

Abstract
The intracellular pathogen Francisella tularensis is the causative agent of tularemia, a zoonosis that can affect humans with potentially lethal consequences. Essential to Francisella virulence is its ability to survive and proliferate within phagocytes through phagosomal escape and cytosolic replication. Francisella spp. encode a variety of acid phosphatases, whose roles in phagosomal escape and virulence have been documented yet remain controversial. Here we have examined in the highly virulent (type A) F. tularensis strain Schu S4 the pathogenic roles of three distinct acid phosphatases, AcpA, AcpB, and AcpC, that are most conserved between Francisella subspecies. Neither the deletion of acpA nor the combination of acpA, acpB, and acpC deletions affected the phagosomal escape or cytosolic growth of Schu S4 in murine and human macrophages, despite decreases in acid phosphatase activities by as much as 95%. Furthermore, none of these mutants were affected in their ability to cause lethality in mice upon intranasal inoculation. Hence, the acid phosphatases AcpA, AcpB, and AcpC do not contribute to intracellular pathogenesis and do not play a major role in the virulence of type A Francisella strains.
The Gram-negative bacterium Francisella tularensis is a highly infectious, facultative intracellular pathogen that causes tularemia, a widespread zoonosis affecting humans. Human tularemia is a fulminant disease that can be contracted by exposure to as few as 10 bacteria, the pneumonic form of which can lead to mortality rates as high as 25% if untreated (35). Three subspecies of F. tularensis, Francisella tularensis subsp. tularensis (type A), Francisella tularensis subsp. holarctica (type B), and Francisella tularensis subsp. mediasiatica, are recognized, among which strains of the first two subspecies can cause tularemia in humans (15). While type B strains are geographically distributed all over the northern hemisphere, the highly virulent type A strains are restricted to North America and account for the most-severe cases of the disease. Francisella novicida, a species of low virulence in humans but high virulence in rodents, has been used extensively as a surrogate model of F. tularensis pathogenesis, based on the assumption that it uses conserved virulence mechanisms (4, 7, 8, 19, 23, 25-29, 31, 41-45, 47). As a facultative intracellular pathogen, F. tularensis is capable of infecting and proliferating in a variety of host cell types, including hepatocytes, epithelial cells, and mononuclear phagocytes (15). Macrophages constitute an important target for infection in vivo (21), and the pathogenesis of F. tularensis depends on the bacterium's ability to survive and replicate within these host cells (15). Upon phagocytosis, Francisella ensures its effective survival and proliferation via rapid phagosomal escape followed by extensive replication in the cytosol (11, 14, 20, 42), thereby segregating itself from the degradative endosomal system and its associated bactericidal activities. Phagosomal escape is a tightly regulated process whose efficiency depends on conditions encountered within the early phagosome (12, 41), such as vacuolar acidification, although some controversy remains as to whether Francisella-containing phagosomes are significantly acidified prior to membrane disruption (13). Regardless of such discrepancies, phagosomal escape is an essential step in Francisella intracellular pathogenesis, since it is a prerequisite for cytosolic replication. Indeed, Francisella mutants that are defective in phagosomal escape do not grow intracellularly and are attenuated in vivo (6, 24, 43-45), and a belated phagosomal escape delays intracellular proliferation of the highly virulent type A strain Schu S4 (12).
Much effort has focused on identifying bacterial factors that contribute to phagosomal escape. Several genes located within a 30-kb chromosomal locus known as the Francisella pathogenicity island (FPI) (31) are required for proper phagosomal escape of F. novicida (43, 44) and the attenuated F. tularensis subsp. holarctica live vaccine strain (LVS) (6, 24), since transposon insertions or targeted deletions in iglC, iglD, and pdpA affect the translocation of the mutants to the cytosol. Based on the homology of some FPI proteins with components of type VI secretion systems in other pathogens (30, 36), the FPI likely encodes a secretion apparatus that is required for phagosomal disruption. Yet a true understanding of FPI functions and the characterization of actual Francisella effectors of phagosomal escape are lacking. In addition to the FPI, Mohapatra et al. have recently reported for F. novicida that the acid phosphatases AcpA, AcpB, AcpC, and Hap are required for phagosomal escape and virulence in mice (27, 29). Acid phosphatases, which are ubiquitous in nature and hydrolyze phosphomonoesters at acidic pHs, have been associated with the survival of intracellular parasites within phagocytes through inhibition of the respiratory burst (1, 3, 9, 22, 37-40), suggesting that they act as virulence factors. In Francisella, a prominent role was established for AcpA, an unusual, respiratory-burst-inhibiting enzyme exemplifying a novel family of acid phosphatases (18, 37). AcpA accounts for most of the acid phosphatase and phospholipase activities in the outer membrane fraction of F. novicida (29). These reports assigned acid phosphatases a role in phagosomal escape yet contradicted a previous study by Baron et al., who concluded that AcpA was not required for the intracellular growth or virulence of F. novicida (4). While the acpA mutants were constructed differently in these studies, the acid phosphatase activity associated with AcpA was abolished in both situations. A proposed explanation for these conflicting results was that the truncated AcpA generated by Baron et al. remained functional as a phospholipase C (37), an activity that would be required for phagosomal escape and virulence (27). Yet this hypothesis has not been tested, leaving the role of AcpA in Francisella virulence a controversial matter.
All studies of Francisella acid phosphatases have been carried out with F. novicida (4, 27, 29, 37), raising the question of significance with regard to the virulent F. tularensis subspecies. In particular, recent whole-genome comparisons between F. novicida and the different Francisella tularensis subspecies have highlighted important intervening sequence (IS)-mediated genome rearrangements in F. tularensis subsp. holarctica and F. tularensis subsp. tularensis strains relative to F. novicida (10). Such rearrangements have disrupted large numbers of open reading frames (ORFs), thereby creating pseudogenes (10) and likely inactivating many functions in virulent F. tularensis strains. For example, Mohapatra et al. (29) have reported that the virulent type A strain Schu S4 is missing a homolog of one of the two hap genes (FTN_0022) present in F. novicida, raising the question of conservation of acid phosphatase-encoding genes in virulent strains. Because phagosomal escape is an essential stage of the Francisella intracellular cycle that is common to F. novicida and F. tularensis, we have postulated that factors required to promote this process must be conserved between these organisms. Here we have compared acid phosphatase-encoding genes in F. novicida and virulent F. tularensis subspecies, and we have generated deletion mutants of the most conserved genes in Schu S4 in order to test their role in the phagosomal escape and pathogenesis of the highly virulent F. tularensis subspecies. We demonstrate that most acid-phosphatase-encoding genes are disrupted in virulent strains and that the most conserved loci are not required for phagosomal escape and virulence.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The prototypic type A virulent strain, F. tularensis subsp. tularensis Schu S4, was obtained from Rick Lyons (University of New Mexico, Albuquerque, NM). The Schu S4 ΔfevR mutant (ΔFTT0383) has been described previously (46). Bacteria were grown on modified Mueller-Hinton (MMH) agar plates (Mueller-Hinton medium supplemented with 0.1% glucose, 0.025% ferric pyrophosphate, and 2% IsoVitaleX [Becton Dickinson, Cockeysville, MD]) for 3 days at 37°C under 7% CO2. For allelic replacement, MMH medium was supplemented either with 10 μg/ml kanamycin (Invitrogen, Carlsbad, CA) or with 8% sucrose (Sigma, St Louis, MO). All manipulations of F. tularensis strain Schu S4 and its derivatives were performed in a biosafety level-3 facility according to standard operating procedures approved by the Rocky Mountain Laboratories Institutional Biosafety Committee.
Construction of in-frame deletion mutants of Schu S4.
In-frame deletion mutants of Schu S4 were generated using the SacB-assisted allelic replacement suicide vector pJC84, as described previously (46). Each deletion was designed to preserve the integrity of the downstream gene(s) (see Fig. 2) and to avoid any polar effect of the deletion. To engineer in-frame deletions of either acpA (the FTT0221 locus), acpB (the FTT0156 locus), or acpC (the FTT0620 locus), 5′ hemifragments, containing upstream regions and the start codon of each gene, and 3′ hemifragments, containing the last 6 codons of each gene and downstream regions, were generated by PCR amplification (Table 1). Both hemifragments were fused by overlap extension PCR, cloned into pCR2.1 TOPO (Invitrogen), and fully sequenced. The resulting fragments contained residual ORFs of the last 6 codons of acpA, acpB, and acpC, respectively, and were subcloned into pJC84 using the BamHI and SalI sites introduced by PCR primers (Table 1) to produce pJC84ΔacpA, pJC84ΔacpB, and pJC84ΔacpC, respectively.
FIG. 2.

Construction of ΔacpA and ΔacpABC Schu S4 mutants. (A) Schematic representation of the acpA, acpB, and acpC loci in the Schu S4 chromosome before and after allelic replacement using pJC84ΔacpA, pJC84ΔacpB, and pJC84ΔacpC, respectively (see Materials and Methods). Dashed lines indicate the chromosomal regions flanking the respective loci that were used for allelic replacement. The allelic replacement was designed to preserve the integrity of the ybgK gene, located immediately downstream of acpA; of the licB gene, located immediately downstream of acpB; and of the tdk gene, located immediately downstream of acpC. (B) PCR confirmation of acpA (top), acpB (center), and acpC (bottom) deletions from the correct Schu S4 chromosomal loci in the ΔacpA and ΔacpABC mutants. Both wild-type and deleted amplified regions are indicated. (C) Acid phosphatase (AP) activities in Schu S4 and acp mutants. Whole-cell lysates were generated from bacterial cultures and assayed for AP activity as described in Materials and Methods. Values are means ± standard deviations for three independent experiments. Asterisks indicate statistically significant differences (P < 0.001) from wild-type activity by a two-tailed Student t test.
TABLE 1.
Primers used in this study
| Primer function and name | Sequence (5′-3′)a | Nature of amplicon |
|---|---|---|
| pJC84 chromosomal detection | ||
| JC420 | CTAGCTAGCAGGAGACATGAACGATGAACATC | 1.5-kb internal fragment of sacB |
| JC427 | GGGACGTCGGATTCACCTTTATGTTGATAAG | 1.5-kb internal fragment of sacB |
| JC428 | GGGACGTCGATTAAGCATTGGTAACTGTCAGACC | 900-bp fragment of the pJC84 backbone |
| JC589 | ATCAGCTCACTCAAAGGCGG | 900-bp fragment of the pJC84 backbone |
| Deletion of acpA | ||
| JC700 | TGGATCCTCTGTAACCACATTAGTATC | 1,137 bp upstream and start codon of acpA |
| JC701 | GTTTAATTTATCCACTACCATATGATACCTTTAGTTGT | 1,137 bp upstream and start codon of acpA |
| JC702 | TTTGTCGACTGCTAATACCGAGAGGTCAGCC | Last 6 codons of acpA and 1,006 bp downstream |
| JC703 | GTAGTGGATAAATTAAACTAAA | Last 6 codons of acpA and 1,006 bp downstream |
| JC704 | TCTCCTAACTAACTTATTTTTG | ΔacpA |
| JC705 | GAAGAGGCAATAAGCGAAAATAC | ΔacpA |
| JC706 | GTTCCAACTTATCGCTCCAAG | ΔacpA within the FTT0221 locus |
| JC707 | GGAGTGGTTGCTGAATATGC | ΔacpA within the FTT0221 locus |
| Deletion of acpB | ||
| JC732 | TGGATCCGCTATGATTAGTAAATCTAGC | 1,035 bp upstream and start codon of acpB |
| JC733 | AATCTTGAGTTCTCACCCATAGAATTATTTTAGACCT | 1,035 bp upstream and start codon of acpB |
| JC734 | TTGGTCGACACATAACTTGTAGTATGAACT | Last 6 codons of acpB and 989 bp downstream |
| JC735 | GGTGAGAACTCAAGATTTTAG | Last 6 codons of acpB and 989 bp downstream |
| JC736 | GATAAATATCCAAGAGCTGAG | ΔacpB |
| JC737 | CGATTACGATAAGTTGTGCTA | ΔacpB |
| JC738 | GAGATCGGTACAGCTATTGCTT | ΔacpB within the FTT0156 locus |
| JC739 | CAAGTAGCATAAATCATCGCG | ΔacpB within the FTT0156 locus |
| Deletion of acpC | ||
| JC724 | TGGATCCTCATCTCTGGAAGTCTATGA | 1,254 bp upstream and start codon of acpC |
| JC725 | GCCAGCTGCCATACATCATTGATGTTTCTAAGTTCTT | 1,254 bp upstream and start codon of acpC |
| JC726 | TTGGTCGACACTATCCTTCTTTAGTTGCT | Last 6 codons of acpC and 635 bp downstream |
| JC727 | ATGTATGGCAGCTGGCAATAA | Last 6 codons of acpC and 635 bp downstream |
| JC728 | ACATTCTCTGGCGTGGTGAG | ΔacpC |
| JC729 | CCAGCATCCATAGCCGAATA | ΔacpC |
| JC730 | TGGCTCTGGTAGATAATCA | ΔacpC within the FTT0620 locus |
| JC731 | TTACTACTCAGGCGTTGAAAC | ΔacpC within the FTT0620 locus |
Underlined sequences denote either the BamHI (GGATCC) or the SalI (GTCGAC) restriction site used for cloning into pJC84.
For allelic replacement in the chromosome of Schu S4, electrocompetent bacteria were prepared and electroporated with recombinant pJC84 plasmid DNA as previously described (46). Kanamycin-resistant merodiploid colonies were tested for integration of the allelic replacement plasmid, using colony PCR with primers JC420 and JC427 (to amplify a 1.5-kb internal fragment of sacB) or primers JC589 and JC428 (to amplify a 900-bp fragment of the pJC84 backbone). Independent clones were then subjected to sucrose counterselection as previously described (46) in order to isolate clones that had undergone allelic replacement. The presence of the deleted allele and allelic replacement within the correct chromosomal locus were verified by PCR using primers JC704 and JC705 and primers JC706 and JC707, respectively, for the acpA deletion, primers JC736 and JC737 and primers JC738 and JC739, respectively, for the acpB deletion, primers JC728 and JC729 and primers JC730 and JC731, respectively, for the acpC deletion, and primers JC420 and JC427 for the loss of the sacB gene (Table 1). Independent clones carrying the correct in-frame deletion in either acpA, acpB, or acpC were isolated and used for further studies. Multiple deletion mutants were generated by repeating the allelic replacement procedure on single (ΔacpA) or double (ΔacpAB) deletion mutants.
Acid phosphatase assay.
To measure acid phosphatase activity associated with either Schu S4 or its isogenic acid phosphatase mutants, strains were grown overnight at 37°C in MMH broth under shaking conditions, and cultures were normalized based on readings of optical density at 600 nm (OD600). Bacteria were collected by centrifugation at 6,000 × g and 4°C for 10 min, and the pellets were washed twice with phosphate-buffered saline (PBS) and resuspended in 1/100 of the original culture volume in PBS. Cells were transferred to 2 ml Lysing Matrix B FastPrep tubes (Qbiogene, Carlsbad, CA) and were lysed through nine cycles of 45 s each at a speed of 6.5 m/s with a FastPrep 120 instrument (Qbiogene); tubes were placed on ice for 1.5 min between each cycle. Lysates were collected and centrifuged at 6,000 × g and 4°C for 10 min to remove any unbroken cells and cell debris. Total-protein concentrations were determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific Inc., Waltham, MA), and samples were normalized for protein content before acid phosphatase activity was measured by the release of p-nitrophenol (pNP) from p-nitrophenylphosphate (2) using an acid phosphatase assay kit (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's instructions. Measurements were expressed as acid phosphatase activity units per milligram of total protein, where 1 U of specific activity is defined as that which yields 1 nM pNP per min at 37°C and pH 4.8. Results were expressed as percentages of the phosphatase activity measured for the wild-type strain.
Macrophage culture and infection.
To generate murine bone marrow-derived macrophages (muBMMs), bone marrow cells were isolated from femurs of 6- to 10-week-old C57BL/6J female mice (Jackson Laboratories, Bar Harbor, ME), differentiated into macrophages as described previously (12), and replated in 24-well cell culture-treated plates at a density of 1 × 105 macrophages/well. Human monocyte-derived macrophages (MDMs) were generated from peripheral blood monocytes subjected to apheresis and enriched by density centrifugation using Ficoll-Paque (GE Healthcare) and by negative selection using the Dynabeads Untouched Human Monocytes kit (Invitrogen) according to the manufacturer's instructions. Mononuclear cells were seeded at a density of 4 × 105/well in RPMI medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 1% nonessential amino acids (NEAA; Invitrogen), and 50 ng/ml recombinant human macrophage colony-stimulating factor (M-CSF; PeproTech Inc., Rocky Hill, NJ). The medium was replenished on day 3 and day 6 of culture, and MDMs were used for infections on day 7. Human blood cells were collected from anonymous volunteers under a protocol reviewed and approved by the NIH Institutional Biosafety Committee. Immediately prior to infection of macrophages, a few colonies from a freshly streaked plate were resuspended in MMH broth, and the OD600 was measured to estimate bacterial numbers. Bacteria were diluted to the appropriate multiplicity of infection (MOI) in either muBMM or MDM medium, and 0.5 ml of the bacterial suspension was added to chilled cells. Macrophage infections were then performed as described previously (12) at an applied MOI of 25 to 100, depending on the analysis performed.
Determination of intracellular bacterial growth.
The intracellular growth of Schu S4 and its derivatives was monitored by determining the number of CFU recovered from lysed macrophages, as described previously (12). The number of viable intracellular bacteria per well was determined in triplicate for each time point.
Immunofluorescence microscopy.
BMMs grown on 12-mm-diameter glass coverslips in 24-well plates were infected at an MOI of 25 and processed for immunofluorescence labeling as described previously (12). The primary antibodies used were mouse anti-F. tularensis lipopolysaccharide (United States Biological, Swampscott, MA), rat anti-mouse LAMP-1 (clone 1D4B, developed by J. T. August and obtained from the Developmental Studies Hybridoma Bank [a national resource developed under the auspices of the NICHD and maintained by the Department of Biological Sciences, University of Iowa, Iowa City, IA 52242]). The secondary antibodies were Alexa Fluor 488-conjugated donkey anti-mouse and Alexa Fluor 568-conjugated donkey anti-rat antibodies (Invitrogen). To quantify the escape of Francisella from its initial phagosome, phagosomal integrity assays were performed as described previously (12). Samples were observed on a Carl Zeiss Axio Imager epifluorescence microscope equipped with a Plan-Apochromat 63× objective (numerical aperture, 1.4) for quantitative analysis or on a Carl Zeiss LSM 710 confocal laser scanning microscope for image acquisition. Confocal images of 1,024 by 1,024 pixels were acquired and assembled using Adobe Photoshop CS3.
Transmission electron microscopy.
BMMs on 12-mm-diameter Aclar coverslips were infected at an MOI of 100 and processed as described previously (12). Sections were viewed in a Hitachi H7500 transmission electron microscope at 80 kV. Images were acquired with a Hamamatsu 2,000-by-2,000-pixel bottom-mount digital camera (Advanced Microscopy Techniques, Danvers, MA) and were assembled in Adobe Photoshop CS3.
Animal infections.
Groups of 10 BALB/cJ mice, 6 to 8 weeks old (Jackson Laboratories), were infected with wild-type F. tularensis Schu S4 or the indicated mutant strains via the intranasal route for survival studies. Immediately prior to infection, a stock vial of bacteria was thawed and serially diluted in PBS to the appropriate bacterial density. Mice were anesthetized intraperitoneally with 100 μl of a solution containing 12.5 mg/ml ketamine plus 3.8 mg/ml xylazine. For intranasal infections, approximately 10 CFU in 25 μl of PBS was administered to the nares of each mouse. Actual doses were confirmed by plating the inoculum on MMH agar plates. Animals were monitored twice daily for signs of morbidity and were euthanized when moribund. All animal infections were performed at biosafety level 3 and were approved by the Rocky Mountain Laboratories Animal Care and Use Committee.
Nucleotide sequence accession numbers.
The NCBI reference sequence (RefSeq) numbers of the Francisella acp and hap genes are as follows: for U112 acpA, YP_897755; for Schu S4 acpA, YP_169276; for FSC200 acpA, ZP_02275294; for U112 acpB, YP_899174; for Schu S4 acpB, YP_169222; for FSC200 acpB, ZP_02274312; for U112 acpC, YP_898702; for Schu S4 acpC, YP_169641; for FSC200 acpC, ZP_02275790 and ZP_02275789; for U112 acpD, YP_898327; for Schu S4 acpD, YP_169785 and YP_170416; for FSC200 acpD, ZP_02274752 and ZP_02275415; for U112 hapA, YP_898596; for Schu S4 hapA, YP_170045; for FSC200 hapA, ZP_02275376; for U112 hapB, YP_897687; and for FSC200 hapB, ZP_02274253.
RESULTS
Degeneration of acid phosphatase-encoding genes in virulent strains.
To evaluate the contribution of acid phosphatases to the virulence of Schu S4, we first examined the conservation of the genetic loci encoding such enzymes in the annotated genome sequences of F. novicida strain U112, F. tularensis subsp. tularensis strains Schu S4 and FSC033, and F. tularensis subsp. holarctica strain FSC200 by using the ERGO Genome Analysis Suite (Integrated Genomics, Chicago, IL) (34). Analysis of the U112 genome identified the acid phosphatase-encoding genes acpA (FTN_0090), acpB (FTN_1556), and acpC (FTN_1061) (Fig. 1), a PAP2 superfamily phosphoesterase-encoding gene named acpD (FTN_0681) (Fig. 1), and the two histidine acid phosphatase-encoding genes hapA (FTN_0954) and hapB (FTN_0022), the latter of which shares 41% amino acid identity with the Legionella pneumophila major acid phosphatase Map (16). The acpA locus was conserved between the U112 and Schu S4 genomes (Fig. 1A), with the deduced AcpA proteins sharing 98.1% identity (see Fig. S1 in the supplemental material), while the acpA loci of FSC200 (see Fig. S1) and two other F. tularensis subsp. holarctica strains (data not shown) encoded an AcpA protein missing the N-terminal 23 amino acid residues and therefore lacking a potential Sec-dependent signal peptide identified using SignalP, version 3.0 (5). The acpB locus was highly conserved between the U112, Schu S4, and FSC200 genomes, with 99% identity between the deduced amino acid sequences (Fig. 1B; see also Fig. S1 in the supplemental material), underscoring a potentially important function of this gene. Like that of acpA, the acpC locus was highly conserved between the U112 (FTN_1061) and Schu S4 (FTT0620) genomes, with 96.4% identity at the protein level, but was disrupted into two small ORFs in FSC200 (Fig. 1C; see also Fig. S1 in the supplemental material). A similar genetic degeneration was observed in two other F. tularensis subsp. holarctica genomes (data not shown), indicating that acpC is a pseudogene in type B strains. Compared to that in U112, the hapA gene in Schu S4 (FTT1064) was truncated through an ISftu1-mediated chromosomal rearrangement (Fig. 1D; see also Fig. S1 in the supplemental material), generating, instead of a 402-amino-acid protein, a 196-amino-acid protein that lacks the C-terminal catalytic histidine (His309) residue typical of this family of histidine acid phosphatases (32, 33). Through similar rearrangements, hapA in FSC200 was also truncated to generate a 351-amino-acid residue protein. Hence, though present in virulent strains (29), hapA is likely a pseudogene. hapB (FTN_0022) was found to be intact in FSC200 but disrupted into two ORFs in the type B strain FSC022 (data not shown) and the type A strain FSC033 (Fig. 1E). Although hapB is not annotated in the Schu S4 genome, sequences with 100% identity with the FSC033 ORFs were found in the Schu S4 genome sequence, indicating that hapB is present yet disrupted in type A strains, but not absent as was previously reported (29). We found an additional, undescribed PAP2 superfamily acid phosphatase, named AcpD, encoded by the U112 locus FTN_0681 (Fig. 1F; see also Fig. S1 in the supplemental material), raising the possibility of additional acid phosphatase activity in F. novicida. However, acpD homologs in both Schu S4 and FSC200 were disrupted into two pseudogenes (FTT0778 and FTT1480 in Schu S4 [Fig. 1F]) by obvious ISftu1-mediated chromosomal rearrangements, where the two gene remnants were located in distant regions of the Schu S4 or FSC200 chromosome (Fig. 1F). Hence, though intact in F. novicida, acpD is disrupted in virulent type A and type B strains of Francisella. Taken together, this analysis highlighted a lack of conservation of acid phosphatase-encoding genes between F. novicida and the virulent F. tularensis subspecies, where most of these genes have been converted into pseudogenes in at least one virulent subspecies through genome reduction events (10). Given the previously documented roles of AcpA, AcpB, and AcpC in the phagosomal escape and virulence of F. novicida (27, 29), we nonetheless examined the roles of all three of these genes in the intracellular trafficking and virulence of Schu S4.
FIG. 1.

Comparative genetic organizations of acid phosphatase-encoding loci in F. novicida, F. tularensis subsp. tularensis, and F. tularensis subsp. holarctica. Shown are the genetic organizations of the acpA (A), acpB (B), acpC (C), hapA (D), hapB (E), and acpD (F) loci in F. novicida U112, F. tularensis subsp. tularensis Schu S4 (A through D and F) or FSC033 (E), and F. tularensis subsp. holarctica FSC200. (A) The acpA gene (shaded arrow) is highly conserved between subspecies yet lacks the first 23 codons, including a putative secretion signal peptide (small filled rectangle), in F. tularensis subsp. holarctica strains. (B) The acpB gene (shaded arrow) is highly conserved between subspecies. (C) The acpC gene (shaded arrow) is conserved between the F. novicida and F. tularensis subsp. tularensis strains but is disrupted (filled arrows) in the F. tularensis subsp. holarctica strain. (D) The hapA gene (FTN_0954) (shaded arrow) is truncated (filled arrows) by ISftu1-mediated chromosomal rearrangements in both F. tularensis subsp. tularensis and F. tularensis subsp. holarctica strains. (E) The hapB gene (FTN_0022) (shaded arrow) is intact in F. novicida and F. tularensis subsp. holarctica strains but is a pseudogene (filled arrows) in the F. tularensis subsp. tularensis strain. (F) The acpD gene is intact (FTN_0681) in the F. novicida strain but is disrupted (filled arrows) by ISftu1-mediated chromosomal rearrangements in both F. tularensis subsp. tularensis and F. tularensis subsp. holarctica strains.
Deletion of the acpABC genes in Schu S4 abolishes its acid phosphatase activity.
To address the roles of AcpA, AcpB, and AcpC in the virulence of Schu S4, we generated either single in-frame, unmarked deletions of acpA, acpB, and acpC or combinations of those deletions (Fig. 2A). As shown in Fig. 2B for the Schu S4 ΔacpA and Schu S4 ΔacpA ΔacpB ΔacpC (referred to below as ΔacpABC) mutants, all deletions were confirmed by PCR amplification of the deleted gene(s) within the proper chromosomal locus. Compared to the parental strain, none of the mutants generated showed any growth defect in culture in MMH broth (data not shown), indicating that acp deletions do not affect the ability of Schu S4 to replicate in a synthetic growth medium. Deletion of acpA decreased the total acid phosphatase activity of Schu S4 by 95% (Fig. 2C), consistent with previous reports on F. novicida (4, 27, 29), arguing that AcpA accounts for most acid phosphatase activities in Francisella. Deletion of acpB or acpC by itself did not dramatically affect acid phosphatase activity levels (Fig. 2C), suggesting that these proteins express only marginal, if any, acid phosphatase activities. In agreement with these results, the total acid phosphatase activity of the ΔacpABC triple deletion mutant was decreased to a level comparable to that of the ΔacpA mutant (Fig. 2C). All three genes were transcribed on MMH plates (46; also unpublished data), suggesting that the lack of detectable acid phosphatase activity associated with AcpB and AcpC is not due to a lack of expression. Taken together, these results demonstrate that the acp deletion mutants displayed significantly reduced acid phosphatase activity, mostly via deletion of acpA.
AcpABC are not required for the intracellular cycle of Schu S4.
To determine whether the acid phosphatases AcpA, AcpB, and AcpC play a role in the intracellular pathogenesis of Schu S4, we first compared the intracellular growth of either the Schu S4 ΔacpA or the Schu S4 ΔacpABC mutant with that of the parental Schu S4 strain in either murine bone marrow-derived macrophages (muBMMs) or human blood monocyte-derived macrophages (MDMs). Over a time course of 12 h, which typically encompasses phagosomal escape and cytosolic replication (46), numbers of viable intracellular ΔacpA and ΔacpABC mutants were indistinguishable from numbers of viable intracellular Schu S4 (Fig. 3A and B), indicating that these deletions do not affect the ability of Schu S4 to proliferate within murine or human macrophages. Under these experimental conditions, both muBMMs and MDMs were capable of controlling an attenuated strain of Francisella, since a fevR deletion mutant (7, 46) was unable to survive intracellularly (Fig. 3).
FIG. 3.
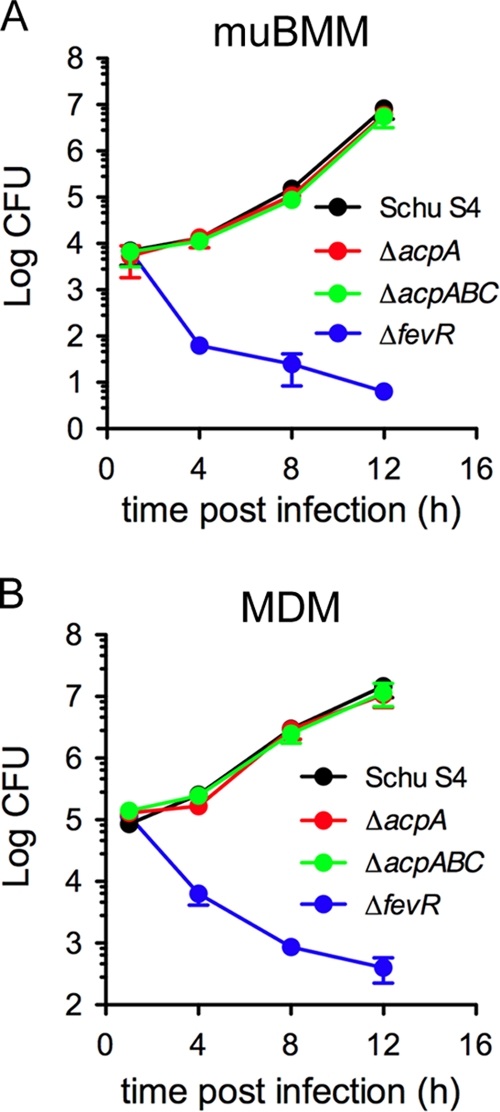
Deletion of acp genes in Schu S4 does not affect intracellular growth in murine or human macrophages. muBMMs or MDMs were infected with either the Schu S4, Schu S4 ΔacpA, Schu S4 ΔacpABC, or Schu S4 ΔfevR strain, and viable intracellular bacteria were enumerated as CFU in a gentamicin protection assay. Growth curves of wild-type and mutant Schu S4 strains in muBMMs (A) and MDMs (B) are shown. In each case, results are representative of two independent experiments performed in triplicate.
To further examine the intracellular behavior of the acp mutants, we monitored their intracellular trafficking in muBMMs by using fluorescence confocal and transmission electron microscopy. At 1 h postinfection (p.i.), neither Schu S4 nor the ΔacpA or ΔacpABC mutant was surrounded by LAMP-1-positive membranes (Fig. 4A, B, and C), suggesting they all had disrupted their phagosomal membranes. Electron microscopy showed that, like Schu S4, neither the ΔacpA nor the ΔacpABC mutant was surrounded by phagosomal membranes at this time point, demonstrating efficient phagosomal membrane disruption at the ultrastructural level (Fig. 4A, B, and C). To quantitatively evaluate phagosomal escape, muBMMs infected with either Schu S4, Schu S4 ΔacpA, Schu S4 ΔacpABC, or the phagosomal escape-deficient strain Schu S4 ΔfevR (46) were subjected to a phagosomal integrity assay. While less than 20% of intracellular ΔfevR bacteria were cytosolic during the first 4 h p.i., consistent with our previous results (46), the percentages of cytosolic ΔacpA and ΔacpABC mutants paralleled those of wild-type Schu S4 (Fig. 4G), demonstrating that these mutations did not affect the efficiency or the kinetics of phagosomal disruption and bacterial release into the cytosol. At 8 h p.i., both the ΔacpA and ΔacpABC mutants showed clear patterns of cytosolic replication similar to those of Schu S4 (Fig. 4D, E, and F), confirming intracellular proliferation. Taken together, our results clearly show a normal intracellular cycle of acid phosphatase-deficient bacteria, ruling out a role for these acid phosphatases in the intracellular pathogenesis of Schu S4.
FIG. 4.

Intracellular trafficking of Schu S4 is not affected by deletion of the acp genes. muBMMs were infected with strain Schu S4, Schu S4 ΔacpA, or Schu S4 ΔacpABC and were processed for either immunofluorescence or transmission electron microscopy. (A to F) Representative confocal fluorescence or electron micrographs of muBMMs at 1 h (A to C) or 8 h (D to F) after infection with either Schu S4 (A and D), Schu S4 ΔacpA (B and E), or Schu S4 ΔacpABC (C and F). Bacteria appear green; LAMP-1 appears red. White arrows on the confocal micrograph insets indicate bacteria that are not surrounded by LAMP-1-positive membranes. Black arrows on the electron micrograph insets indicate a lack of phagosomal membranes. Bars, 10 and 2 μm for confocal microscopy panels and insets, respectively; 0.5 and 0.2 μm for electron microscopy panels and insets, respectively. (G) Phagosomal escape kinetics of strains Schu S4, Schu S4 ΔacpA, Schu S4 ΔacpABC, and Schu S4 ΔfevR in muBMMs. Macrophages were infected with individual strains, and were processed for a phagosomal integrity assay, as described in Materials and Methods. Values are means ± standard deviations for three independent experiments.
AcpABC are not required for in vivo virulence of Schu S4.
Although AcpA, AcpB, and AcpC are not required for Schu S4 intracellular pathogenesis, there remained the possibility of a role for these phosphatases in the overall virulence of Francisella in vivo. To address this question, we infected BALB/cJ mice intranasally with either Schu S4, Schu S4 ΔacpA, or the Schu S4 ΔacpABC triple mutant and monitored their survival. In this infection model, Schu S4 mutants either defective in phagosomal escape or impaired in cytosolic proliferation fail to cause any lethality (46). Regardless of the infecting strain, mice from all groups became moribund and had to be euthanized by day 4 p.i. (Fig. 5), indicating that deletion of either acpA alone or acpA, acpB, and acpC together does not affect the virulence of Schu S4 in vivo.
FIG. 5.
Deletion of the acp genes does not affect the virulence of Schu S4. Survival curves of BALB/cJ mice infected intranasally with either Schu S4 or its isogenic ΔacpA or ΔacpABC mutant are shown. Inocula were 11 (Schu S4), 15 (Schu S4 ΔacpA), and 11 (Schu S4 ΔacpABC) CFU.
DISCUSSION
Despite the discovery of the FPI and its documented role in the phagosomal escape of Francisella species, very little is known about other molecular determinants of the intracellular pathogenesis of this bacterium. Recently, Mohapatra et al. have examined the role of the acid phosphatases AcpA, AcpB, AcpC, and Hap in pathogenesis and concluded that these proteins are required for the phagosomal escape, and hence for the intracellular growth, of F. novicida (27, 29). This contradicted a previous report of a lack of a role for AcpA in intracellular growth and virulence in the same organism (4). In our efforts to identify pathogenic determinants of the prototypical type A strain Schu S4, we sought to examine the roles played by acid phosphatase-encoding genes. Because phagosomal escape is crucial to the pathogenesis of all Francisella species, we postulated that bacterial factors required for this process are highly conserved, including acid phosphatase-encoding genes, based on their role in phagosomal disruption in F. novicida (27, 29). Here, however, we show poor conservation of these bacterial determinants between F. novicida and the virulent F. tularensis subsp. tularensis and F. tularensis subsp. holarctica strains. While acpD, hapA, and hapB are pseudogenes in F. tularensis subsp. tularensis strains, acpA is truncated at its 5′ end, and acpC, acpD, and hapA are pseudogenes, in F. tularensis subsp. holarctica strains. Degeneration of these genes in the virulent subspecies argues for a lack of selective pressure in the context of Francisella pathogenesis and hence for a nonessential role, if any. To confirm that such disrupted genes do not express any residual virulence-related functions and to exclude any possibility of reconstitution of a functional enzyme through the expression of the two pseudogene fragments (Fig. 1F; see also Fig. S1 in the supplemental material), we deleted FTT0778 (which is homologous to the 5′ portion of F. novicida acpD) in either Schu S4 or Schu S4 ΔacpABC. Regardless of the strain background, we observed no decrease in acid phosphatase activity and no defect in the infection cycle or virulence in mice (data not shown). These results confirm the nonfunctionality of acpD remnants in virulent strains and argue against a contribution of genes that have been subject to genome reduction to Francisella virulence.
We nonetheless deleted in Schu S4 the acid phosphatase-encoding genes that are most conserved (acpA, acpB, acpC) between F. novicida and most virulent F. tularensis subspecies, but the resulting mutants showed no defect in intracellular pathogenesis. Additionally, the ability of these mutants to cause lethality in mice was not impaired. Although it remains possible that acpA, acpB, and acpC, or one of these genes, play minor roles in the disease process that were not revealed in our intranasal infection model, our results indicate that these genes are not major determinants of Schu S4 virulence. Given its prominent acid phosphatase activity (Fig. 2C) (29), its demonstrated phospholipase C activity (37), and its putative secretion signal, AcpA appeared a likely candidate for a role in phagosomal membrane disruption, yet the phagosomal escape of the ΔacpA mutant of Schu S4 was unaffected. Additionally, the deletion at the 5′ end of acpA in F. tularensis subsp. holarctica strains has removed any putative secretion signal (see Fig. S1 in the supplemental material), yet this does not prevent type B strains from efficiently disrupting their early phagosome (11), excluding a yet to be demonstrated secretion of AcpA as a requirement for phagosomal degradation. Neither AcpB nor AcpC contributed in a significant manner to the total acid phosphatase activity of Schu S4, nor were they involved in phagosomal escape or virulence. While AcpA has been extensively characterized as an enzyme (4, 17, 18, 37), the classification of AcpB and AcpC as HAD family acid phosphatases is based on Pfam domain analyses, and their actual molecular functions remain to be evaluated.
Taken together, our results rule out a role of acid phosphatases in the phagosomal escape and overall pathogenesis of highly virulent F. tularensis subspecies. These findings are therefore consistent with the study of F. novicida AcpA by Baron et al. (4) but contrast with the conclusions of Mohapatra et al. (27, 29). Although one could invoke differential requirements for bacterial virulence factors between murine macrophages (our main model in this study) and human monocytes (used in the study by Mohapatra et al.) to explain these different conclusions, we did not uncover any defect of the Schu S4 acp mutants in primary human macrophages either, ruling out this possibility. While additional studies are needed to reconcile previous reports on the roles of acid phosphatases in F. novicida, our findings that Acp proteins do not play a role in the virulence of type A strains call for a reconsideration of the generalized conclusion that acid phosphatases are required for the phagosomal escape of Francisella. More generally, the assumption that findings generated using F. novicida hold true for the more virulent F. tularensis subspecies should be viewed with caution and confirmed experimentally.
Supplementary Material
Acknowledgments
We are grateful to Leigh Knodler and Audrey Chong for critical reading of the manuscript and to Katy Bosio for assistance with animal experiments.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Editor: R. P. Morrison
Footnotes
Published ahead of print on 26 October 2009.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Aragon, V., S. Kurtz, and N. P. Cianciotto. 2001. Legionella pneumophila major acid phosphatase and its role in intracellular infection. Infect. Immun. 69:177-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aragon, V., S. Kurtz, A. Flieger, B. Neumeister, and N. P. Cianciotto. 2000. Secreted enzymatic activities of wild-type and pilD-deficient Legionella pneumophila. Infect. Immun. 68:1855-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baca, O. G., M. J. Roman, R. H. Glew, R. F. Christner, J. E. Buhler, and A. S. Aragon. 1993. Acid phosphatase activity in Coxiella burnetii: a possible virulence factor. Infect. Immun. 61:4232-4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baron, G. S., T. J. Reilly, and F. E. Nano. 1999. The respiratory burst-inhibiting acid phosphatase AcpA is not essential for the intramacrophage growth or virulence of Francisella novicida. FEMS Microbiol. Lett. 176:85-90. [DOI] [PubMed] [Google Scholar]
- 5.Bendtsen, J. D., H. Nielsen, G. von Heijne, and S. Brunak. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783-795. [DOI] [PubMed] [Google Scholar]
- 6.Bönquist, L., H. Lindgren, I. Golovliov, T. Guina, and A. Sjostedt. 2008. MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect. Immun. 76:3502-3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brotcke, A., and D. M. Monack. 2008. Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect. Immun. 76:3473-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brotcke, A., D. S. Weiss, C. C. Kim, P. Chain, S. Malfatti, E. Garcia, and D. M. Monack. 2006. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun. 74:6642-6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burtnick, M., A. Bolton, P. Brett, D. Watanabe, and D. Woods. 2001. Identification of the acid phosphatase (acpA) gene homologues in pathogenic and non-pathogenic Burkholderia spp. facilitates TnphoA mutagenesis. Microbiology 147:111-120. [DOI] [PubMed] [Google Scholar]
- 10.Champion, M. D., Q. Zeng, E. B. Nix, F. E. Nano, P. Keim, C. D. Kodira, M. Borowsky, S. Young, M. Koehrsen, R. Engels, M. Pearson, C. Howarth, L. Larson, J. White, L. Alvarado, M. Forsman, S. W. Bearden, A. Sjostedt, R. Titball, S. L. Michell, B. Birren, and J. Galagan. 2009. Comparative genomic characterization of Francisella tularensis strains belonging to low and high virulence subspecies. PLoS Pathog. 5:e1000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Checroun, C., T. D. Wehrly, E. R. Fischer, S. F. Hayes, and J. Celli. 2006. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U. S. A. 103:14578-14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong, A., T. D. Wehrly, V. Nair, E. R. Fischer, J. R. Barker, K. E. Klose, and J. Celli. 2008. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity Island protein expression. Infect. Immun. 76:5488-5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clemens, D. L., B. Y. Lee, and M. A. Horwitz. 2009. Francisella tularensis phagosomal escape does not require acidification of the phagosome. Infect. Immun. 77:1757-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clemens, D. L., B. Y. Lee, and M. A. Horwitz. 2004. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72:3204-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellis, J., P. C. Oyston, M. Green, and R. W. Titball. 2002. Tularemia. Clin. Microbiol. Rev. 15:631-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felts, R. L., T. J. Reilly, M. J. Calcutt, and J. J. Tanner. 2006. Crystallization of a newly discovered histidine acid phosphatase from Francisella tularensis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62:32-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felts, R. L., T. J. Reilly, and J. J. Tanner. 2005. Crystallization of AcpA, a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. Biochim. Biophys. Acta 1752:107-110. [DOI] [PubMed] [Google Scholar]
- 18.Felts, R. L., T. J. Reilly, and J. J. Tanner. 2006. Structure of Francisella tularensis AcpA: prototype of a unique superfamily of acid phosphatases and phospholipases C. J. Biol. Chem. 281:30289-30298. [DOI] [PubMed] [Google Scholar]
- 19.Gallagher, L. A., E. Ramage, M. A. Jacobs, R. Kaul, M. Brittnacher, and C. Manoil. 2007. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc. Natl. Acad. Sci. U. S. A. 104:1009-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golovliov, I., V. Baranov, Z. Krocova, H. Kovarova, and A. Sjostedt. 2003. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 71:5940-5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall, J. D., M. D. Woolard, B. M. Gunn, R. R. Craven, S. Taft-Benz, J. A. Frelinger, and T. H. Kawula. 2008. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 76:5843-5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jungnitz, H., N. P. West, M. J. Walker, G. S. Chhatwal, and C. A. Guzman. 1998. A second two-component regulatory system of Bordetella bronchiseptica required for bacterial resistance to oxidative stress, production of acid phosphatase, and in vivo persistence. Infect. Immun. 66:4640-4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lauriano, C. M., J. R. Barker, S. S. Yoon, F. E. Nano, B. P. Arulanandam, D. J. Hassett, and K. E. Klose. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc. Natl. Acad. Sci. U. S. A. 101:4246-4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindgren, H., I. Golovliov, V. Baranov, R. K. Ernst, M. Telepnev, and A. Sjostedt. 2004. Factors affecting the escape of Francisella tularensis from the phagolysosome. J. Med. Microbiol. 53:953-958. [DOI] [PubMed] [Google Scholar]
- 25.Ludu, J. S., O. M. de Bruin, B. N. Duplantis, C. L. Schmerk, A. Y. Chou, K. L. Elkins, and F. E. Nano. 2008. The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J. Bacteriol. 190:4584-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mariathasan, S., D. S. Weiss, V. M. Dixit, and D. M. Monack. 2005. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202:1043-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohapatra, N. P., A. Balagopal, S. Soni, L. S. Schlesinger, and J. S. Gunn. 2007. AcpA is a Francisella acid phosphatase that affects intramacrophage survival and virulence. Infect. Immun. 75:390-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohapatra, N. P., S. Soni, B. L. Bell, R. Warren, R. K. Ernst, A. Muszynski, R. W. Carlson, and J. S. Gunn. 2007. Identification of an orphan response regulator required for Francisella virulence and transcription of pathogenicity island genes. Infect. Immun. 75:3305-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohapatra, N. P., S. Soni, T. J. Reilly, J. Liu, K. E. Klose, and J. S. Gunn. 2008. The combined deletion of four Francisella acid phosphatases attenuates virulence and macrophage vacuolar escape. Infect. Immun. 76:3690-3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mougous, J. D., M. E. Cuff, S. Raunser, A. Shen, M. Zhou, C. A. Gifford, A. L. Goodman, G. Joachimiak, C. L. Ordonez, S. Lory, T. Walz, A. Joachimiak, and J. J. Mekalanos. 2006. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science (New York) 312:1526-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nano, F. E., N. Zhang, S. C. Cowley, K. E. Klose, K. K. Cheung, M. J. Roberts, J. S. Ludu, G. W. Letendre, A. I. Meierovics, G. Stephens, and K. L. Elkins. 2004. A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 186:6430-6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostanin, K., E. H. Harms, P. E. Stevis, R. Kuciel, M. M. Zhou, and R. L. Van Etten. 1992. Overexpression, site-directed mutagenesis, and mechanism of Escherichia coli acid phosphatase. J. Biol. Chem. 267:22830-22836. [PubMed] [Google Scholar]
- 33.Ostanin, K., A. Saeed, and R. L. Van Etten. 1994. Heterologous expression of human prostatic acid phosphatase and site-directed mutagenesis of the enzyme active site. J. Biol. Chem. 269:8971-8978. [PubMed] [Google Scholar]
- 34.Overbeek, R., N. Larsen, T. Walunas, M. D'Souza, G. Pusch, E. Selkov, Jr., K. Liolios, V. Joukov, D. Kaznadzey, I. Anderson, A. Bhattacharyya, H. Burd, W. Gardner, P. Hanke, V. Kapatral, N. Mikhailova, O. Vasieva, A. Osterman, V. Vonstein, M. Fonstein, N. Ivanova, and N. Kyrpides. 2003. The ERGO genome analysis and discovery system. Nucleic Acids Res. 31:164-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oyston, P. C., A. Sjostedt, and R. W. Titball. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2:967-978. [DOI] [PubMed] [Google Scholar]
- 36.Pukatzki, S., A. T. Ma, D. Sturtevant, B. Krastins, D. Sarracino, W. C. Nelson, J. F. Heidelberg, and J. J. Mekalanos. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. U. S. A. 103:1528-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reilly, T. J., G. S. Baron, F. E. Nano, and M. S. Kuhlenschmidt. 1996. Characterization and sequencing of a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. J. Biol. Chem. 271:10973-10983. [DOI] [PubMed] [Google Scholar]
- 38.Remaley, A. T., R. H. Glew, D. B. Kuhns, R. E. Basford, A. S. Waggoner, L. A. Ernst, and M. Pope. 1985. Leishmania donovani: surface membrane acid phosphatase blocks neutrophil oxidative metabolite production. Exp. Parasitol. 60:331-341. [DOI] [PubMed] [Google Scholar]
- 39.Saha, A. K., J. N. Dowling, K. L. LaMarco, S. Das, A. T. Remaley, N. Olomu, M. T. Pope, and R. H. Glew. 1985. Properties of an acid phosphatase from Legionella micdadei which blocks superoxide anion production by human neutrophils. Arch. Biochem. Biophys. 243:150-160. [DOI] [PubMed] [Google Scholar]
- 40.Saleh, M. T., and J. T. Belisle. 2000. Secretion of an acid phosphatase (SapM) by Mycobacterium tuberculosis that is similar to eukaryotic acid phosphatases. J. Bacteriol. 182:6850-6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santic, M., R. Asare, I. Skrobonja, S. Jones, and Y. Abu Kwaik. 2008. Acquisition of the vATPase proton pump and phagosome acidification is essential for escape of Francisella tularensis into the macrophage cytosol. Infect. Immun. 76:2671-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santic, M., M. Molmeret, and Y. Abu Kwaik. 2005. Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell. Microbiol. 7:957-967. [DOI] [PubMed] [Google Scholar]
- 43.Santic, M., M. Molmeret, K. E. Klose, S. Jones, and Y. A. Kwaik. 2005. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell. Microbiol. 7:969-979. [DOI] [PubMed] [Google Scholar]
- 44.Schmerk, C. L., B. N. Duplantis, P. L. Howard, and F. E. Nano. 2009. A Francisella novicida pdpA mutant exhibits limited intracellular replication and remains associated with the lysosomal marker LAMP-1. Microbiology 155:1498-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmerk, C. L., B. N. Duplantis, D. Wang, R. D. Burke, A. Y. Chou, K. L. Elkins, J. S. Ludu, and F. E. Nano. 2009. Characterization of the pathogenicity island protein PdpA and its role in the virulence of Francisella novicida. Microbiology 155:1489-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wehrly, T. D., A. Chong, K. Virtaneva, D. E. Sturdevant, R. Child, J. A. Edwards, D. Brouwer, V. Nair, E. R. Fischer, L. Wicke, A. J. Curda, J. J. Kupko III, C. Martens, D. D. Crane, C. M. Bosio, S. F. Porcella, and J. Celli. 2009. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell. Microbiol. 11:1128-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weiss, D. S., A. Brotcke, T. Henry, J. J. Margolis, K. Chan, and D. M. Monack. 2007. In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U. S. A. 104:6037-6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.