

Abstract
Numerous prions (infectious proteins) have been identified in yeast that result from the conversion of soluble proteins into β-sheet-rich amyloid-like protein aggregates. Yeast prion formation is driven primarily by amino acid composition. However, yeast prion domains are generally lacking in the bulky hydrophobic residues most strongly associated with amyloid formation and are instead enriched in glutamines and asparagines. Glutamine/asparagine-rich domains are thought to be involved in both disease-related and beneficial amyloid formation. These domains are overrepresented in eukaryotic genomes, but predictive methods have not yet been developed to efficiently distinguish between prion and nonprion glutamine/asparagine-rich domains. We have developed a novel in vivo assay to quantitatively assess how composition affects prion formation. Using our results, we have defined the compositional features that promote prion formation, allowing us to accurately distinguish between glutamine/asparagine-rich domains that can form prion-like aggregates and those that cannot. Additionally, our results explain why traditional amyloid prediction algorithms fail to accurately predict amyloid formation by the glutamine/asparagine-rich yeast prion domains.
Amyloid fibers are associated with a large number of neurodegenerative diseases and systemic amyloidoses. Amyloid fibrils are rich in a cross-beta quaternary structure in which β-strands are perpendicular to the long axis of the fibril (8).
[URE3] and [PSI+] are the prion (infectious protein) forms of the Saccharomyces cerevisiae proteins Ure2 and Sup35, respectively (61). Formation of both prions involves conversion of the native proteins into an infectious, amyloid form. Ure2 and Sup35 have served as powerful model systems for examining the basis for amyloid formation and propagation. Both proteins possess a well-ordered functional domain responsible for the normal function of the protein, while a functionally and structurally separate glutamine/asparagine (Q/N)-rich intrinsically disordered domain is necessary and sufficient for prion aggregation and propagation (4, 26, 27, 52, 53). Both proteins can form multiple prion variants, which are distinguished by the efficiency of prion propagation and by the precise structure of the amyloid core (14, 54).
Five other prion proteins have also been identified in yeast: Rnq1 (13, 46), Swi1 (15), Cyc8 (33), Mca1 (30), and Mot3 (1). Numerous other proteins, including New1, contain domains that show prion activity when inserted in place of the Sup35 prion-forming domain (PFD) (1, 42). Each of these prion proteins contains a Q/N-rich PFD. Similar Q/N-rich domains are overrepresented in eukaryotic genomes (28), raising the intriguing possibility that prion-like structural conversions by Q/N-rich domains may be common in other eukaryotes. However, we currently have little ability to predict whether a given Q/N-rich domain can form prions.
A variety of algorithms have been developed to predict a peptide's propensity to form amyloid fibrils based on its amino acid sequence, including BETASCAN (6), TANGO (17), Zyggregator (51), SALSA (62), and PASTA (55). These algorithms have been successful at identifying regions prone to amyloid aggregation and predicting the effects of mutations on aggregation propensity for many amyloid-forming proteins. However, they have generally been quite ineffective for Q/N-rich amyloid domains such as the yeast PFDs. For example, using the statistical mechanics-based algorithm TANGO (17), which predicts aggregation propensity based on a peptide's physicochemical properties, Linding et al. found that the Sup35 and Ure2 PFDs both completely lack predicted β-aggregation nuclei (24). Similarly, yeast PFDs are generally lacking in the hydrophobic residues predicted by algorithms such as Zyggregator to nucleate amyloid formation.
Why are these algorithms so effective for many amyloid-forming proteins but not for yeast PFDs? For most amyloid proteins, amyloid formation is driven by short hydrophobic protein stretches, and increased hydrophobicity is correlated with an increased amyloid aggregation propensity (34). In contrast, the yeast PFDs are all highly polar domains, due largely to the high concentration of Q/N residues and the lack of hydrophobic residues. High Q/N content is clearly not a requirement for a domain to act as a prion in yeast, since neither the mammalian prion protein PrP nor the Podospora anserina prion protein HET-s is Q/N rich, yet fragments from both proteins can act as prions in yeast (49, 50). However, the significant compositional differences between the yeast PFDs and most other amyloid/prion proteins suggest that there may be two distinct classes of amyloid-forming proteins driven by different types of interactions. Specifically, Q/N residues, which are predicted to have a relatively low amyloid propensity in the context of hydrophobic amyloid domains (34), may promote amyloid formation when present at sufficiently high density. Stacking of Q/N residues to form polar zippers has been proposed to stabilize amyloid fibrils (35). Consistent with this hypothesis, mutational studies of Sup35 indicate that Q/N residues are critical for driving [PSI+] formation (12), and expanded poly-Q or poly-N tracts are sufficient to drive amyloid aggregation (36, 63). Therefore, this paper examines the sequence features that allow the polar, Q/N-rich yeast PFDs to form prions.
Mutational studies of the PFDs of Ure2 and Sup35 have shown that amino acid composition is the predominant feature driving prion formation (40, 41). Due to the unique compositional biases observed in the yeast PFDs, algorithms have been developed to identify potential PFDs based solely on amino acid composition (19, 28, 42). These algorithms are designed to produce a list of potential prion proteins that meet a specific set of criteria (such as high Q/N content) but are not able to predict the prion propensity of each member of the list or to predict the effects of mutations on prion formation. A recent study by Alberti et al. was the first to systematically test whether compositional similarity to known PFDs is sufficient to distinguish between Q/N-rich proteins that form prions and those that do not. They developed a hidden Markov model to identify domains that are compositionally similar to known PFDs and then analyzed the 100 highest-scoring Q/N-rich domains in a series of in vivo and in vitro assays (1). Remarkably, they discovered 18 proteins with prion-like activity in all assays. However, an equal number, including some of the domains with greatest compositional similarity to known PFDs, showed no prion-like activity.
This inability to distinguish between Q/N-rich proteins that form prions and those that do not might seem to suggest that amino acid composition is not an accurate predictor of prion propensity. However, an alternative explanation is that known yeast PFDs are not an ideal training set for a composition-based prediction algorithm, since yeast prions are likely not optimized for maximal prion propensity. It is unclear whether yeast prion formation is a beneficial phenomenon providing a mechanism to regulate protein activity or a detrimental phenomenon analogous to human amyloid disease. [PSI+] can increase resistance to certain stress conditions (56), but the failure to observe [PSI+] in wild yeast strains (29) argues that beneficial [PSI+] formation is at most a rare event. If yeast prions are diseases, the PFDs certainly would not be optimized for maximum prion potential. If prion formation is a beneficial event allowing for rapid conversion between active and inactive states, the prion potential of the PFD would be optimized such that the frequencies of prion formation and loss would yield the optimal balance of prion and nonprion cells (25). Thus, specific residues might be excluded from yeast PFDs either because they inhibit prion formation or because they too strongly promote prion formation; bioinformatic analysis can reveal which residues are excluded from yeast PFDs but not why they are excluded. Accurate prediction of prion propensity requires understanding which deviations from known prion-forming compositions will promote prion formation and which will inhibit.
We have therefore developed the first in vivo method to quantitatively determine the prion propensity for each amino acid in the context of a Q/N-rich PFD. As expected, we found proline and charged residues to be strongly inhibitory to prion formation; but surprisingly, despite being largely underrepresented in yeast PFDs, hydrophobic residues strongly promoted prion formation. Furthermore, although Q/N residues dominate yeast PFDs, prion propensity appears relatively insensitive to the exact number of Q/N residues. Using these data, we were able to distinguish with approximately 90% accuracy between Q/N-rich domains that can form prion-like aggregates and those that cannot. These experiments provide the first detailed insight into the compositional requirements for yeast prion formation and illuminate the different methods by which Q/N- and non-Q/N-rich amyloidogenic proteins aggregate.
MATERIALS AND METHODS
Strains and media.
Standard yeast media and methods were used, as described previously (44), except that yeast extract-peptone-dextrose (YPD) contained 0.5% yeast extract instead of the standard 1%. In all experiments, yeast were grown at 30°C. All experiments were performed with Saccharomyces cerevisiae strain 780-1D/pJ533 (47). This strain's genotype is α kar1-1 SUQ5 ade2-1 his3 leu2 trp1 ura3 sup35::KanMx [PSI+] [PIN+]; pJ533 expresses SUP35 from a URA3 plasmid as the sole copy of SUP35 in the cell.
PFD truncation mapping.
SUP35-27 deletions were generated by a two-step PCR procedure in which the regions N-terminal and C-terminal to the site of deletion were amplified in separate reactions. Products of these reactions were combined and reamplified with EDR259 (CCAAAGCTCCCATTGCTTCTG) and EDR262 (GCATCAGCACTGGTAACATTGG). To insert the final PCR products into yeast under the control of the SUP35 promoter, PCR products were cotransformed with BamHI/HindIII-cut pJ526 (cen LEU2; from Dan Masison, National Institutes of Health [41]) into yeast strain 780-1D/pJ533 and selected on synthetic complete medium lacking leucine (SC−Leu). Transformants were spotted onto 5-fluoroorotic acid (5-FOA) containing medium to select for loss of pJ533.
Creating mutant libraries.
Degenerate oligonucleotides were used to randomly mutate regions of the SUP35-27 PFD. Nucleotides 115 to 138 were mutated in library 1 and nucleotides 163 to 186 in library 2. Primers EDR1003 (GGGTTACCGTATTGGTTG GCGTAGTTGTAVNNVNNVNNVNNVNNVNNVNNVNNTTGTTGCTGTTGCCCGTATTGGTTGTTATTATAGCCGCTTCC) for library 1 and EDR1121 (GGACGTTGATACTGTTGTTGTTGTGACTGTTGACCGTTTCCVNNVNNVNNVNNVNNVNNVNNVNNACCGTATTGGTTGGCGTAGTTGTAACCTCCAGC) for library 2, made by Invitrogen, were antisense, containing degenerate segments such that the reverse complement encoded a 25% mix of each nucleotide at positions 1 and 2 of each mutated codon and a 33.3% mix of C, G, and T at the third position. The 5′ and 3′ ends of EDR1003 and EDR1121 contained regions of homology to SUP35-27. These primers were paired with EDR259 to amplify the N-terminal region of SUP35-27. In a second PCR, a primer complementary to the nondegenerate 5′ region of EDR1003 or EDR1121 (EDR1007 [CAACTACGCCAACCAATACGGTAACCC] or EDR672 [GGAAACGGTCAACAGTCACAACAACAACAGTATCAACGTCCCCAG TATAACCAGTACTACCAAGCTCAGAATAATCAACCTCAGGGTTTC], respectively) was paired with EDR262 to amplify the C-terminal side of the SUP35-27. Products of these reactions were combined and reamplified with the outer primers. The final PCR products were cotransformed with BamHI/HindIII-cut pJ526 into yeast strain 780-1D/pJ533 and selected on SD−Leu. Transformants were spotted onto 5-FOA-containing medium to select for loss of pJ533.
Screening for [PSI+] clones.
Library mutants that grew on 5-FOA were then stamped onto synthetic complete medium lacking adenine (SC−Ade) and YPD and grown for 3 to 5 days at 30°C. Only isolates that were red when grown on YPD and did not grow on SC−Ade were pooled into minilibraries (∼50 colonies each). Minilibraries were plated on SC−Ade at concentrations of 106 and 105 cells per plate and grown for 5 days at 30°C. To test curability, Ade+ colonies were grown on YPD and on YPD plus 4 mM GdHCl and then restreaked on YPD to test for loss of the Ade+ phenotype. Clones in which the Ade+ phenotype was stable and curable were sequenced.
Analysis of prion-forming libraries.
Hydrophobicities (39), α-helix propensities (22), and β-sheet propensities (48) were calculated using previously reported scales. Because neither the α-helix propensity nor β-sheet propensity scales contained values for proline, we set the proline α-helix and β-sheet propensities equal to 1 to account for the known ability of proline to disrupt α-helices and β-sheets.
Compositions of yeast prion domains.
For each yeast PFD, the odds ratio for each amino acid (ORPFD) was calculated as
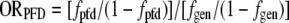 |
(1) |
where fpfd is the fraction of residues in the PFD that are the indicated amino acid and fgen is the fraction of codons within all predicted open reading frames in the yeast genome that code for the amino acid. Codon frequencies are from the Saccharomyces Genome Database (http://www.yeastgenome.org/). For the plotting of ORPFD versus the observed odds ratio (ORobs), for amino acids that were completely absent from the PFD, fpfd was set as 0.5 divided by the length of the PFD to avoid zero values in the logarithm.
Calculating prion propensity and disorder.
Proteins were scanned using an 11- or 41-amino-acid window size. Prion propensities were calculated as the sum of ln (ORobs) across the window. When clusters of prolines (defined as two or more prolines separated by no more than one intervening residue) were present within a window, only the first proline in the cluster was counted in the prion propensity calculations. For plotting, the position of the window was defined based on the central amino acid. For windows near the termini of proteins (such that there are fewer than 20 amino acids on the C- or N-terminal side of the central amino acid), the denominator was adjusted accordingly in calculating the average values. In averaging consecutive windows, for windows near the termini, windows of less than 41 amino acids were weighted in the average according to their length. Randomly selected proteins were chosen from all annotated open reading frames using the Excel software program's random number generating function. Randomly selected open reading frames are RPS17A, YGR235C, YPR1, NIP100, ERG12, ARP10, YAR003W, ECM7, YNL083W, YLR247C, YDR275W, YOR087W, SEC8, ALG6, YBR226C, ROM1, MAL33, MYO3, SFI1, and YJL039C.
Distribution of proline residues.
If proline residues were randomly distributed in a sequence, the probability that one of the two residues immediately preceding or following a given proline would be another proline is equal to 1 minus the prevalence of nonproline residues within the remainder of the sequence raised to the fourth power. The expected number of clustered prolines within each group (prion, prion-like, and nonprion) was calculated as the sum of the predicted number of clustered prolines for each member of the group. Using chi-square analysis, this predicted value was compared to the observed fraction of prolines for which one of the two subsequent residues was also a proline.
RESULTS
Mapping the Sup35-27 prion domain.
Randomizing the order of the amino acids in the Sup35 PFD while maintaining amino acid composition does not prevent [PSI+] formation (41). We used one of these scrambled versions of Sup35, Sup35-27, as a template for mutagenesis. Sup35-27 was chosen for three reasons. First, wild-type Sup35 very rarely forms prions without overexpression, making it difficult to isolate prion-forming clones upon mutagenesis. Sup35-27 forms prions de novo with greater efficiency than wild-type Sup35, allowing for isolation of a broader range of prion-forming clones. Second, any specific prion-promoting primary sequence elements or any binding sites within the PFD for interacting proteins were likely disrupted by randomization, simplifying interpretation of the results of our library screen. Finally, solid-state nuclear magnetic resonance suggests that Sup35-27 and wild-type Sup35 form fibrils that are structurally similar (45).
To identify ideal regions of the Sup35-27 PFD to target for mutagenesis, we mapped the prion-promoting regions of the PFD through deletion analysis. Prion formation was detected by monitoring nonsense suppression of the ade2-1 allele (11). ade2-1 mutants are unable to grow without adenine and form red colonies when grown in the presence of limiting adenine. Sup35 is a translation termination factor. [PSI+] formation inactivates Sup35, resulting in increased read-through of stop codons (23), allowing ade2-1 [PSI+] cells to grow without adenine and form white colonies on limiting adenine.
SUP35-27 carrying various deletions was expressed as the sole copy of SUP35 in the cell, and Ade+ colony formation was monitored. To confirm that Ade+ colony formation was a result of [PSI+] formation, we tested individual Ade+ colonies to determine whether the Ade+ phenotype was curable by guanidine. Growth on medium containing low concentrations of guanidine cures [PSI+] (57) by inhibiting Hsp104p (18, 20). We found that 20 amino acids could be removed from either end of the PFD while retaining [PSI+] formation (Fig. 1). Deletion analysis of the PFD showed various levels of importance for different regions within the prion core (Fig. 1B), with the region of amino acids 31 to 50 being particularly sensitive to deletion.
FIG. 1.

Deletion mapping of the Sup35-27p PFD. (A) Sequence of Sup35-27, with regions mutated in libraries 1 and 2 shown in bold italics. (B) Ten-amino-acid segments were deleted from the Sup35-27p PFD. Deletion mutants were expressed as the sole copy of SUP35 in the cell. All mutants produced stable curable prions. “+++” indicates that more than 4 Ade+ colonies per 106 cells were detected; “++” indicates 1 to 4 Ade+ colonies per 106 cells; “+” indicates fewer than 1 Ade+ colony per 106 cells.
Random mutagenesis of SUP35-27.
Based on our deletion data, we targeted amino acids 31 to 50 of Sup35-27 for random mutagenesis. In preliminary experiments, we tested the optimal size of the mutated region. Mutagenesis of 12 amino acids almost entirely eliminated prion formation, while mutagenesis of either four or eight amino acids still allowed an easily detectable level of prion formation (data not shown). Because a larger region of mutagenesis would provide more data and increase the stringency of selection, eight amino acids were mutated in all subsequent experiments. Amino acids 39 to 46 of the PFD were targeted for random mutagenesis because these residues lie within the region that seems critical for prion formation and because the composition of this region is fairly representative of the Sup35 PFD.
We used an oligonucleotide-based mutagenesis approach. An oligonucleotide was designed that annealed to the regions flanking codons 39 to 46 but in which the codons 39 to 46 were replaced with the sequence (NNB)8, where N represents any of the four nucleotides and B represents any nucleotide except adenine. Excluding adenine from the final position prevents insertion of two of the three stop codons without excluding any amino acids. This oligonucleotide was used to build a library of randomly mutated versions of SUP35-27. The library was transformed into yeast cells in which the sole copy of SUP35 was expressed from a plasmid. Using plasmid shuffling, wild-type SUP35 was replaced with the random library.
To remove any Sup35 mutants that might result in nonsense suppression, each clone was screened for Sup35 activity using the ade2-1 allele. All library clones were spotted onto medium containing limiting adenine and onto medium lacking adenine. Colonies that grew red on limiting adenine and did not grow without adenine were pooled. To prevent rare strong prion-forming clones from dominating selection, clones were pooled into minilibraries consisting of approximately 50 clones. With this size of minilibrary, we were able to isolate a single prion-forming clone from about half of the minilibraries. SUP35 was sequenced from individual library clones prior to prion selection to generate a naive library data set (Table 1).
TABLE 1.
Mutated sequences from selected and unselected libraries
| Library 1a |
Library 2b |
||
|---|---|---|---|
| [PSI+] isolate | Naive library | [PSI+] isolate | Naive library |
| VNIFPYYN | TDPWVPHP | FANHAHWV | SVSDHTNP |
| VTSGSYNT | NPEVPNAN | GTTYAPLF | KGRVSGPE |
| ASNIVMNC | THHSHTLP | WNAFSTYS | ATSPVPRH |
| AHTTNMIV | YLPFMDTP | HTVHHIYP | YEYSPLQH |
| YNCSVNML | PPIVKPRT | LNTFPHSY | TMTDLPYL |
| FSIYMPYK | VDDRHMFS | DIMTNNAE | ESILWASQ |
| LLVHSNAI | CKSVCNFD | SQDYSSYD | FTRAKSRT |
| WGARQFNI | GISTRSQE | CINTGLWL | TTSYHPEL |
| VTTDILAM | VSLSKNRL | HLHMSMLS | VAHCRHPL |
| RRDYLTRF | LRDPDTCS | DRHYFAGS | SSTLLDPK |
| STVICGVI | RKATDLFP | GGPIFNTK | IETHFTLS |
| IHFWPRAP | TAYVRHID | SFMAVETR | APHGLGPT |
| HSNVSVIH | DRYKGKPH | TWDGIGYR | RCSDSQGV |
| TWAPIMVY | DPNAALVF | SPPFETSP | VHHDPVST |
| MFQHGIGV | HIHPLFIH | GVNTHTSY | HPIMSSLS |
| TRIWNFSG | TLARRDPP | SIHMRVSS | LGPVHYRN |
| YHSVEFRI | PNASGIHY | HNDRTAFM | SMHNGTHR |
| TTVNHHFN | ADSASNAS | PQNQTWAD | DGPTYDWT |
| GSLSLQYF | NGPAYPLA | PDYFFHPT | PYKAATRN |
| IFDIANHS | SVNPALYR | HVPSPAHQ | PTYNDPST |
| LQPCYCSR | SGVSTAVR | DSDHHFWP | LSQSYVQE |
| MLSSNFIH | LNRITLRN | TSNTIIRA | YDSGTPPK |
| SSGPLNFI | IVPRNVNC | DCLGYPGL | SQQRFNPT |
| CLSPAECR | NISPFSKD | SMHNGTHR | HRDNCRTR |
| QFVARVFR | MTQNPHIF | ESILWASQ | PPQAVYPP |
| LKSVITWN | LSARPLGH | PRLTNHSS | QHASGRDG |
| SVHVNSTS | LGNPTFHY | FWMQRNSC | QTRFYGIH |
| AQDSHPDI | SFSYVTFP | QTTTAIHA | |
| NNPQYLFK | CQINWRTA | PHEAVSSC | |
| DERPWCPE | GPPFPGQN | RRHYAPSI | |
| GPTMNNRD | VASWASVG | KYMYHANM | |
| THRHNKHR | YREGDNLW | LADSNTPR | |
| KGSPSTPT | HTLVFNDR | SLAAPRDN | |
| EAPSKSAQ | FWIDGSAD | ||
| RPERRSNP | DRHYFAGS | ||
| ICWHTEPY | IRTHMSSK | ||
| CIKHINSI | ARNMTRYL | ||
| PVPSSSQP | RAYDILPV | ||
| GANSAITN | NEDPGTDT | ||
| SHLWRRNR | SRSIRYDN | ||
| DSHTGTPR | SQDYSSYD | ||
| STVPPPHH | |||
| VNCARGTA | |||
| QVASQNGR | |||
| SSNKFMHT | |||
| GFTKALPG | |||
| ALSSRQWS | |||
| IDKNLMSH | |||
| CFLRSYMG | |||
| VALIPKTA | |||
| HNLANHSH | |||
| KMTTNTKH | |||
Library 1, mutated amino acids 39 to 46 of the Sup35-27 PFD.
Library 2, mutated amino acids 55 to 62 of the Sup35-27 PFD.
Minilibraries were plated on medium lacking adenine to select for [PSI+] formation. To distinguish Ade+ cells resulting from [PSI+] formation from those resulting from DNA mutation, we tested individual Ade+ colonies to determine whether the Ade+ phenotype was curable by guanidine. Cells were grown on YPD with and without guanidine and then restreaked on YPD to test for loss of the Ade+ phenotype (data not shown). SUP35 was sequenced from cells that stably maintained the Ade+ phenotype on YPD but lost it after growth on YPD plus guanidine. We isolated 27 such stable prion isolates from an initial library of 3,016 clones (Table 1).
Compositional biases among the prion-forming isolates.
For each amino acid, the observed odds ratio (ORobs), representing the degree of over/underrepresentation of the amino acid within the prion-forming isolates, was determined (Table 2). ORobs was defined as
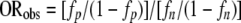 |
(2) |
where fp is the per-residue frequency of the amino acid among the prion-forming isolates and fn is the per-residue frequency of the amino acid among the naive library.
TABLE 2.
Library 1 amino acid representation
| Amino acid(s) | Frequencya |
Odds ratiob | ln (ORobs) | P valuec | |
|---|---|---|---|---|---|
| Selected [PSI+] library | Unselected naive library | ||||
| Phenylalanine (F) | 0.075 | 0.032 | 2.31 | 0.84 | 0.040 |
| Isoleucine (I) | 0.102 | 0.045 | 2.26 | 0.81 | 0.015 |
| Valine (V) | 0.102 | 0.045 | 2.26 | 0.81 | 0.015 |
| Tyrosine (Y) | 0.054 | 0.025 | 2.18 | 0.78 | 0.099 |
| Methionine (M) | 0.038 | 0.020 | 1.96 | 0.67 | 0.19 |
| Tryptophan (W) | 0.024 | 0.012 | 1.95 | 0.67 | 0.32 |
| Cysteine (C) | 0.033 | 0.022 | 1.52 | 0.42 | 0.43 |
| Serine (S) | 0.125 | 0.109 | 1.14 | 0.13 | 0.68 |
| Asparagine (N) | 0.096 | 0.089 | 1.08 | 0.080 | 0.88 |
| Glutamine (Q) | 0.024 | 0.022 | 1.07 | 0.069 | 1.00 |
| Glycine (G) | 0.038 | 0.040 | 0.96 | −0.039 | 1.00 |
| Leucine (L) | 0.059 | 0.061 | 0.96 | −0.040 | 1.00 |
| Threonine (T) | 0.069 | 0.078 | 0.89 | −0.12 | 0.75 |
| Histidine (H) | 0.059 | 0.078 | 0.76 | −0.28 | 0.50 |
| Alanine (A) | 0.042 | 0.072 | 0.67 | −0.40 | 0.38 |
| Arganine (R) | 0.054 | 0.081 | 0.67 | −0.41 | 0.31 |
| Glutamic acid (E) | 0.009 | 0.017 | 0.55 | −0.61 | 0.51 |
| Proline (P) | 0.038 | 0.127 | 0.30 | −1.20d | 0.002 |
| Aspartic acid (D) | 0.014 | 0.051 | 0.28 | −1.28 | 0.041 |
| Lysine (K) | 0.009 | 0.045 | 0.21 | −1.58 | 0.028 |
| Groups | |||||
| Aromatic (FWY) | 0.144 | 0.067 | 2.32 | 0.002 | |
| Hydrophobic (FILMV) | 0.347 | 0.195 | 2.20 | 3.0 × 10−05 | |
| Charged (DEKR) | 0.083 | 0.183 | 0.41 | 8.8 × 10−04 | |
| Positive (KR) | 0.060 | 0.118 | 0.48 | 0.024 | |
| Negative (DE) | 0.023 | 0.065 | 0.34 | 0.034 | |
| Polar (NQHST) | 0.343 | 0.346 | 0.98 | 0.92 | |
| Q/N | 0.111 | 0.103 | 1.08 | 0.79 | |
[PSI+] values represent the frequency of occurrence of the amino acid among the prion-forming isolates; naive values represent the frequency of occurrence of the amino acid among the unselected clones.
Odds ratios (ORobs) were calculated using equation 2.
P value is based on the 2-tailed Fisher exact probability test.
When multiple consecutive prolines are separated by less than one residue, a value of zero is used for ln (ORobs) for each proline after the first in the cluster.
A statistically significant (P < 0.05) overrepresentation of Phe, Ile, and Val was seen among the prion-forming isolates, and a statistically significant bias was seen against Asp, Lys, and Pro (Table 2). Other, more subtle biases were seen that were not statistically significant due to the limits of sample size. Grouping of similar amino acids allows detection of more subtle biases by effectively increasing the sample size. We observed a strong bias in favor of nonpolar amino acids and aromatic amino acids among the prion-forming library, while charged residues were underrepresented (Table 2). Surprisingly, Q/N residues were not statistically significantly overrepresented.
Yeast PFDs are biased toward the most amyloidogenic disorder-promoting residues.
Consistent with our experimental data, charged residues are strongly underrepresented in yeast PFDs; however, in contrast with our data, hydrophobic residues are also strongly underrepresented and Q/N residues are highly overrepresented in yeast PFDs (19, 42). Consequently, there is almost no correlation between the amino acids that most strongly promote prion formation and the compositions of the Ure2, Sup35, and Rnq1 PFDs (Fig. 2A to C). A similar lack of correlation is seen for the other newly discovered PFDs (Mot3, Mca1, Swi1, and Cyc8; data not shown), although the exact boundaries of these PFDs have not been clearly defined, complicating their analysis.
FIG. 2.

The Ure2, Sup35, and Rnq1 PFDs are biased toward amyloidogenic disorder-promoting residues. (A to C) Relationship between the degree to which an amino acid promotes prion formation and the amino acid's prevalence within yeast PFDs. ln (ORobs) (from Table 2) was plotted versus ln (ORPFD) (as calculated in equation 1) for the prion domain from Ure2 (A), Sup35 (B), or Rnq1 (C). (D to F) Analysis only of the disorder-prone amino acids Lys, Pro, Gly, Arg, Asn, Gln, Ser, Glu, and Asp.
Intrinsic disorder seems to explain the disconnect between the residues that promote prion formation and those that are actually present in PFDs. A key feature of the yeast PFDs is that they are intrinsically disordered (37, 43). When we consider only those residues most strongly associated with intrinsic disorder—Lys, Pro, Gly, Arg, Asn, Gln, Ser, Glu, and Asp (58)—there is excellent correlation between our experimentally determined prion propensities and the compositions of the yeast PFDs (Fig. 2D to F). These disorder-promoting residues are highly overrepresented in the Sup35, Rnq1, and Ure2 PFDs (P < 0.0001 for each PFD), accounting for 75%, 76%, and 79% of the amino acids in the PFDs, respectively, and are similarly overrepresented in all other known yeast PFDs (P < 0.01 for each PFD). Thus, the yeast PFDs are biased toward residues that balance disorder propensity and prion propensity.
Characteristics that promote prion formation.
The aggregation propensity for mutants of various non-Q/N-rich amyloidogenic polypeptides is positively correlated with hydrophobicity and β-sheet propensity and negatively correlated with charge and α-helix propensity (34). Consistent with these predictions, hydrophobicity (39) and β-sheet propensity (48) were significantly greater among the prion-forming clones than among the naive library (for the β-sheet propensity scale, lower values represent increased β-sheet propensity), and the absolute value of the net charge was significantly lower (Table 3). Surprisingly, α-helix propensity (22) was modestly greater among the prion-forming clones. However, this can be attributed almost entirely to a bias against prolines among the prion-forming clones; when prolines are excluded from the calculation, the average per-residue α-helix propensity is statistically indistinguishable between the naive and prion-forming libraries.
TABLE 3.
Physical properties of prion-forming isolates from library 1
| Property | Mean value ± SEM for naive library | Mean value ± SEM for prion-forming isolates | P value |
|---|---|---|---|
| Hydrophobicitya | −4.30 ± 0.72 | 0.02 ± 0.75 | 1.8 × 10−4 |
| β-Sheet propensityb | 3.18 ± 0.10 | 2.34 ± 0.09 | 3.5 × 10−7 |
| α-Helix propensityc | 1.35 ± 0.17 | 0.73 ± 0.18 | 0.028 |
| Charged | 0.88 ± 0.11 | 0.44 ± 0.12 | 0.016 |
From reference 39. Higher values represent greater hydrophobicities. For each naive or prion-forming isolate, the sum of the hydrophobicities of each residue within the mutagenized region was calculated. Data are the average sum per construct for the respective libraries. Standard errors are indicated.
From reference 48. Higher values represent lower β-sheet propensities.
From reference 22. Higher values represent lower α-helix propensities.
The absolute value of the net charge of the mutated region.
We examined the degree to which over/underrepresentation of each amino acid among the prion-forming isolates could be explained based on the physical properties of the amino acid. For each amino acid, the natural log of the amino acid's odds ratio was plotted as a function of various physical properties. Hydrophobicity and β-sheet propensity were both positively correlated with ln (ORobs) (Fig. 3A and B). In contrast, no significant correlation was seen between an amino acid's α-helix propensity and its odds ratio (Fig. 3C).
FIG. 3.

Relationship between the properties of an amino acid and its prevalence among library 1 prion-forming isolates. ln (ORobs) for each amino acid in library 1 (from Table 2) plotted versus hydrophobicity (A), β-sheet propensity (B) (lower values represent greater β-sheet propensities), α-helix propensity (C) (lower values represent greater α-helix propensities), or ln (ORest) (D). P values were calculated by Spearman's rank correlation. Error bars represent 95% confidence intervals.
Assuming that the effects of hydrophobicity and β-sheet propensity are additive, we combined these properties for each amino acid to predict an estimated odds ratio (ORest):
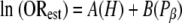 |
(3) |
where H and Pβ are the hydrophobicity and β-sheet propensity of the amino acid, respectively. As a starting estimate for A and B, we used the slopes of Fig. 3A and B (0.25 and −2.26, respectively). This function was used to calculate the ORest for each amino acid. The estimated odds ratio showed a strong correlation with the observed odds ratio (r2 = 0.74) (Fig. 3D). The observed slope was 0.71; it is not surprising that it would be less than 1, since hydrophobicity and β-sheet propensity are not truly independent. Based on the curve fit, A and B in equation (3) were modified, yielding the following equation, which predicts the prion propensity of each amino acid within experimental error (Fig. 3D):
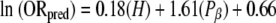 |
(4) |
Similar, but weaker, biases are seen at a second position.
To determine whether different regions of the PFD show different amino acid biases, we generated a second library (library 2) targeted to amino acids 55 to 62—a region that appears to be less critical for [PSI+] formation based on our deletion mapping. Using the same methods as for library 1, we screened 1,033 clones and found 33 capable of forming prions—a success rate of 3.19%, versus 0.895% for library 1. This nearly 4-fold increase in the prion formation rate highlights the lesser importance of amino acids 55 to 62 for prion formation.
Both individual amino acids and groups of amino acids show general biases in library 2 (Table 4) similar to those in library 1 (Table 2). There was a strong correlation between the odds ratios for each amino acid for library 1 and library 2 (Fig. 4A). This is even more apparent when amino acids are considered in groups (hydrophobic, charged, polar, and aromatic), where the correlation plot of the odds ratios for the two libraries has an r2 value of 0.98 (Fig. 4B). For both plots, the slope is approximately 0.65, indicating that although similar amino acids are selected for among the prion-forming clones in the two libraries, the strength of selection is stronger for library 1 than for library 2.
TABLE 4.
Library 2 amino acid representation
| Amino acid(s) | Frequencya |
Odds ratiob | P valuec | |
|---|---|---|---|---|
| Selected [PSI+] library | Unselected naive library | |||
| Tryptophan (W) | 0.042 | 0.009 | 4.71 | 0.013 |
| Phenylalanine (F) | 0.064 | 0.021 | 3.16 | 0.011 |
| Asparagine (N) | 0.068 | 0.040 | 1.77 | 0.14 |
| Methionine (M) | 0.030 | 0.021 | 1.43 | 0.60 |
| Glycine (G) | 0.057 | 0.046 | 1.26 | 0.58 |
| Isoleucine (I) | 0.038 | 0.030 | 1.25 | 0.65 |
| Cysteine (C) | 0.015 | 0.012 | 1.25 | 1.00 |
| Histidine (H) | 0.080 | 0.070 | 1.15 | 0.75 |
| Valine (V) | 0.038 | 0.037 | 1.04 | 1.00 |
| Glutamic acid (E) | 0.023 | 0.024 | 0.93 | 1.00 |
| Serine (S) | 0.110 | 0.119 | 0.91 | 0.80 |
| Threonine (T) | 0.087 | 0.095 | 0.91 | 0.78 |
| Alanine (A) | 0.057 | 0.064 | 0.88 | 0.73 |
| Leucine (L) | 0.045 | 0.052 | 0.87 | 0.85 |
| Glutamine (Q) | 0.030 | 0.037 | 0.82 | 0.82 |
| Tyrosine (Y) | 0.049 | 0.064 | 0.76 | 0.48 |
| Proline (P) | 0.072 | 0.095 | 0.74 | 0.37 |
| Aspartic acid (D) | 0.045 | 0.067 | 0.66 | 0.29 |
| Arginine (R) | 0.045 | 0.076 | 0.58 | 0.17 |
| Lysine (K) | 0.004 | 0.021 | 0.17 | 0.081 |
| Groups | ||||
| Aromatic (FWY) | 0.155 | 0.095 | 1.76 | 0.031 |
| Hydrophobic (FILMV) | 0.216 | 0.162 | 1.43 | 0.11 |
| Charged (DEKR) | 0.117 | 0.189 | 0.57 | 0.023 |
| Positive (KR) | 0.049 | 0.098 | 0.48 | 0.029 |
| Negative (DE) | 0.068 | 0.091 | 0.73 | 0.36 |
| Polar (NQHST) | 0.375 | 0.360 | 1.07 | 0.73 |
| Q/N | 0.098 | 0.076 | 1.32 | 0.38 |
The [PSI+] value represents the frequency of occurrence of the amino acid among the prion-forming isolates; the naive value represents the frequency of occurrence of the amino acid among the unselected clones.
Odds ratios were calculated using equation 2.
P value is based on the 2-tailed Fisher exact probability test.
FIG. 4.

Library 2 shows biases similar to but weaker than those of library 1. ln (ORobs) from library 1 was plotted versus ln (ORobs) from library 2 for each amino acid (A) or for groups of amino acids (B). Hydrophobic residues were defined as Phe, Ile, Leu, Met, and Val. Polar amino acids were defined as Ser, Thr, His, Gln, and Asn. Charged amino acids are Asp, Glu, Lys, and Arg. Aromatic amino acids were defined as Trp, Tyr, and Phe. Error bars represent 95% confidence intervals.
Identification of regions sensitive to mutagenesis.
We hypothesized that the differential sensitivity to mutation seen in the regions targeted for mutagenesis in libraries 1 and 2 could be explained by differences in the prion propensity of the two regions. We scanned the Sup35-27 PFD using a window size of 11 amino acids. For each window, we calculated the predicted prion potential as the sum of the ln (ORobs) (Fig. 5A), based on the experimentally obtained values from library 1 (Table 2). These plots predict the region targeted in library 1 to have relatively high prion propensity and the region targeted in library 2 to have low prion propensity. These results are consistent with the greater sensitivity to deletion (Fig. 1) and stringency of selection (Fig. 4) seen for library 1 than for library 2.
FIG. 5.
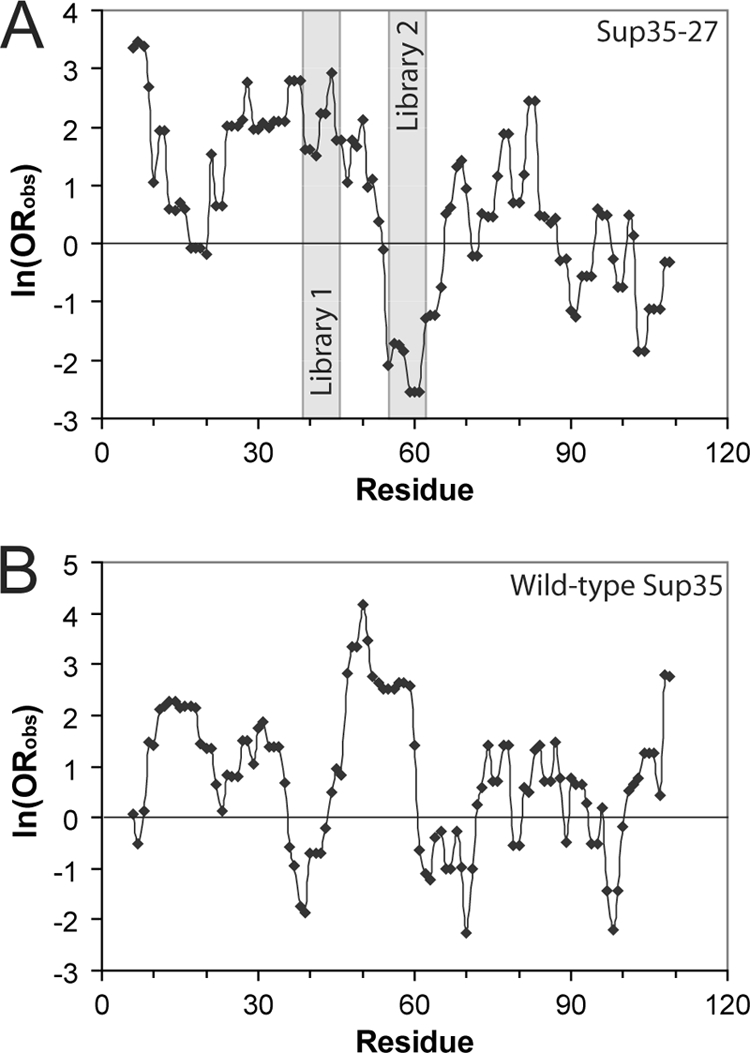
Predicted prion-prone regions. The PFD of Sup35-27 (A) or wild-type Sup35 (B) was scanned using an 11-amino-acid window size. At each position within the prion domains, the sum of ln (ORobs) for the indicated amino acid and the five amino acids on either side was calculated to determine the prion propensity of the window. Regions mutated in libraries 1 and 2 are indicated.
Similar analysis of the wild-type Sup35 PFD reveals two peaks in prion potential, spanning amino acids 8 to 35 and 44 to 61 (Fig. 5B). This nicely coincides with analysis of Sup35 showing that (i) mutations that block [PSI+] propagation specifically localize to amino acids 8 to 34 of the PFD (12); (ii) the amyloid core, as defined by hydrogen-deuterium exchange, spans either the first 40 or first 70 amino acids of the PFD, depending on the structural variant analyzed (54); (iii) the minimal fragment required to efficiently induce [PSI+] formation is amino acids 1 to 64 (32); and (iv) depending on the prion variant, amino acids 7 to 21, 9 to 37, or 5 to 52 are critical to the integrity of the Sup35 amyloid core (7).
Predicting prion propensity based on composition.
A key question is whether results from mutagenesis of small regions can be extrapolated to predict prion formation by larger PFDs. Scanning of 20 randomly selected non-Q/N-rich proteins using an 11-amino-acid window size (Fig. 6A, red circles) shows that regions with prion propensity equal to or greater than that of the Sup35 and Ure2 PFDs (Fig. 6A, blue circles) are common. Although many of these regions are predicted using the FoldIndex software program (38) to be natively folded (in contrast to the intrinsically disordered Sup35 and Ure2 PFDs), within these randomly selected proteins are disordered regions with a prion propensity comparable to that of the Sup35 and Ure2 PFDs (Fig. 6A). Therefore, scanning with an 11-amino-acid window size did not allow effective identification of these PFDs.
FIG. 6.

Predicting prion propensity for Q/N-rich domains. (A and B) Prion propensity and FoldIndex order predictions for 20 randomly selected open reading frames (red) (see Materials and Methods for gene names) and the PFDs of Sup35p and Ure2p (blue). Proteins/PFDs were scanned using an 11-amino-acid (A) or 41-amino-acid (B) window size. For each window, the predicted prion propensity (calculated as the average ln (ORobs) across the window) versus the predicted FoldIndex order propensity (where negative values are associated with disorder) was plotted. (C and D) Prion propensity and order prediction for Q/N-rich domains with prion-like activity (circles), for Q/N-rich domains shown by Alberti et al. (1) to lack prion-like activity in four prion assays (shaded triangles), for the HET-s PFD (blue square), and for human PrP (yellow square). Q/N-rich domains with prion-like activity include both known PFDs (shaded circles) and domains shown by Alberti et al. to have prion-like activity in four prion assays (circles with red slashes). For each potential PFD, the average prion propensity and average disorder are plotted for the 41 consecutive 41-amino-acid windows with maximum average predicted prion propensity (C) or the 11-amino-acid window with maximum prion propensity (D). The region of the graph identified as prion prone in panel C is shaded.
We hypothesized that yeast PFDs would be characterized by extended regions of disorder and a high prion propensity. Therefore, to reduce noise in scanning of entire proteins, we expanded our window size to 41 amino acids, which roughly correlates with the minimal fragment required to induce yeast prion formation (41). When the same 20 randomly selected proteins were scanned using a 41-amino-acid window size, regions of high prion propensity were consistently predicted to have a high FoldIndex order propensity; by contrast, the Sup35 and Ure2 PFDs are predicted to have a positive prion propensity and a negative FoldIndex (Fig. 6B). Thus, these PFDs are unique in having extended segments that are both prion prone and intrinsically disordered.
To determine whether a combination of prion propensity and disorder could be used to distinguish between prion-forming and non-prion-forming Q/N-rich domains, we utilized the massive data set generated by Alberti et al. (1). They tested the 100 proteins with greatest compositional similarity to the Sup35, Ure2, Rnq1, and New1p PFDs (Mot3, Cyc8, Mca1, and Swi1 had not yet been published) in four different assays for prion-like activity. All of these domains were highly enriched in Q/N residues. Eighteen proteins showed prion-like activity in all assays (including four known prion proteins; Mot3, Cyc8, and Mca1 scored positive only in a subset of the tests), while 18 did not show activity in any of the assays. We scanned each of these 36 potential PFDs, as well as each of the known PFDs, using a 41-amino-acid window size, calculating the average FoldIndex order propensity and the prion propensity, with one modification. In domains with prion-like activity, when proline residues are present, they disproportionately occur in clusters; in contrast, such clusters are underrepresented in Q/N-rich domains that lack prion-like activity (Table 5). Likewise, Alberti et al. reported that the spacing of proline residues affected the prion propensity, with more-dispersed spacing correlating with a decreased prion propensity (1). This makes sense; because prolines are known β-sheet breakers (9, 10), a cluster of prolines will disrupt the potential for β-sheet formation only at a single location, while the same number of proline residues dispersed throughout a sequence would result in multiple separate locations where β-strands could be disrupted. Therefore, multiple consecutive prolines were treated as a single proline in our prion propensity calculations.
TABLE 5.
Clustering of prolines in prion-forming Q/N-rich domains
| Group | No. of prolines in clustersa |
P valuee | |
|---|---|---|---|
| Predictedb | Observed | ||
| Prion-like domainsc | 50.8 | 68 | 0.0054 |
| Non-prion Q/N domainsd | 39.0 | 25 | 0.0073 |
That is, prolines for which one of the two residues following the proline or one of the two residues preceding it is also a proline.
Predictions are based on the total number of proline residues within each potential PFD, assuming that the proline residues are randomly distributed.
Includes both the 18 Q/N-rich domains shown by Alberti et al. to have prion-like activity in four different assays (1) and the other known yeast PFDs (Mot3, Cyc8, and Mca1).
The 18 Q/N-rich domains shown by Alberti et al. to lack prion-like activity in all four assays (1).
P value is calculated by chi-square analysis.
The profiles of the domains found to form prion-like aggregates were generally different from those that did not show prion activity. All of the known PFDs except that of Cyc8 have extended regions that are predicted to be both disordered and prion prone (Fig. 7A, blue circles), while similar regions are extremely rare in the nonprion domains (Fig. 7A, red circles). Similarly, each of the domains shown by Alberti et al. to have prion-like activity in all four assays (1) had multiple consecutive windows that were both disordered and prion prone (Fig. 7B); by contrast, such regions are rare in peptides that lack prion activity (Fig. 7C).
FIG. 7.

Prion propensity maps. (A) Scanning of known yeast prion proteins. Each of the yeast prion proteins was scanned using a 41-amino-acid window size, calculating for each window the average FoldIndex order propensity and prion propensity. Prion propensities were calculated based on the average ln (ORobs) for each amino acid in the window. The prion domains of each protein are shown in blue and the nonprion domains in red. (B) Scan of Q/N-rich domains tested by Alberti et al. that showed prion-like activity in all assays. (C) Scan of Q/N-rich domains tested by Alberti et al. that lacked prion-like activity in all assays. (D) Scan of the HET-s PFD and the human prion protein PrP.
To determine whether the presence of consecutive prion-prone windows could be used to distinguish between prion-forming and non-prion-forming Q/N domains, we identified for each peptide the 41 consecutive 41-amino-acid windows that had the maximum average predicted prion propensity. By averaging 41 consecutive windows, we are effectively calculating prion propensity for 81 consecutive amino acids (40 on each side of a central residue); however, because the central residue is present in each 41-amino-acid window while outer residues are present only in a subset of the windows, the weighting of each residue is inversely proportional to its distance from the central residue. Therefore, this method incorporates the idea that the sequence requirements for prion formation are more flexible further from the core of the PFD. When the prion propensity for the optimal region of each peptide was plotted versus the average FoldIndex value, a clear difference was seen between those peptides that showed prion-like activity and those that did not (Fig. 6C). If the criterion for a PFD is defined as a region of negative FoldIndex order propensity that has an average prion propensity greater than 0.05 (the shaded area in Fig. 6C), 17 of the 18 peptides lacking prion-like activity (shaded triangles) are properly scored as nonprion; in contrast, 16 of 18 domains shown by Alberti et al. to have prion-like activity in all four assays (circles with slashes through them) and six of the seven known yeast PFDs (shaded circles) are correctly scored as prion positive. Additionally, none of the randomly selected proteins from Fig. 6A had any regions that score as prion prone by these criteria. Although the mammalian prion protein PrP does contain a short region that has negative FoldIndex order propensity and positive predicted prion propensity spanning windows centered on residues 143 to 174 (Fig. 7D), neither PrP nor the Podospora anserina prion protein HET-s scores as prion prone by our criteria (Fig. 6C). This is not surprising, since neither is particularly Q/N rich and therefore they are likely to be more accurately predicted by algorithms designed for non-Q/N-rich amyloid proteins.
When potential PFDs were scanned using an 11-amino-acid window size, with the window of maximum prion propensity plotted for each protein, there is considerable overlap between those proteins that showed prion-like activity and those that did not (Fig. 6D). Therefore, yeast PFDs are characterized not by short, highly prion-prone segments but instead by large disordered regions of modest prion propensity.
Compositional similarity to known PFDs is a poor predictor of a Q/N-rich domain's prion propensity.
Various algorithms to identify novel PFDs based on compositional similarity to known PFDs have been designed to look for some combination of the following: (i) high Q/N content (19, 28, 42, 46), (ii) a bias against charged residues (19, 28, 42), (iii) a bias against hydrophobic residues (19), and (iv) subsidiary biases for glycine, serine, or tyrosine (19). However, when the Q/N-rich proteins with and without prion-like activity are compared, little or no difference is seen for any of these groups of amino acids (Fig. 8A). Therefore, although these biases have proven effective for identifying prion candidates (42, 46), they are not sufficient to discriminate among these candidates.
FIG. 8.

Ranking of Q/N-rich domains based on composition. (A) Box-and-whiskers plot of the prevalence of various amino acids among each of the domains shown by Alberti et al. (1) to have prion-like activity in four prion assays, the domains that lacked prion-like activity in all four prion assays, and the known PFDs. (B) Plot of compositional similarity to known PFDs versus prion-like activity. Alberti et al. (1) ranked the 100 domains with greatest compositional similarity to known yeast PFDs and then tested each domain for prion-like activity in four assays. Prion-like activity was scored on a scale from 0 to 10 (with 10 reflecting full prion-like activity in all four assays). Domains that were not testable in one or more assays are excluded. Known PFDs are indicated with open diamonds. Rank correlation and P values were calculated by Spearman's rank analysis. (C) Plot of predicted prion propensity versus prion-like activity. For each of the domains tested by Alberti et al., prion propensity was calculated as the 41 consecutive 41-amino-acid windows with maximum average predicted prion propensity that also had a negative FoldIndex order propensity.
Likewise, although the hidden Markov model of Alberti et al. was extremely effective at identifying prion candidates, it was not effective at predicting which of the top-ranking candidates would have prion-like activity. There was modest correlation between a peptide's ranking for compositional similarity to known PFDs and its prion-like activity, as scored by Alberti et al. based on performance in their four assays (Fig. 8B). The 50 domains with the greatest compositional similarities to known prions showed higher average prion-like activity than those that ranked 51 to 100. However, this algorithm was completely ineffective at ranking these top 50 proteins. Among the 50 highest-scoring domains, there was essentially no correlation between ranking and prion-like activity (correlation coefficient = 0.015 and P = 0.96 by Spearman's rank analysis).
Our algorithm was much more effective at discriminating among these potential PFDs. We scanned each of the 100 domains using a 41-amino-acid window size, identifying for each domain the 41 consecutive 41-amino-acid windows that had the greatest predicted prion propensity while also having an average negative FoldIndex score (i.e., the most prion-prone disordered region). There was a strong correlation between predicted prion propensity using our algorithm and prion activity (Fig. 8C). Our top 15 ranked domains (from Pub1, Nrp1, Rnq1, New1, YBR016W, YPL184C, Jsn1, Lsm4, Ngr1, Ure2, Cbk1, Mot3, Sok2, YBL081W, and Sup35) had an average score of 8.1 on the 10-point prion-like activity scale of Alberti et al., with all but one scoring at least 5 out of 10 (the one exception, Jsn1, scored 4 out of 10). Additionally, all six domains that scored 10 out of 10 ranked in our top 15. In contrast, the 15 domains with the greatest compositional similarities to known PFDs (as ranked by Alberti et al., excluding domains that were not testable in one or more experiments) had an average score of 5.3; this group included only three of the proteins that scored 10 out of 10 and included six that scored below 5 out of 10. Together, these data demonstrate that our algorithm is much more effective at predicting prion-like activities of Q/N-rich proteins than algorithms that solely utilize compositional similarity to known yeast PFDs.
DISCUSSION
Although algorithms designed to identify proteins that are compositionally similar to known PFDs have been effective at identifying prion candidates (1, 19, 28, 42, 46), none of these algorithms has proven effective at distinguishing between Q/N-rich proteins that can form prions and those that cannot. Our results explain the basis for this failure. The major flaw in composition-based searches is that they are unable to predict how deviations from the observed biases in known PFDs will affect prion propensity. For example, charged and hydrophobic residues are both underrepresented in yeast PFDs. However, our data suggest very different reasons for these biases; charged residues inhibit prion formation, while hydrophobic residues too strongly promote prion formation and/or order. Accurate prediction of prion propensity requires understanding which deviations from known prion-forming compositions will promote prion formation and which will inhibit it.
To allow accurate prediction of prion propensity, we have developed a method of measuring the prion propensities of individual amino acids in vivo. Our mutagenesis uncovered three significant findings: a strong bias against prolines and charged residues, a strong bias for hydrophobic residues, and no significant bias for or against Q/N residues. The bias against charged residues is consistent with findings of previous bioinformatic and mutational analyses (12, 19). However, the other two findings were quite surprising. Q/N residues are highly overrepresented in yeast PFDs (19, 28). Similarly, although bioinformatic and mutational analyses suggest that tyrosines may promote prion formation or propagation (2, 19), hydrophobic residues in general are highly underrepresented in yeast PFDs (19, 28).
Consequently, there was almost no correlation between the experimentally determined prion propensity for a given amino acid and its prevalence within the known PFDs. Thus, we hypothesize that the yeast PFDs are not optimized for maximum intrinsic amyloid propensity but instead that their compositions reflect a balance of intrinsic disorder and prion propensity. While the presence of a small number of additional hydrophobic residues within the Sup35-27 PFD promotes prion formation, larger numbers would likely lead to hydrophobic collapse, potentially inhibiting prion formation or leading to nonspecific aggregation. Therefore, although the effects of individual mutations within a Q/N-rich domain can be accurately predicted based on hydrophobicity and β-sheet propensity (equation 4), it is necessary to consider order propensity when examining the composition of entire domains. This hypothesis provides a possible explanation for the prevalence of Qs and Ns in the yeast PFDs. Qs and Ns have a relatively high prion propensity compared to other disorder-promoting residues. Alternatively, there may be a threshold number and/or density of Q/N residues required for prion formation, and above this threshold small changes in the number of Q/N residues may exert only a subtle effect.
Based on the apparent importance of intrinsic disorder for yeast PFDs, we propose that amyloid proteins can be divided into three broad classes. First are proteins such as transthyretin (21). These proteins have highly amyloid-prone regions that are usually involved in stable interactions within the folded structure of the protein. As seen in Fig. 6A, regions of high amyloid propensity are common but are generally found in regions predicted to be ordered. For these proteins, native state stability will largely determine amyloid propensity (21). This may explain why a recent genome-wide analysis of Escherichia coli proteins found little correlation between the aggregation propensity and the content of hydrophobic residues (31); hydrophobic residues will have competing effects, increasing the intrinsic aggregation propensity while also preventing aggregation by stabilizing the native fold of the protein. Second are non-Q/N-rich disordered peptides such as Aβ, where amyloid formation is driven by short, highly amyloidogenic nucleation domains (3, 34). Third are the Q/N-rich amyloid proteins. Rather than having a high concentration of strongly amyloidogenic (but also order-promoting) residues, the yeast PFDs form prions by excluding order-promoting residues and by having large stretches with a high concentration of the most amyloidogenic disorder-promoting residues. Thus, yeast PFDs do not have to overcome native-state stability to form prions, explaining their efficient prion formation despite their relatively low prion propensity.
Differences between classes of amyloid proteins explain why most amyloid prediction algorithms (developed based on non-Q/N-rich proteins) fail to predict amyloid formation by Q/N-rich proteins, such as the yeast PFDs or expanded polyglutamine tracts. These algorithms are designed to identify the short, highly amyloid-prone segments thought to characterize most amyloid domains (16), not the large disordered segments with modest prion propensity that characterize yeast PFDs.
Although our results support a dominant role for amino acid composition in driving prion formation, they also point to subtle effects of primary sequence. Our algorithm directly incorporates one primary sequence feature: proline patterns. Excluding proline patterning from our algorithm modestly reduces the correlation between the predicted prion propensity and prion-like activity in Fig. 8C from 0.73 to 0.70 (data not shown); however, we are still able to distinguish between Q/N-rich domains that show prion-like activity and those that do not with a high degree of accuracy, and only one of the potential PFDs in Fig. 6C changes from being scored as prion positive to prion negative or vice versa—Mca1, which is scored as barely negative without proline patterning and barely positive with it (data not shown). Therefore, 34 of 39 potential PFDs in Fig. 6C are correctly scored based on composition alone.
Because our algorithm allows for reasonably accurate predictions based solely on composition, examination of outlier proteins not accurately predicted by our algorithm may reveal other primary sequence features that affect prion formation. For example, the only known PFD wrongly categorized as nonprion by our algorithm, Cyc8, contains the repeat sequence (AQ)22. Alternating polar and nonpolar residues have been shown to promote amyloid formation (5, 59, 60), although alanine has not generally been included among the nonpolar residues when such patterns are considered. Therefore, further studies will be needed to address the role of such repeat sequences in prion formation and potentially to find ways to incorporate such patterns into our algorithm.
Finally, a remarkable finding of Alberti et al. was that among the 100 potential PFDs that they tested, although asparagines were highly enriched among the domains that showed prion-like activity, glutamines were more prevalent in nonaggregating domains (1). The simplest explanation for this difference is that asparagines are more prion prone than glutamines. However, our data point to an alternative reason why asparagine residues may appear to be more positively correlated with prion propensity. Consistent with the experimental findings of Alberti et al., among the 100 potential PFDs there is a strong positive correlation between our predicted prion propensity and asparagine content but a modest negative correlation between predicted prion propensity and glutamine content (Fig. 9A and B). Because glutamine and asparagine have nearly identical prion propensities in our formula, the significant difference in predicted prion propensity between Q-rich and N-rich domains must result from some feature of the tested domains other than Q/N content. Indeed, it results largely from differences in the prevalence of prolines in Q-rich versus N-rich sequences. Among the tested sequences, glutamine content is strongly correlated with proline content (Fig. 9C) while asparagine content is negatively correlated with proline content (Fig. 9D). Thus, although asparagine may be modestly more prion prone than glutamine (asparagine was modestly favored in library 2), the observed differences between glutamines and asparagines at least in part reflect an artifact of the context in which these residues are found among the tested sequences.
FIG. 9.

Among domains that are compositionally similar to known yeast PFDs, high glutamine content is correlated with high proline content. (A) For each of the 100 domains tested by Alberti et al., glutamine content was plotted versus the predicted prion propensity (calculated as for Fig. 6C). Rank correlation and P values were calculated by Spearman's rank analysis. (B) Asparagine content plotted versus predicted prion propensity. (C) Glutamine content plotted versus proline content. (D) Asparagine content plotted versus proline content.
Overall, our data provide detailed insight into how amino acid composition affects prion formation by Q/N-rich domains. By highlighting the critical role for intrinsic disorder in yeast prion formation, our data explain the discrepancies between the compositions of the yeast PFDs and the amino acids thought to promote amyloid formation. Our data will allow genome scanning to accurately identify novel PFDs and to identify critical nucleating domains within known PFDs.
Acknowledgments
We thank Reed Wickner, Olve Peersen, P. Shing Ho, Frank Shewmaker, Kevin Hatcher-Ross, and members of the Ross laboratory for helpful discussion and Nathan Liss, Holly Hamilton, and Daniel Lee for their tireless help in screening thousands of colonies.
This work was funded in part by an American Heart Association grant (0735321N) to E.D.R.
Footnotes
Published ahead of print on 2 November 2009.
REFERENCES
- 1.Alberti, S., R. Halfmann, O. King, A. Kapila, and S. Lindquist. 2009. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137:146-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrov, I. M., A. B. Vishnevskaya, M. D. Ter-Avanesyan, and V. V. Kushnirov. 2008. Appearance and propagation of polyglutamine-based amyloids in yeast: tyrosine residues enable polymer fragmentation. J. Biol. Chem. 283:15185-15192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balbach, J. J., Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed, and R. Tycko. 2000. Amyloid fibril formation by A beta 16-22, a seven-residue fragment of the Alzheimer's beta-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 39:13748-13759. [DOI] [PubMed] [Google Scholar]
- 4.Bradley, M. E., and S. W. Liebman. 2004. The Sup35 domains required for maintenance of weak, strong or undifferentiated yeast [PSI+] prions. Mol. Microbiol. 51:1649-1659. [DOI] [PubMed] [Google Scholar]
- 5.Broome, B. M., and M. H. Hecht. 2000. Nature disfavors sequences of alternating polar and non-polar amino acids: implications for amyloidogenesis. J. Mol. Biol. 296:961-968. [DOI] [PubMed] [Google Scholar]
- 6.Bryan, A. W., Jr., M. Menke, L. J. Cowen, S. L. Lindquist, and B. Berger. 2009. BETASCAN: probable beta-amyloids identified by pairwise probabilistic analysis. PLoS Comput. Biol. 5:e1000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, H. Y., J. Y. Lin, H. C. Lee, H. L. Wang, and C. Y. King. 2008. Strain-specific sequences required for yeast [PSI+] prion propagation. Proc. Natl. Acad. Sci. U. S. A. 105:13345-13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiti, F., and C. M. Dobson. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75:333-366. [DOI] [PubMed] [Google Scholar]
- 9.Chou, P. Y., and G. D. Fasman. 1974. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry 13:211-222. [DOI] [PubMed] [Google Scholar]
- 10.Chou, P. Y., and G. D. Fasman. 1978. Empirical predictions of protein conformation. Annu. Rev. Biochem. 47:251-276. [DOI] [PubMed] [Google Scholar]
- 11.Cox, B. S. 1965. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 26:211-232. [DOI] [PubMed] [Google Scholar]
- 12.DePace, A. H., A. Santoso, P. Hillner, and J. S. Weissman. 1998. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93:1241-1252. [DOI] [PubMed] [Google Scholar]
- 13.Derkatch, I. L., M. E. Bradley, J. Y. Hong, and S. W. Liebman. 2001. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell 106:171-182. [DOI] [PubMed] [Google Scholar]
- 14.Derkatch, I. L., Y. O. Chernoff, V. V. Kushnirov, S. G. Inge-Vechtomov, and S. W. Liebman. 1996. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144:1375-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du, Z., K. W. Park, H. Yu, Q. Fan, and L. Li. 2008. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 40:460-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteras-Chopo, A., L. Serrano, and M. L. de la Paz. 2005. The amyloid stretch hypothesis: recruiting proteins toward the dark side. Proc. Natl. Acad. Sci. U. S. A. 102:16672-16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Escamilla, A. M., F. Rousseau, J. Schymkowitz, and L. Serrano. 2004. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 22:1302-1306. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira, P. C., F. Ness, S. R. Edwards, B. S. Cox, and M. F. Tuite. 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 40:1357-1369. [DOI] [PubMed] [Google Scholar]
- 19.Harrison, P. M., and M. Gerstein. 2003. A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol. 4:R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung, G., and D. C. Masison. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr. Microbiol. 43:7-10. [DOI] [PubMed] [Google Scholar]
- 21.Kelly, J. W. 1998. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 8:101-106. [DOI] [PubMed] [Google Scholar]
- 22.Koehl, P., and M. Levitt. 1999. Structure-based conformational preferences of amino acids. Proc. Natl. Acad. Sci. U. S. A. 96:12524-12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kushnirov, V. V., and M. D. Ter-Avanesyan. 1998. Structure and replication of yeast prions. Cell 94:13-16. [DOI] [PubMed] [Google Scholar]
- 24.Linding, R., J. Schymkowitz, F. Rousseau, F. Diella, and L. Serrano. 2004. A comparative study of the relationship between protein structure and [beta]-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 342:345-353. [DOI] [PubMed] [Google Scholar]
- 25.Masel, J., and C. K. Griswold. 2009. The strength of selection against the yeast prion [PSI+]. Genetics 181:1057-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masison, D. C., M. L. Maddelein, and R. B. Wickner. 1997. The prion model for [URE3] of yeast: spontaneous generation and requirements for propagation. Proc. Natl. Acad. Sci. U. S. A. 94:12503-12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masison, D. C., and R. B. Wickner. 1995. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 270:93-95. [DOI] [PubMed] [Google Scholar]
- 28.Michelitsch, M. D., and J. S. Weissman. 2000. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. U. S. A. 97:11910-11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayashiki, T., C. P. Kurtzman, H. K. Edskes, and R. B. Wickner. 2005. Yeast prions [URE3] and [PSI+] are diseases. Proc. Natl. Acad. Sci. U. S. A. 102:10575-10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nemecek, J., T. Nakayashiki, and R. B. Wickner. 2009. A prion of yeast metacaspase homolog (Mca1p) detected by a genetic screen. Proc. Natl. Acad. Sci. U. S. A. 106:1892-1896. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Niwa, T., B. W. Ying, K. Saito, W. Jin, S. Takada, T. Ueda, and H. Taguchi. 2009. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc. Natl. Acad. Sci. U. S. A. 106:4201-4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osherovich, L. Z., B. S. Cox, M. F. Tuite, and J. S. Weissman. 2004. Dissection and design of yeast prions. PLoS Biol. 2:E86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel, B. K., J. Gavin-Smyth, and S. W. Liebman. 2009. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat. Cell Biol. 11:344-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pawar, A. P., K. F. Dubay, J. Zurdo, F. Chiti, M. Vendruscolo, and C. M. Dobson. 2005. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 350:379-392. [DOI] [PubMed] [Google Scholar]
- 35.Perutz, M. F., B. J. Pope, D. Owen, E. E. Wanker, and E. Scherzinger. 2002. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc. Natl. Acad. Sci. U. S. A. 99:5596-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters, T. W., and M. Huang. 2007. Protein aggregation and polyasparagine-mediated cellular toxicity in Saccharomyces cerevisiae. Prion 1:144-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pierce, M. M., U. Baxa, A. C. Steven, A. Bax, and R. B. Wickner. 2005. Is the prion domain of soluble Ure2p unstructured? Biochemistry 44:321-328. [DOI] [PubMed] [Google Scholar]
- 38.Prilusky, J., C. E. Felder, T. Zeev-Ben-Mordehai, E. H. Rydberg, O. Man, J. S. Beckmann, I. Silman, and J. L. Sussman. 2005. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21:3435-3438. [DOI] [PubMed] [Google Scholar]
- 39.Roseman, M. A. 1988. Hydrophilicity of polar amino acid side-chains is markedly reduced by flanking peptide bonds. J. Mol. Biol. 200:513-522. [DOI] [PubMed] [Google Scholar]
- 40.Ross, E. D., U. Baxa, and R. B. Wickner. 2004. Scrambled prion domains form prions and amyloid. Mol. Cell. Biol. 24:7206-7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross, E. D., H. K. Edskes, M. J. Terry, and R. B. Wickner. 2005. Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. U. S. A. 102:12825-12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santoso, A., P. Chien, L. Z. Osherovich, and J. S. Weissman. 2000. Molecular basis of a yeast prion species barrier. Cell 100:277-288. [DOI] [PubMed] [Google Scholar]
- 43.Serio, T. R., A. G. Cashikar, A. S. Kowal, G. J. Sawicki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf, and S. L. Lindquist. 2000. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289:1317-1321. [DOI] [PubMed] [Google Scholar]
- 44.Sherman, F. 1991. Getting started with yeast. Methods Enzymol. 194:3-21. [DOI] [PubMed] [Google Scholar]
- 45.Shewmaker, F., E. D. Ross, R. Tycko, and R. B. Wickner. 2008. Amyloids of shuffled prion domains that form prions have a parallel in-register beta-sheet structure. Biochemistry 47:4000-4007. [DOI] [PubMed] [Google Scholar]
- 46.Sondheimer, N., and S. Lindquist. 2000. Rnq1: an epigenetic modifier of protein function in yeast. Mol. Cell 5:163-172. [DOI] [PubMed] [Google Scholar]
- 47.Song, Y., Y. X. Wu, G. Jung, Y. Tutar, E. Eisenberg, L. E. Greene, and D. C. Masison. 2005. Role for Hsp70 chaperone in Saccharomyces cerevisiae prion seed replication. Eukaryot. Cell 4:289-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Street, A. G., and S. L. Mayo. 1999. Intrinsic beta-sheet propensities result from van der Waals interactions between side chains and the local backbone. Proc. Natl. Acad. Sci. U. S. A. 96:9074-9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taneja, V., M. L. Maddelein, N. Talarek, S. J. Saupe, and S. W. Liebman. 2007. A non-Q/N-rich prion domain of a foreign prion, [Het-s], can propagate as a prion in yeast. Mol. Cell 27:67-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tank, E. M., D. A. Harris, A. A. Desai, and H. L. True. 2007. Prion protein repeat expansion results in increased aggregation and reveals phenotypic variability. Mol. Cell. Biol. 27:5445-5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tartaglia, G. G., A. P. Pawar, S. Campioni, C. M. Dobson, F. Chiti, and M. Vendruscolo. 2008. Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 380:425-436. [DOI] [PubMed] [Google Scholar]
- 52.Ter-Avanesyan, M. D., A. R. Dagkesamanskaya, V. V. Kushnirov, and V. N. Smirnov. 1994. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 137:671-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ter-Avanesyan, M. D., V. V. Kushnirov, A. R. Dagkesamanskaya, S. A. Didichenko, Y. O. Chernoff, S. G. Inge-Vechtomov, and V. N. Smirnov. 1993. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 7:683-692. [DOI] [PubMed] [Google Scholar]
- 54.Toyama, B. H., M. J. Kelly, J. D. Gross, and J. S. Weissman. 2007. The structural basis of yeast prion strain variants. Nature 449:233-237. [DOI] [PubMed] [Google Scholar]
- 55.Trovato, A., F. Seno, and S. C. Tosatto. 2007. The PASTA server for protein aggregation prediction. Protein Eng. Des. Sel. 20:521-523. [DOI] [PubMed] [Google Scholar]
- 56.True, H. L., and S. L. Lindquist. 2000. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407:477-483. [DOI] [PubMed] [Google Scholar]
- 57.Tuite, M. F., C. R. Mundy, and B. S. Cox. 1981. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics 98:691-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weathers, E. A., M. E. Paulaitis, T. B. Woolf, and J. H. Hoh. 2004. Reduced amino acid alphabet is sufficient to accurately recognize intrinsically disordered protein. FEBS Lett. 576:348-352. [DOI] [PubMed] [Google Scholar]
- 59.West, M. W., and M. H. Hecht. 1995. Binary patterning of polar and nonpolar amino acids in the sequences and structures of native proteins. Protein Sci. 4:2032-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.West, M. W., W. Wang, J. Patterson, J. D. Mancias, J. R. Beasley, and M. H. Hecht. 1999. De novo amyloid proteins from designed combinatorial libraries. Proc. Natl. Acad. Sci. U. S. A. 96:11211-11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wickner, R. B. 1994. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264:566-569. [DOI] [PubMed] [Google Scholar]
- 62.Zibaee, S., O. S. Makin, M. Goedert, and L. C. Serpell. 2007. A simple algorithm locates beta-strands in the amyloid fibril core of alpha-synuclein, Abeta, and tau using the amino acid sequence alone. Protein Sci. 16:906-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zoghbi, H. Y., and H. T. Orr. 2000. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 23:217-247. [DOI] [PubMed] [Google Scholar]