DOI] [PubMed] [Google Scholar]
- Bisle B, Schmidt A, Scheibe B, Klein C, Tebbe A, Kellermann J, Siedler F, Pfeiffer F, Lottspeich F, Oesterhelt D. Quantitative profiling of the membrane proteome in a Halophilic archaeon. Mol Cell Proteomics. 2006;5:1543–1558. doi: 10.1074/mcp.M600106-MCP200. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brock TD, Brock KM, Belley RT, Weiss RL. Sulfolobus: a new genus of sulphur-oxidizing bacteria living at low pH and high temperature. Arch Microbiol. 1972;84:54–68. doi: 10.1007/BF00408082. [DOI] [PubMed] [Google Scholar]
- Buchanan CL, Connaris H, Danson MJ, Reeve CD, Hough DW. An extremely thermostable aldolase from Sulfolobus solfataricus with specificity for non-phosphorylated substrates. Biochem J. 1999;343:563–570. doi: 10.1042/0264-6021:3430563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens CP, Waesche A (1999) Codon bias-adjusted BL21 derivatives for protein expression strategies. Newsletter 12(2):49–51
- Cava F, Hidalgo A, Berenguer J. Thermus thermophilus as biological model. Extremophiles. 2009;13:213–231. doi: 10.1007/s00792-009-0226-6. [DOI] [PubMed] [Google Scholar]
- Drengstig T, Ueda HR, Ruoff P (2008) Predicting perfect adaptation motifs in reaction kinetic networks. J Phys Chem B 112(51):16752–16758 [DOI] [PubMed]
- Elferink MGL, Albers SV, Konings WN, Driessen AJM. Sugar transport in Sulfolobus solfataricus is mediated by two families of binding protein dependent ABC transporters. Mol Microbiol. 2001;39:1494–1503. doi: 10.1046/j.1365-2958.2001.02336.x. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Ettema TJG, Ahmed H, Geerling ACM, Van der Oost J, Siebers B. The non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN) of Sulfolobus solfataricus: a key-enzyme of the semi-phosphorylative branch of the Entner–Doudoroff pathway. Extremophiles. 2008;12:75–88. doi: 10.1007/s00792-007-0082-1. [DOI] [PubMed] [Google Scholar]
- Fischer F, Wolters D, Rögner M, Poetsch A. Toward the complete membrane proteome: high coverage of integral membrane proteins through transmembrane peptide detection. Mol Cell Proteomics. 2006;5:444–453. doi: 10.1074/mcp.M500234-MCP200. [DOI] [PubMed] [Google Scholar]
- Fröls S, Gordon PM, Panlilio MA, Schleper C, Sensen CW. Elucidating the transcription cycle of the UV-inducible hyperthermophilic archaeal virus SSV1 by DNA microarrays. Virology. 2007;365:48–59. doi: 10.1016/j.virol.2007.03.033. [DOI] [PubMed] [Google Scholar]
- Goncalves LG, Lamosa P, Huber R, Santos H. Di-myo-inositol phosphate and novel UDP-sugars accumulate in the extreme hyperthermophile Pyrolobus fumarii. Extremophiles. 2008;12(3):383–389. doi: 10.1007/s00792-008-0143-0. [DOI] [PubMed] [Google Scholar]
- Grogan DW. Phenotypic characterization of the archaebacterial genus Sulfolobus: comparison of five wild-type strains. J Bacteriol. 1989;171:6710–6719. doi: 10.1128/jb.171.12.6710-6719.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Jeffery CJ, Bahnson BJ, Chien W, Ringe D, Petsko GA. Crystal structure of rabbit phosphoglucose isomerase, a glycolytic enzyme that moonlights as neuroleukin, autocrine motility factor, and differentiation mediator. Biochemistry. 2000;39(5):955–964. doi: 10.1021/bi991604m. [DOI] [PubMed] [Google Scholar]
- Jonuscheit M, Martusewitsch E, Stedman KM, Schleper C. A reporter gene system for the hyperthermophilic archaeon Sulfolobus solfataricus based on a selectable and integrative shuttler vector. Mol Microbiol. 2003;48(5):1241–1252. doi: 10.1046/j.1365-2958.2003.03509.x. [DOI] [PubMed] [Google Scholar]
- Kagawa HK, Yaoi T, Brocchieri L, McMillan RA, Alton T, Trent JD. The composition, structure and stability of a group II chaperonin are temperature regulated in a hyperthermophilic archaeon. Mol Microbiol. 2003;48(1):143–156. doi: 10.1046/j.1365-2958.2003.03418.x. [DOI] [PubMed] [Google Scholar]
- Kelman Z, White MF. Archaeal DNA replication and repair. Curr Opin Microbiol. 2005;8(6):669–676. doi: 10.1016/j.mib.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Kim S, Lee SB. Identification and characterization of Sulfolobus solfataricusd-gluconate dehydratase: a key enzyme in the non-phosphorylated Entner–Doudoroff pathway. Biochem J. 2005;387:271–280. doi: 10.1042/BJ20041053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee SB. Catalytic promiscuity in dihydroxy-acid dehydratase from the thermoacidophilic archaeon Sulfolobus solfataricus. J Biochem. 2006;139:591–596. doi: 10.1093/jb/mvj057. [DOI] [PubMed] [Google Scholar]
- Lamble HJ, Heyer NI, Bull SD, Hough DW, Danson M. Metabolic pathway promiscuity in the archaeon Sulfolobus solfataricus revealed by studies on glucose dehydrogenase and 2-keto-3-deoxygluconate aldolase. J Biol Chem. 2003;278(36):34066–34072. doi: 10.1074/jbc.M305818200. [DOI] [PubMed] [Google Scholar]
- Lamble HJ, Theodossis A, Milburn CC, Taylor GL, Bull SD, Hough DW, Danson M. Promiscuity in the part-phosphorylative Entner–Doudoroff pathway of the archaeon Sulfolobus solfataricus. FEBS Lett. 2005;579:6865–6869. doi: 10.1016/j.febslet.2005.11.028. [DOI] [PubMed] [Google Scholar]
- Markowitz VM, Szeto E, Palaniappan K, Grechkin Y, Chu K, Chen IM, Dubchak I, Anderson I, Lykidis A, Mavromatis K, Ivanova NN, Kyrpides NC. The integrated microbial genomes (IMG) system in 2007: data content and analysis tool extensions. Nucleic Acids Res. 2008;36 (Database issue):D528–D533. doi: 10.1093/nar/gkm846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martusewitsch E, Sensen C, Schleper C. High spontaneous mutation rate in the hyperthermophilic archaeaon Sulfolobus solfataricus is mediated by transposable elements. J Bacteriol. 2000;182:2574–2581. doi: 10.1128/JB.182.9.2574-2581.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Tomita M, Ishihama Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J Proteome Res. 2008;7:731–740. doi: 10.1021/pr700658q. [DOI] [PubMed] [Google Scholar]
- Ni XY, Drengstig T, Ruoff P (2009) The control of the controller: molecular mechanisms for robust perfect adaptation and temperature compensation Biophys J (accepted) [DOI] [PMC free article] [PubMed]
- Olivier BG, Snoep JL. Web-based kinetic modelling using JWS Online. Bioinformatics. 2004;20:2143–2144. doi: 10.1093/bioinformatics/bth200. [DOI] [PubMed] [Google Scholar]
- Pham TK, Sierocinski P, van der Oost J, Wright PC (2009) Quantitative proteomic analysis of Sulfolobus solfataricus membrane proteins. J Proteome Res (submitted) [DOI] [PubMed]
- Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002;32 Suppl:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Redder P, She Q, Garrett RA. Non-autonomous mobile elements in the Crenarchaeon Sulfolobus solfataricus. J Mol Biol. 2001;306:1–6. doi: 10.1006/jmbi.2000.4377. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Creti R, Londei P. In vitro translation of archaeal natural mRNAs at high temperature. FEMS Microbiol Lett. 1993;107:89–94. doi: 10.1111/j.1574-6968.1993.tb06009.x. [DOI] [Google Scholar]
- Schleper C, Kubo K, Zillig W. The particle SSV1 from the extremely thermophilic archaeon Sulfolobus is a virus: demonstration of infectivity and of transfection with viral DNA. Proc Natl Acad Sci USA. 1992;89:7645–7649. doi: 10.1073/pnas.89.16.7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleper C, Röder R, Singer T, Zillig W. An insertion element of the extremely thermophilic archaeon Sulfolobus solfataricus transposes into the endogenous b-galactosidase gene. Mol Gen Genet. 1994;243:91–96. doi: 10.1007/BF00283880. [DOI] [PubMed] [Google Scholar]
- She Q, Singh RK, Confalonieri F, Zivanovic Y, Allard G, Awayez MJ, Chan-Weiher CCY, Groth Clausen I, Curtis B-A, De Moors A, Erauso G, Fletcher C, Gordon PMK, Heikamp-de Jong I, Jeffries AC, Kozera CJ, Medina N, Peng X, Thi-Ngoc HP, Redder P, Schenk ME, Theriault C, Tolstrup N, Charlebois RL, Doolittle WF, Duguet M, Gaasterland T, Garrett RA, Ragan MA, Sensen CW, Van der Oost J. The complete genome of the crenarchaeon Sulfolobus solfataricus P2. PNAS. 2001;98(14):7835–7840. doi: 10.1073/pnas.141222098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherawat M, Tolan DR, Allen KN. Structure of a rabbit muscle fructose-1, 6-bisphosphate aldolase A dimer variant. Acta Crystallogr D Biol Crystallogr. 2008;64(5):543–550. doi: 10.1107/S0907444908004976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorko R, Osipiuk J, Stetter KO. Glycogen-bound polyphosphate kinase from the archaebacterium Sulfolobus acidocaldarius. J Bacteriol. 1989;171:5162–5164. doi: 10.1128/jb.171.9.5162-5164.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders APL, Walther J, Peter S, Kinnman I, de Vos MGJ, van de Werken HJG, Brouns SJJ, van der Oost J, Wright PC. Reconstruction of central carbon metabolism in Sulfolobus solafatricus using a two-dimensional gel electrophoresis map, stable isotope labelling and DNA microarray analysis. Proteomics. 2006;6(15):1518–1529. doi: 10.1002/pmic.200402070. [DOI] [PubMed] [Google Scholar]
- Snoep JL, Westerhoff HV (2005) From isolation to integration, a systems biology approach for building the silicon cell. In: Westerhoff HV, Alberghina L (eds) Topics in current genetics (series): systems biology: definitions and perspectives, vol 13. Springer, Berlin, pp 13–30
- Strelkov S, von Elstermann M, Schomburg D. Comprehensive analysis of metabolites in Corynebacterium glutamicum by gas chromatography/mass spectrometry. Biol Chem. 2004;385:853–861. doi: 10.1515/BC.2004.111. [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffat BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Tusher V, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. PNAS. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Oost J, Siebers B (2007) The glycolytic pathways of Archaea: evolution by tinkering. In: Garrett RA, Klenk H-P (eds) Archaea: evolution, physiology and molecular biology, vol 22, pp 247–260. Blackwell, Malden
- Verhees CH, Kengen SW, Tuininga JE, Schut GJ, Adams MWW, De Vos WM, Van der Oost J. The unique features of glycolytic pathways in Archaea. Biochem J. 2003;375:231–246. doi: 10.1042/BJ20021472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhees CH, Kengen SW, Tuininga JE, Schut GJ, Adams MWW, De Vos WM, Van der Oost J (2004) The unique features of glycolytic pathways in Archaea. Biochem J 377:819–822 (Erratum) [DOI] [PMC free article] [PubMed]
- Wagner M, Berkner S, Ajon M, Driessen AJM, Lipps G, Albers SV. Expanding and understanding the genetic toolbox of the hyperthermophilic genus Sulfolobus. Biochem Soc Trans. 2009;37:97–101. doi: 10.1042/BST0370097. [DOI] [PubMed] [Google Scholar]
- Weinberg MV, Schut GJ, Brehm S, Datta S, Adams MW. Cold shock of a hyperthermophilic archaeon: Pyrococcus furiosus exhibits multiple responses to a suboptimal growth temperature with a key role for membrane-bound glycoproteins. J Bacteriol. 2005;187(1):336–348. doi: 10.1128/JB.187.1.336-348.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernersson R, Nielsen HB (2005) OligoWiz 2.0—integrating sequence feature annotation into design of microarray probes. Nucleic Acids Res 33:W611–W615 [DOI] [PMC free article] [PubMed]
- Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Wilson D, Charoensawan V, Kummerfeld SK, Teichmann SA. DBD—taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 2008;36 (database issue):D88–D92. doi: 10.1093/nar/gkm964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthington P, Hoang V, Perez-Pomares F, Blum P. Targeted disruption of the alpha-amylase gene in the hyperthermophilic archaeon Sulfolobus solfataricus. J Bacteriol. 2003;185:482–488. doi: 10.1128/JB.185.2.482-488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wang S, Bai J, Shi L, Li D, Xu Z, Niu Y, Lu J, Bao Q. ArchaeaTF: an integrated database of putative transcription factors in Archaea. Genomics. 2008;91(1):102–107. doi: 10.1016/j.ygeno.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Yang IV, Chen E, Hasseman JP, Liang W, Frank BC, Wang S, Sharov W, Saeed AI, White J, Li J, Lee Yeatman TJ, Quackenbush J. Within the fold: assessing differential expression measures and reproducibility in microarray assays. Genome Biol. 2002;3:0062.1–0062.12. doi: 10.1186/gb-2002-3-11-research0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaparty M, Zaigler A, Stamme C, Soppa J, Hensel R, Siebers B. DNA microarray analysis of the central carbohydrate metabolism: glycolytic/gluconeogenic carbon switch in the hyperthermophilic Crenarchaeum Thermoproteus tenax. J Bacteriol. 2008;190(6):2231–2238. doi: 10.1128/JB.01524-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillig W, Stetter KO, Wunderl S, Schulz W, Priess H, Scholz I. The Sulfolobus-”Caldariella” Group: taxonomy on the basis of the structure of DNA-dependent RNA polymerases. Arch Microbiol. 1980;125:259–269. doi: 10.1007/BF00446886. [DOI] [Google Scholar]
- Zolghadr B, Weber S, Szabó Z, Driessen AJM, Albers SV (2007) Identification of a system required for the functional surface localization of sugar binding protein with class III signal peptides in Sulfolobus solfataricus. Mol Microbiol 64:795–806 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.