

Abstract
SF2575 1 is a tetracycline polyketide produced by Streptomyces sp. SF2575 and displays exceptionally potent anticancer activity towards a broad range of cancer cell lines. The structure of SF2575 is characterized by a highly substituted tetracycline aglycon. The modifications include methylation of the C-6 and C-12a hydroxyl groups, acylation of the 4-(S)-hydroxyl with salicylic acid, C-glycosylation of the C-9 of the D-ring with d-olivose and further acylation of the C4′-hydroxyl of d-olivose with the unusual angelic acid. Understanding the biosynthesis of SF2575 can therefore expand the repertoire of enzymes that can modify tetracyclines, and facilitate engineered biosynthesis of SF2575 analogs. In this study, we identified, sequenced and functionally analyzed the ssf biosynthetic gene cluster which contains 40 putative open reading frames. Genes encoding enzymes that can assemble the tetracycline aglycon, as well as installing these unique structural features are found in the gene cluster. Biosynthetic intermediates were isolated from the SF2575 culture extract to suggest the order of pendant groups addition is C-9 glycosylation, C-4 salicylation and O-4′ angelycylation. Using in vitro assays, two enzymes that are responsible for C-4 acylation of salicylic acid were identified. These enzymes include an ATP-dependent salicylyl-CoA ligase SsfL1 and a putative GDSL family acyltransferase SsfX3, both of which were shown to have relaxed substrate specificity towards substituted benzoic acids. Since the salicylic acid moiety is critically important for the anticancer properties of SF2575, verification of the activities of SsfL1 and SsfX3 sets the stage for biosynthetic modification of the C-4 group towards structural-activity relationship studies of SF2575. Using heterologous biosynthesis in Streptomyces lividans, we also determined that biosynthesis of the SF2575 tetracycline aglycon 8 parallels that of oxytetracycline 4 and diverges after the assembly of 4-keto-anhydrotetracycline 51. The minimal ssf polyketide synthase together with the amidotransferase SsfD produced the amidated decaketide backbone that is required for the formation of 2-naphthacenecarboxamide skeleton. Additional enzymes, such as cyclases, C-6 methyltransferase and C-4/C-12a dihydroxylase were functionally reconstituted.
Keywords: topoisomerase, polyketide, salicylate, biosynthesis, acyltransferase
Bacterial aromatic polyketide natural products comprise a group of molecules that displays diverse structures and bioactivities yet share common biosynthetic origins. The poly-β-ketone backbone is synthesized by a minimal polyketide synthase (PKS) consisting of a ketosynthase (KS or KSα), a chain-length factor (CLF or KSβ) and an acyl carrier protein (ACP)1. Additional tailoring enzymes are required to fix the regioselectivity of sequential cyclization steps, and perform a multitude of decorating reactions to afford the various polycyclic bioactive compounds. A small, yet extremely important group of aromatic polyketides is the tetracycline family. Tetracyclines are characterized by their linearly fused four-ring structure and a heavily oxidized 2-naphthacenecarboxamide carbon skeleton. Other common features include the planar phenyldiketone arrangement and the tricarbonylmethane moiety in the A ring. This family is exemplified by the well-known tetracyclines chlorotetracycline 5 and oxytetracycline 42 (Scheme 1). Discovered in 1948 from Streptomyces aureofaciens3 and in 1950 from Streptomyces rimosus4, respectively, these compounds were commercialized as broad spectrum antibiotics and used successfully until bacterial resistance prompted the need for the creation of analogs to combat drug resistant strains5. The natural tetracycline scaffold has since been used to create second and third generation tetracyclines semisynthetically. The third generation tetracycline tigecycline gained FDA approval in 20056, 7, demonstrating the continued utility of the tetracycline scaffold. Remarkably, since the initial discovery of 4 and 5 over sixty years ago, there have been only a few natural tetracyclines discovered. Among these are SF25758, 9 1 and related compounds TAN-1518A 2 and TAN-1518B 310, chelocardin11 and dactylocycline12. Each of these compounds contains the tetracycline core and exhibits novel structural features not observed in 4 or 5. Given the rarity of tetracycline natural products, understanding the biosynthesis of these newly discovered compounds may provide a route to the generation of new tetracycline antibiotics through the use of combinatorial biosynthesis and metabolic engineering strategies.
Scheme 1.

SF2575 was first isolated by Hatsu et. al. from the producing strain Streptomyces sp. SF25759. The 2-naphthacenecarboxamide carbon framework of 1 is identical to that of 4 and 5. In addition, there are a number of unique tailoring modifications decorating the tetracyclic aglycon including two methoxy groups at C-6 and C-12a, a 4-(S)-salicylate that replaces the more common 4-(R)-dimethylamine substituent, a C-9 C-glycoside of 2-6-dideoxy-arabino-hexopyranose (d-olivose), and acylation of the O-4′ of d-olivose with (Z)-2-methyl-2-butenoate (angelate). The angelate and salicylate moieties are unusual polyketide modifications which add to the intrigue of the biosynthesis of 1. In addition to its unique structure, 1 also introduces potent anticancer bioactivity to the tetracycline family. Screening by Hatsu et. al. demonstrated that 1 has very weak antibiotic activity, which is most likely due to, among others, methylation of the C-12a hydroxyl group that disrupts interactions with the bacterial 30S ribosomal subunit13. SF2575 was, however, found to have exceptionally potent anticancer activity both through an in vitro cytotoxicity assay with P388 leukemia cells, and during an in vivo assay using mouse xenografts9. More recently, a 60-cell line screening by the National Cancer Institute demonstrated the potent activity of 1 against nearly all types of cancer cell lines tested, resulting in an average IC50 value of 11.2 nM. The mechanism of action of several closely related SF2575 analogs, 2 and 3 has been identified as inhibition of DNA topoisomerase I10. As these structures are nearly identical to that of 1, it is likely that this family of compounds shares a common molecular target. As key enzymes during DNA replication, both DNA topoisomerase I and II are known targets for current anticancer therapies in clinical use such as doxorubicin, a topoisomerase II poison14, and camptothecin derivatives which target topoisomerase I15. Elucidation of the SF2575 biosynthetic pathway can therefore be useful toward future structure-activity relationship (SAR) studies and engineered biosynthesis of new anticancer compounds.
In this report we have identified and sequenced the ssf gene cluster responsible for the biosynthesis of 1. This ssf gene cluster is only the third tetracycline family gene cluster to be identified and sequenced following chlorotetracycline16 and oxytetracycline17, 18. The genetic information offers valuable opportunities to further enhance our understanding of tetracycline biosynthesis, and to investigate an entirely new set of tetracycline tailoring modifications that distinguish 1 from the previously studied 4 and 5. Bioinformatic analysis of the gene cluster along with identification of the possible biosynthetic intermediates from S. sp. SF2575 fermentation extract led to the assembly of a putative biosynthetic pathway for 1. We have identified key tailoring enzymes involved in the attachment of salicylate to the tetracycline aglycon and verified their function through in vitro assays. Additionally, we have reconstituted the early portions of the ssf pathway in a heterologous host and demonstrated the biosynthesis of the tetracycline core of 1 parallels the biosynthetic pathway of 418-20.
Results and Discussion
Identification of SF2575 intermediates from crude extract
Comparing the core structures of 1 and 4, biosynthesis of the naphthacenecarboxamide carbon frameworks of 1 is predicted to parallel that of the oxytetracycline (oxy) biosynthetic pathway17, 18 and may share a number of common intermediates. As shown in Scheme 2, the putative aglycon is likely to be 8, which exhibits the typical features of a tetracycline except replacement of 4-(R)-dimethylamine with 4-(S)-hydroxyl, inversion of stereochemistry at C-6 and O-methylations at C-6 and C-12a. Downstream tailoring of 1 is therefore likely to occur following assembly of 8. In order to decipher the timing of the three unique post-PKS tailoring reactions leading to the biosynthesis of 1: glycosylation with d-olivose 14, O-4′ acylation of angelic acid and C-4 acylation of salicylic acid 19, crude extract of S. sp. SF2575 culture was prepared and analyzed on HPLC and LCMS to try to identify any potential stable intermediates that may be present. S. sp. SF2575 culture was grown on solid Bennette's media for 10 days at 30°C. A sample was extracted each day starting with day 4 to analyze the metabolic profile. Using selected ion monitoring, two potential intermediates identified were 6 ([M+H]+ at m/z 576, RT = 18.3 min) and 7 ([M+H]+ at m/z 696, RT = 26.8 min), in addition to the parent compound 1 ([M+H]+ at m/z 778, RT = 34.2 min) (Figure 1). The UV spectrum of each compound was similar to that of 1 with a characteristic tetracycline λmax at 358 nm, and a λmax at 302 nm for 1 and 7 characteristic of the salicylate. The UV spectrum of 6 lacks this contribution at 302 nm resulting in a smooth peak at 358 nm, and is indicative of the loss of salicylate compared to 7 as suggested by the molecular weight difference. To confirm the identity of these compounds as shown in Figure 1, authentic standards were prepared from base hydrolysis of purified 1 as described by Hatsu et. al.8 Treatment of 1 with 0.5 M NaOH led to the hydrolysis of the angelate and afforded 7, while complete conversion to 6 was obtained by treating 1 with 1.0 M NaOH for 15 hours. Following purification of 6 and 7 from the base hydrolysis reactions, proton NMR and HRMS were used to confirm the structures by comparison to published data (Supporting Information)8. These prepared samples were then used as standards to verify the identities of 6 and 7 in the fermentation extract of S. sp. SF2575 by HPLC retention time, mass fragmentation pattern and UV spectra.
Scheme 2.

Figure 1.
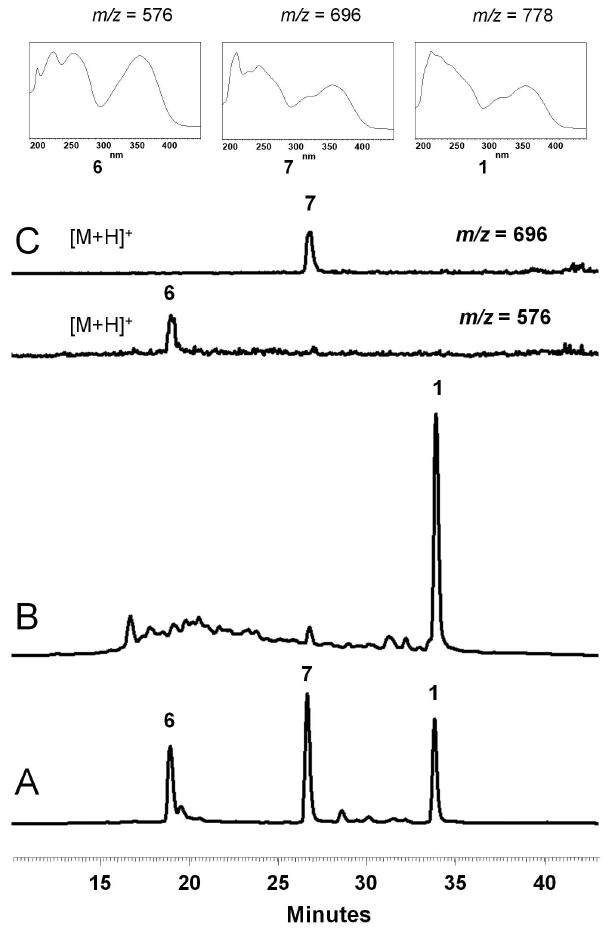
Production of 1 and biosynthetic intermediates by S. sp. SF2575. The data were collected on the Shimadzu LC-MS. (A) LC trace (358 nm) for a mixture of standards 6, 7 and 1. (B) LC trace (358 nm) of the extract after 7 days growth on solid Bennette's media. (C) Selected ion monitoring was used to confirm the identification of 7 ([M+H]+ m/z = 696) and 6 ([M+H]+ m/z = 576). The UV spectra of these compounds are shown for comparison.
Other potential intermediates, such as 6 acylated with angelate, or 8 acylated with salicylate, were not detected in the crude extracts using selected ion monitoring by LC-MS. This evidence, along with the positive detection of 6 and 7 were used to construct a proposed aglycon tailoring pathway of 1 as shown in Scheme 2. Therefore, the likely order of the tailoring modifications starts with C-glycosylation of 8 with 14 that results in 6, followed by attachment of salicylic acid 19 at C-4 to produce 7, and capped by the acylation of O-4′ of 7 with angelate to produce the final product 1. It is noteworthy that each of these isolated intermediates has already been O-methylated at the C-12a and C-6 hydroxyl groups, which indicates that these modifications likely take place early in the biosynthesis, providing further support for 8 as an advanced intermediate. While 8 was not identified in the fermentation extract, this could be due either to the potential instability of 8 or the rate of the glycosylation reaction which may not permit the accumulation of 8 in detectable quantities.
Bioactivity of SF2575 and putative biosynthetic intermediates
Since the potent antiproliferative activity of 1 is new to the tetracycline family, we sought to gain insight to the structural features of 1 that may contribute to this activity. The angelate and salicylate modifications of 1 are unique structural features among tetracyclines and bacterial aromatic polyketides. To probe whether these moieties are the “warheads” that contribute to the antitumor activities of 1, we performed in vitro cytotoxicity assays with Nalm-6 pre-B cells using 6, 7 and 1 (Supporting Figure S5). While the parent compound 1 had a potent IC50 of 8.8 nM, removal of the angelate resulted in a significantly attenuated IC50 of 327 nM for 7. Further hydrolysis of the salicylate as in 6 led to an additional 15-fold decrease of potency with an IC50 of 5.2 μM. Similar trends were observed when other cell lines, including MCF-7, HeLa, and M249 were subjected to the cytotoxicity assays (Supporting Figure S5). Hence, both pendant groups are critically important for the bioactivity of 1 and are attractive targets for SAR studies. Identification of the enzymes responsible for these additions may therefore provide useful biosynthetic approaches towards functionalizing these positions, especially considering the densely functionalized tetracycline core may be difficult to assess synthetically.
Identification and sequencing of the ssf biosynthetic gene cluster
To construct a blueprint of SF2575 biosynthesis, the ssf gene cluster was identified by screening a cosmid library prepared from S. sp. SF2575 genomic DNA using degenerative primers published by Wawrik et. al. for screening the type II polyketide KSα gene.21 Several PCR products from different cosmids were sequenced and were found to encode an identical KS fragment, which indicates that there is likely only one aromatic PKS gene cluster present in the S. sp. SF2575 genome. Three overlapping cosmids were sequenced by a combination of shotgun sequencing and primer walking (Figure 2). The cluster spans a 47.2 kb region and contains 40 ORFs putatively involved in SF2575 biosynthesis, including the essential KSα-KSβ heterodimer for synthesis of the polyketide backbone. To confirm that this cluster is responsible for SF2575 biosynthesis, the ssfB gene encoding KSβ was inactivated by an insertion of a kanamycin resistance gene (aphII) through a double crossover recombination. Disruption of this gene resulted in complete loss of SF2575 production (Supporting Figure S6), hence confirming the link between 1 and the sequenced ssf gene cluster.
Figure 2.
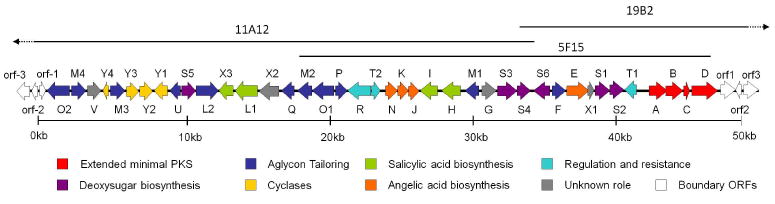
Organization of the ssf biosynthetic gene cluster. Genes are categorized according to their proposed role. The gene cluster spans 47.2 kb and contains 40 ORFs. Details of proposed functions are shown in Table 1.
A map of the ssf gene cluster is shown in Figure 2. The putative boundaries are determined based on sequence analysis and biosynthetic logic. The ssf genes identified along with their proposed function are listed in Table 1. Represented within are genes likely responsible for the biosynthesis of the aglycon 8, pendant groups 14, 19 and 25, as well as regulation and self-resistance. Based on the putative functions of these genes, the proposed biosynthetic pathways of 8 and the subsequent tailoring of 8 to 1 are shown in Scheme 4 and Scheme 2, respectively.
Table 1.
Putatively assigned functions of enzymes encode d in the ssf brosynthetic gene cluster
| Name | Closest homolog | Putative Role | Size | Identity/ Similarity | Accession number |
|---|---|---|---|---|---|
| SsfA | OxyA | Ketosynthase | 45 kDa | 76% / 86% | CAA80985 |
| SsfB | OxyB | Chain length factor | 43 kDa | 64% / 77% | AAZ78326 |
| SsfC | OxyC | Acyl carrier protein | 9.5 kDa | 54% / 73% | AAZ78327 |
| SsfD | OxyD | Amidotransferase | 68 kDa | 68% / 79% | AAZ78328 |
| SsfE | ChlJ | Carboxytransferase | 58 kDa | 81% / 89% | AAZ77684 |
| SsfF | Npun_F4742 | Ketoreductase | 34 kDa | 55% / 70% | ACC83094 |
| SsfG | ChlB3 | Ketosynthase III | 36 kDa | 37% / 56% | DQ116941 |
| SsfH | MbtI | Salicylate synthase | 48 kDa | 42% / 57% | Q7D785 |
| SsfI | PlmI | DAHP-7-phosphate synthase | 45 kDa | 56% / 67% | AY354515 |
| SsfJ | SAV_2786 | Enoyl CoA hydratase/isomerase | 28 kDa | 61% / 75% | BAC70497 |
| SsfK | Athe_1156 | 3-Ketoacyl-(ACP) reductase | 26 kDa | 38% / 59% | ACM60257 |
| SsfL1 | SdgA | Salicylyl-AMP ligase | 58kDa | 75% / 84% | BAC78380 |
| SsfL2 | OxyH | Acyl-CoA ligase | 56 kDa | 51% / 67% | DQ143963 |
| SsfM1 | OxyT | O-methyltransferase | 36 kDa | 48% / 62% | DQ143963 |
| SsfM2 | DauK | O-methyltransferase | 35 kDa | 38% / 53% | AAB16938 |
| SsfM3 | MtmMII | Methyltransferase | 37 kDa | 38% / 55% | CAK50790 |
| SsfM4 | OxyF | C-methyltransferase | 37 kDa | 51% / 68% | AAZ78330 |
| SsfN | PpzT | Ketosynthase III | 31 kDa | 46% / 63% | CAX48662 |
| SsfO1 | OxyS | Oxygenase | 55 kDa | 42% / 54% | AAZ78342 |
| SsfO2 | OxyL | Oxygenase | 59 kDa | 57% / 70% | AAZ78335 |
| SsfP | CmmQ | Dehydrogenase | 29 kDa | 49%/54% | YP_118212 |
| SsfQ | Lct52 | SAM synthetase | 43 kDa | 84% / 90% | ABX71135 |
| SsfR | Krad_0275 | MFS transporter | 58 kDa | 33% / 51% | ABS01765 |
| SsfS1 | StrD | dTDP glucose synthase | 39 kDa | 72% / 83% | BAG22759 |
| SsfS2 | CalS3 | Glucose-4,6-dehydratase | 36 kDa | 69% / 80% | AAM94770 |
| SsfS3 | Sim20 | Hexose-2,3-dehydratase | 53 kDa | 58% / 72% | AF322256 |
| SsfS4 | ChlC4 | Hexose-3-ketoreductase | 33 kDa | 49% / 59% | AAZ77681 |
| SsfS5 | NanG4 | Hexose-4-ketoreductase | 37 kDa | 42% / 53% | AAP42863 |
| SsfS6 | HedJ | Glycosyltransferase | 41 kDa | 40% / 55% | AAP85354 |
| SsfT1 | SnorA | Regulation | 31 kDa | 44% / 61% | CAA12016 |
| SsfT2 | SAML0351 | Regulation (TetR family) | 23 kDa | 48% / 62% | CAJ89338 |
| SsfU | OxyJ | Ketoreductase | 27 kDa | 76% / 86% | AAZ78333 |
| SsfV | ZhuC | Acyltransferase | 34 kDa | 54% / 64% | AAG30190 |
| SsfX1 | ChlI | Unknown function | 8.5 kDa | 50% / 60% | AAZ77683 |
| SsfX2 | RemJ | Carbohydrate kinase | 36 kDa | 64% / 72% | CAE51179 |
| SsfX3 | AviX9 | Acyltransferase | 40 kDa | 48% / 61% | AAK83171 |
| SsfY1 | OxyK | Aromatase/Cyclase | 34 kDa | 57% / 67% | AAZ78334 |
| SsfY2 | NcnE | Cyclase | 33 kDa | 56% / 68% | AAD20271 |
| SsfY3 | CmmQ | Cyclase | 31 kDa | 29% / 42% | CAE17552 |
| SsfY4 | OxyI | Cyclase | 13 kDa | 66% / 76% | AAZ78332 |
| Orf-1 | Hypothetical protein SGR_5601 | Unknown | 12 kDa | 84%/94% | BAG22430 |
| Orf-2 | SSEG_08767 | FeS assembly protein | 17 kDa | 89%/96% | EDY55253 |
| Orf-3 | SSEG_03816 | Cysteine desulferase | 46 kDa | 90%/94% | EDY65440 |
| Orf1 | Hypothetical conserved protein | Unknown | 66 kDa | 60%/69% | EEP13034 |
| Orf2 | Hypothetical protein SCO3803 | Unknown | 14 kDa | 78%/88% | NP_627922 |
| Orf3 | SSEG_08979 | AraC Transcriptional Regulator | 35 kDa | 82%/90% | EDY56197 |
Scheme 4.

Genes putatively involved in the biosynthesis of d-olivose 14
Deoxysugar decoration is an important tailoring modification of bacterial secondary metabolites and is usually essential for the bioactivity of the natural products22. The C-9 of 1 is modified with a C-glycoside d-olivose, which is also found among other polyketides such as urdamycin A23 and simocyclinone24. Putative enzymes encoded in the ssf gene cluster that can convert glucose to NDP-d-olivose are SsfS1 (NDP-glucose synthase), SsfS2 (4′,6′-dehydratase), SsfS3 (2′,3′-dehydratase), SsfS4 (C-3′ reductase), and SsfS5 (C-4′ reductase) (Scheme 2). The proposed pathway includes the unstable intermediate 12 which is reduced at the 3′ position by SsfS4.25 NDP-d-olivose is subsequently transferred to the C-9 of the aglycon 8 by a C-glycosyltransferase to yield 6. C-glycosyltransferases are more rare than O-glycosyltransferases, and are found in a number of aromatic PKS biosynthetic gene clusters such as urdamycin23, hedamycin26, and simocyclinone24. The putative ssf glycosyltransferase, SsfS6, has high sequence homology to HedJ, a C-glycosyltransferase from the hedamycin biosynthetic pathway26. Since SsfS6 is the first glycosyltransferase that can modify the tetracycline scaffold, it can potentially be a useful enzyme towards increasing the structural diversity of the D-ring, and in particular the C-9 position, of tetracycline compounds.
Genes putatively involved in the biosynthesis of salicylate 19
The C-4 salicylate substitution in 1 is unusual not just among tetracyclines but quite rare among aromatic polyketides. One known example is that of thermorubin, in which the salicylate moiety is thought to be incorporated as a starter unit27. A related modification is the addition of a 6-methylsalicylic acid which is typically synthesized via a dedicated type I PKS, such as those found in the chlorothricin28 or polyketomycin29 pathways. Sequence analysis of ssf gene cluster revealed a likely mechanism of salicylate 19 synthesis from the shikimate pathway (Scheme 2), similar to that utilized in the biosynthesis of siderophores yersinobactin30 and mycobactin31. SsfH was found to be homologous to the salicylate synthase genes Irp930 and MtbI31 from yersinobactin and mycobactin biosynthesis respectively, which convert chorismate 18, a byproduct of the shikimate pathway, to 19 in a two step reaction that proceeds through an isochorismate intermediate30. In addition, the ssf gene cluster contains ssfI which encodes 3-deoxy-d-arabino-heptulosonate-7-phosphate (DAHP) synthase that condenses erythrose-4-phosphate 16 and phosphoenolpyruvate 15 into DAHP 1732, which is the first step in the shikimate pathway. Genes encoding the remainder of the shikimate pathway that converts 17 to 18 are absent from the ssf gene cluster and are likely shared with the endogenous metabolism of S. sp. SF2575. The ssfI gene is therefore likely an extra copy dedicated for the biosynthesis of 1 and serves to direct carbon flow through the shikimate pathway when SF2575 biosynthesis is induced.
Genes involved in the transfer of salicylate to C-4
Attachment of 19 to 6 is proposed to occur through an ATP-dependent activation of 19 to salicylyl-CoA 20 by a salicylyl-CoA ligase, and subsequent transfer catalyzed by an acyltransferase. SsfL1 has high sequence homology to NcsB233 and MdpB234, which catalyze formation of the CoA-ester of naphthoic acid and 3,6-dimethylsalicylic acid in the enediyne biosynthetic pathways of neocarzinostatin and maduropeptin, respectively. To verify that SsfL1 is involved in the activation of 19, the enzyme was expressed from E. coli BL21(DE3) as an N-terminal hexahistidine tag fusion protein and purified using Ni-NTA chromatography (∼5 mg/L, Supporting Figure S7). ATP and 19 were added to SsfL1 and the release of pyrophosphate (PPi) was monitored spectrophotometrically at 340 nm using a coupled assay that oxidizes NADH. As shown in Figure 3A, significant PPi release was observed only in the presence of SsfL1, ATP and 19. To demonstrate the CoA ligase properties of SsfL1, enzymatic synthesis of 20 was performed by introducing CoA into the assay. After overnight incubation, a new compound consistent with the retention time of 20 was purified from the reaction mixture by HPLC and mass analysis confirmed the identity ([M+H]+ at m/z = 888, Supporting Figure S8).
Figure 3.
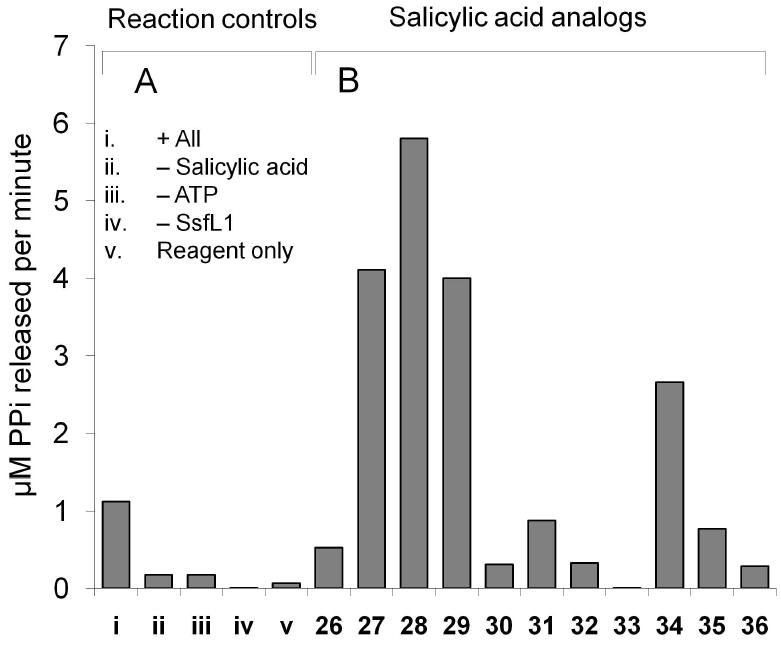
In vitro assay of SsfL1 activity and substrate specificity. (A) Synthesis of salicylyl-AMP as indicated by the release of PPi in the presence of ATP, salicylate 19 and the salicylyl-CoA ligase SsfL1. i) all reaction components; ii) no 19; iii) no ATP; iv) no SsfL1; and v) pyrophosphate reagent only. (B) Utilization of substituted benzoic acids by SsfL1 as indicated by PPi release assay. The structures of compounds indicated by number here are shown in Scheme 3.
For the ligation of 20 to 6, no clear enzyme candidate emerged during the bioinformatic analysis. One of the initially unassigned enzymes, SsfX3, has high sequence homology to only one enzyme from a published natural product biosynthetic pathway, which is AviX9 from the avilamycin gene cluster (56%/44% similarity/identity). However, the role of AviX9 in avilamycin biosynthesis has not been identified35. Several proteins with lower homology to SsfX3 were reported as hypothetical proteins in the GDSL lipase/acyl-hydrolase family (PF00657). Characteristic features of this class of acyltransfer enzymes are i) the active site motif GDSL; ii) the lack of a nucleophilic elbow formed by the GXSXG motif of typical hydrolases; and iii) a flexible substrate binding pocket36, 37. Despite the low sequence homology, residues strictly conserved in this family (Ser, Gly, Asn, His)36 can be predicted to be S174, G209, N236 and H338 in SsfX3. Residues which form the catalytic triad were also identified by sequence homology and are predicted to be S174, D333 and H338. Therefore, SsfX3 was considered as a potential acyltransferase that may catalyze the acyltransfer of either 20 or an activated form of angelate during the biosynthesis of 1.
To probe the potential role of SsfX3, we first assayed the reverse hydrolysis reaction using different products. 1 was treated with purified SsfX3 overnight and the assay mixture was analyzed by LC-MS. The result showed the presence the parent compound 1 and a second compound with mass corresponding to the loss of 19 ([M+H]+ at m/z = 658, Supporting Figure S9). A control reaction without SsfX3 did not afford this compound, as determined by selected ion monitoring. The UV of this compound showed λmax of 358 nm and the loss of the shoulder at 302 nm, consistent with the hydrolysis of 19. Despite the low conversion, this result was the first indication that SsfX3 may be involved in C-4 salicylation. To examine if the intermediate 7 may be the endogenous product of SsfX3, we repeated the same hydrolysis reaction using purified 7. Analysis of the product mixture showed that nearly all 7 was converted to 6 in the presence of SsfX3, while no 6 was recovered in the absence of the enzyme (Supporting Figure S9). To confirm the SsfX3-catalyzed acyltransfer reaction, an in vitro assay was performed in which SsfX3 and SsfL1 were added to 6 with 19, ATP and free CoA. The reaction was incubated for 30 minutes at room temperature and analyzed by HPLC (Figure 4A). The reaction containing all components showed nearly quantitative conversion of 6 to 7 (Figure 4A, trace iii). Omission of any of the enzyme, substrate, or cofactor component of the assay did not lead to the synthesis of 7, as shown in Figure 4A traces iv-viii. Together, these results confirmed that SsfX3 is responsible for the conversion of 6 to 7 using 20 as the acyl donor.
Figure 4.
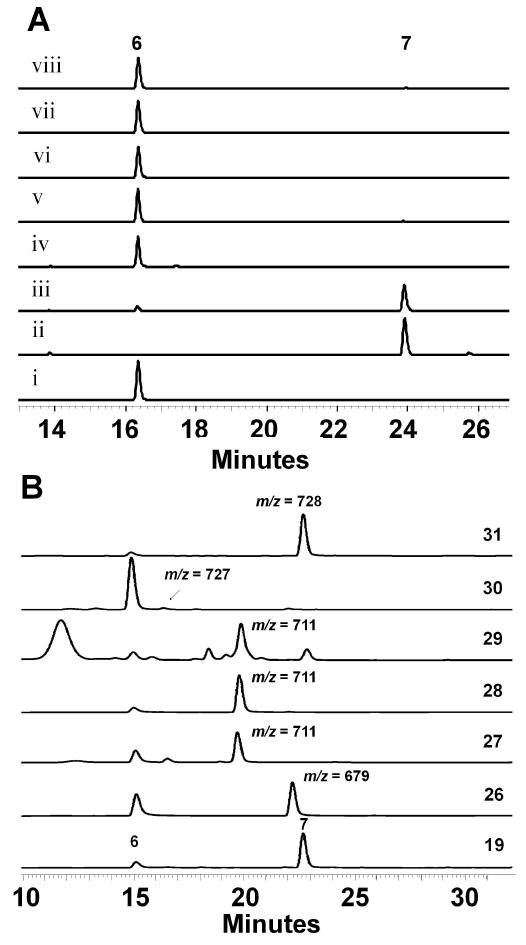
In vitro assay of SsfX3 activity and substrate specificity. (A) The tandem actions of SsfL1 and SsfX3 transfer 19 to the aglycon substrate 6 to yield 7. The assays are performed in 50 mM HEPES, pH 7.9 and 10 mM MgCl2. i) the semisynthetic 6 standard; ii) the semisynthetic 7 standard; iii) Complete reaction containing 50 mM HEPES pH 7.9, 10 mM MgCl2, 2 mM ATP, 2 mM free CoA, 2 mM 19, 20 μM 6, 1.5 μM SsfX3 and 15 μM SsfL1. Control reactions were performed as iii) with the following exclusions: iv) no SsfL1; v) no ATP; vi) no 19; vii) no SsfX3; and viii) no CoA. The reactions were examined with HPLC (358 nm). (B) Synthesis of analogs of 7 using salicylic acid analogs, SsfL1 and SsfX3. All reactions were performed at 25 °C for 30 minutes, extracted with organic solvent and analyzed by LC-MS (358 nm, positive ionization).
Substrate specificity of SsfL1 and SsfX3
Exploiting the substrate tolerance of SsfX3 and SsfL1 may be useful towards diversifying the C-4 functionality of 1. To probe this, differently substituted benzoic acids (Scheme 3) were tested for activation and acyltransfer by SsfL1 and SsfX3, respectively. Since SsfL1 is the initial gatekeeper in these reactions, the different substrates were first examined for recognition by SsfL1 using the PPi release assay as described above (Figure 3B). Interestingly, nearly all the substrates examined showed levels of PPi release above background (except 33), demonstrating SsfL1 has relaxed substrate specificity. The 2-OH substituent is clearly not essential for binding, as benzoic acid 26, 2-chlorobenzoic acid 31 and 2-methoxy benzoic acid 35 all supported similar rates of pyrophosphate release. Surprisingly, three dihydroxybenzoic acids (27-29), as well as 4-aminosalicylic acid 34 were significantly better substrates than 19, indicating substantial plasticity in the binding pocket of SsfL1.
Scheme 3.

Acyltransferases from the GSDL family are often found to have broad substrate specificity, which is proposed to be the result of a flexible substrate binding pocket.36 Therefore, SsfX3 may be similarly tolerant and use different aryl-CoAs as substrates. Given nearly all substrates shown in Scheme 3 can be activated by SsfL1, we tested whether new variants of 7 can be synthesized in a coupled assay using both SsfL1 and SsfX3. To do so, 2 mM of each analog was combined with SsfL1 (15 μM), and SsfX3 (1.5 μM) in an in vitro assay containing 2 mM free CoA, 2 mM ATP, 10 mM MgCl2, and 20 μM 6 in 50 mM HEPES buffer pH 7.9. The reaction was incubated at room temperature for 30 minutes and analyzed by LCMS for emergence of analogs of 7 (Figure 4B). The CoA-activated forms of the dihydroxybenzoic acids 27-29 were readily recognized by SsfX3 and led to the synthesis of new analogs of 7. This demonstrates that the additional hydroxyl group is well tolerated at all three positions and this finding may be used to produce more hydrophilic derivatives of 1. However, the trihydroxybenzoic acid 30, which was a poor substrate in the SsfL1 activation assay, only afforded trace amounts of the adduct in the coupled assay as revealed by selected ion monitoring. The coupled assay also reveals SsfL1 and SsfX3 have significant differences in substrate tolerance. Whereas 34 and 35 were each well-activated by SsfL1, neither compound was transacylated to 6 by SsfX3. To test if SsfX3 can accept membrane permeable aryl thioesters, we mixed benzoyl-S-cysteamine (benzoyl-SNAC) with 6 and SsfX3. The resulting extract showed that the C-4 benzoyl analog of 7 was synthesized in similar yield as that using 26 and CoA in the coupled-enzyme system. This demonstrated SsfX3 also has relaxed substrate tolerance towards the acyl carrier. Taken together, these results indicate SsfL1 and SsfX3 are attractive enzymes in the biocatalytic synthesis of analogs of 1 for structural-activity relationship studies. Mutasynthesis of 1 containing different substituents at C-4 may also be accomplished by inactivating the endogenous salicylic biosynthetic pathways, such as deletion of ssfH.
Genes putatively involved in the biosynthesis of angelyl-CoA 25
Little has been reported on the biosynthesis of angelic acid among bacterial natural products. Angelate is commonly found to be associated with plant metabolites and is biosynthesized from the isoleucine catabolic pathway38, 39. No genes were found in the ssf cluster that indicates a plant-like pathway for this unusual substitution in 1. Instead, a set of genes encoding enzymes involved in the formation and tailoring of a short chain acyl group was found. These genes include SsfE (carboxytransferase), SsfJ (enoyl-CoA hydratase/isomerase), SsfN (KSIII), and SsfK (3-ketoacyl-ACP reductase). These genes resemble those found in fatty acid biosynthesis and may be responsible for synthesizing angelyl-CoA 25 from a propionyl-CoA 21, or other short chain acyl building block. One possible pathway is shown in Scheme 2.
SsfE contains a carboxyltransferase domain (PF01039) and is homologous to biotin-dependent methylmalonyl-CoA decarboxylase and propionyl-CoA carboxylase. Several of these homologs are present in polyketide gene clusters such as chlorothricin28 (ChlJ), jadomycin40 (JadN), simocyclinone24 (SimA12), medermycin41 (med-ORF22) and others. One possible role for SsfE is to synthesize methylmalonyl-CoA 22 from 21. The next set of steps in the biosynthesis of 25 are proposed to catalyzed by ssfK, ssfN and ssfJ, which are cotranscribed as a tricistronic cassette on the same operon. SsfN is homologous to KS III enzymes such as FabH, which catalyzes the condensation between acetyl-CoA and malonyl-ACP to initiate fatty acid biosynthesis42. FabH homologs have been found in a number of polyketide gene clusters such as in R112843, frenolicin43, and daunorubicin44 to catalyze the decarboxylative condensation of short acyl groups. SsfN may therefore catalyze the condensation of 22 with acetyl-CoA to form 2-methyl-acetoacetyl-CoA 23. β-ketoreduction of 23 catalyzed by SsfK, which is homologous to 3-oxoacyl-ACP reductases such as FabG45, yields 3-hydroxyl-2-methyl-butyryl-CoA 24. Stereospecific dehydration of 24 by SsfJ, which is a member of the enoyl-CoA hydratase/isomerase family (PF00378), results in 25. As an alternative start to this pathway, SsfN may directly condense malonyl-CoA with acetyl-CoA to form acetoacetyl-CoA. The α-methyl group may then be incorporated by SsfM3, which is homologous to C-methyltransferases, to afford 23.
Transfer of 25 to 7 is predicted to be catalyzed by an acyltransferase using similar mechanisms as for other acylated deoxysugars present in chromomycin46 or mannopeptimycin47. Those belong to a large acyltransferase family (PF01757) that contains proteins with O-acetyltransferase activity, including the deoxyhexose O-acyltransferases MdmB48. Unexpectedly, no similar acyltransferase is present in the ssf gene cluster. One acyltransferase homolog, SsfV, however, remains unassigned. SsfV has homologs in many type II polyketide gene clusters including OxyP from the oxy pathway and ZhuC from the R1128 pathway. A Pfam homology search revealed that it contains an acyltransferase domain (PF00698) from a family of proteins that includes bacterial malonyl-CoA-ACP transacylase (MAT). In vitro experiments with homolog ZhuC showed that it did indeed have malonyl-CoA ACP transacylase activity, however it was much slower than endogenous MAT from the fatty acid biosynthetic pathway49, 50. Additional in vitro studies demonstrated that ZhuC could efficiently hydrolyze acetyl and propionyl primed ACPs and therefore acts to ensure that the R1128 PKS is primed with the correct medium chain length acyl-ACPs50. Similarly, SsfV may be involved in ensuring the biosynthesis of 1 is initiated with malonamyl starter unit and is hence unlikely to be involved in angelate attachment.
The ssf gene cluster encodes another KS III homolog SsfG, which resembles ChlB3 and ChlB6 from the chlorothricin biosynthetic cluster28. The role of these genes in the processing and attachment of the 5-chloro-6-methyl-O-methylsalicylic acid during chlorothricin biosynthesis has recently been published51. ChlB3 is utilized to transfer 6-methylsalicylic acid from the type I PKS ChlB1 to a discrete ACP for further tailoring. ChlB6 is shown to transfer the tailored 5-chloro-6-methyl-O-methylsalicylic acid to d-olivose of desmethylsalicyl chlorothricin to form chlorothricin51. Considering SsfG displays high sequence similarity to ChlB6 (52%) and the same deoxysugar substrate d-olivose is involved, SsfG therefore likely catalyzes the attachment of angelate in the ssf pathway. Instead of using a discrete ACP as the acyl carrier, the CoA activated angelate 25 may directly serve as the substrate of SsfG.
Genes encoding enzymes that synthesize the polyketide backbone 40
The ssf minimal PKS consists of SsfA (KSα), SsfB (KSβ or CLF), and SsfC (ACP) that are highly homologous to the oxy minimal PKS18. The amidotransferase SsfD, which is responsible for producing the malonamate starter unit unique to tetracyclines, is found adjacent to the ssf minimal PKS. The C-2 amidated starter unit is one of the signature moieties of tetracyclines, and the revelation of SsfD as an OxyD homolog with 68% sequence identity was one of the earliest convincing pieces of evidence that this gene cluster indeed encodes a tetracycline compound. Like OxyD, SsfD is highly similar to class II asparagines synthases that utilize ATP to convert aspartate to asparagine using glutamine as the nitrogen source52. Zhang and coworkers demonstrated the “extended minimal PKS”, which includes OxyABCD, is sufficient for producing the amidated polyketide chain 38.18 In addition, SsfU, which is 76% identical to OxyJ, is predicted to be the C-8 (or C-9* using biosynthetic carbon numbering as shown in Scheme 4) ketoreductase that regioselectively reduces the nascent backbone to yield 4020.
To verify the function of the ssf minimal PKS, genes encoding SsfA, SsfB and SsfC, SsfD and SsfU were placed into a pRM5-derived Streptomyces-E. coli shuttle vector53 (Table 2). Shuttle plasmid pLP27 was subsequently transformed into Streptomyces lividans strain K4-11454. The resulting organic extract was analyzed by HPLC and is shown in Figure 5A. The product profile of K4-114/pLP27 was indistinguishable from that of K4-114/pWJ35, which contained the corresponding enzymes from the oxy biosynthetic pathway18. By comparison to an authentic standard, the major product of the extract (RT = 13.5 min) was confirmed to be the isoquinolone 42 (20 mg/L), which can form via the spontaneous cyclization of the C-9* reduced amidated polyketide 4018. We further investigated whether the oxy and ssf extended minimal PKS components are functionally interchangeable. Three additional shuttle vectors were prepared with a combination of oxy and ssf genes as shown in Table 2 and transformed into K4-114. Each of these host/vectors combinations were capable of producing 42 in similar quantities as K4-114/pLP27, indicating that each of the ssf components are functionally compatible and equivalent to their oxy counterparts. These results confirm i) the extended ssf minimal PKS (SsfABCD) is capable of synthesizing the full length amidated polyketide precursor 38; ii) the hypothesis that the carbon backbone of 1 is biosynthesized via a tetracycline-like pathway; and iii) SsfU as the C-9* KR that affords 40. Also detected from these extracts was the acetate-primed product RM20b 41 (RT = 17.7 min), consistent with the previous report of oxy minimal PKS being able to initiate polyketide biosynthesis with the acetate starter unit55.
Table 2.
Plasmids and major metabolites produced by S. lividans K4-114 transformed with each plasmid
| Plasmid | ssf Genes | oxy Genes | Major Products |
|---|---|---|---|
| pLP24 | ssfD | oxyABCJ | 41,42 |
| pLP25 | ssfDC | oxyABJ | 41,42 |
| pLP26 | ssfABD | oxyCJ | 41,42 |
| pLP27 | ssfABCDU | 41,42 | |
| pLP27/pLP77 | ssfABCDUY1 | 44 | |
| pLP27/pLP113 | ssfABCDUY1Y2M4 | 46 | |
| pLP27/pLP115 | ssfABCDUY1Y2M4Y4 | 46 | |
| pLP27/pLP118 | ssfABCDUY1Y2M4Y4Y3 | 46 | |
| PLP27/pLP126 | ssfABCDUY1Y2M4L2 | 50 | |
| pLP36 | ssfO2 | oxyABCDJKNF | 52 |
| pLP75 | ssfO2 | oxyABCDJKNFQT | 57 |
Figure 5.
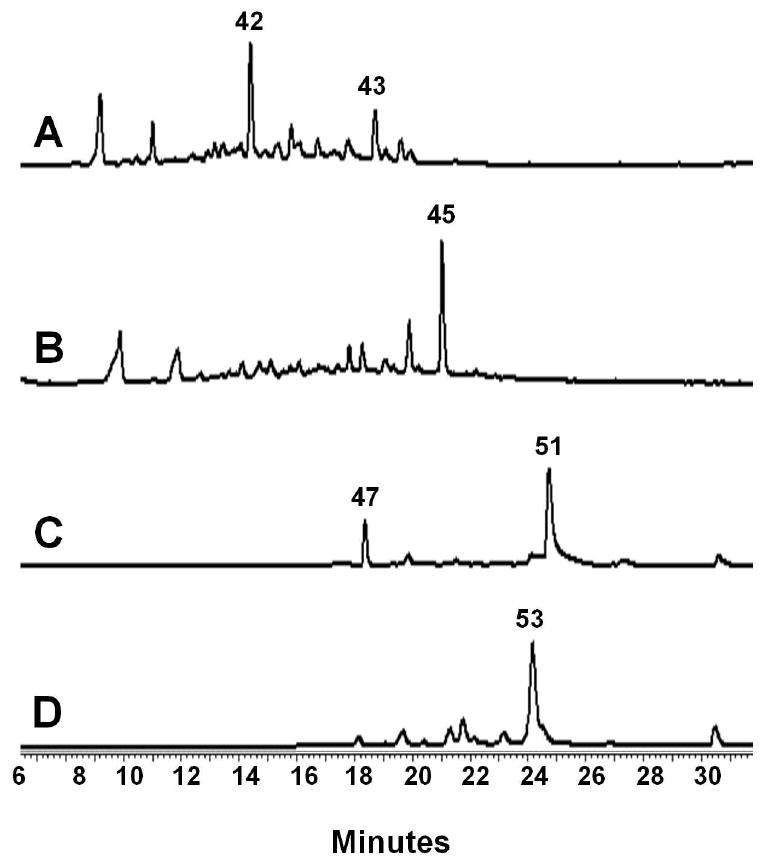
Reconstitution of tetracycline intermediates using ssf genes expressed in S. lividans K4-114. (A) HPLC analysis (245 nm) of the K4-114/pLP27 extract shows the amidated, reduced polyketide 42 is the major product, confirming the biosynthesis of the polyketide backbone 40 by SsfABCD and the subsequent C-9* reduction by SsfU. (B) HPLC analysis (253 nm) of the K4-114/pLP27/pLP77 extract shows addition of the putative cyclase SsfY1 leads to complete cyclization and aromatization of the D and formation of the shunt benzopyrone 44. (C) HPLC analysis (430 nm) of K4-114/pLP27/pLP126 extract shows 50, the oxidized form of 49, as the dominant product. Biosynthesis of 49 using entirely ssf genes (SsfABCDUY1Y2M4L2) indicates the tetracycline nature of the ssf biosynthetic pathway. (D) HPLC analysis (395 nm) of K4-114/pLP36 extract confirms the function of SsfO2 as the oxygenase that dihydroxylated C-4 and C-12a of 49. The resulting product 51 undergoes spontaneous degradation to afford the observed product 52.
Genes encoding enzymes that synthesize 6-methylpretetramid 49
Sequential cyclizations of the four rings are the next steps following biosynthesis of the full length polyketide chain. In the oxy cluster three cyclases were identified based on sequence homology, however only two were shown to be required to yield the intermediate pretetramid 4720. Diverging from the oxy cluster, the ssf gene cluster contains four genes that have homology to cyclases. SsfY1 is highly similar to OxyK (67% sequence similarity) and is predicted to be responsible for D ring cyclization to afford 43. SsfY4 is similar to OxyI, itself similar to fourth ring cyclase MtmX from the mithramycin pathway. However OxyI was found to be unnecessary in the biosynthesis of 4720. Although SsfY2 has low sequence homology (25% similarity and 14.5% identity) to the second ring cyclase (OxyN) from the oxy pathway, it displays stronger homology to second ring cyclases from benzoisochromanequinone (BIQ) pathways such as granaticin (68% similarity, 56% identity)56 and medermycin (66% similarity, 55% identity)41. SsfY2 is therefore predicted to be the second ring cyclase in the ssf pathway. Intriguingly, an additional cyclase, SsfY3, also displays sequence similarity to first ring cyclase/aromatases, with 42% similarity to CmmQ46 and 39% similarity to OxyK. Completion of the biosynthesis of 49 requires C-6 methylation of 47, which is catalyzed by the OxyF homolog SsfM4.
To deduce the role, or lack thereof, of each of these cyclases in the biosynthesis of 49, we heterologously expressed different combinations of cyclases found in the ssf gene cluster with SsfABCDU. We first constructed a pSET152-derived integrating vector57 containing ssfY1 and cotransformed the resulting plasmid pLP77 with pLP27 into K4-114. We detected the complete disappearance of 42 in the extract and emergence of a single major product that corresponds to the benzopyrone WJ78 44 (Figure 5B), which is the shunt product of the amidated polyketide intermediate 43 that has been regioselectively cyclized at the D-ring58. Cotransformation of K4-114 with pLP27 and an integrating vector containing ssfY3, did not lead to the recovery of 44, indicating SsfY3 cannot substitute for SsfY1 and likely serves a different role in the biosynthesis of 1.
We next set out to reconstitute the biosynthesis of 49 in K4-114 using entirely ssf genes. SsfM4 was chosen to be the likely C-6 methyltransferase because of its high sequence homology to OxyF. The integrating vector pLP113 bearing ssfY1Y2M4 was cotransformed with pLP27 and resulting extract was intensely yellow and contained the anthraquinone carboxylic acid WJ83Q2a 46. This confirmed SsfY2 was responsible for the cyclization of the second (or C) ring, while the third ring is likely spontaneously cyclized. However, to our surprise, the expected tetracyclic product 49, which can be isolated in the oxidized form WJ119 50, was not found. This observation is different from what was observed for the oxy pathway, in which the equivalent set of oxy enzymes yielded 5020. To determine if the ssf pathway requires the additional cyclases found in its gene cluster towards the formation of 47, pLP115 and pLP118 were constructed, which contained in addition to genes encoded in pLP113, ssfY4, and ssfY3Y4, respectively. K4-114 contransformed with pLP27 and these constructs continued to produce 46 only, suggesting that for the ssf pathway, different enzyme(s) may be needed to facilitate cyclization of the fourth ring. We tested this possibility by inserting additional ssf genes juxtaposed to the ssf cyclase genes in the cluster into K4-114 and examined the metabolic profiles. Fortuitously, upon insertion of ssfL2 into pLP113 to afford pLP126, and cotransformation of pLP27 and pLP126 into K4-114, the formation of 50 was detected as shown in Figure 5C. SsfL2 has homology to long-chain fatty acyl-CoA ligases as well as putative acyl-CoA ligases from several aromatic polyketide pathways such as CmmLII from the chromomycin gene cluster46, PokL from the polyketomycin gene cluster29, MtmL from the mithramycin gene cluster59, as well as OxyH18. The role of these enzymes in their respective biosynthetic pathways has not been determined and has been suggested to function as an accessory protein46. Interestingly, biosynthesis of these compounds all require the equivalent Claisen cyclization of the A rings. The mechanism of SsfL2 in the heterologous biosynthesis of 49 with ssf PKS components is yet to be determined.
Tailoring enzymes putatively involved in the production of the aglycon 8
During biosynthesis of 4, the ring A of 49 is doubly hydroxylated by OxyL at C-12a and C-4 to produce the cyclohexenone containing 51, which is unstable and degrades into observed product WJ135 5219. SsfO2, which is found at the boundary of the ssf gene cluster, is an OxyL homolog and is most likely to perform this role in the ssf pathway. Compound 51 is likely the last common intermediate between the oxy and the ssf biosynthetic pathways. In the oxy pathway, reductive amination of the C-4 ketone group by OxyQ to yield 4-amino-ATC 56 and subsequent dimethylation by OxyT afford the stable intermediate anyhydrotetracycline 5719. The C-4 position in 1 lacks the dimethylamino group and is instead acylated with a salicylic acid group. This requires ketoreduction of 51 at C-4 to produce 4-hydroxyl-ATC 53 for the downstream salicylation reaction.
To determine whether this hydroxyl is directly installed by SsfO2 or whether an additional ketoreduction is required, the oxyL gene was replaced with ssfO2 in the shuttle plasmid pWJ135, which contains all the oxy genes required to synthesize 51 (Table 2)19. K4-114 transformed with the resulting plasmid pLP36 produced the tricyclic ketone 52 (Figure 5D), which is a spontaneously degraded product of 51 as observed previously19. To further confirm the product of SsfO2 contains the C-4 ketone functionality as in 51, we examined if SsfO2 can complement the role of OxyL in the biosynthesis of 57, which requires reductive amination of the C-4 ketone in 51. The genes oxyQ and oxyT were introduced to pLP36 to afford pLP75 (Table 2). As expected, K4-114/pLP75 produced ∼ 10 mg/L of 57 as the dominant product, demonstrating that OxyL and SsfL2 catalyze the identical transformation of 49 to 51. These results support the proposal that a dedicated C-4 ketoreductase is required to reduce 51 to 53. SsfF is a ketoreductase with sequence homology to NADPH-dependent oxidoreductases with specificity for aromatic and nonpolar substrates (PF00248), and is therefore assigned as a candidate to catalyze this step.
The last tailoring enzyme in the ssf pathway that shares resemblances to enzymes found in oxy pathway is SsfO1 which has 54% similarity to OxyS. OxyS is known to be responsible for the C-6 hydroxylation of 57 to form 5a, 11a- dehydrotetracycline 5860, 61, and SsfO1 therefore likely plays the parallel role to oxidize the aromatic C-ring in 53 to 54. SsfO1, however, most likely catalyzes the C-6 hydroxylation with opposite stereochemistry as OxyS, based on X-ray crystallographic structure of 18. Two O-methyltransferases are then needed to methylate the C-6 and the C-12a hydroxyl groups. Three methyltransferases (other than the C-6 methyltransferase SsfM4) are present in the ssf gene cluster. SsfM1 and SsfM2 bear strong resemblance to O-methyltransferases, while SsfM3 is similar to both O-methyltransferases and C-methyltransferases. The C12a-OH methylation modification can take place either prior to SsfO1 oxidation (using either 51 or 53 as substrate), or after C-6 oxidation (as shown in Scheme 4). The gene encoding SsfM2 is transcribed from the same operon that encodes ssfO1 and is therefore the likely C-6 O-methyltransferase. In the same operon is the gene ssfQ, which encodes an S-adenosylmethionine (SAM) synthases and likely serves as an auxiliary role to produce sufficient SAM for the methyltransfer reactions. It is known that 6-deoxy analogs of tetracyclines, such as doxycycline, have enhanced stability toward acid and base degradation62. The C-6-OH methylation may have a similar stabilizing effect on 1. Once identified, the C-6 O-methyltransferase may potentially be useful to generate C-6-methoxytetracycline analogs that have enhanced stability.
The last step in the biosynthesis of 4 is the reduction of 5a,11a-dehydrooxytetracycline 59 to 4. This reductive modification required for the biosynthesis of 4 and 5 has been postulated to involve a gene located outside their respective gene clusters63. The tchA gene encodes a flavin dependent oxidoreductase with reported cofactor 7,8-didemethyl-8-hydroxy-5-deazariboflavin (FO) and has been identified in S. aureofaciens to be involved for the biosynthesis of 564. An identical reduction step is also needed to complete the biosynthesis of aglycon 8, from for example, 55. One candidate for this reaction in the ssf gene cluster is a putative dehydrogenase SsfP. SsfP shares 54% sequence similarity to a putative F420-dependent methylenetetrahydromethanopterin reductase, but shares no sequence resemblance to TchA. Experimental evidence will be needed to determine if this reduction step is catalyzed by an enzyme encoded in the ssf gene cluster such as SsfP or if an external gene is required as in chlorotetracycline biosynthesis.
Genes encoding regulatory and resistance determinants
The ssf gene cluster encodes two regulatory proteins, SsfT1 and SsfT2; and a resistance protein SsfR. SsfT2 is predicted to be a TetR family65 repressor protein with a DNA-binding helix-turn-helix motif at the N-terminus. SsfR is an efflux protein from the Major Facilitator Superfamily (MFS) class of ATP independent transporters which utilize a chemiosmotic gradient to transport small molecules (PF07690)66. The ssfT2 gene is located directly adjacent to ssfR with opposite polarity. Therefore the transcription of ssfR may be controlled by SsfT2 in a similar fashion as the TetR/TetA pair65. The second regulatory protein SsfT1 is a positive regulator in the ssf gene cluster, and is predicted to be a member of the pathway-specific Streptomyces antibiotic regulatory protein (SARP) family of transcriptional activators67. These activators have been the subject of metabolic engineering efforts as evidenced by the recent publication on the fredericamycin pathway which demonstrated a greater than 5 fold increase in antibiotic yield by overexpression of FdmR168.
Conclusions
We have identified the ssf biosynthetic gene cluster and biochemically characterized key steps in the biosynthesis of 1. Biosynthesis of 1 from the aglycon 8 requires a new set of tetracycline tailoring enzymes including the C-9 glycosyltransferase SsfS6 and the C-4 salicylyl transferase SsfX3. In vitro assays with SsfL1 and SsfX3 have shown that both enzymes have relaxed substrate flexibility toward different aromatic substrates, a feature that was exploited in the chemoenzymatic synthesis of several analogs of 7. Using heterologous biosynthesis we have also verified the tetracycline nature of the gene cluster by reconstituting the biosynthesis of key intermediates. The identification of the ssf gene cluster and formulation of a putative biosynthetic pathway will therefore enable engineered biosynthesis of new tetracycline analogs.
Materials and Methods
Bacterial strains and DNA manipulation
Streptomyces sp. SF2575 was obtained from Meiji Co. E. coli XL-1 Blue (Stratagene) was used for the manipulation of plasmid DNA. E. coli strain BL21(DE3) was used for protein expression. Fosmids 11A12 and 5F15 were used as templates to amplify DNA by PCR. PCR fragments were cloned into pCR-Blunt vector obtained from Invitrogen and sequenced (Laragen). All restriction enzymes were purchased from New England Biolabs. PCR reagents for pfx DNA polymerase were purchased from Invitrogen. PCR Primers are listed in Table S1. S. lividans strain K4-11454 was used at a host for the transformation of shuttle vector constructs containing ssf genes. Protoplast preparation and PEG-mediated DNA transformation techniques were performed as published by Hopwood et al69.
Spectroscopic analysis
NMR of compounds was obtained on a Bruker ARX400 at the University of California Department of Chemistry and Biochemistry NMR facility. Accurate high resolution mass spectrometry was performed at the University of California Riverside Mass Spectrometry Facility. HPLC spectra were obtained on a Beckman Gold HPLC using a reverse phase C18 column (Alltech Apollo C18 5μ column; 250mm × 4.6mm) and a linear gradient of 5% CH3CN in water (0.1% trifluoroacentic acid [TFA]) to 95% CH3CN in water (0.1% TFA) in 30 minutes at a flow rate of 1 ml/min. LC-MS spectra were obtained on a Shimadzu 2010 EV Liquid Chromatography Mass Spectrometer using positive and negative electrospray ionization and a Phenomenex Luna 5μ 2.0 × 100 mm C18 reverse-phase column. Samples were separated on a linear gradient of 5 to 95% CH3CN in water (0.1% formic acid) for 30 minutes at a flow rate of 0.1 ml/min unless otherwise noted.
Cultivation of Streptomyces sp. SF2575 and purification of 1
Streptomyces sp. SF2575 was cultured in R5 media for subculture and seed culture. Modified Bennett's agar (per 1 L, beef extract 1 g, bacto-peptone 2 g, yeast extract 1 g, glucose 10 g, agar 20 g) was used as a production media. For isolation of 1, Streptomyces sp. SF2575 was pre-cultured in 50 ml of R5 liquid media in a 250 ml baffled Erlenmeyer flask at 30 °C and 250 rpm for 3 days. 1 ml of pre-cultures were inoculated on plates (4 L) of modified Bennett's agar in 150mm × 10mm Petri dishes and incubated at 30 °C for 8 days. The cultured agar was harvested and extracted with the same volume of ethyl acetate twice. Organic solvent was dried in vacuo to obtain 875.2 mg of oily crude extract. The crude extract was fractionated with open silica column (200-430 mesh, 50mmΦ × 300mm) using a methanol and chloroform gradient. Fractions containing 1 were collected and purified by preparative HPLC to obtain 22 mg of 1. HPLC purification was performed using HPLC an Alltech Alltima reverse phase column (5μm, 10 × 250 mm) with an isocratic gradient of 75% CH3CN in water (0.1% TFA).
Preparation of authentic standards 6 and 7
7 was prepared by a two hour incubation of a 10 mg/ml solution of 1 with 0.5 M NaOH. Complete conversion of 1 to 6 was obtained by incubation of a 10 mg/ml solution of 1 in 1.0 M NaOH for 15 hours. To isolate 6 and 7, the reaction mixture for each was adjusted to pH 7 by addition of HCl and the products were extracted with ethyl acetate/acetic acid (99%/1%). Following the evaporation of the solvent, the product was resuspended in methanol and purified by HPLC (Alltech Alltima reverse phase column 5μm, 10×250mm) with an isocratic gradient of 78% CH3CN in water (0.1% TFA). Purified 6 and 7 were characterized by LC/MS, HRMS and proton NMR (see Supplementary Information).
Bioactivity assays
Cell proliferation was determined by (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay for adherent HeLa, MCF-7, and M249 cells. 5,000 cells per well (4,000 HeLa) were plated in 100 μl media in a 96-well plate and allowed to incubate for 18 hours. Following incubation, 10 mM stock solution of 1, 6 or 7 solubilized in DMSO was added to media to prepare dilutions containing three times the final concentration. 50 μl of the appropriate dilution was added to each well. Cell proliferation was assayed 72 hours after treatment using CellTiter 96® AQueous OneSolution cell proliferation assay reagent (Promega). Direct cell counting was used for Nalm-6 suspension cells by Vi-CELL™ analyzer (Beckman). 400 μl cells (500,000 cells/ml) were plated in a 24-well plate. 400 μl media containing twice the desired final drug concentration was added after 20 hours. Cells were analyzed 72 hours after treatment. All cells were cultured according to ATCC or DSMZ guidelines. Prism (GraphPad) was used for analysis.
Identification and sequencing of the ssf gene cluster
A fosmid library containing S. sp. SF2575 genomic DNA was prepared using the CopyControl™ Fosmid library production kit from Epicentre. Fosmids were screened using degenerate primers for the KSα gene as reported by Wawrik et al21. Fosmids 5F15, 11A12 and 19B2 were sequenced by a combination of shotgun sequencing and primer walking. ORFs were identified using Frameplot software (http://www.nih.go.jp/∼jun/cgi-bin/frameplot.pl) and functions of the encoded proteins were assigned by sequence similarity using NCBI protein-protein BLAST. Pfam protein family database was also used to identify functional domains.70 The sequence has been deposited in the GenBank database under accession number GQ409537.
Inactivation of ssfB gene
For ssfB (CLF) disruption, approximately 1 kb of the 5′ region of ssfB gene was amplified from genomic DNA of S. sp. SF2575 by PCR using a forward primer KS2-LEK-S which introduces an EcoRI site, and a reverse primer KS2-LEK-A2 which introduces a KpnI site. The PCR product obtained was cloned into pCR-Blunt vector to produce pCR-KSII-LEK. Approximately 1 kb of the 3′ region of ssfB was also amplified by PCR using primer set KS2-RPH-S and KS2-RPH-A to add PstI and HindIII site to its 5′ and 3′ends, respectively. The PCR product obtained was cloned to produce pCR-KSII-RPH. The 1.1 kb of aphII gene was obtained from pFD-neoS plasmid.71 Plasmid pCR-KSII-LEK was digested with EcoRI and KpnI; and plasmid pCR-KSII-RPH was digested with PstI and HindIII. Fragments were ligated together with KpnI-PstI digested aphII gene into plasmid pKC1139 digested with EcoRI and HindIII to produce about 9.5 Kb of pKC-SF2-KSII. The construct was delivered into S. sp. SF2575 by conjugation with E. coli ET12567(pUZ8002). Intergeneric conjugation between E. coli and Streptomyces was performed as described previously with minor modification.57 The transformant was resistance to both apramycin and kanamycin, and was grown in fresh R2YE/kanamycin liquid medium at 37° C for 1–2 days in order to force chromosomal integration of the gene-disruption constructs. The resulting disruption mutant (S. sp. SF2575 ΔssfB) was selected on kanamycin containing plates. Gene disruption in S. SF2575 ΔssfB was confirmed by PCR of total genomic DNA. S. sp. SF2575 ΔssfB did not produce SF2575 or intermediates when grown on Bennett's plates as confirmed by LC-MS analysis.
Expression and purification of recombinant enzymes
SsfX3 and SsfL1 were cloned into a pET28 vector with an N-terminal His tag and expressed in E. coli BL21(DE3) strain. For each enzyme, a 5 ml overnight culture of BL21(DE3) transformed with expression plasmid was grown in LB medium containing 35 mg/L kanamycin. This overnight culture was used to inoculate a 500 mL flask of LB containing 35 mg/L kanamycin. Cultures were grown at 37°C until OD600 of 0.5 at which point IPTG was added to a final concentration of 120 μM to induce protein expression. The induced cultures were further shaken overnight at 16°C.
Protein purification was carried out at 4°C. Cell pellets were resuspended in Buffer A (50 mM Tris-HCl, pH 7.9, 10 mM imidazole and 50 mM NaCl). Cell membrane was disrupted by sonication. Cell lysate was centrifuged at 16,000 rpm and the soluble fraction was collected and incubated with Ni-NTA resin (Qiagen) for 2 hours with gentle rotation. Protein-resin mixture was added to a gravity flow column and buffers containing increasing step gradients of imidazole were applied. The target proteins were eluted in Buffer A containing 250 mM imidazole. A 30 kDa MWCO Amicon filtration column (Millipore) was used for buffer exchange and concentration of the protein solution. Purified enzymes were stored in Buffer B (50 mM Tris-HCl pH 7.9, 2 mM EDTA, 2 mM DTT). Proteins were aliquoted, flash frozen on dry ice and stored at -80°C. Protein concentrations were measured by Bradford assay using bovine serum albumin (BSA) as a standard72.
In vitro assays
PPi assay reagent was purchased from Sigma (P7275) and was performed according to manufacturer instructions. Typical reaction mix includes: 33% PPi reagent, 100 mM Tris-HCl pH 7.5, 2 mM ATP, 2 mM salicylic acid and 10 μM SsfL1. Reaction progress was monitored at 340 nm using a BioTek PowerWave XS plate reader for thirty minutes at room temperature. All in vitro assays containing SsfX3 were performed with the following conditions unless otherwise specified: 50 mM HEPES pH 7.9, 10 mM MgCl2, 2 mM ATP, 2 mM free CoA, 2 mM salicylic acid, 20 μM 6, 1.5 μM SsfX3 and 15 μM SsfL1. Reaction mixture was incubated at room temperature for 30 minutes. A 100 μl reaction was extracted with 200 μl acetonitrile (1% acetic acid). Organic phase was then removed and solvent was evaporated in vacuo. Samples were then resuspended in 30 μl methanol and analyzed by HPLC or LCMS as described in Spectroscopic analysis.
Heterologous biosynthesis of polyketides using S. lividans
S. lividans K4-114 strain54 transformed with different constructs shown in Table 2 were grown on R5 agar plus 50 μg/ml thiostrepton. Cultures were grown at 30°C for approximately one week until colonies were well formed. For HPLC analysis, colonies were streaked out on 40 ml of fresh R5 agar (plus 50 μg/ml thiostrepton) and incubated for 7-10 days until the plate was well pigmented. The agar was then finely chopped and extracted with 40 ml organic solvent (99% ethyl acetate, 1% acetic acid). Sodium sulfate was added to the extract to remove residual water, and the solvent was evaporated and the products were resuspended in 400 μl DMSO. The extracts were analyzed by reverse phase HPLC. Compounds were identified by matching retention time and UV spectra to authentic standards.
Supplementary Material
Acknowledgments
This work is funded by NSF CBET #0545860 and CDMRP W81XWH-08-1-0614to Y. T.; and NIH training grant #1T32GM067555 to L. B. P. The authors thank Dr. Kathleen Sakamoto for advice on the bioactivity assays and providing the Nalm-6 cell line and Prof. Tatiana Segura for the gift of the HeLa, MCF-7, and M249 cell lines. We thank Prof. Michael Jung and Dr. Wenjun Zhang for helpful discussion.
Footnotes
Supporting Information Available: Additional Experimental procedures, and compound characterizations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Nat Prod Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- 2.Chopra I, Roberts M. Microbiol Mol Biol Rev. 2001;65:232–260. doi: 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duggar BM. Ann NY Acad Sci. 1948;51:177–181. doi: 10.1111/j.1749-6632.1948.tb27262.x. [DOI] [PubMed] [Google Scholar]
- 4.Finlay AC, Hobby GL, P'an SY, Regna PP, Routein JB, Seeley DB, Shull GM, Sobin BA, Solomons IA, Vinson JW, Kane JH. Science. 1950;111:85. [Google Scholar]
- 5.Wright GD, Zakeri B. Biochem Cell Biol. 2008;86:124–136. doi: 10.1139/O08-002. [DOI] [PubMed] [Google Scholar]
- 6.Sum PE, Lee VJ, Testa RT, Hlavka JJ, Ellestad GA, Bloom JD, Gluzman Y, Tally FP. J Med Chem. 1994;37:184–188. doi: 10.1021/jm00027a023. [DOI] [PubMed] [Google Scholar]
- 7.Sum PE, Petersen P. Bioorg Med Chem Lett. 1999;9:1459–1462. doi: 10.1016/s0960-894x(99)00216-4. [DOI] [PubMed] [Google Scholar]
- 8.Hatsu M, Sasaki T, Gomi S, Kodama Y, Sezaki M, Inouye S, Kondo S. J Antibiot. 1992;45:325–330. doi: 10.7164/antibiotics.45.325. [DOI] [PubMed] [Google Scholar]
- 9.Hatsu M, Sasaki T, Watabe H, Miyadoh S, Nagasawa M, Shomura T, Sezki M, Inouye S, Kondo S. J Antibiot. 1992;45:320–324. doi: 10.7164/antibiotics.45.320. [DOI] [PubMed] [Google Scholar]
- 10.Horiguchi T, Hayashi K, Tsubotani S, Iinuma S, Harada S, Tanida S. J Antibiot. 1994;47:545–556. doi: 10.7164/antibiotics.47.545. [DOI] [PubMed] [Google Scholar]
- 11.Mitscher LA, Juvarkar JV, Rosenbrook W, Andres WW, Schenck JR, Egan RS. J Am Chem Soc. 1970;92:6070–6071. doi: 10.1021/ja00723a049. [DOI] [PubMed] [Google Scholar]
- 12.Tymiak AA, Aklonis C, Bolgar MS, Kahle AD, Kirsch DR, O'Sullivan J, Porubcan MA, Principe P, Trejo WH. J Org Chem. 1993;58:535–537. [Google Scholar]
- 13.Brodersen DE, Clemons WM, Jr, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 14.Kellner U, Sehested M, Jensen PB, Gieseler F, Rudolph P. Lancet Oncol. 2002;3:235–243. doi: 10.1016/s1470-2045(02)00715-5. [DOI] [PubMed] [Google Scholar]
- 15.Pommier Y. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 16.Ryan MJ. U S Patent 5989903. 1999
- 17.Hunter IS. Tetracyclines. In: Martin JF, editor. Microbial secondary metabolites: biosynthesis, genetics and regulation. Research Signpost; Lucknow, India: 2002. pp. 141–166. [Google Scholar]
- 18.Zhang W, Ames BD, Tsai SC, Tang Y. Appl Environ Microbiol. 2006;72:2573–2580. doi: 10.1128/AEM.72.4.2573-2580.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Watanabe K, Cai X, Jung ME, Tang Y, Zhan J. J Am Chem Soc. 2008;130:6068–6069. doi: 10.1021/ja800951e. [DOI] [PubMed] [Google Scholar]
- 20.Zhang W, Watanabe K, Wang CCC, Tang Y. J Biol Chem. 2007;282:25717–25725. doi: 10.1074/jbc.M703437200. [DOI] [PubMed] [Google Scholar]
- 21.Wawrik B, Kerkhof L, Zylstra GJ, Kukor JJ. Appl Environ Microbiol. 2005;71:2232–2238. doi: 10.1128/AEM.71.5.2232-2238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salas JA, Méndez C. Trends Microbiol. 2007;15:219–232. doi: 10.1016/j.tim.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Faust B, Hoffmeister D, Weitnauer G, Westrich L, Haag S, Schneider P, Decker H, Kunzel E, Rohr J, Bechthold A. Microbiology. 2000;146:147–154. doi: 10.1099/00221287-146-1-147. [DOI] [PubMed] [Google Scholar]
- 24.Trefzer A, Pelzer S, Schimana J, Stockert S, Bihlmaier C, Fiedler HP, Welzel K, Vente A, Bechthold A. Antimicrob Agents Chemother. 2002;46:1174–1182. doi: 10.1128/AAC.46.5.1174-1182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Draeger G, Park SH, Floss HG. J Am Chem Soc. 1999;121:2611–2612. [Google Scholar]
- 26.Bililign T, Hyun CG, Williams JS, Czisny AM, Thorson JS. Chem Biol. 2004;11:959–969. doi: 10.1016/j.chembiol.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 27.Aragozzini F, Craveri R, Maconi E, Ricca GS, Scolastico C. J Chem Soc, Perkins Trans 1. 1988:1865–1867. [Google Scholar]
- 28.Jia XY, Tian ZH, Shao L, Qu XD, Zhao QF, Tang J, Tang GL, Liu W. Chem Biol. 2006;13:575–585. doi: 10.1016/j.chembiol.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Daum M, Peintner I, Linnenbrink A, Frerich A, Weber M, Paululat T, Bechthold A. ChemBioChem. 2009;10:1073–1083. doi: 10.1002/cbic.200800823. [DOI] [PubMed] [Google Scholar]
- 30.Kerbarh O, Ciulli A, Howard NI, Abell C. J Bacteriol. 2005;187:5061–5066. doi: 10.1128/JB.187.15.5061-5066.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zwahlen J, Kolappan S, Zhou R, Kisker C, Tonge PJ. Biochemistry. 2007;46:954–964. doi: 10.1021/bi060852x. [DOI] [PubMed] [Google Scholar]
- 32.Knaggs AR. Nat Prod Rep. 1999;16:525–560. doi: 10.1039/a707501d. [DOI] [PubMed] [Google Scholar]
- 33.Cooke HA, Zhang J, Griffin MA, Nonaka K, Van Lanen SG, Shen B, Bruner SD. J Am Chem Soc. 2007;129:7728–7729. doi: 10.1021/ja071886a. [DOI] [PubMed] [Google Scholar]
- 34.Van Lanen SG, Oh Tj, Liu W, Wendt-Pienkowski E, Shen B. J Am Chem Soc. 2007;129:13082–13094. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weitnauer G, Mühlenweg A, Trefzer A, Hoffmeister D, Süβmuth RD, Jung G, Welzel K, Vente A, Girreser U, Bechthold A. Chem Biol. 2001;8:569–581. doi: 10.1016/s1074-5521(01)00040-0. [DOI] [PubMed] [Google Scholar]
- 36.Akoh CC, Lee GC, Liaw YC, Huang TH, Shaw JF. Prog Lipid Res. 2004;43:534–552. doi: 10.1016/j.plipres.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Upton C, Buckley JT. Trends Biochem Sci. 1995;20:178–179. doi: 10.1016/s0968-0004(00)89002-7. [DOI] [PubMed] [Google Scholar]
- 38.McGaw BA, Woolley JG. Phytochemistry. 1979;18:1647–1649. [Google Scholar]
- 39.Seigler DS. Plant Secondary Metabolism. Kluwer Academic Publishers; Norwell, MA: 1998. p. 759. [Google Scholar]
- 40.Wang L, McVey J, Vining LC. Microbiology. 2001;147:1535–1545. doi: 10.1099/00221287-147-6-1535. [DOI] [PubMed] [Google Scholar]
- 41.Ichinose K, Ozawa M, Itou K, Kunieda K, Ebizuka Y. Microbiology. 2003;149:1633–1645. doi: 10.1099/mic.0.26310-0. [DOI] [PubMed] [Google Scholar]
- 42.Magnuson K, Jackowski S, Rock CO, Cronan JE. Microbiol Rev. 1993;57:522–542. doi: 10.1128/mr.57.3.522-542.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang Y, Lee TS, Kobayashi S, Khosla C. Biochemistry. 2003;42:6588–6595. doi: 10.1021/bi0341962. [DOI] [PubMed] [Google Scholar]
- 44.Grimm A, Madduri K, Ali A, Hutchinson CR. Gene. 1994;151:1–10. doi: 10.1016/0378-1119(94)90625-4. [DOI] [PubMed] [Google Scholar]
- 45.Heath RJ, Rock CO. J Biol Chem. 1996;271:1833–1836. doi: 10.1074/jbc.271.4.1833. [DOI] [PubMed] [Google Scholar]
- 46.Menéndez N, Nur-e-Alam M, Braña AF, Rohr J, Salas JA, Méndez C. Chem Biol. 2004;11:21–32. doi: 10.1016/j.chembiol.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 47.Magarvey NA, Haltli B, He M, Greenstein M, Hucul JA. Antimicrob Agents Chemother. 2006;50:2167–2177. doi: 10.1128/AAC.01545-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hara O, Hutchinson CR. J Bacteriol. 1992;174:5141–5144. doi: 10.1128/jb.174.15.5141-5144.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meadows ES, Khosla C. Biochemistry. 2001;40:14855–14861. doi: 10.1021/bi0113723. [DOI] [PubMed] [Google Scholar]
- 50.Tang Y, Koppisch AT, Khosla C. Biochemistry. 2004;43:9546–9555. doi: 10.1021/bi049157k. [DOI] [PubMed] [Google Scholar]
- 51.He QL, Jia XY, Tang MC, Tian ZH, Tang GL, Liu W. ChemBioChem. 2009;10:813–819. doi: 10.1002/cbic.200800714. [DOI] [PubMed] [Google Scholar]
- 52.Boehlein SK, Richards NG, Schuster SM. J Biol Chem. 1994;269:7450–7457. [PubMed] [Google Scholar]
- 53.McDaniel R, Ebert-Khosla S, Hopwood DA, Khosla C. Science. 1993;262:1546–1550. doi: 10.1126/science.8248802. [DOI] [PubMed] [Google Scholar]
- 54.Ziermann R, Betlach MC. Biotechniques. 1999;26:106–110. doi: 10.2144/99261st05. [DOI] [PubMed] [Google Scholar]
- 55.Fu H, Ebert-Khosla S, Hopwood DA, Khosla C. J Am Chem Soc. 1994;116:6443–6444. [Google Scholar]
- 56.Ichinose K, Bedford DJ, Tornus D, Bechthold A, Bibb MJ, Peter Revill W, Floss HG, Hopwood DA. Chem Biol. 1998;5:647–659. doi: 10.1016/s1074-5521(98)90292-7. [DOI] [PubMed] [Google Scholar]
- 57.Bierman M, Logan R, O'Brien K, Seno ET, Nagaraja Rao R, Schoner BE. Gene. 1992;116:43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- 58.Zhang W, Wilke BI, Zhan J, Watanabe K, Boddy CN, Tang Y. J Am Chem Soc. 2007;129:9304–9305. doi: 10.1021/ja0736919. [DOI] [PubMed] [Google Scholar]
- 59.Prado L, Lombó F, Braña AF, Méndez C, Rohr J, Salas JA. Mol Gen Genet. 1999;261:216–225. doi: 10.1007/s004380050960. [DOI] [PubMed] [Google Scholar]
- 60.Binnie C, Warren M, Butler MJ. J Bacteriol. 1989;171:887–895. doi: 10.1128/jb.171.2.887-895.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vancurová I, Volc J, Flieger M, Neuzil J, Novotná J, Vlach J, Bĕhal V. Biochem J. 1988;253:263–267. doi: 10.1042/bj2530263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brown JR, Ireland DS. Structural Requirements for Tetracycline Activity. In: Garattini S, Goldin A, Hawking F, Kopin IJ, editors. Advances in Pharmacology and Chemotherapy. Vol. 15. Academic Press; New York: 1978. pp. 161–202. [DOI] [PubMed] [Google Scholar]
- 63.Butler MJ, Friend EJ, Hunter IS, Kaczmarek FS, Sugden DA, Warren M. Mol Gen Genet. 1989;215:231–238. doi: 10.1007/BF00339722. [DOI] [PubMed] [Google Scholar]
- 64.Nakano T, Miyake K, Endo H, Dairi T, Mizukami T, Katsumata R. Biosci Biotechnol Biochem. 2004;68:1345–1352. doi: 10.1271/bbb.68.1345. [DOI] [PubMed] [Google Scholar]
- 65.Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R. Microbiol Mol Biol Rev. 2005;69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pao SS, Paulsen IT, Saier MH., Jr Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wietzorrek A, Bibb M. Mol Microbiol. 1997;25:1181–1184. doi: 10.1046/j.1365-2958.1997.5421903.x. [DOI] [PubMed] [Google Scholar]
- 68.Chen Y, Wendt-Pienkowski E, Shen B. J Bacteriol. 2008;190:5587–5596. doi: 10.1128/JB.00592-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. John Innes Foundation; Norwich, UK: 2000. [Google Scholar]
- 70.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer ELL, Bateman A. Nucl Acids Res. 2008;36:D281–288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Denis F, Brzezinski R. FEMS Microbiol Lett. 1991;81:261–264. doi: 10.1016/0378-1097(91)90224-x. [DOI] [PubMed] [Google Scholar]
- 72.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.