

Abstract

An enantioselective approach to the perylenequinone core found in the mold perylenequinone natural products is outlined. Specifically, the first asymmetric syntheses of helical chiral perylenequinones absent any additional stereogenic centers are described. Key elements of the synthetic venture include a catalytic enantioselective biaryl coupling, a PIFA-induced naphthalene hydroxylation, and a palladium-mediated aromatic decarboxylation. Transfer of the binaphthalene axial stereochemistry to the perylenequinone helical stereochemistry proceeded with good fidelity. Furthermore, the resultant perylenequinones were shown to possess sufficient atropisomeric stability to be viable intermediates in the biogenesis of the perylenequinone natural products. This stability supports the use of the helical axis as a stereochemical relay in synthesis of the natural products containing additional stereochemical centers.
Introduction
The parent perylenequinone 1 was first prepared by Zinke in 1929,1 but it was not until 1954 that the correct structure was elucidated (Figure 1).2 This compound was subsequently isolated from the fungus, Daldinia concentrica as blue-black crystals and is the simplest of the naturally occurring perylenequinones.3 Most members of this family of natural products fall into 3 classes (examples of each in Figure 1): (a) C20 compounds without carbon substituents,4 (b) mold perylenequinones containing carbon substituents, and (c) perylenequinones from aphids. Of the three, class B is the most prevalent. Due to their diversity, interesting stereochemistries, and anticancer activities we have undertaken the syntheses of these compounds via dimerization of achiral naphthols.
Figure 1.

Representative Structures of the Perylenequinone Classes.
The archetypical cercosporin 6 (Figure 2) was among the first of the mold perylenequinones to be isolated in 1957.5 The unusual features of this molecule hindered a complete structural determination until the 1970s.6 The majority of the structural details were determined by classical redox quinone chemistry, but the most significant finding was the transformation involving the thermal atropisomerization of (M)-cercosporin into (P)-epi-cercosporin. The opposite helical configuration of the perylenequinone epimers was confirmed by circular dichroism (CD) analysis,6b and the helical nature along with the absolute and relative configuration of 6 was established crystallographically.7 Since the discovery of 6, more than 20 perylenequinones have been isolated as dark red pigments from a variety of fungi within the Ascomycota phylum (Figure 2).8 Correlation with the CD and NMR spectra of 6 has allowed determination of the helical configurations and relative stereochemistries of most of the remaining natural products.
Figure 2.

Mold Perylenequinones 2–17.
Notably, all of the mold perylenequinones (Figure 2) are derivatives of 1 (Figure 1). With the exception of hypomycin B (15), the C4,C4’- or C 5,C5’-phenols are not alkylated. These acidic phenols (pKa ~7.3–8.3)9 are characteristic of the structures and form strong internal hydrogen bonds with the quinone carbonyls.10 Many of the mold perylenequinones are C2-symmetric, having C7,C7’-substitution with centrochiral stereocenters of the same absolute configuration. The remainder (hypocrellin, hypocrellin A–B, shiraiachrome A, elsinochrome B1-B2, hypomycin B11) likely originated from C2-symmetric intermediates.
Regardless of the varied C7,C7’-substitution, all class B perylenequinones contain a conjugated pentacyclic core in a highly oxidized state. Furthermore, the distinct oxygenation pattern at the C2,C2’,C4,C4’,C5,C5’,C6,C6’-centers (Figure 2) is conserved throughout the series. In the past two decades, the construction of this highly oxygenated core en route to the perylenequinone natural products has been examined by several groups.12,13,14,15,16,17,18,19 Prior to these achievements, there had been relatively few synthetic efforts devoted to this family of natural products. As mentioned above, 1 (class A, Figure 1) was first unknowingly prepared by Zinke and coworkers (ca. 1929) from tetranitroperylene 18 upon exposure to sulfuric acid at 140 °C under air.1 Due to the atypical stability of the proposed product 19, Calderbank and coworkers repeated the procedure (Scheme 1),2 and the structure of 4,4’,5,5’-tetraquinone 19 reported by Zinke was correctly reassigned as the parent perylenequinone 1.
Scheme 1.

Previous Synthesis of Parent Perylenequinone 1.
Surprisingly, it was not until 1972 that there was further progress when Weisgraber and Weiss attempted to synthesize the oxygenation pattern present in the mold perylenequinones (Figure 2). The initial assignments of both the coupling product 21 and the subsequent perylenequinone 22 were discovered to be incorrect,20,21 and the final cyclized product was reassigned as dinaphthofuranedione 24 (Scheme 2).
Scheme 2.

Weiss Attempted Synthesis of Perylenequinone 22 (PPA = polyphosphoric acid).
In work by Dallacker and Leidig, a different approach was used in the synthesis of 2,2’,4,4’-tetramethoxyperylenequinone, commencing with formation of bisphenyl substrate 25. Unfortunately, the synthesis was lengthy and lacked the necessary C6,C6’-oxygenation.22
Prior to the report by Broka on the calphostins,14 syntheses of the highly oxidized perylenequinone core were either racemic or yielded optically inactive products. Thus, these early efforts did not address a key architectural element of the perylenequinones – the chiral helical pentacyclic core. Due to steric interactions of the C7,C7’- and C2,C2’-substitution, the pentacyclic core is forced to adopt a chiral helical structure (dihedral angle of 20°). With larger C7,C7’- and C2,C2’-groups, the rate of interconversion of the two possible enantiomeric conformational isomers is slow leading to stable atropisomers. As discussed above, the helicity of most perylenequinones was determined by correlation to cercosporin. In the cases of hypocrellin B 4 and scutiaquinone B 17, there is an absence of helicity in the CD spectra. However, calculations indicate that the lowest energy ground state structures are in fact helical (Scheme 4). Presumably, the double bond of hypocrellin B 4 and the lack of substituents at C2,C7’-positions of scutiaquinone B 17 lower the atropisomerization barrier such that the two helical forms rapid interconvert.23,24
Scheme 4.

Conformational Isomers of Hypocrellin B and Scutiaquinone B.
The helicity of the perylenequinone natural products is not constant. Rather compounds exhibiting both (M)- and (P)-helicity have been isolated, leaving the origin of the helical stereochemistry an interesting and unsolved issue. Since different helical stereochemistries arise from different organisms (hypocrellin from Hypocrella bambusae25 and hypocrellin A from Shiraia bambusicola23), it seems likely that an enzyme-mediated enantioselective dimerization occurs in the biosynthetic routes. The fact that horseradish peroxidase, a metalloporphyrin enzyme, has been found to elicit asymmetric naphthol couplings with good yields and modest enantioselectivities indicates that enzymes are capable of catalyzing this transformation (Eq 1).26 Presumably, the peroxidase catalyzes the production of a stabilized radical to enable the homocoupling.27 It is entirely probable, therefore, that helical chiral perylenequinones, such as 30, absent any centrochiral elements (i.e. R2 not containing further stereocenters) can be produced enzymatically. Such production likely occurs via an axial chiral binaphthalene with transfer of axial stereochemistry to helical stereochemistry in the perylenequinone forming step (Scheme 5).
Scheme 5.

Proposed Enzymatic Perylenequinone Formation.
 |
(1) |
In an examination of prior approaches, the installation of the helical chirality has been the most difficult aspect and thus the focal point of the syntheses.14–17 Poor diastereocontrol in the couplings of chiral C7,C7’-substituted naphthalenes also points away from such a pathway in the biosynthesis (i.e. 38 to 8a–d, Scheme 6).14–17 Thus, we formulated a plan revolving around diketone perylenequinone 33. Notably, perylenequinone 33 is a retrosynthetic intermediate to four different natural product classes: the calphostins (8a–d) and cercosporins (6) via reduction, the hypocrellins (2) via aldol cyclization, and the elsinochromes (10) via oxidative enol coupling. In contrast, there is no direct route from monomer 38 to the hypocrellins (2) and elsinochromes (10); oxidation and loss of the two alcohol stereocenters would be required first. We proposed that helical perylenequinone 33 could be produced from cyclization of axial chiral binaphthalene 34 which would in turn be produced by facile enantioselective oxidative coupling of the corresponding monomer 35. The illustrated monomer 35 is generated in a few simple steps from phenylacetic acid 37, in contrast to the laborious preparations of enantiopure monomers 38.
Scheme 6.

Retrosynthesis of the perylenequinone natural products.
A central element of this synthetic plan is the atropisomeric stability of the key intermediate 33. The calphostins (8a–d) exhibit good atropisomeric stability, but the simple change of introducing a bridging methylidene as in cercosporin (6) abrogates much of this stability with atropisomerization occurring at 37 °C.7 Due to the sterically larger C7,C7’-hydroxypropyl groups in the calphostins (8a–d) compared to the 2-oxopropyl groups of 33, we anticipated that 33 would exhibit lower atropisomeric stability. Since no enantiopure perylenequinones with only the helical stereochemistry as a chiral element had been reported previously, we set out to establish the degree of configurational stability of such species. In this paper, we detail the synthesis of the enantiopure perylenequinone core, addressing installation of the necessary oxygenation pattern as well as the formation and stability of the helical stereochemistry in the absence of other stereochemical substitution.28
Results and Discussion
Retrosynthetic Analysis
With an initial goal of producing an enantiopure perylenequinone possessing only the helical stereochemical component, we elected to undertake the synthesis of analog 39 possessing simple n-propyl groups at the C7, C7’-positions. Based on prior reports, we identified two intermediates (40 and 42) that could lead to the desired perylenequinone 39, both devolving to common chiral biaryl intermediate 41(Scheme 7). Merlic has reported diastereoselective oxidative couplings to either provide the calphostin perylenequinone directly or via a bis-ortho-quinone intermediate,17,29 such as 40 (path a). Similarly, Broka14 and Coleman16 utilized a diastereoselective biaryl coupling to yield an axial chiral biaryl as an intermediate in their calphostin syntheses. The perylenequinone arose from the cyclization of the corresponding 5,5’-bisnaphthol, similar to debenzylated 42 (path b). For both paths a and b, effective stereochemistry transfer of the axial stereochemistry to the ultimate helical stereochemistry was observed.14–17 However, the role of the bulky protected C7,C7’-hydoxypropyl groups in stabilizing this stereochemistry transfer was unknown.
Scheme 7.

Perylenequinone Retrosynthetic Analysis.
Due to both the reported success of Merlic29 and also the precedent for the oxidation of naphthalenes to bis-ortho-quinones,30 we initially chose to pursue path a. We commenced this effort by exploring the ability of our catalyst system in the enantioselective coupling of a series of highly substituted naphthols to provide the required axial chiral precursor 41.28
Monomer Synthesis and Asymmetric Oxidative Coupling
Based on prior methods for the formation of highly substituted naphthols,20a we developed a protocol to generate naphthols 45a–d with high efficiency (Scheme 8, Scheme 9).28 Commencing with simple commercially available phenylacetic acids (43a–c), the requisite naphthols were generated rapidly via acid chloride formation, malonate acylation, and cationic cyclization. After diacetylation, the less hindered C2-acetate was hydrolyzed with NaOMe to provide the desired coupling substrates 45a–c (Scheme 8). The flexibility of this route to quickly generate substrates with a variety of substitution patterns has proven invaluable in identifying optimal substrates for the enantioselective oxidative biaryl coupling and for the perylenequinone syntheses.
Scheme 8.

Synthesis of Naphthol Substrates.
Scheme 9.

Synthesis of C7-Propyl Substrate.
In the formation of biaryl intermediate 41(Scheme 7), the first naphthol proposed was C7-allyl 48 (Scheme 9). The allyl group would provide a platform both to intermediate 41 (Scheme 7) and also to introduce functional groups present in the natural products. The synthesis began with an allylation of the hydroxyl and carboxyl groups of commercially available 46 followed by a Claisen rearrangement and methylation of the revealed phenol (Scheme 9). Unfortunately, all attempts to form the desired 48 from 47 were unsuccessful, most likely due to incompatibility of the allyl group with the strongly acidic cyclization conditions. In light of this result, the hydrogenation of 47 was performed to yield the inert propyl derivative. After saponification to supply 49, the naphthol 45d was successfully prepared using the optimized conditions for 45a–c.
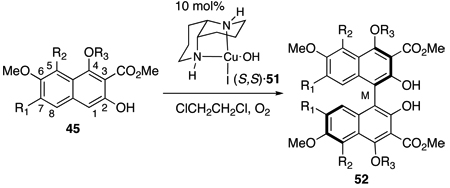
In previous efforts, we have assessed the versatility of our diaza-cis-decalin catalyst 51 in catalytic asymmetric oxidative naphthol couplings.28,18,19,31 Table 1 outlines the significant results pertinent to this study.32 Notably, an electron-withdrawing, coordinating group at C3 enhances selectivity,31 presumably by via coordination to the copper complex together with the C2-phenol. The effects of the substrate oxidation potential are illustrated in entries 1–3 (Table 1),28 where the electron rich nature of the trismethoxy substrate (entry 2) led to lower optical purities due to rapid atropisomerization of the product 52e via a further oxidation. Replacement of the donating C4-methoxy group with an electron-withdrawing acetoxy group (entry 3) reduced the oxidation potential and furnished the pentasubstituted naphthalene 52b in high enantioselectivity. The introduction of an additional electron-donating group (C5-methoxy) counteracted the effect of the C4-acetoxy group and facilitated enantiomerization of 52c (entry 4).28 Additionally, the steric gearing effects prevented the substrate from adopting the conformation needed for catalyst coordination, explaining the slow conversion as well as the low enantioselectivity. With these results in hand, it was straightforward to design substrates with a suitable oxidation potential (sufficiently electron rich for the substrate to oxidize, but not too electron rich to allow product oxidation) to obtain high reactivity and selectivity. For example, 52a,b,d (entries 1, 3, 5) furnish functionalized binaphthols in high yield and enantioselectivity. The M-helicity of these products was assigned based on trends observed with related substrates; in the over 30 cases where the stereochemistry has been assigned via correlation, crystallization, or calculation, the S,S-catalyst provides the M-helicity and the R,R-catalyst the P-helicity.31 Significantly, the enantioenriched naphthol 52d for this investigation is generated with the necessary functionalization at the C2,C3,C4,C7-centers.
Table 1.
Enantioselective Biaryl Coupling with Diaza-cis-decalin (S,S)·51.
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Product | Yield, ee |
| 1 | H | H | Ac | a | 71%, 86% ee |
| 2 | OMe | H | Me | e | 81%, 35% ee (P) |
| 3 | OMe | H | Ac | b | 72%, 90% ee (P)a |
| 4 | OMe | OMe | Ac | c | 41%, 27% ee |
| 5 | n-propyl | H | Ac | d | 85%, 87% ee |
(R,R)·51 catalyst complex was used.
Perylenequinone via a bis-ortho-Quinone
Following the success of the enantioselective biaryl couplings, investigations begun into transforming biaryl 52d, arising from 45d, to perylenequinone 39 via bis-ortho-quinone 40 (Scheme 7). As previously mentioned, there is precedent for oxidation of naphthalenes with a phenol at the position peri to the reaction center using various oxidants.30 Our efforts commenced with the synthesis of model biaryl 53, containing the necessary C4,C4’-phenols, but possessing C7,C7’-methoxy groups instead of propyl groups. Direct benzylation of the C2,C2’-phenols to generate 53 was unsuccessful, a trend that was observed throughout the series. Apparently, the C2,C2’-positions are relatively hindered, which slows alkylation significantly. On the other hand, Mitsunobu reaction generally proceeded well on these centers and was used here to install the C2,C2’-benzyl ethers. Subsequent hydrolysis of the C4,C4’-acetates afforded 53 (Scheme 10). After screening a number of oxidants, the hypervalent iodine reagent bis(trifluoroacetoxy)iodobenzene [PIFA = PhI(O2CCF3)2] following McKillop’s protocol was found to generate the desired bis-ortho-quinone 54 as confirmed by the crystal structure.
Scheme 10.

Synthesis of Bis-ortho-quinone 54 (ORTEP drawing with 30% probability thermal ellipsoids).
With the model bis-ortho-quinone 54 in hand, we next examined formation of the perylenequinone core. Merlic has reported the transformation of a similar bis-ortho-quinone 55 to perylenequinone 56 using trifluoroacetic acid (TFA) under O2 (Eq 2).17 When applied to methylated 57, this procedure yielded only decomposition products (Eq 3). We attributed this result to the differences between the two ortho-quinones (55 vs. 57), specifically the presence of C2,C2’-benzyl ether, C3,C3’-ester, and C7,C7’-methyl ether groups in 57.
 |
(2) |
 |
(3) |
Reasoning that the C7,C7’-methoxy groups of 57 (Eq 3) changed the electronic characteristics of the ortho-quinone relative to 55 (Eq 2), we moved to biaryl 59 (Scheme 11) with C7,C7-n-propyl groups as well as C2,C2’-methoxy groups to more closely approximate Merlic’s system (Eq 2) in formation of the pentacyclic core. Coupling of 45d with CuCl(OH)TMEDA under O2 provided large quantities of the racemic biaryl 52d, which we employed to develop a viable route to 39. Biaryl 52d was treated with NaH and MeI to methylate the C2,C2’-phenols. As has been seen throughout the series, alkylation of the hindered C2,C2’-phenols is difficult and this transformation required the use of anhydrous DMF33 to succeed. Subsequent hydrolysis of C4,C4’-acetates efficiently afforded 59 (Scheme 11). Oxidation utilizing PIFA yielded the desired bis-ortho-quinone 60 in quantitative yield, but this compound was unstable to chromatography or prolonged exposure to air. Immediate methylation of the C4,C4’-phenols with n-Bu4NF and MeI provided the stable 61, which could be purified. Surprisingly, 61 could not be isomerized to the perylenequinone 62 and only decomposition occurred following the procedure developed by Merlic. Presumably, the isomerization requires one of the ortho-quinone moieties to reduce so that a nucleophilic phenol can undergo a transannular addition to the remaining electrophilic ortho-quinone. Subsequent tautomerization and air oxidation would then furnish the perylenequinone. In an attempt to reproduce this manifold, bis-ortho-quinone 61 was treated under conditions to induce reduction (NaBH4 and O2 or DBU and O2); however, only decomposition was observed.
Scheme 11.

Attempted Synthesis of C7,C7’-Bispropyl Perylenequinone.
The presence of the C3,C3’-methyl ester groups accounts for the last major difference between the previously isomerized bis-ortho-quinone 55 (Eq 2) and the structures 57 (Eq 3) and 61 (Scheme 11). The electron withdrawing nature of the methyl esters may destabilize the perylenequinone 62 and/or interfere with the isomerization. To test the hypothesis, decarboxylation of the biaryl was examined. To generate a biaryl that would be stable to saponification and decarboxylation, the previously formed bis-ortho-quinone 61 (Scheme 11) was reduced with sodium dithionite (Na2S2O4) and methylated (Scheme 12). Unfortunately, mixtures of the desired octamethyl ether 63, partially-reduced hexamethyl ether 63a and starting material 61 were obtained with the highest yield of 46% for 63. Saponification of 63 proceeded smoothly with aqueous LiOH in refluxing dioxane, to afford the bisacid. Subsequent decarboxylation with Cu-quinoline at 180 °C furnished 64 in a modest 30% yield over two steps (Scheme 12). Thereafter, treatment of 64 with BCl3 in an attempt to selectively cleave the C5,C5’-methyl ethers resulted in a mixture of demethylated compounds, which upon treatment with K3Fe(CN)6 yielded only a complex mixture from which none of the desired 65 was isolated. Fortunately, removal of the C5,C5’-methyl ethers was not necessary for oxidation and direct exposure of 64 to ceric ammonium nitrate (CAN) afforded the oxidized bis-ortho-quinone 40 (Scheme 12). Longer reaction times in the CAN oxidation did not afford the desired perylenequinone 65, and all attempts to isomerize 40 to 65 led only to decomposition. This result was surprising due to the close structural similarity of 40 (Scheme 12) to 55 (eq 2).
Scheme 12.

Decarboxylation and Attempted Synthesis of a Perylenequinone.
Since an examination of the transformation proposed by Hauser and Merlic15,17 indicated that a direct isomerization of 40 to 65 (Scheme 12) is not viable without a reducing agent, protocols involving a reduction were examined. Strong precedent for such an approach is seen in the work of Lown13 and Zhang12 on the syntheses of a perylenequinones from bis-ortho-quinones via a two-step reduction-oxidation sequence (FeCl2, then FeCl3; SO2-H2O, then FeCl3). Due to the length of the synthetic route needed to obtain large quantities of bis-ortho-quinone 40, 61 was used to examine these new isomerization procedures. Treatment of 61 with the reducing agent Na2S2O4 followed by FeCl3 afforded a complex mixture that was determined to contain the undesired isomer 61 as the major product as well as the desired product 62(Scheme 13).34
Scheme 13.

Two-step Synthesis of Perylenequinone from Bis-ortho-quinone.
With confidence that the synthesis of perylenequinone 62 via this route could be optimized the reactions were examined with enantioenriched 52d to verify the optical purity of the synthetic intermediates. Formation of enantioenriched 59 (70% ee) following the racemic synthesis seen in Scheme 11, permitted the PIFA-mediated oxidation of the optically active intermediate to bis-ortho-quinone 61 in 70% yield (Eq 4). Disappointingly, when evaluated by both optical rotation and HPLC, compound 61 was found to be racemic.
 |
(4) |
The mechanism of the oxidation reaction was studied to provide an explanation of the atropisomerization. The use of PIFA and phenyliodonium diacetate [PIDA = PhI(OAc)2] for phenolic oxidation has been well reviewed,35 and notably, biphenol 67, upon oxidation leads to the extended quinone 68 (Eq 5).36 When only two equivalents of PIFA (7.5 equiv used in Eq 4) were used in the oxidation of 59, a binaphthone (69, Figure 3) was isolated as a dark green material from the reaction mixture. In our assignment of 69, the propyl groups are placed anti to each other because the 1H NMR spectrum does not indicate any restricted rotation about the propyl groups (i.e., diastereotopic protons), which is seen in the 1H NMR spectra of perylenequinones where the propyl groups are in close proximity. Similar to 69, the highly strained bianthrone 70 has been reported in the literature.37,38 Interestingly, bianthrone 70 was found to equilibrate between twisted and stack conformations (Figure 3). Analogously, it is clear that 69 is a chiral compound, but the facile interconversion of the twisted and stacked conformations provides a ready racemization pathway (Figure 4).
Figure 3.

Strained Extended Quinone Structures.
Figure 4.

Proposed Racemization Pathway of 69.
 |
(5) |
The use of the less reactive PIDA allowed a full evaluation of the oxidation pathway of 59 (Scheme 14). While all efforts to optimize formation of binaphthone 69 with PIFA as the oxidant proved to be unsuccessful, PIDA allows 69 to be isolated in a 91% yield (Scheme 14) with minimal amounts of decomposition during the reaction. The treatment of 69 with two equivalents of PIFA yielded racemic 60, strongly supporting the intermediacy of 69 during the PIFA-oxidation of 59 to bis-ortho-quinone 61 (Eq 4).
Scheme 14.

Isolated Intermediate in the Oxidation to bis-ortho-Quinone 60.
Based upon the above evidence, an oxidative mechanism for conversion of binaphthol 59 to ortho-quinone 60 is proposed in Scheme 15. One equivalent of PIFA oxidizes 59 to the binaphthone 69 via adduct 71. The trifluoroacetic acid liberated from the PIFA protonates the C4-carbonyl, promoting conjugate addition of H2O to the C5-center. After tautomerization, another oxidation forms the binaphthone 74. A second conjugate addition of H2O at the C5'-position is followed by tautomerization to intermediate 75 that can then be oxidized twice further. Final hydrolysis with the excess H2O present of the alkylated carbonyls of 76 furnishes bis-ortho-quinone 60. This mechanism requires four equivalents of an oxidant to form 60 from 59, which is consistent with the amount employed in Eq 4.
Scheme 15.

Proposed Mechanism for Bis-ortho-quinone Formation.
Perylenequinone via an Oxidized Binaphthalene
Due to the racemization of biaryl 59 during the formation of bis-ortho-quinone 60, path a (Scheme 7) was not a viable route to enantiopure 39. Shifting to path b, we examined the formation of 39 via an advanced biaryl intermediate 42, containing C5,C5’- benzyl ethers (Scheme 7). The first undertaking was the hydroxylation of the C5,C5’-centers to provide 42 from available precursor 41. Based upon the above results (Scheme 15), the C4,C4’-phenols needed to be masked as ethers during this oxidation to prevent formation of binaphthone 69. Thus, an oxidation was required that did not rely on hydroxyl activation in oxidizing the C5,C5’-centers. One such method is the oxygenation of aryl lithiums with electrophilic oxygen sources. Since the requisite binaphthalene (see 82 below) is very electron rich and the C5,C5’-centers are hindered, a naphthalene model system 78 was utilized to explore the stability and reactivity of the corresponding C5-lithium species. Model 78 was prepared by C5-bromination of 77 followed by hydrolysis of the C2,C4-acetates and methylation to provide trimethyl ether 78. With tert-butyllithium, lithium halogen exchange provided intermediate 79 as confirmed by quenching with D2O which generated the corresponding C5-deuterated arene and minimal amounts of C3-tert-butyl ketone. Unfortunately, reaction with a series of oxidants and electrophiles (oxygen gas, Davis oxaziridine, lithium salt of tert-butyl hydroperoxide, and B(OMe)3)39 was unsuccessful, which was attributed to the hindered nature of the C5 center. Given this challenge, an alternate aryl hydroxylation reaction was sought.
Notably, Kita has utilized PIFA to induce the nucleophilic substitution of phenyl ethers to afford functionalized arenes.40,41 His studies indicate that the reaction proceeds by a single-electron transfer (SET) resulting in the formation of a radical cation [ArH•+] as the reactive intermediate (81, Eq 6).40 This mechanism contrasts with PIFA-oxidations described above (Scheme 10, Scheme 11, Scheme 14, and Scheme 15; eq 4), which proceeded via phenolic C4,C4’-hydroxyl activation (Scheme 15). Based on the reported success of the acetoxy group as a nucleophile in the PIFA-induced SET pathway, we set out to synthesize an intermediate containing the necessary perylenequinone oxygenation in this manner. The greatest concern in this transformation was the regioselection in the addition to the radical cation. In considering the radical cation, many resonance structures are possible, but the two that are pertinent (cations at unsubstituted positions where reaction will allow regeneration of aromaticity via deprotonation) are illustrated in Scheme 17. The stabilization of the cations is expected to be similar at C5 and C8. However, the radical would need to reside at C6 or C7 to maintain the greatest amount of conjugation in the system. The C6-methoxy would be expected to stabilize radical character at the C6-position to a greater degree. Thus, resonance structure 82b is expected to predominate leading to reaction at the C5-center. In addition, the C5-position is less hindered by the neighboring C4,C6-methoxys than the C8-position with larger flanking groups (C1-aryl group and C7-propyl group).
Scheme 17.

Regioselection in the PIFA-mediated Hydroxylation.
 |
(6) |
The requisite substrate to test this hypothesis was prepared from 52d using reagent grade DMF during methylation to conveniently prepare hexamethyl ether 82 in one step.33 After initial C2,C2’-methylation, the small amount of water in the DMF was converted to NaOH, cleaving the C4,C4’-acetates; subsequent methylation afforded 82 (Scheme 18). Treatment of 82 with PIFA and TMSOAc in (CF3)2CHOH afforded a mixture of compounds, from which bisphenol 87 and the C5,C5’-bistrifluoroacetate 86 were isolated. The reaction also proceeded well without TMSOAc; presumably, trifluoroacetate from the PIFA is sufficiently nucleophilic to trap the radical cation. When the reaction mixture (without TMSOAc) was treated with aqueous NaOH, the bistrifluoroacetate intermediate 86 was efficiently cleaved furnishing 87 in 89% yield (Scheme 18). However, there was erosion in the optical purity, from 86% ee in 82 to 68–70% ee in bisnaphthol 87. We propose that this attrition arises from racemization of radical cation or cation intermediates where the atropisomerization barrier is lower.28 To mitigate this effect, hexafluoroisopropanol was replaced with the less polar trifluoroethanol, which should be sufficiently polar to permit the initial SET to the radical cation but will reduce its lifetime. Pleasingly, bisnaphthol 87 was obtained with improved enantiopurity (82% ee) but in a diminished 51% yield.42
Scheme 18.

PIFA-mediated Hydroxylation.
The oxidative cyclization of 87 with MnO2 proceeded smoothly to yield perylenequinone 88,16 representing the first enantioselective synthesis of a perylenequinone with the helical chirality as the only stereochemical element. Since the optical purity of 88 could not be measured directly, the C4,C4’-methyl ethers were selectively removed using MgI2 to provide 89.15,17 An HPLC assay verified the stereochemical integrity of the axial to helical chirality transfer (Scheme 19).
Scheme 19.

Synthesis of Optically Active Perylenequinone 89.
Decarboxylation and Atropisomeric Stability
This initial result on the atropisomeric stability of a perylenequinone with no stereogenic elements aside from the helical axis was encouraging. However, it was unclear that the corresponding compound lacking the C3,C3’-ester groups, which were needed as outlined in the retrosynthesis (see 33 in Scheme 6), would possess the same degree of atropisomeric stability. Removal of the C3,C3’-ester groups alleviates steric gearing interactions thereby decreasing the effective size of the C2,C2’-groups so that they might more easily slide by one another, thereby lower the atropisomerization barrier.
Thus, the synthesis of 39, which contains the necessary substitution pattern of the natural products perylenequinone core, was our next goal (Scheme 20). To accomplish this goal, 87 was treated with benzyl bromide and NaH to mask the reactive C5,C5’-phenols. Hydrolysis was affected by LiOH to supply the diacid 90. Disappointingly, the high temperatures of conventional aromatic decarboxylation protocols, such as Cu-quinoline, caused not only atropisomerization of the biaryl but also significant decomposition resulting in modest yields of 42 (31% after optimization). As a consequence we developed a palladium decarboxylation protocol involving stoichiometric palladium(II) trifluoroacetate and silver carbonate followed by the reduction of the resultant aryl palladium species. The described procedure worked well on these systems and supplied the key intermediate 42 in 87% yield without racemization.19,43 Further examination of this strategy lead to the development of a single-step decarboxylation using catalytic palladium and excess of trifluoroacetic acid as the proton source to provide a variety arenes in good yield.43 After hydrogenolysis of bisbenzyl ether 42, the axial chiral bisphenol was oxidized by MnO2 to afford the helical chiral perylenequinone as a bright red resin (Scheme 20). The directed, selective cleavage of the C4,C4’-methyl ethers was accomplished by MgI2 to complete the first synthesis of optically active perylenequinone 39.44,45 Pleasingly, perylenequinone 39 was also found to exhibit considerable atropisomeric stability with an atropisomerization half-life of 4 d at 60 °C in benzene.19
Scheme 20.

Palladium-Mediated Decarboxylation and Synthesis of Optically Active 39.
Summary
As summarized in Scheme 21, the final route to 39 proceeded in 18 steps and a 5.4% overall yield (92% average yield per step). The key steps in this synthesis were an enantioselective biaryl coupling, a PIFA-induced phenol formation, and a palladium-mediated aromatic decarboxylation. The development and optimization of these transformations was crucial not only to this investigation but also for the syntheses of the perylenequinone natural products detailed in the subsequent papers in this series.
Scheme 21.

Enantioselective Total Synthesis of (M)-39.
Conclusions
The synthesis of 39 provided the first helical chiral perylenequinone containing the natural product substitution pattern but absent the conventional stereogenic C7,C7’-substitution. The central issues addressed here including generation of the necessary oxygenation pattern in a complex binaphthol and formation of the helical stereochemistry. Importantly, such perylenequinones were shown to possess sufficient atropisomeric stability to be viable intermediates in the biogenesis of the perylenequinone natural products. Furthermore, this stability supports their use as key intermediates in biomimetic total synthesis ventures (see 33 in the retrosynthesis in Scheme 6). Thus, these discoveries laid the groundwork for the total syntheses of (+)-calphostin D (8d) and (+)-phleichrome (9)19 and the first total syntheses of cercosporin (6)19 and hypocrellin A (ent-2)18, which are described in the succeeding papers in this series. Importantly, the versatility of this strategy enabled selective and convergent synthesis of all stereoisomeric combinations permitting comparison of their biological effects for the first time.19
Experimental Section
2-(4-Methoxy-3-propylphenyl)acetic acid (49)
To a solution of 47 (1.0 g, 4.0 mmol) in MeOH (50 mL) was added 10% Pd/C (106 mg) and the mixture was stirred under H2 (1 atm). After 2 d, the reaction was filtered through Celite, rinsing with CH2Cl2. This solution was filtered through SiO2 and the solvent was evaporated to yield the dipropyl product as an oil (1.0 g, 100%): 1H NMR (360 MHz, CDCl3) δ 0.90–0.97 (m, 6H), 1.57–1.67 (m, 4H), 2.57 (t, J = 7.5 Hz, 2H), 3.54 (s, 2H), 3.80 (s, 3H), 4.05 (t, J = 6.5 Hz, 2H), 6.78 (d, J = 8.1 Hz, 1H), 7.06 (m, 2H).
A mixture of propyl ester (329 mg, 1.31 mmol) and 1 N NaOH (5 mL, 5 mmol) in THF (5 mL) was stirred 16 h. The reaction was diluted with 1 N NaOH and hexanes, and the aqueous phase was extracted with hexanes. The aqueous phase was then acidified with 1 N HCl and extracted with EtOAc. The organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield 49 as a white solid (250 mg, 92%): mp 58–60 °C; IR (thin film) 3076 (br), 3003, 2961, 1710, 1251 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.94 (t, J = 7.4 Hz, 3H), 1.56–1.62 (m, 2H), 2.56 (t, J = 7.6 Hz, 2H), 3.57 (s, 2H), 3.80 (s, 3H), 6.79 (d, J = 8.2 Hz, 1H), 7.03 (s, 1H), 7.07 (d, J = 8.3 Hz, 1H), 11.0 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 178.5, 157.0, 131.8, 131.3, 128.0, 125.3, 110.8, 55.8, 40.7, 32.6, 23.3, 14.5; HRMS (CI): [M+] calcd for C12H16O3, 208.1099; found, 208.1104.
Methyl 1,3-dihydroxy-7-methoxy-6-propyl-2-naphthoate (50)
To a solution of acid 49 (6.4 g, 30.7 mmol) in PhH (30 mL) was added SOCl2 (9.0 mL, 123 mmol). The reaction was heated to reflux under Ar for 2 h. The solvent was evaporated in vacuo to yield a brown oil. To a suspension of NaH (60 % in mineral oil, 3.7 g, 92.1 mmol) in THF (200 mL) under Ar was added dimethyl malonate (10.5 mL, 92.1 mmol). After stirring for 30 min, the mixture was chilled in an ice-H2O bath and a solution of the above acid chloride in THF (50 mL) was added. The reaction was stirred for 3 h with warming to room temperature. The mixture was poured into a mixture of ice and 1 N HCl and extracted into EtOAc. The organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated. The oil was filtered through SiO2, rinsing with 0–20% EtOAc/hexanes. The solvent was evaporated and the oil was dissolved in MeSO3H (55 mL), and P2O5 (5.0 g) was added. After stirring under Ar for 3 h, the mixture was poured over ice-H2O and filtered. The yellow solid was dissolved in CH2Cl2 and was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield 50 (6.9 g, 77%) that was carried on to the next step without further purification: IR (thin film) 3443, 2957, 1670, 1644, 1252, 1217, 1158 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.99 (t, J = 7.3 Hz, 3H), 1.64–1.71 (m, 2H), 2.69 (t, J = 7.4 Hz, 2H), 3.92 (s, 3H), 4.10 (s, 3H), 6.71 (s, 1H), 7.29 (s, 1H), 7.43 (s, 1H), 8.63 (br s, 1H), 11.32 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 171.0, 160.4, 155.2, 152.4, 138.5, 133.7, 126.7, 118.9, 102.4, 101.2, 97.2, 55.7, 53.3, 33.3, 23.0, 14.5; HRMS (CI): [M+] calcd for C16H18O5, 290.1154; found, 290.1156.
Methyl 1-acetoxy-3-hydroxy-7-methoxy-6-propyl-2-naphthoate (45d)
A solution of naphthol 50 (6.0 g, 20.7 mmol), Ac2O (60 mL) and pyridine (60 mL) was stirred for 3 h. The reaction mixture was poured over ice-1 N HCl and stirred 30 min. The solid was filtered, rinsing with H2O, and dried by pulling air through the filter cake to yield the diacetate (6.9 g, 89%): IR (thin film) 2955, 1776, 1724, 1316, 1195 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.97 (t, J = 7.3 Hz, 3H), 1.64–1.69 (m, 2H), 2.32 (s, 3H), 2.44 (s, 3H), 2.72 (t, J = 7.4 Hz, 2H), 3.90 (s, 3H), 3.91 (s, 3H), 7.01 (s, 1H), 7.37 (s, 1H), 7.52 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 170.0, 169.2, 164.6, 158.0, 146.4, 143.8, 137.1, 130.6, 128.2, 125.6, 118.4, 116.5, 99.5, 55.7, 52.8, 33.1, 22.9, 21.3, 21.2, 14.4; HRMS (ESI) calcd for C20H22O7 (M+) 374.1366, found 374.1364.
To a solution of this acetate (2.5 g, 6.7 mmol) in MeOH (150 mL) and CH2Cl2 (15 mL) under Ar was added K2CO3 (913 mg, 6.6 mmol). After stirring for 15 min, the green reaction mixture was poured over ice-1 N HCl. The aqueous phase was extracted with CH2Cl2 and the organic extracts were washed with brine, dried (Na2SO4), and the solvent was evaporated. Purification was accomplished with chromatography (10–20% EtOAc/hexanes) to yield product 45d as a yellow solid (1.7 g, 76%): mp 121.5–125.5 °C; IR (thin film) 3155, 2957, 1773,46 1675, 1223, 1197 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.98 (t. J = 7.4 Hz, 3H), 1.65–1.69 (m, 2H), 2.48 (s, 3H), 2.69 (t, J = 7.5 Hz, 2H), 3.89 (s, 3H), 4.00 (s, 3H), 6.91 (s, 1H), 7.17 (s, 1H), 7.39 (s, 1H), 10.48 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 169.9, 169.6, 156.3, 155.2, 147.6, 138.5, 133.8, 127.0, 121.6, 110.1, 107.2, 99.1, 55.5, 53.4, 33.2, 22.9, 21.3, 14.4; HRMS (CI) calcd for C18H20O6 (M+) 332.1260, found 332.1250.
General Procedure for the Oxidative Biaryl Coupling
To a solution of the 2-naphthol substrate dissolved in the appropriate solvent was added the copper catalyst (10 mol%). The mixture was sonicated to yield a clear green, blue, purple, or red solution, which was stirred under O2 (1 atm) at the indicated temperature and time. After cooling, the reaction mixture was diluted with CH2Cl2 and washed with 1 N HCl. The aqueous phase was back extracted with CH2Cl2 and the combined organic solutions were dried over Na2SO4. Filtration and concentration afforded the crude product. Purification was accomplished by SiO2 chromatography.
(M)-Dimethyl 4,4'-diacetoxy-2,2'-dihydroxy-6,6'-dimethoxy-7,7'-dipropyl-1,1'-binaphthyl-3,3'- dicarboxylate (52d)
A mixture of naphthol 45d (103 mg, 0.309 mmol), 4 Å molecular sieves (86 mg) and CuI-(S,S-51) (11.4 mg, 0.0329 mmol) in MeCN (1.5 mL) was stirred at room temperature under an O2 atmosphere for 24 h. The reaction was quenched with 1 N HCl, and the aqueous phase was extracted with EtOAc. The organic extracts were washed with H2O and brine, dried (Na2SO4), and filtered through SiO2 with 10% MeOH/CH2Cl2. Purification was accomplished via chromatography to yield the product as a resin (86.7 mg, 85%) in 87% ee: +20.9 (c 0.23, MeOH, 87% ee); IR (thin film) 3130, 2958, 1770, 1672, 1234, 1195 cm−1; 1H NMR (300 MHz, CDCl3) δ 0.79 (t, J = 7.4 Hz, 6H), 1.39–1.44 (m, 4H), 2.44–2.55 (m, 10H), 3.90 (s, 6H), 4.01 (s, 6H), 6.91 (s, 2H), 7.05 (s, 2H), 10.64 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 170.1, 169.7, 156.3, 152.9, 147.8, 138.7, 133.2, 125.8, 121.8, 115.4, 107.3, 99.7, 55.5, 53.4, 33.5, 23.2, 21.9, 14.3; HRMS (ESI) calcd for C36H38O12Na (MNa+) 685.2261, found 685.2276; CSP HPLC (Chiralpak AD, 1.0 mL/min, 70:30 hexanes:iPrOH) tR(S) = 10.5 min, tR(R) = 13.3 min.
Dimethyl 2,2'-bis(benzyloxy)-4,4'-dihydroxy-7,7'-dimethoxy-5,5',6,6'-tetraoxo-5,5',6,6'- tetrahydro-1,1'-binaphthyl-3,3'-dicarboxylate (54)
Bisphenol 53(51 mg, 0.07 mmol, 74% ee) was dissolved in MeCN:CH2Cl2:H2O (6:1:1, 8 mL) and PhI(OCOCF3)2 (300 mg, 0.7 mmol) was added. After 2 h at room temperature, the deep red solution was diluted with CH2Cl2 and washed with H2O. Concentration afforded an oil which was purified by SiO2 chromatography (2–3.5% MeOH/ CH2Cl2) to provide 54 as a red solid (45 mg, 88%): IR (thin film) 3683, 2954, 1733, 1693, 1636, 1608, 1274, 1261, 1227, 1115 cm−1; 1H NMR (500 MHz, CD2Cl2/d6-DMSO) δ 3.60 (s, 6H), 3.99 (s, 6H), 4.84 (d, J = 9.0 Hz, 2H), 5.08 (d, J = 9.0 Hz, 2H), 5.84 (s, 2OH), 7.09 (m, 4H), 7.24 (m, 6H), 12.9 (s, 2OH); 13C NMR (125 MHz, CD2Cl2/d6-DMSO) δ 179.8, 175.3, 165.4, 163.6, 152.8, 137.1, 135.6, 128.5, 128.4, 128.1, 127.7, 126.7, 119.7, 115.3, 109.2, 109.1, 76.2, 56.0, 53.0; HRMS (ESI) cald for C40H30O14Na (MNa+) 757.1558, found 757.1533.
Dimethyl 2,2',4,4'-tetramethoxy-5,5',6,6'-tetraoxo-7,7'-dipropyl-5,5',6,6'-tetrahydro-1,1'-binaphthyl-3,3'-dicarboxylate (61)
To a solution of binaphthol 59 (230 mg, 0.379 mmol) in MeCN (50 mL), CH2Cl2 (20 mL), and H2O (10 mL) was added PhI(OC(O)CF3)2 (1.2 g, 2.8 mmol). The dark blue reaction mixture was stirred for 45 min, then diluted with H2O and EtOAc. The phases were separated and the aqueous phase was washed with EtOAc. The organic phase was extracted with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield 60 as an orange solid that was carried on to the next step without further purification: 1H NMR (500 MHz, CDCl3) δ 0.87 (t, J = 7.3 Hz, 6H), 1.39–1.44 (m, 4H), 2.32 (t, J = 6.7 Hz, 4H), 3.80 (s, 6H), 3.99 (s, 6H), 6.60 (s, 2H), 12.8 (br s, 2H); CSP HPLC (Chiralpak AD, 1.0 mL/min, 70:30 hexanes:iPrOH) tR(S) = 12.3 min, tR(R) = 22.2 min.
To a solution of ortho-quinone (0.379 mmol) and MeI (0.20 mL, 3.2 mmol) in THF (30 mL) was added TBAF (1 M in THF, 1.5 mL, 1.5 mmol). The black reaction mixture was stirred for 16 h, then diluted with EtOAc and H2O. The phases were separated and the aqueous phase was washed with EtOAc. The organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield an orange solid. Purification by chromatography (30% EtOAc/hexanes) yielded product 61 as an orange solid (168 mg, 70%): mp 95–97 °C; IR (thin film) 2955, 1737, 1667, 1224 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.87 (t, J = 7.3 Hz, 6H), 1.41 (m, 4H), 2.31 (m, 4H), 3.71 (s, 6H), 3.94 (s, 6H), 3.97 (s, 6H), 6.71 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 180.8, 177.3, 165.7, 163.6, 161.1, 143.0, 138.0, 137.0, 122.9, 122.7, 118.9, 63.8, 60.8, 53.5, 32.0, 21.9, 14.0; HRMS (ESI) calcd for C34H34O12Na (MNa+) 657.1948, found 657.1936; CSP HPLC (Chiralpak AD, 1.0 mL/min, 70:30 hexanes:iPrOH) tR(S) = 14.7 min, tR(R) = 36.3 min.
2,2',4,4'-Tetramethoxy-7,7'-dipropyl-1,1'-binaphthyl-5,5',6,6'-tetraone (40)
To a solution of 64 (3.0 mg, 0.0052 mmol) in MeCN (0.5 mL) was added H2O (1 drop) and CAN (31 mg, 0.057 mmol). The red reaction mixture was stirred for 10 min, then diluted with H2O and extracted with EtOAc. The organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield 40 as an orange oil that was not stable to further purification: 1H NMR (500 MHz, CDCl3) δ 0.83 (t, J = 7.4 Hz, 6H), 1.35–1.40 (m, 4H), 2.26 (t, J = 6.4 Hz, 4H), 3.83 (s, 6H), 4.08 (s, 6H), 6.52 (s, 2H), 6.69 (s, 2H).
2,2’,6,6’-Tetramethoxy-4,4’-diox-7,7’-dipropyl-4H,4’H-[1,1’]binaphthalenylidene-3,3’- dicarboxylic acid dimethyl ester (69)
A mixture of MeCN (7 mL), CH2Cl2 (3 mL), and H2O (1 mL) was degassed with Ar for 15 min. Naphthol 59 (32.3 mg, 0.0532 mmol) was added followed by PIDA (44.2 mg, 0.118 mmol). The reaction mixture turned a dark blue color and was stirred for 1 h. The reaction mixture was diluted with EtOAc and H2O, the phases were partitioned, and the organic layer was washed with H2O and brine. After drying (Na2SO4) the solvent was evaporated. Purification was accomplished with chromatography to yield 29 mg (91%) of a dark green resin that was not stable in solution over extended periods of time: IR (thin film) 2957, 1737, 1586, 1227 cm−1; 1H NMR (500 MHz, CD2Cl2) δ 0.86 (t, J = 7.4 Hz, 6H), 1.47–1.50 (m, 2H), 2.52 (m, 4H), 3.58 (s, 6H), 3.90 (s, 6H), 3.97 (s, 6H), 7.34, (s, 2H), 7.50 (s, 2H); HRMS (ES) calcd for C34H37O10 (MH+) 605.2387, found 605.2412. Bianthrone 69 was unstable in solution over time precluding 13C NMR characterization.
(M)-Dimethyl 2,2',4,4',6,6'-hexamethoxy-7,7'-dipropyl-1,1'-binaphthyl-3,3'-dicarboxylate (82)
To a solution of binaphthol (M)-52d (70 mg, 0.11 mmol) in DMF (2 mL) under an Ar atmosphere was added NaH (60%, 82 mg, 2.1 mmol). The mixture was stirred for 10 min, then CH3I (0.2 mL, 3.2 mmol) was added. The reaction was stirred at room temperature for 18 h. The yellow mixture was quenched with H2O and the aqueous phase was extracted with EtOAc. The organics were washed with brine, dried (MgSO4), and the solvent was evaporated in vacuo to yield a yellow oil. Purification was accomplished via chromatography (25% EtOAc/hexanes) to yield 82 as a resin (52 mg, 75%): – 26.4 (c 0.22, MeOH, 86% ee); IR (thin film) 2956, 1731, 1592, 1493, 1454, 1224, 1085, 1015, 733 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.74 (t, J = 7.3 Hz, 6H), 1.37–1.41 (m, 4H), 2.44–2.49 (m, 4H), 3.31 (s, 6H), 3.95 (s, 6H), 3.97 (s, 6H), 4.13 (s, 6H), 6.90 (s, 2H), 7.40 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 167.9, 156.7, 153.6, 151.8, 135.5, 130.8, 127.1, 124.9, 120.4, 120.2, 100.2, 63.1, 62.3, 55.8, 52.9, 33.2, 23.2, 14.2; HRMS (ESI) calcd for C36H42O10Na (MNa+) 657.2676, found 657.2666.
(M)-Dimethyl 5,5'-dihydroxy-2,2',4,4',6,6'-hexamethoxy-7,7'-dipropyl-1,1'-binaphthyl-3,3'- dicarboxylate (87)
To a solution of binaphthalene 82 (131 mg, 0.269 mmol) in CF3CH2OH (10 mL) under an Ar atmosphere was added PhI(OC(O)CF3)2 (199 mg, 0.463 mmol). After stirring the purple mixture for 30 min, the solvent was evaporated in vacuo. The resultant mixture was treated with a mixture of 1 N NaOH (0.1 mL, 0.1 mmol) in H2O/THF/EtOH (1:1:1, v:v:v) for 30 min, followed by acidification with 1 N HCl. The aqueous phase was extracted with EtOAc, the organics were washed with brine, dried (Na2SO4), and the solvent was evaporated. Purification was accomplished by chromatography (25% EtOAc/hexanes) to yield product 87 as a yellow resin (70 mg, 51%, 82% ee): –12.1 (c 0.22, MeOH, 76% ee); IR (film) 3369, 2957, 1732, 1311 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.74 (t, J = 7.2 Hz, 6H), 1.35–1.42 (m, 4H), 2.42–2.51 (m, 4H), 3.32 (s, 6H), 3.93 (s, 6H), 3.98 (s, 6H), 4.16 (s, 6H), 6.42 (s, 2H), 9.24 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 167.1, 154.4, 152.6, 146.0, 142.6, 139.8, 132.9, 120.8, 119.2, 117.6, 114.2, 64.6, 62.3, 60.9, 53.2, 33.2, 23.9, 14.2; HRMS (ESI) calcd for C36H42O12Na (MNa+) 689.2574, found 689.2599; CSP HPLC (Chiralpak AD, 1.0 mL/min, 98:2 hexanes:i-PrOH) tR(S) = 12.3 min, tR(R) = 15.6 min.
(M)-5,5'-Bis(benzyloxy)-2,2',4,4',6,6'-hexamethoxy-7,7'-dipropyl-1,1'-binaphthyl-3,3'- dicarboxylic acid (90)
To a solution of 68% ee binaphthol 87 (37 mg, 0.055 mmol) and benzyl bromide (46 µL, 0.38 mmol) in DMF (1.0 mL) under an Ar atmosphere was added 60% NaH (12.5 mg, 0.312 mmol). The dark green mixture was stirred for 15 h, quenched with H2O, and washed with EtOAc. The organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield an orange oil. Purification was accomplished by chromatography (15% EtOAc/hexanes) to yield the bisbenzyl ether (36 mg, 78%) as a yellow resin: –24.7 (c 1.0, CHCl3, 68% ee); IR (thin film) 2939, 1735, 1330, 1094 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.75 (t, J = 7.4 Hz, 6H), 1.39–1.43 (m, 4H), 2.49–2.54 (m, 4H), 3.39 (s, 6H), 3.95 (s, 6H), 3.98 (s, 6H), 4.00 (s, 6H), 5.09 (m, 4H), 6.76 (s, 2H), 7.38 (t, J = 7.3 Hz, 2H), 7.44 (t, J = 7.3 Hz, 4H), 7.63 (d, J = 7.0 Hz, 4H); 13C NMR (125 MHz, CDCl3) δ 167.8, 154.4, 152.6, 146.8, 138.9, 138.4, 133.8, 129.2, 128.8, 128.3, 127.4, 123.1, 120.7, 120.3, 114.2, 77.1, 64.6, 62.3, 60.9, 53.2, 33.2, 23.9, 14.2; HRMS (ESI) calcd for C50H54O12Na (MNa+) 869.3513, found 869.3541.
To a suspension of the bisbenzyl ether (163 mg, 0.192 mmol) in dioxane (8 mL) and H2O (9 mL) was added LiOH·H2O (402 mg, 9.57 mmol), and the mixture was heated to reflux for 24 h. The brown mixture was cooled to room temperature and diluted with H2O. The aqueous phase was washed with EtOAc, and this organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to recover 73 mg (0.086 mmol, 45%) of starting material. The aqueous phase was acidified with 1 N HCl, extracted with EtOAc, and the organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield the diacid 90 (81 mg, 93% based on recovered starting material) as an orange resin that was used for the next step without further purification: – 29.1° (c 1.0, CHCl3, 82% ee); IR (thin film) 2937, 1736, 1707, 1327, 1107 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.76 (t, J = 7.3 Hz, 6H), 1.40–1.44 (m, 4H), 2.50–2.54 (m, 4H), 3.44 (s, 6H), 3.72 (s, 6H), 3.98 (s, 6H), 3.99 (s, 6H), 5.11 (dd, J = 9.8, 14.9 Hz, 4H), 6.80 (s, 2H), 7.36 (t, J = 7.3 Hz, 2H), 7.43 (t, J = 7.3 Hz, 4H), 7.64 (d, J = 7.0 Hz, 4H); 13C NMR (125 MHz, CDCl3) δ 172.5, 155.2, 152.4, 150.7, 146.9, 139.3, 138.2, 134.5, 129.3, 128.8, 128.3, 122.9, 122.1, 120.6, 120.1, 67.5, 65.2, 62.4, 61.7, 33.2, 23.9, 14.2; HRMS (ESI) calcd for C48H50O12Na (MNa+) 841.3200, found 841.3240.
(M)-5,5'-Bis(benzyloxy)-2,2',4,4',6,6'-hexamethoxy-7,7'-dipropyl-1,1'-binaphthyl (42)
To a solution of diacid 90 (26.7 mg, 0.0326 mmol) in DMSO-DMF (0.3 mL) was added Pd(OC(O)CF3)2 (22.8 mg, 0.0739 mmol). The mixture was heated in a 90 °C oil bath for 1 h. After cooling the brown reaction mixture to room temperature, 1 N HCl was added, and the mixture was extracted with EtOAc. The organic phase was washed with H2O and brine, dried (Na2SO4), and the solvent was evaporated to yield a brown oil. This material was dissolved in THF (1.0 mL) and stirred under a H2 atmosphere for 5 min. After filtration through Celite, purification was accomplished via chromatography (20% EtOAc/heanes) to yield 42 (20.7 mg, 87%) as a resin: 0 (c 1.0, CHCl3, 70% ee)*; IR (thin film) 2933, 1338 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.75 (t, J = 7.2 Hz, 6H), 1.35–1.42 (m, 4H), 2.46–2.53 (m, 4H), 3.74 (s, 6H), 3.92 (s, 6H), 3.99 (s, 6H), 5.10 (m, 4H), 6.64 (s, 2H), 6.76 (s, 2H), 7.37 (t, J = 7.3 Hz, 2H), 7.43 (t, J = 7.3 Hz, 4H), 7.62 (d, J = 7.0 Hz, 4H); 13C NMR (125 MHz, CDCl3) δ 157.1, 154.9, 148.8, 147.2, 138.9, 137.0, 133.7, 129.0, 128.5, 127.8, 122.1, 116.5, 112.5, 95.9, 76.2, 61.4, 57.5, 56.1, 32.8, 23.6, 14.0; HRMS (ESI) calcd for C46H50O8Na (MNa+) 753.3403, found 753.3427.
*This compound was re-synthesized but the optical rotation was still 0. The compound was carried on to the next step and enantioselectivity confirmed by CSP HPLC (69% ee).
4,9-Dihydroxy-2,6,7,11-tetramethoxy-1,12-dipropylperylene-3,10-dione (39)
To a solution of binaphthalene 42 (6.4 mg, 0.0088 mmol) in MeOH (0.3 mL) and THF (0.3 mL) was added 10% Pd/C (2 mg). The mixture was stirred under an atmosphere of H2 for 3 h. The reaction was filtered through Celite, rinsing with MeOH and CH2Cl2. The solvents were evaporated to yield an unstable yellow oil (69% ee): 1H NMR (500 MHz, CDCl3) δ 0.76 (t, J = 7.3 Hz, 6H), 1.38–1.43 (m, 4H), 2.43–2.47 (m, 4H), 3.69 (s, 6H), 3.89 (s, 6H), 4.16 (s, 6H), 6.37 (s, 2H), 6.70 (s, 2H), 9.28 (s, 2H); CSP HPLC (Chiralpak AD, 1.0 mL/min, 98:2 hexanes:i-PrOH) tR(S) = 37.9 min, tR(R) = 45.2 min.
To a solution of bisnaphthol (11 mg, 0.020 mmol) in Et2O (1 mL) was added MnO2 (60 mg, 0.69 mmol). The mixture was stirred for 30 min, filtered through Celite, and the solvent was evaporated to yield a red resin, which was chromatographed (2.5% MeOH/CH2Cl2) to yield the perylenequinone as a red resin: –410 (c 0.021, MeOH, 69% ee); 1H NMR (500 MHz, CDCl3) δ 0.54 (t, J = 7.3 Hz, 6H), 0.96–0.99 (m, 2H), 1.18–1.22 (m, 2H), 2.48–2.51 (m, 2H), 3.08–3.11 (m, 2H), 4.05 (s, 6H), 4.11 (s, 6H), 4.17 (s, 6H), 6.78 (s, 2H).
To a solution of the above perylenequinone product (12 mg, 0.0221 mmol) in THF (2 mL) under an argon atmosphere was added a solution of MgI2 in Et2O (0.07 M, 665 µL, 0.0464 mmol). The dark purple mixture was stirred 10 min (until the mixture turns from purple to black), diluted with EtOAc, washed with saturated aq NH4Cl, and dried (Na2SO4). Concentration yielded a red residue, which was chromatographed (2.5% MeOH/CH2Cl2) to yield product 39 as a red resin (10 mg, 87% (69% for the three steps)): 1H NMR (500 MHz, CDCl3) δ 0.52 (t, J = 7.2 Hz, 6H), 0.88 (m, 2H), 1.25 (m, 2H), 2.76 (m, 2H), 3.33 (m, 2H), 4.09 (s, 6H), 4.21 (s, 6H), 6.59 (s, 2H), 15.82 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 14.1, 24.3, 35.0, 56.3, 61.1, 101.1, 105.7, 116.5, 126.2, 127.5, 140.5, 151.0, 166.5, 174.1, 176.8; IR (film) 3383, 2927, 2858, 1607, 1460, 1274, 1213 cm−1; HRMS (ES) calcd for C30H31O8 (MH+) 519.2019, found 519.2036; See Supporting Information for CD spectrum; CSP HPLC (Chiralpak AD-H, 0.5 mL/min, 95:5 hexanes:i-PrOH) tR (P) = 15.9 min, tR (M) = 17.1 min.
Supplementary Material
Scheme 3.

Dallacker Synthesis of Perylenequinone 27.
Scheme 16.

Attempted C5-Oxidation of a naphthalene model system.
Acknowledgements
We are grateful to the NIH (CA-109164) and NSF (CHE-0911713) for financial support. Partial instrumentation support was provided by the NIH for MS (1S10RR023444) and NMR (1S10RR022442). We thank 3D Pharmaceuticals (C.A.M.), Novartis (B.J.M.), and the Division of Organic Chemistry of the American Chemical Society (C.A.M., B.J.M.) for graduate fellowships. We thank Feng Gai for assistance with CD measurements.
Footnotes
Dedicated to the memory of Ralph Hirschmann.
Supporting Information Available Additional experimental descriptions, NMR spectra, and crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zinke A, Hirsch W, Brozek E. Monatsch. Chem. 1929;51:205–220. [Google Scholar]
- 2.Calderbank A, Johnson AW, Todd AR. J. Chem. Soc. 1954:1285–1289. [Google Scholar]
- 3.Anderson JM, Murray J. Chem. Ind. 1956:376–376. [Google Scholar]
- 4.This class also includes some partially reduced compounds from the molds of Alternaria and Stemphylium.
- 5.Kuyama S, Tamura T. J. Am. Chem. Soc. 1957;79:5725–5726. [Google Scholar]
- 6.a) Lousberg RJJ, Weiss U, Salemink CA, Arnone A, Merlini L, Nasini G. Chem. Commun. 1971:1463–1464. [Google Scholar]; b) Yamazaki S, Ogawa T. Agr. Biol. Chem. 1972;36:1707–1718. [Google Scholar]
- 7.Nasini G, Merlini L, Andreetti GD, Bocelli G, Sgarabotto P. Tetrahedron. 1982;38:2787–2796. [Google Scholar]
- 8.For a review, see: Daub ME, Herrero S, Chung K-R. FEMS Microbiol. Lett. 2005;252:197–206. doi: 10.1016/j.femsle.2005.08.033. and references therein.
- 9.Hongyu Z, Zhiyi Z. Chinese Sci. Bull. 1997;42:2005–2009. [Google Scholar]
- 10.The chelation allows for keto-enol tautomerization; see reference 21.
- 11.a) Liu WZ. M.D. thesis. Yunnan University; 2000. “Fermentation and Structure Determination of Hypomycin B”. [Google Scholar]; b) Liu WZ, Chen YT, Xie JL. J. Yunnan Univ., Nat. Sci. Ed. 2000;22:389–391. [Google Scholar]; c) Liu WZ, Ma LY, Li C, Chen YT, Xie JL. Acta Pharmacol. Sin. 2001;36:313–314. [PubMed] [Google Scholar]
- 12.Chao C, Zhang P. Tetrahedron Lett. 1988;29:225–226. [Google Scholar]
- 13.Diwu Z, Lown JW. Tetrahedron. 1992;48:45–54. [Google Scholar]; b) Liu J, Diwu Z, Lown JW. Tetrahedron. 1993;49:10785–10792. [Google Scholar]
- 14.Broka CA. Tetrahedron Lett. 1991;32:859–862. [Google Scholar]
- 15.Hauser FM, Sengupta D, Corlett SA. J. Org. Chem. 1994;59:1967–1969. [Google Scholar]
- 16.a) Coleman RS, Grant EB. J. Am. Chem. Soc. 1994;116:8795–8796. [Google Scholar]; b) Coleman RS, Grant EB. J. Am. Chem. Soc. 1995;117:10889–10904. [Google Scholar]
- 17.a) Merlic CA, Aldrich CC, Albaneze-Walker J, Saghatelian A. J. Am. Chem. Soc. 2000;122:3224–3225. doi: 10.1021/ja994313+. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Merlic CA, Aldrich CC, Albaneze-Walker J, Saghatelian A, Mammen J. J. Org. Chem. 2001;66:1297–1309. doi: 10.1021/jo0014663. [DOI] [PubMed] [Google Scholar]
- 18.O’Brien EM, Morgan BJ, Kozlowski MC. Angew. Chem. Int. Ed. 2008;47:6877–6880. doi: 10.1002/anie.200800734. [DOI] [PubMed] [Google Scholar]
- 19.Morgan BJ, Dey S, Johnson SW, Kozlowski MC. J. Am. Chem. Soc. 2009;131:9413–9425. doi: 10.1021/ja902324j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Weisgraber KH, Weiss U. J. Chem. Soc. Perkin Trans. 1. 1972:83–88. doi: 10.1039/p19720000083. [DOI] [PubMed] [Google Scholar]; b) Weiss U, Fales HM, Weisgraber KH. Liebigs Ann. Chem. 1979:914–919. [Google Scholar]; c) Fales HM, Weiss U, Jaouni T. Liebigs Ann. Chem. 1983:367–371. [Google Scholar]
- 21.The proposed structure of 22 is the keto-enol tautomer of the mold perylenequinones seen in Figure 2, with exception of hypomycin B, 15. The perylenequinones actually exist as a mixture of the two tautomers, though the predominance of each tautomeric form, is different for each natural product: Arnone A, Merlini L, Mondelli R, Nasini G, Ragg E, Scaglioni L, Weiss U. J. Chem. Soc. Perkin Trans. 2. 1993:1447–1454..
- 22.Dallacker F, Leidig H. Chem. Ber. 1979;112:2672–2679. [Google Scholar]
- 23.Kishi T, Tahara S, Tsuda M, Tanaka C, Takahashi S. Planta Med. 1991;57:376–379. doi: 10.1055/s-2006-960121. [DOI] [PubMed] [Google Scholar]
- 24.Ayers S, Zink DL, Mohn K, Powell JS, Brown CM, Murphy T, Brand R, Pretorius S, Stevenson D, Thompson D, Singh SB. J. Nat. Prod. 2007;70:425–427. doi: 10.1021/np0604937. [DOI] [PubMed] [Google Scholar]
- 25.Chen WS, Chen YT, Wang XY, Friedrichs E, Puff H, Breitmaier E. Liebigs Ann. Chem. 1981:1880–1885. [Google Scholar]
- 26.Sridhar M, Vadivel SK, Bhalerao UT. Tetrahedron Lett. 1997;38:5695–5696. [Google Scholar]
- 27.Feringa B, Wynberg H. Bioorg. Chem. 1978;7:397–408. [Google Scholar]
- 28.For a preliminary communication of this work, see: Mulrooney CA, Li X, DiVirgilio ES, Kozlowski MC. J. Am. Chem. Soc. 2003;125:6856–6857. doi: 10.1021/ja027745k..
- 29.In prior work, Zhang (reference 12) and Lown (reference 13) have shown similar transformations in the racemic syntheses of perylenequinones. Hauser (reference 15) also utilized dimerization of an ortho-quinone, but found the pathway from the bis-ortho-quinone to the perylenequinone to be unproductive.
- 30.Broadhurst MJ, Hassal CH, Thomas GJ. J. Chem. Soc., Perkin Trans. 1. 1982:2239–2248. [Google Scholar]
- 31.a) Li X, Yang J, Kozlowski MC. Org. Lett. 2001;3:1137–1140. doi: 10.1021/ol015595x. [DOI] [PubMed] [Google Scholar]; b) Kozlowski MC, Li X, Carroll PJ, Xu Z. Organometallics. 2002;21:4513–4522. [Google Scholar]; c) Xie X, Phuan P-W, Kozlowski MC. Angew. Chem., Int. Ed. 2003;42:2168–2170. doi: 10.1002/anie.200250325. [DOI] [PubMed] [Google Scholar]; d) Li X, Hewgley JB, Mulrooney C, Yang J, Kozlowski MC. J. Org. Chem. 2003;68:5500–5511. doi: 10.1021/jo0340206. [DOI] [PubMed] [Google Scholar]; e) Morgan BJ, Xie X, Phuan P-W, Kozlowski MC. J. Org. Chem. 2007;72:6171–6182. doi: 10.1021/jo070636+. [DOI] [PubMed] [Google Scholar]; f) DiVirgilio ES, Dugan EC, Mulrooney CA, Kozlowski MC. Org. Lett. 2007;9:385–388. doi: 10.1021/ol062468y. [DOI] [PubMed] [Google Scholar]; g) Kozlowski MC, Dugan EC, DiVirgilio ES, Maksimenka K, Bringmann G. Adv. Synth. Catal. 2007;349:583–594. [Google Scholar]; h) Hewgley JB, Stahl SS, Kozlowski MC. J. Am. Chem. Soc. 2008;130:12232–12233. doi: 10.1021/ja804570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Further, investigations of the biaryl coupling reaction are examined in the subsequent paper in this series.
- 33.In a later synthesis involving substrate 52d, wet DMF is used as the solvent. These conditions cleave the acetates (presumably due to NaOH formed from trace H2O combined with the NaH) revealing a tetraphenol, which is exhaustively alkylated with the MeI to form a tetramethyl ether (82, Scheme 18).
- 34.Compound 62 proved unstable to SiO2 chromatography, but a small portion was purified by semi-preparative reverse-phase HPLC to confirm the structure via 1H NMR spectroscopy.
- 35.a) Stang PJ, Zhdankin VV. Chem. Rev. 1996;96:1123–1178. doi: 10.1021/cr940424+. [DOI] [PubMed] [Google Scholar]; b) Moriarty RM, Prakash O. Acc. Chem. Res. 1986;19:244–250. [Google Scholar]; c) Pohnert G. J. Prakt. Chem. 2000;342:731–734. [Google Scholar]
- 36.Pelter A, Ward RS. Tetrahedron. 2001;57:273–282. [Google Scholar]
- 37.The dinaphthofuranedione 24 in Scheme 2 (reference 20) is also similar to the strained extended quinone structure of 69.
- 38.a) Kral A, Laatsch H. Z. Naturforsch. 1993;48b:1401–1407. [Google Scholar]; b) Tapuhi Y, Kalisky O, Agranat I. J. Chem. Org. 1979;44:1949–1952. [Google Scholar]; c) Evans DH, Fitch A. J. Am. Chem. Soc. 1984;106:3039–3041. [Google Scholar]
- 39.a) Parker KA, Koziski KA. J. Org. Chem. 1987;52:674–676. [Google Scholar]; b) Davis FA, Wei J, Sheppard AC, Gubernick S. Tetrahedron Lett. 1987;28:5115–5118. [Google Scholar]; c) Green K. J. Org. Chem. 1991;56:4325–4326. [Google Scholar]
- 40.Kita Y, Tohma H, Hatanaka K, Takada T, Fujita S, Mitoh S, Sakurai H, Oka S. J. Am. Chem. Soc. 1994;116:3684–3691. [Google Scholar]
- 41.Arisawa M, Ramesh NG, Nakajima M, Tohma H, Kita Y. J. Org. Chem. 2001;66:59–65. doi: 10.1021/jo000953f. [DOI] [PubMed] [Google Scholar]
- 42.Improvements to the hydroxylation reaction were later developed, mitigating the loss of yield and enantioenrichment. As seen in the subsequent papers in this series on more complex intermediates, no erosion of enantioenrichment was observed when NaOAc was used to quench the reagents after oxidation. The NaOAc was added prior to solvent evaporation and subsequent NaOH treatment removes excess oxidant and TFA that can cause degradation.
- 43.Dickstein JS, Mulrooney CA, O’Brien EM, Morgan BJ, Kozlowski MC. Org. Lett. 2007;9:2441–2444. doi: 10.1021/ol070749f. [DOI] [PubMed] [Google Scholar]
- 44.Merlic, in reference 17, synthesized perylenequinone 39 as the racemate via the dimerization an ortho-quinone, but no logical entry to the enantioenriched variant is available with such a route.
- 45.Following this work, the synthesis of 39 evolved further, culminating in a 16 step protocol with 4.2% overall yield of 98% ee material. The evolution is described briefly in reference 19 and in detail in the third paper in this series.
- 46.Pouchert CJ, Behnke J, editors. The Aldrich Library of 13C and 1H FTNMR Spectra. Ed.1. U. S. A.: Aldrich Chemical Co., Inc.; 1993. The high wavenumber of the C=O in the acetate groups has been observed in simple acetate protected phenols. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.