

Abstract
Altered hepatic lipid homeostasis, hepatocellular injury, and inflammation are features of nonalcoholic steatohepatitis, which contributes significantly to liver-related morbidity and mortality in the Western population. A collection of inflammatory mediators have been implicated in the pathogenesis of steatohepatitis in mouse models. However, the pathways essential for coordination and amplification of hepatic inflammation and injury caused by steatosis are not completely understood. We tested the hypothesis that tissue factor (TF)-dependent thrombin generation and the thrombin receptor protease activated receptor-1 (PAR-1) contribute to liver inflammation induced by steatosis in mice. Wild-type C57Bl/6J mice fed a diet deficient in methionine and choline for 2 weeks manifested steatohepatitis characterized by increased serum alanine aminotransferase activity, macrovesicular hepatic steatosis, hepatic inflammatory gene expression, and lobular inflammation. Steatohepatitis progression was associated with thrombin generation and hepatic fibrin deposition. Coagulation cascade activation was significantly reduced in low TF mice, which express 1% of normal TF levels. Hepatic triglyceride accumulation was not affected in low TF mice or PAR-1-deficient mice. In contrast, biomarkers of hepatocellular injury, inflammatory gene induction, and hepatic accumulation of macrophages and neutrophils were greatly reduced by TF-deficiency and PAR-1-deficiency. The results suggest that TF-dependent thrombin generation and activation of PAR-1 amplify hepatic inflammation and injury during the pathogenesis of steatohepatitis.
Non-alcoholic fatty liver disease (NAFLD) is increasingly appreciated as a hepatic feature of the metabolic syndrome. NAFLD may occur in 25% of the Western population and altered hepatic function increases the risk for developing diseases including diabetes and atherosclerosis.1,2 The progression of simple hepatic steatosis to the more severe nonalcoholic steatohepatitis (NASH) contributes significantly to liver-related morbidity and mortality.3 Requisite histological features of NASH include macrovesicular hepatic steatosis, evidence of hepatocellular injury, and lobular inflammation.4 In a subset of patients with chronic steatohepatitis, stellate cell activation coordinates a fibrogenic response causing fibrosis and cirrhosis.5 Of importance, the mechanisms required for the progression of hepatic inflammation during steatohepatitis are not completely understood.
Animal models used to define mechanisms of steatohepatitis have used genetic and dietary modification to induce various features of the disease.2 In particular, feeding mice a diet deficient in methionine and choline (MCD diet) is an established model to study the progression of steatohepatitis and has been extensively used to study mechanisms of hepatic inflammation and fibrosis. Rodents fed an MCD diet for 2 weeks manifest a defect in hepatic β oxidation resulting in accumulation of triglyceride and the induction of steatohepatitis.2,6,7 Prolonged feeding (>4 weeks) of the MCD diet activates hepatic stellate cells and increases collagen expression and deposition in the liver. Utilization of the MCD diet model has revealed the contribution of hepatic triglyceride,8 various inflammatory mediators,9,10 nuclear receptors,11,12 and signaling pathways13 in the manifestation of steatohepatitis.
An important physiological process disrupted by chronic liver disease is blood coagulation. Several studies have indicated that the progression of liver disease is associated with altered blood coagulation.14 For example, steatosis in patients with the metabolic syndrome is associated with a shift in the balance of procoagulant and antifibrinolytic factors favoring coagulation.15–17 This links the progression of NAFLD with increased risk of thrombotic complications associated with vascular disease and the metabolic syndrome. However, it is not clear whether the altered coagulation impacts progression of the liver pathology in patients with NAFLD or NASH.
The coagulation cascade is initiated by tissue factor (TF), the transmembrane receptor for coagulation factor VIIa.18 TF is expressed by the normal liver,19 albeit at much lower levels compared with other tissues (eg, lung, heart).20 Of importance, potent inducers of TF expression such as bacterial lipopolysaccharide and pro-inflammatory cytokines (eg, tumor necrosis factor [TNF]α, monocyte chemoattractant protein [MCP]-1) are linked to the pathogenesis of NAFLD and NASH in humans and animal models.21–24 TF-dependent coagulation cascade activation leads to generation of the serine protease thrombin, which cleaves circulating fibrinogen to form fibrin. Thrombin also elicits intracellular signaling by activating the G-protein coupled receptor protease activated receptor-1 (PAR-1).25 This TF–PAR-1 pathway has been shown to increase inflammation in other models of tissue injury.26–29 However, the contribution of both TF and PAR-1 to coagulation and inflammation during steatohepatitis has not been determined.
To this end, we characterized the procoagulant response associated with steatohepatitis induced in mice by a MCD diet. Furthermore, we used mice expressing 1% of normal TF levels (ie, low TF mice30 and PAR-1-deficient mice31 to test the hypothesis that TF-dependent thrombin generation contributes to the pathogenesis of murine steatohepatitis by activating PAR-1.
Materials and Methods
Mice
All mice were used between 8 and 12 weeks of age. Fourteen male, wild-type congenic C57Bl/6J mice were purchased from The Jackson Laboratory, (Bar Harbor, ME). For studies examining the effect of either TF-deficiency or PAR-1-deficiency, each experiment was uniquely controlled with age-matched mice on a similar genetic background. Seventeen male low TF mice (mTF−/−hTF+ mice,30 and 17 heterozygous littermate control mice (mTF+/−hTF+ mice) backcrossed 6 generations onto a C57Bl/6J background (kindly provided by Dr. Nigel Mackman, University of North Carolina-Chapel Hill) were used for these studies. The expression of a human TF (hTF) transgene at 1% of normal levels rescues the embryonic lethality of murine TF deficiency in the low TF mice.30 Heterozygous mice (mTF+/−hTF+ mice) generated by crossing male low TF mice (mTF−/−hTF+ mice) with female heterozygous mice (mTF+/−hTF+ mice) were used as control mice for all studies with the low TF mice. PAR-1−/− mice31 backcrossed eight generations onto a C57Bl/6J background were maintained by homozygous breeding. PAR-1+/+ mice backcrossed eight generations onto a C57Bl/6J background were maintained by homozygous breeding and used as wild-type control mice for all studies with PAR-1−/− mice. Fourteen PAR-1+/+ mice and 12 PAR-1−/− mice were used for these studies. Mice were maintained in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care at the University of Kansas Medical Center. Mice were housed at an ambient temperature of 22°C with alternating 12-hour light/dark cycles, and allowed water and rodent chow ad libitum (Teklad 8604; Harlan, Indianapolis, IN) before feeding custom diets. All animal procedures were performed according to the guidelines of the American Association for Laboratory Animal Science and were approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee.
Dietary Model of Nonalcoholic Steatohepatitis
The rodent diet deficient in methionine and choline (MCD diet, 518810) and an identical diet sufficient in methionine and choline (control diet, 518754) were prepared by Dyets, Inc. (Bethlehem, PA). Mice were fed either control diet or MCD diet ad libitum for 2 weeks. Mice were then anesthetized using isoflurane and blood was collected from the retro-orbital venous plexus, allowed to clot, and serum was collected by centrifugation. Blood was then collected from the caudal vena cava into sodium citrate (final concentration, 0.38%) and subjected to centrifugation for the collection of plasma. Sections of liver from the left lateral lobe were fixed in 10% neutral-buffered formalin for 24 hours, and then embedded in paraffin. The right medial lobe was affixed to a cork with optimal cutting temperature compound and immersed for 3 minutes in liquid nitrogen-chilled isopentane. The remaining liver was snap frozen in liquid nitrogen.
Histopathology and Clinical Chemistry
Formalin-fixed livers were sectioned at 5 μm, stained with H&E, and evaluated by light microscopy. The serum activity of alanine aminotransferase (ALT) was determined using a commercially available reagent (Thermo Fisher, Waltham, MA).
Immunohistochemistry and Immunofluorescence
Frozen livers were sectioned at 8 μm for each type of staining. Immunofluorescent staining for insoluble fibrin was performed as described previously32 using a rabbit anti-human fibrinogen antibody (Dako, Carpinteria, CA, A0080). The primary antibody was detected by the addition of goat anti-rabbit IgG conjugated to Alexa 488 (Invitrogen, Carlsbad, CA, A11008). Slides were then washed and counterstained with 4,6-diamidino-2-phenylindole (Invitrogen). For neutrophil immunohistochemistry, sections were fixed in 4% neutral-buffered formalin for 10 minutes, washed with PBS, pH 7.4, comprised of 137 mmol/L NaCl, 2.7 mmol/L KCl, and 10 mmol/L phosphate buffer, then endogenous peroxidase quenched by incubating the sections for 30 minutes in methanol containing 3% hydrogen peroxide. The sections were subsequently washed with PBS and blocked for 1 hour at room temperature with 10% rabbit serum diluted in PBS. Sections were then incubated with a rat anti-mouse neutrophil antibody (Serotec, Raleigh, NC; MCA771G) diluted 1:100 in block buffer for 2 hours at room temperature. The primary antibody was detected using a Vectastain ELITE ABC kit specific for rat IgG and ImmPACT DAB substrate according to the manufacturers protocol (Vector Laboratories, Burlingame, CA). Sections were counterstained briefly with Hematoxylin QS (Vector Laboratories), dehydrated and coverslipped. Staining for macrophages was performed using a similar protocol with rat anti-mouse CD68 and rat anti-mouse F4/80 each diluted 1:100 in block buffer as primary antibodies (Serotec; MCA1957GA, MCA497GA). The slides were visualized using an Olympus BX41 microscope (Olympus, Lake Success, NY). Images were captured using an Olympus DP70.
The average number of neutrophil foci (>5 clustered neutrophils) was determined in 20 randomly selected ×200 fields. The average number of macrophages was determined in 10 randomly selected ×400 fields. The evaluator was blinded to both treatment and genotype.
Hepatic Triglyceride Determination
Hepatic triglyceride content was determined as described previously.33 In brief, 100 mg of frozen liver tissue was homogenized in 1 ml of buffer containing 18 mmol/L Tris (pH 7.5), 300 mmol/L mannitol, 50 mmol/L EGTA, and 0.1 mmol/L phenylmethylsulfonyl fluoride. Five hundred microliters of homogenate was mixed with 4 ml of chloroform/methanol (2:1) and incubated overnight at room temperature with occasional shaking. Subsequently, 1 ml of H2O was added to this solution, vortexed, and subjected to centrifugation for 5 minutes at 3000 × g. The lower lipid phase was then collected and concentrated by vacuum. The lipid pellets were dissolved in a mixture of 270 μl isopropanol and 30 μl of Triton X-100 and analyzed spectrophotometrically for triglycerides (600 nm) using commercially available reagents (Wako, Richmond, VA).
Plasma Fibrinogen and Thrombin-Antithrombin Determination
Thrombin-antithrombin (TAT) levels in plasma were determined using a commercial enzyme-linked immunosorbent assay kit (Siemens Health care Diagnostics, Deerfield, IL). Plasma fibrinogen levels were determined using a commercial kit (Siemens Health care Diagnostics) and a STart4 Coagulation Analyzer (Diagnostica-Stago, Parsippany, NJ).
Fibrin Western Blotting
Fibrin extraction from snap frozen liver samples and western blotting for fibrin was performed as we have described previously.19 Levels of fibrin in 100 mg frozen liver were determined using the 59D8 antibody (kindly provided by Dr. Charles Esmon, Oklahoma Medical Research Foundation), which specifically detects fibrin formed by thrombin cleavage.34
RNA Isolation, cDNA Synthesis, and Real-Time PCR
Total RNA was isolated from 100 mg of snap-frozen liver using TRI reagent (Molecular Research Center, Inc., Cincinnati, OH) according to the manufacturer's protocol. One microgram of RNA was used for the synthesis of cDNA using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) and MyCycler thermal cycler (Bio-Rad). Levels of TNFα, cyclooxygenase (COX)-2, MCP-1 (Ccl2), macrophage inflammatory protein (MIP)-2 (Cxcl2), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA were determined using TaqMan gene expression assays from Applied Biosystems and TaqMan gene expression master mix (Applied Biosystems) on an ABI Prism 7300 sequence detection system (Applied Biosystems). The expression of each gene was normalized relative to GAPDH expression levels, and relative expression level determined using the comparative Ct method.
Statistics
Comparison of two groups was performed using Student's t-test. Comparison of three or more groups was performed using one-way analysis of variance and Student-Newman-Keuls posthoc test. The criterion for statistical significance was P < 0.05.
Results
MCD Diet-Induced Steatohepatitis and Coagulation Cascade Activation
First, we used wild-type congenic C57Bl/6J mice to characterize the procoagulant response associated with MCD diet-induced steatohepatitis. Compared with mice fed the control diet, mice fed the MCD diet for 2 weeks developed macrovesicular hepatic steatosis in the liver (Figure 1, A–B). Triglyceride levels increased in livers of mice fed the MCD diet, as compared with control diet (Figure 1D). Steatosis was accompanied by lobular and focal accumulation of inflammatory cells (Figure 1, B–C). In agreement with these histological changes, serum ALT activity increased significantly in mice fed the MCD diet, as compared with mice fed the control diet (Figure 1E). Moreover, levels of the mRNAs encoding the pro-inflammatory genes TNFα, MCP-1, MIP-2, and COX-2 were increased in livers of mice fed the MCD diet (Figure 1F). Macrophage accumulation (27 ± 3 vs. 62 ± 9, mean ± SEM per High Power Field) and neutrophil accumulation (0 ± 0 vs. 3 ± 0.7, mean ± SEM neutrophil foci per High Power Field) increased in the livers of mice fed the MCD diet compared with control diet. In agreement with previous studies,6,7 the data indicate that feeding mice the MCD diet for 2 weeks induces steatosis, hepatocellular injury, and liver inflammation.
Figure 1.
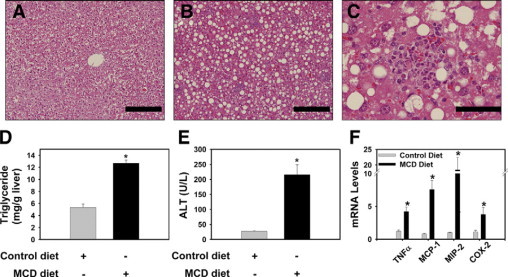
MCD diet-induced steatohepatitis in wild-type mice. Wild-type congenic C57Bl/6J mice were fed either control diet or MCD diet for 14 days. A: Representative photomicrograph of H&E-stained liver section from a mouse fed control diet showing normal hepatic architecture and glycogen accumulation. B: Representative photomicrograph of H&E-stained liver section from a mouse fed the MCD diet showing substantial macrovesicular hepatic steatosis and marked focal inflammatory cell accumulation. For (A−B), scale bar = 20 μm. C: Representative photomicrograph showing a typical foci of inflammatory cells in a liver from a mouse fed the MCD diet. For (C), scale bar = 5 μm. Hepatic triglyceride levels (D), serum ALT activity (E), and hepatic levels of inflammatory gene mRNAs (F) were determined. Data are expressed as mean ± SEM. n = 4 to 5 mice per group. Significantly different from mice fed the control diet, *P < 0.05.
Plasma TAT levels were significantly increased in wild-type congenic C57Bl/6J mice fed the MCD diet for 2 weeks, indicating activation of the coagulation cascade (Figure 2A). Consistent with increased thrombin generation, plasma fibrinogen levels were significantly decreased in mice fed the MCD diet (Figure 2B). Hepatic expression of the fibrinogen α, β, and γ mRNAs was not significantly altered (data not shown). Immunostaining revealed fibrin deposition in the hepatic sinusoids of mice fed the MCD diet, with the minimal fibrin staining in livers of mice fed the control diet generally restricted to the intima of the large vessels (Figure 2, C–D). This result was confirmed by Western blotting for fibrin using the monoclonal 59D8 antibody, which detects fibrin generated by thrombin cleavage (Figure 2E). Together, these results indicate that MCD diet-induced steatohepatitis is associated with thrombin generation in the liver.
Figure 2.
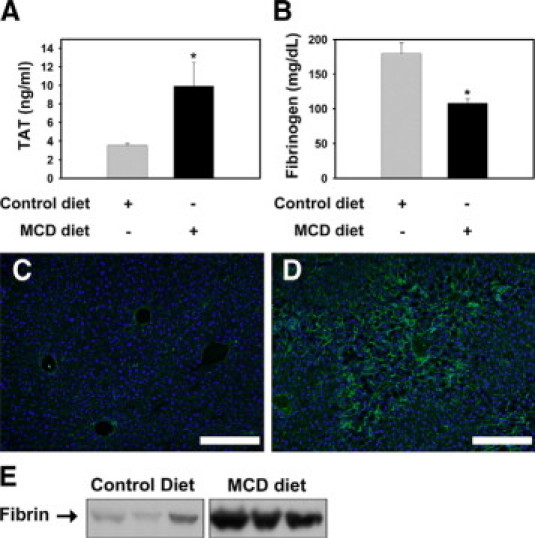
Procoagulant response associated with MCD diet-induced steatohepatitis in wild-type mice. A: Plasma thrombin-antithrombin (TAT) and (B) fibrinogen levels were determined in wild-type congenic C57BL/6J mice fed control diet or MCD diet for 14 days. Data are expressed as mean ± SEM, n = 5 to 9 mice per group. Significantly different from mice fed the control diet, *P < 0.05. Representative photomicrographs showing fibrin staining (green) in liver sections counterstained with 4,6-diamidino-2-phenylindole from mice fed control diet (C) or the MCD diet (D). Scale bar = 20 μm. E: Representative Western blot showing fibrin levels in liver extracts determined using the 59D8 monoclonal antibody. The bands shown were scanned digitally from the same film.
Effect of TF-Deficiency on Thrombin Generation, Hepatic Triglyceride Accumulation and Serum ALT Activity in Mice Fed the MCD Diet
Next, we used the low TF mice (mTF−/−hTF+ and heterozygous control mice (mTF+/−hTF+ mice) to elucidate the role of TF in the pathogenesis of steatohepatitis. Plasma TAT levels were significantly reduced in low TF mice fed the MCD diet, as compared with heterozygous control mice fed the MCD diet (Figure 3A), indicating that TF-dependent thrombin generation accompanies the progression of steatohepatitis. However, plasma TAT levels in low TF mice fed the MCD diet remained elevated above those observed in low TF mice fed the control diet. This suggests that either the low level of human TF expressed by the low TF mice contributes to thrombin generation or that thrombin generation also occurs in part, in a TF-independent manner in the MCD diet model. Hepatic triglyceride levels increased similarly in heterozygous control mice and low TF mice fed the MCD diet (Figure 3B). Serum ALT activity was increased in heterozygous control mice fed the MCD diet, but was significantly reduced in low TF mice fed the MCD diet (Figure 3C), albeit not to levels observed in low TF mice fed the control diet. This suggests that the TF-deficiency attenuates, but does not prevent the hepatocellular injury.
Figure 3.
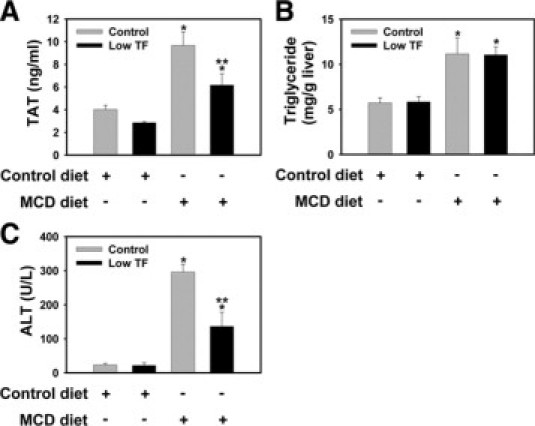
Effect of TF-deficiency on thrombin generation, hepatocellular injury, and hepatic triglyceride accumulation in MCD diet-induced steatohepatitis. Heterozygous control mice (mTF+/−hTF+ mice) and low TF mice (mTF−/−hTF+ mice) (see Materials and Methods) were fed control diet or the MCD diet for 14 days. A: Plasma thrombin-antithrombin (TAT) levels. B: Hepatic triglyceride levels. C: Serum alanine aminotransferase (ALT) activity. n = 5 to 11 mice per group. Data are expressed as mean ± SEM. Significantly different from the same mice fed control diet, *P < 0.05. Significantly different from heterozygous control mice fed the MCD diet, **P < 0.05.
TF-Deficiency Reduces Pro-Inflammatory Gene Induction and Hepatic Inflammation in Mice Fed the MCD Diet
TNFα, MCP-1, MIP-2, and COX-2 mRNA levels were significantly increased in the livers of heterozygous control mice fed the MCD diet (Figure 4A). The degree of mRNA induction for each of these genes was significantly reduced in low TF mice fed the MCD diet (Figure 4A). TNFα and MIP-2 proteins did not achieve detectable levels in the plasma (not shown). In agreement with expression of MCP-1 mRNA, plasma MCP-1 levels were increased in heterozygous control mice fed the MCD diet, but not in the low TF mice (Table 1). In agreement with the increase in hepatic triglyceride levels, the degree of steatosis was similar in livers from low TF mice and heterozygous control mice fed the MCD diet (Figure 4B). Hepatic macrophage and neutrophil accumulation increased significantly in heterozygous control mice fed the MCD diet (Figure 4, C and D). Of importance, as compared with the heterozygous control mice, the extent of macrophage and neutrophil accumulation was significantly reduced in low TF mice fed the MCD diet (Figure 4, C and D).
Figure 4.
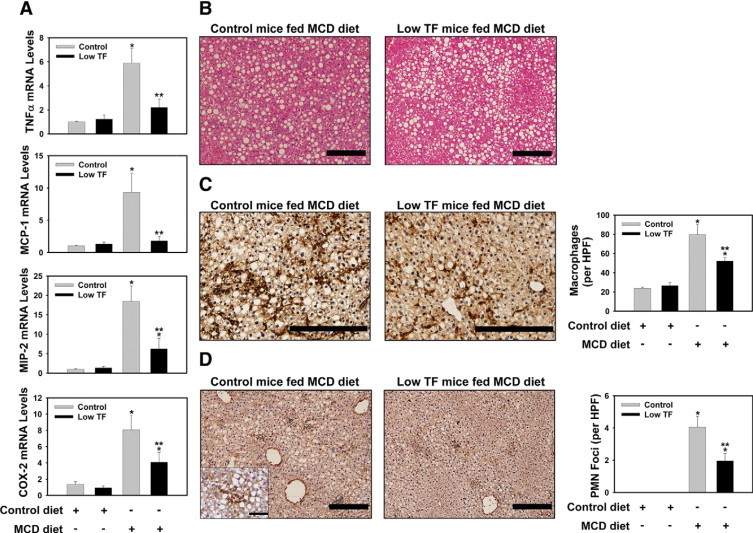
Effect of TF-deficiency on MCD diet-induced hepatic pro-inflammatory gene expression and inflammatory cell accumulation. Heterozygous control mice (mTF+/-hTF+ mice) and low TF mice (mTF−/−hTF+ mice) (see Materials and Methods) were fed control diet or the MCD diet for 14 days. Total RNA was isolated from whole liver. The levels of (A) TNFα mRNA, MCP-1 mRNA, MIP-2 mRNA, and COX-2 mRNA were determined by real-time PCR. B: Representative photomicrographs showing H&E-stained liver sections from a heterozygous control mouse and low TF mouse fed the MCD diet. Scale bar = 20 μm. C: Representative photomicrographs showing combined F4/80 and CD68 immunohistochemical staining for macrophages (dark brown) in liver sections from a heterozygous control mouse and low TF mouse fed the MCD diet. The average number of macrophages per ×400 field was determined. Scale bar = 20 μm. D: Representative photomicrographs showing immunohistochemical staining for neutrophils (dark brown) in liver sections from a heterozygous control mouse and low TF mouse fed the MCD diet. Scale bar = 20 μm. The high magnification (×1000) inset shows a representative neutrophil foci. For the inset, Scale bar = 5 μm. (The average number of neutrophil foci (>5 clustered neutrophils) per ×200 field was determined. n = 5 to 11 mice per group. Data are expressed as mean ± SEM. Significantly different from the same mice fed control diet, *P < 0.05. Significantly different from heterozygous control mice fed the MCD diet, **P < 0.05.
Table 1.
Effect of TF or PAR-1 Deficiency on Plasma MCP-1 Concentration in Mice Fed the MCD Diet
| Plasma MCP-1 (pg/ml) |
||
|---|---|---|
| Mice | Control diet | MCD diet |
| Heterozygous control mice (mTF+/−hTF+ mice) | 20.1 ± 3.9 | 62.8 ± 10.0* |
| Low TF mice (mTF−/−hTF+ mice) | 18.8 ± 4.3 | 32.7 ± 7.6† |
| Wild-type mice (PAR-1+/+ mice) | 20.8 ± 6.6 | 57.9 ± 8.8* |
| PAR-1-deficient mice (PAR-1−/− mice) | 17.2 ± 2.8 | 23.4 ± 5.3† |
Heterozygous control mice (mTF+/−hTF+ mice) and low TF mice (mTF−/−hTF+ mice), and wild-type control mice (PAR-1+/+ mice) and PAR-1−/− mice (see Materials and Methods) were fed control diet or the MCD diet for 14 days. n = 5 to 11 mice per group. Data are expressed as mean± SEM. *Significantly different from the same mice fed control diet. †Significantly different from respective control mice fed the MCD diet.
PAR-1 Contributes to Hepatic Injury and Inflammation Induced by the MCD Diet in Mice
One mechanism whereby TF could contribute to hepatic injury and inflammation in steatohepatitis is by generating thrombin that activates PAR-1, which is expressed constitutively by nonparenchymal cells in the liver.26,35 To evaluate the role of PAR-1 in pathogenesis of steatohepatitis we used PAR-1−/− mice and wild-type control mice (PAR-1+/+ mice, see Materials and Methods). Interestingly, we found that PAR-1 mRNA expression was also increased in the livers of mice fed the MCD diet (not shown). Analogous to our finding that TF-deficiency reduced serum ALT activity without impacting steatosis, PAR-1-deficiency significantly reduced serum ALT activity, but not hepatic triglyceride accumulation in mice fed the MCD diet (Figure 5, A–B). Moreover, MCD diet induction of mRNAs encoding TNFα, MCP-1, MIP-2, and COX-2 was significantly reduced in PAR-1−/− mice compared with wild-type control mice (PAR-1+/+ mice) (Figure 6A). In contrast to wild-type control mice, plasma MCP-1 levels were significantly lower in PAR-1−/− mice fed the MCD diet (Table 1). The degree of steatosis was similar in livers from wild-type control mice and PAR-1−/− mice fed the MCD diet (Figure 6B). Hepatic macrophage and neutrophil accumulation increased significantly in the livers of wild-type control mice (PAR-1+/+ mice) fed the MCD diet (Figure 6, C and D). Of importance, as compared with the wild-type control mice, the extent of macrophage and neutrophil accumulation was significantly reduced in PAR-1−/− mice fed the MCD diet (Figure 6, C and D).
Figure 5.
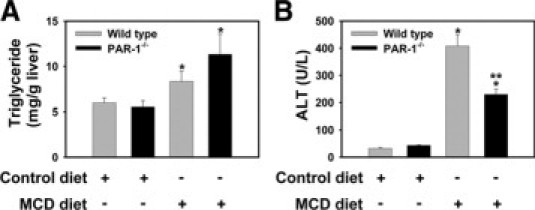
Effect of PAR-1-deficiency on hepatocellular injury and hepatic triglyceride accumulation in MCD diet-induced steatohepatitis. Wild-type control mice (PAR-1+/+ mice) and PAR-1−/− mice (see Materials and Methods) were fed control diet or the MCD diet for 14 days. A: Hepatic triglyceride levels. B: Serum alanine aminotransferase (ALT) activity. n = 4 to 11 mice per group. Data are expressed as mean ± SEM. Significantly different from the same mice fed control diet, *P < 0.05. Significantly different from wild-type control mice fed the MCD diet, **P < 0.05.
Figure 6.
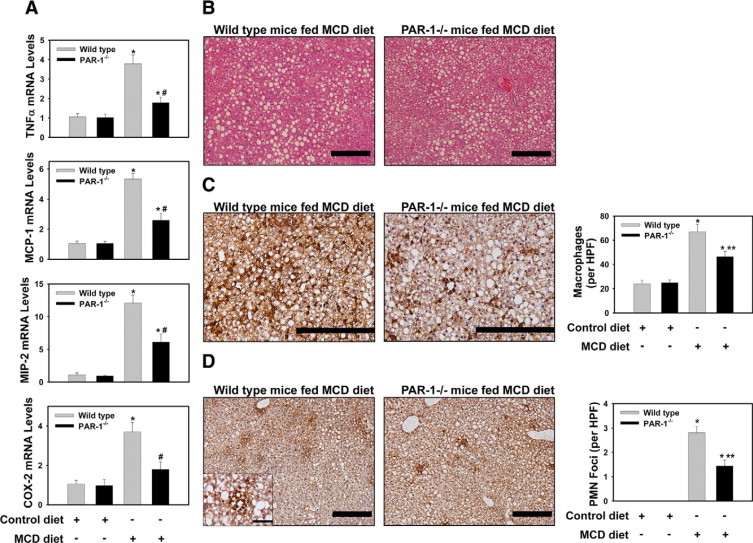
Effect of PAR-1 deficiency on MCD diet-induced hepatic pro-inflammatory gene expression and inflammatory cell accumulation. Wild-type control mice (PAR-1+/+ mice) and PAR-1−/− mice (see Materials and Methods) were fed control diet or the MCD diet for 14 days. Total RNA was isolated from whole liver. The levels of (A) TNFα mRNA, MCP-1 mRNA, MIP-2 mRNA, and COX-2 mRNA were determined by real-time PCR. (B) Representative photomicrographs showing H&E-stained liver sections from a wild-type control mouse and PAR-1−/− mouse fed the MCD diet. Scale bar = 20 μm. C: Representative photomicrographs showing combined F4/80 and CD68 immunohistochemical staining for macrophages (dark brown) in liver sections from a wild-type control mouse and PAR-1−/− mouse fed the MCD diet. Scale bar = 20 μm. The average number of macrophages per 400X field was determined. D: Representative photomicrographs showing immunohistochemical staining for neutrophils (dark brown) in liver sections from a wild-type control mouse and PAR-1−/− mouse fed the MCD diet. Scale bar = 20 μm. The high magnification (×1000) inset shows a representative neutrophil foci. For the inset, the scale bar = 5 μm. The average number of neutrophil foci (>5 clustered neutrophils) per ×200 field was determined. n = 5 to 11 mice per group. Data are expressed as mean ± SEM. Significantly different from the same mice fed control diet, *P < 0.05. Significantly different from wild-type control mice fed the MCD diet, **P < 0.05.
Discussion
Studies in patients suggest that liver steatosis and steatohepatitis are associated with perturbed coagulation. In agreement with this observation, we found that TF-dependent thrombin generation occurred in mice fed the lipogenic MCD diet. Reducing TF expression in all cells did not impact hepatic lipid accumulation. In contrast, the TF-deficiency reduced serum ALT activity, inflammatory gene induction and inflammatory cell accumulation in livers of mice fed the MCD diet. Of importance, the effect of PAR-1 deficiency mirrored the effect of TF-deficiency on steatohepatitis, as indicated by reduced hepatocellular injury and inflammation in PAR-1−/− mice fed the MCD diet. Taken together, the results suggest that TF-dependent thrombin generation and subsequent activation of PAR-1 amplifies liver injury and inflammation during steatohepatitis (Figure 7).
Figure 7.
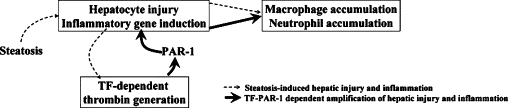
Proposed mechanism of TF−PAR-1-dependent amplification of hepatocellular injury and inflammation during steatohepatitis. Hepatic lipid accumulation induces modest hepatocyte injury and inflammation that induce TF-dependent coagulation cascade activation and thrombin generation in the liver sinusoids. Thrombin cleaves and activates PAR-1 expressed by nonparenchymal cells, which amplifies expression of numerous pro-inflammatory genes and promotes exaggerated inflammatory cell accumulation and hepatic injury.
The expression of TF by hepatocytes or nonparenchymal cells in the liver sinusoid likely presents a substantial regulatory challenge for the hepatic microcirculation. Hepatocytes synthesize numerous circulating coagulation factors and fenestration of the sinusoidal endothelium allows for exposure of the hepatocytes to components of the plasma. Interestingly, we found that TF mRNA levels and activity increased slightly in livers of mice fed the MCD diet (not shown), suggesting that steatohepatitis induces a procoagulant state in the liver. Indeed, the MCD diet-induced steatohepatitis was associated with TF-dependent thrombin generation as indicated by increased plasma TAT levels and decreased plasma fibrinogen levels. This points to activation of the extrinsic pathway of coagulation during the progression of steatohepatitis, although we cannot exclude amplification of coagulation by the intrinsic pathway. Taken together, the results suggest that a modest TF-dependent procoagulant state accompanies the progression of MCD diet-induced steatohepatitis in mice.
The cellular distribution of TF in the liver has not been completely characterized. TF expression by one or more cell types during steatohepatitis is plausible. For example, isolated human hepatocytes have been shown to express TF,36 although the regulation of TF expression and activity in these cells is not understood. Hepatic nonparenchymal cells including Kupffer cells, stellate cells, and endothelial cells can be induced to express TF.19,37,38 Moreover, the expression of TF by extrahepatic cells, such as monocytes or platelets, which accumulate in the liver during steatohepatitis could contribute to overall liver TF procoagulant activity.39,40 Further studies are necessary to determine the mechanism whereby TF activity increases in the livers of mice fed the MCD diet.
Patients with severe liver disease are often at risk of bleeding as a consequence of failed synthesis of coagulation factors by the injured liver. In contrast, the progression of liver disease, such as steatohepatitis, may be accompanied by a more modest procoagulant state that modifies the disease pathogenesis. For instance, plasma levels of coagulation factor VII and plasminogen activator inhibitor-1 and the hepatic expression of coagulation factor XIII are increased in patients with NAFLD.17,41,42 However, to date the role of coagulation in steatohepatitis has not been addressed. We found that a genetic deficiency in TF did not impact hepatic triglyceride accumulation in mice fed the MCD diet, suggesting that TF-induced coagulation is an effect secondary to steatosis in this model. Indeed, TF-deficiency is unlikely to directly impact steatosis in this model, which is likely driven by failed triglyceride metabolism and export from the liver.43 However, TF induction or activation secondary to simple steatosis (or mild inflammation) may coordinate the activation of additional pathways that determine the ultimate severity of the hepatic inflammation and transition to steatohepatitis. Indeed, TF-deficiency reduced serum ALT activity and decreased liver inflammation in mice fed the lipogenic MCD diet. This suggests that TF-induced coagulation is an essential regulator of the hepatic inflammatory response resulting from steatosis in the MCD diet model of steatohepatitis (Figure 7).
Cross talk between inflammation and coagulation proteases is integral in multiple disease processes.44 Considerable evidence implicates thrombin activation of PAR-1 as a critical point of communication between coagulation and inflammation. Indeed, TF-dependent thrombin generation and PAR-1 activation promote inflammation in models of tissue injury including kidney ischemia-reperfusion, glomerulonephritis, lipopolysaccharide-induced liver injury, and bleomycin-induced lung injury.26–29 PAR-1 is expressed by hepatic nonparenchymal cells including Kupffer cells, endothelial cells, and stellate cells.26,35 PAR-1 activation on hepatic stellate cells induces cytokine expression.37 Moreover, PAR-1 activation has been shown to elicit neutrophil activation in lipopolysaccharide-primed, isolated, and perfused livers.26 Of importance, mimicking the effect of TF-deficiency on steatohepatitis, MCD diet-induced hepatic inflammation and injury were reduced in PAR-1−/− mice without effect on triglyceride accumulation. Taken together, the results suggest that TF-dependent thrombin generation leads to activation of PAR-1 on nonparenchymal cells that promote hepatic inflammation and injury.
One mechanism whereby the TF−PAR-1 pathway could promote hepatic inflammation is through coordinated induction of genes that further modify inflammatory cell accumulation and activation. For example, the induction of TNFα was reduced in low TF mice and PAR-1−/− mice fed the MCD diet. Several studies implicate TNFα as a mediator of liver inflammation in mice fed the MCD diet,45–47 although this is not the case for all studies.9 Similarly, TF and PAR-1 contributed to hepatic expression of COX-2, which has been shown to contribute to hepatic inflammatory cell accumulation in mice fed the MCD diet.10,48 Hepatic expression of the neutrophil chemokine MIP-2 involved both TF and PAR-1 in mice fed the MCD diet. However, the role of the CXC chemokines and neutrophils in the MCD diet model of steatohepatitis is not completely understood. Hepatic neutrophil accumulation has been observed both in murine models of steatohepatitis49 and in livers from patients with steatohepatitis,50 and neutrophil activation may contribute to oxidative stress in this disease.50
Of particular interest was the effect of TF and PAR-1 deficiency on expression of the chemokine MCP-1. Our data indicate that TF and PAR-1 are essential for induction of MCP-1 mRNA in the liver and increased MCP-1 protein levels in the plasma of mice fed the MCD diet. In patients with NAFLD or NASH, hepatic MCP-1 mRNA expression and plasma MCP-1 levels are increased23,42 and MCP-1 has been shown to contribute to monocyte/macrophage accumulation in liver in multiple models.51–54 Indeed, in livers from patients with chronic liver disease, MCP-1 expression correlated with monocytic cell infiltration.55
Rodent models of NASH typically use genetic- or diet-induced hepatic lipid accumulation to produce the various sequelae of NASH.2 The MCD diet model of steatohepatitis is well established and provides substantial opportunity to investigate mechanisms of hepatic inflammation and hepatocyte injury as requisite features of steatohepatitis progression. However, unlike the clinical condition, mice fed the MCD diet lose weight56 and do not develop insulin resistance.2 In contrast, recent studies have shown that induction of hyperlipidemia by a Western diet induces obesity and insulin resistance, as well as steatohepatitis and fibrosis in rodents.57,58 Although the time required to observe requisite histopathological changes increases to >4 months, these models represent important correlates to the development of NAFLD and NASH in the human population. TF-dependent coagulation and PAR-1-dependent inflammation have not yet been investigated in these models. However, it is important to note that in other models of NASH, including the genetically obese leptin-deficient and leptin receptor-deficient mice, TF expression is increased in the liver.59 Additional evaluation of the roles of both TF and PAR-1 in models of NASH associated with insulin resistance is ongoing in our laboratory.
In summary, we found that steatohepatitis induced by feeding mice the MCD diet is associated with TF-dependent coagulation. Deficiency in either TF or the thrombin receptor PAR-1 reduced the severity of liver inflammation. The data indicate that TF and PAR-1 contribute to the hepatic expression of key inflammatory mediators and are important regulators of both macrophage and neutrophil accumulation during steatohepatitis.
Acknowledgements
We thank Huina Cai and Noriko Esterly for outstanding technical assistance and Dr. Bryan Copple for critical reading of the manuscript.
Footnotes
Supported by American Heart Association Scientist Development grant 0835121G (J.P.L.) and by the National Institutes of Health P20 RR021940 (J.P.L.) and RO1DK081343 (G.L.G.).
References
- 1.Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649–1657. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 2.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 3.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 4.Kleiner DE, Brunt EM, Van NM, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 5.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koppe SW, Sahai A, Malladi P, Whitington PF, Green RM. Pentoxifylline attenuates steatohepatitis induced by the methionine choline deficient diet. J Hepatol. 2004;41:592–598. doi: 10.1016/j.jhep.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 7.Sahai A, Malladi P, Melin-Aldana H, Green RM, Whitington PF. Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am J Physiol Gastrointest Liver Physiol. 2004;287:G264–G273. doi: 10.1152/ajpgi.00002.2004. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 9.Dela Pena A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663–1674. doi: 10.1053/j.gastro.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Yu J, Ip E, Dela PA, Hou JY, Sesha J, Pera N, Hall P, Kirsch R, Leclercq I, Farrell GC. COX-2 induction in mice with experimental nutritional steatohepatitis: role as pro-inflammatory mediator. Hepatology. 2006;43:826–836. doi: 10.1002/hep.21108. [DOI] [PubMed] [Google Scholar]
- 11.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 12.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 13.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 14.Northup PG, Sundaram V, Fallon MB, Reddy KR, Balogun RA, Sanyal AJ, Anstee QM, Hoffman MR, Ikura Y, Caldwell SH. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost. 2008;6:2–9. doi: 10.1111/j.1538-7836.2007.02772.x. [DOI] [PubMed] [Google Scholar]
- 15.Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis, and thrombosis. Thromb Haemost. 2008;99:995–1000. doi: 10.1160/TH07-11-0682. [DOI] [PubMed] [Google Scholar]
- 16.Cigolini M, Targher G, Agostino G, Tonoli M, Muggeo M, De SG. Liver steatosis and its relation to plasma haemostatic factors in apparently healthy men–role of the metabolic syndrome. Thromb Haemost. 1996;76:69–73. [PubMed] [Google Scholar]
- 17.Kotronen A, Yki-Jarvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- 18.Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 19.Luyendyk JP, Cantor GH, Kirchhofer D, Mackman N, Copple BL, Wang R. Tissue factor-dependent coagulation contributes to alpha-naphthylisothiocyanate-induced cholestatic liver injury in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G840–G849. doi: 10.1152/ajpgi.90639.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackman N, Sawdey MS, Keeton MR, Loskutoff DJ. Murine tissue factor gene expression in vivo: Tissue and cell specificity and regulation by lipopolysaccharide. Am J Pathol. 1993;143:76–84. [PMC free article] [PubMed] [Google Scholar]
- 21.Abiru S, Migita K, Maeda Y, Daikoku M, Ito M, Ohata K, Nagaoka S, Matsumoto T, Takii Y, Kusumoto K, Nakamura M, Komori A, Yano K, Yatsuhashi H, Eguchi K, Ishibashi H. Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int. 2006;26:39–45. doi: 10.1111/j.1478-3231.2005.01191.x. [DOI] [PubMed] [Google Scholar]
- 22.Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, Keshavarzian A. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008;28:1026–1033. doi: 10.1111/j.1478-3231.2008.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haukeland JW, Damas JK, Konopski Z, Loberg EM, Haaland T, Goverud I, Torjesen PA, Birkeland K, Bjoro K, Aukrust P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44:1167–1174. doi: 10.1016/j.jhep.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Kudo H, Takahara T, Yata Y, Kawai K, Zhang W, Sugiyama T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 2009;51:168–175. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 25.Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci USA. 1999;96:11023–11027. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Copple BL, Moulin F, Hanumegowda UM, Ganey PE, Roth RA. Thrombin and protease-activated receptor-1 agonists promote lipopolysaccharide-induced hepatocellular injury in perfused livers. J Pharmacol Exp Ther. 2003;305:417–425. doi: 10.1124/jpet.102.046391. [DOI] [PubMed] [Google Scholar]
- 27.Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med. 2000;191:455–461. doi: 10.1084/jem.191.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olman MA, Mackman N, Gladson CL, Moser KM, Loskutoff DJ. Changes in procoagulant and fibrinolytic gene expression during bleomycin-induced lung injury in the mouse. J Clin Invest. 1995;96:1621–1630. doi: 10.1172/JCI118201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sevastos J, Kennedy SE, Davis DR, Sam M, Peake PW, Charlesworth JA, Mackman N, Erlich JH. Tissue factor deficiency and PAR-1 deficiency are protective against renal ischaemia reperfusion injury. Blood. 2007;109:577–583. doi: 10.1182/blood-2006-03-008870. [DOI] [PubMed] [Google Scholar]
- 30.Parry GCN, Erlich JH, Carmeliet P, Luther T, Mackman N. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest. 1998;101:560–569. doi: 10.1172/JCI814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Connolly AJ, Suh DY, Hunt TK, Coughlin SR. Mice lacking the thrombin receptor. PAR1, have normal skin wound healing. Am J Pathol. 1997;151:1199–1204. [PMC free article] [PubMed] [Google Scholar]
- 32.Ganey PE, Luyendyk JP, Newport SW, Eagle TM, Maddox JF, Mackman N, Roth RA. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46:1177–1186. doi: 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, Esterly N, Guo GL, Klaassen CD. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J Pharmacol Exp Ther. 2008;325:655–664. doi: 10.1124/jpet.107.135822. [DOI] [PubMed] [Google Scholar]
- 34.Weiler-Guettler H, Christie PD, Beeler DL, Healy AM, Hancock WW, Rayburn H, Edelberg JM, Rosenberg RD. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J Clin Invest. 1998;101:1983–1991. doi: 10.1172/JCI2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaca MD, Zhou X, Benyon RC. Regulation of hepatic stellate cell proliferation and collagen synthesis by proteinase-activated receptors. J Hepatol. 2002;36:362–369. doi: 10.1016/s0168-8278(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 36.Stephenne X, Vosters O, Najimi M, Beuneu C, Dung KN, Wijns W, Goldman M, Sokal EM. Tissue factor-dependent procoagulant activity of isolated human hepatocytes: relevance to liver cell transplantation. Liver Transpl. 2007;13:599–606. doi: 10.1002/lt.21128. [DOI] [PubMed] [Google Scholar]
- 37.Fiorucci S, Antonelli E, Distrutti E, Severino B, Fiorentina R, Baldoni M, Caliendo G, Santagada V, Morelli A, Cirino G. PAR1 antagonism protects against experimental liver fibrosis. Role of proteinase receptors in stellate cell activation. Hepatology. 2004;39:365–375. doi: 10.1002/hep.20054. [DOI] [PubMed] [Google Scholar]
- 38.Mochida S, Arai M, Ohno A, Yamanobe F, Ishikawa K, Matsui A, Maruyama I, Kato H, Fujiwara K. Deranged blood coagulation equilibrium as a factor of massive liver necrosis following endotoxin administration in partially hepatectomized rats. Hepatology. 1999;29:1532–1540. doi: 10.1002/hep.510290533. [DOI] [PubMed] [Google Scholar]
- 39.Mackman N. Regulation of tissue factor gene expression in human monocytic and endothelial cells. Haemostasis. 1996;26:17–20. doi: 10.1159/000217234. [DOI] [PubMed] [Google Scholar]
- 40.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, Tilley RE, Rondina MT, Harris EM, Kraiss LW, Mackman N, Zimmerman GA, Weyrich AS. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med. 2006;203:2433–2440. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edens MA, Kuipers F, Stolk RP. Non-alcoholic fatty liver disease is associated with cardiovascular disease risk markers. Obes Rev. 2009;10:412–419. doi: 10.1111/j.1467-789X.2009.00594.x. [DOI] [PubMed] [Google Scholar]
- 42.Greco D, Kotronen A, Westerbacka J, Puig O, Arkkila P, Kiviluoto T, Laitinen S, Kolak M, Fisher RM, Hamsten A, Auvinen P, Yki-Jarvinen H. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1281–G1287. doi: 10.1152/ajpgi.00074.2008. [DOI] [PubMed] [Google Scholar]
- 43.Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem. 1988;263:2998–3004. [PubMed] [Google Scholar]
- 44.Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- 45.Kudo H, Takahara T, Yata Y, Kawai K, Zhang W, Sugiyama T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 2009;51:168–175. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 46.Koca SS, Bahcecioglu IH, Poyrazoglu OK, Ozercan IH, Sahin K, Ustundag B. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation. 2008;31:91–98. doi: 10.1007/s10753-007-9053-z. [DOI] [PubMed] [Google Scholar]
- 47.Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y, Han JY, Kato S, Shimoda M, Oike Y, Tomizawa M, Makino S, Ohkura T, Saito H, Kumagai N, Nagata H, Ishii H, Hibi T. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. 2006;55:415–424. doi: 10.1136/gut.2005.071118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu J, Wu CW, Chu ES, Hui AY, Cheng AS, Go MY, Ching AK, Chui YL, Chan HL, Sung JJ. Elucidation of the role of COX-2 in liver fibrogenesis using transgenic mice. Biochem Biophys Res Commun. 2008;372:571–577. doi: 10.1016/j.bbrc.2008.05.069. [DOI] [PubMed] [Google Scholar]
- 49.Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M, Renner EL, Clavien PA. Serotonin mediates oxidative stress and mitochondrial toxicity in a murine model of nonalcoholic steatohepatitis. Gastroenterology. 2007;133:608–618. doi: 10.1053/j.gastro.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 50.Ikura Y, Ohsawa M, Suekane T, Fukushima H, Itabe H, Jomura H, Nishiguchi S, Inoue T, Naruko T, Ehara S, Kawada N, Arakawa T, Ueda M. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: impact on disease progression. Hepatology. 2006;43:506–514. doi: 10.1002/hep.21070. [DOI] [PubMed] [Google Scholar]
- 51.Dambach DM, Watson LM, Gray KR, Durham SK, Laskin DL. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology. 2002;35:1093–1103. doi: 10.1053/jhep.2002.33162. [DOI] [PubMed] [Google Scholar]
- 52.Hildebrand F, Hubbard WJ, Choudhry MA, Frink M, Pape HC, Kunkel SL, Chaudry IH. Kupffer cells and their mediators: the culprits in producing distant organ damage after trauma-hemorrhage. Am J Pathol. 2006;169:784–794. doi: 10.2353/ajpath.2006.060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hokeness KL, Kuziel WA, Biron CA, Salazar-Mather TP. Monocyte chemoattractant protein-1 and CCR2 interactions are required for IFN-alpha/beta-induced inflammatory responses and antiviral defense in liver. J Immunol. 2005;174:1549–1556. doi: 10.4049/jimmunol.174.3.1549. [DOI] [PubMed] [Google Scholar]
- 54.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2 (gene targeting/monocyte chemoattractant protein 1/trafficking/peritonitis/granuloma) Proc Natl Acad Sci USA. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M, Romanelli RG, Laffi G, Gentilini P. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol. 1998;152:423–430. [PMC free article] [PubMed] [Google Scholar]
- 56.Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, Turner SM, Badger TM, Pitas RE, Maher JJ. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res. 2006;47:2280–2290. doi: 10.1194/jlr.M600198-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.DeLeve LD, Wang X, Kanel GC, Atkinson R, McCuskey RS. Prevention of hepatic fibrosis in a murine model of metabolic syndrome with non-alcoholic steatohepatitis. Am J Pathol. 2008;173:993–1001. doi: 10.2353/ajpath.2008.070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu ZJ, Fan JG, Ding XD, Qiao L, Wang GL. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Dig Dis Sci. 2009 doi: 10.1007/s10620-009-0815-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samad F, Pandey M, Loskutoff DJ. Regulation of tissue factor gene expression in obesity. Blood. 2001;98:3353–3358. doi: 10.1182/blood.v98.12.3353. [DOI] [PubMed] [Google Scholar]