

Abstract
A rapid, water-soluble enzyme-mediated radical chain initiation system involving glucose oxidase and Fe+2 generated hydrogels within minutes at 25°C and in ambient oxygen. The initiation components were evaluated for their effect on polymerization rates of hydroxyethyl acrylate-poly(ethylene glycol)575 diacrylate comonomer solutions using near-infrared spectroscopy. Increasing glucose concentration increased polymerization rates until reaching a rate plateau above 1 × 10−3M of glucose. A square root dependence of the initial polymerization rate on Fe+2 concentration was observed between 1.0 × 10−4M and 5.0 × 10−4M of Fe+2 whereupon excess Fe+2 reduced final acrylate conversions. The glucose oxidase-mediated initiation system was employed for encapsulation of fibroblasts (NIH3T3s) into a poly(ethylene glycol) tetra-acrylate (Mn~20,000) hydrogel scaffold demonstrating 96% (±3%) viability at 24 hours post-encapsulation. This first use of enzyme-mediated redox radical chain initiation for cellular encapsulation demonstrates polymerization of hydrogels in situ with kinetic control, minimal oxygen inhibition issues and utilization of low initiator concentrations.
Keywords: PEG-hydrogel, cell encapsulation, radical chain polymerization, redox initiation, glucose oxidase, enzyme
Introduction
A hydrophilic and crosslinked hydrogel, that prevents dissolution while promoting water uptake, facilitates biomedical application including drug delivery, biosensor fabrication and tissue engineering.1 One prominent tissue regenerative approach encapsulates cells into supportive three-dimensional hydrogel scaffolds to guide cellular adhesion, molecular signaling, proliferation and, ultimately, tissue formation.2–4 The homogenous distribution of cells and biomolecules in the hydrogel precursor solution permits the delivery to any targeted site, allowing morphologic customization of the polymerized gel. A realization of such hydrogel cellular scaffolds requires a gelation process wherein the precursor solution is water-soluble; the polymerization conditions are mild; the cure time is readily tailored for a particular application; and the gel components are all cytocompatible.
The use of radical chain polymerization of high molecular weight poly(ethylene glycol) (PEG) vinyl monomers (i.e., macromers) often meets these demands and further permits molecular design of mechanical properties, swelling behavior and bioactive molecule incorporation. Although photoinitiation is frequently employed for generation of these cell-laden scaffolds,5–8 photopolymerization limitations associated with cure depth and shadow effects have prompted investigations into successful light independent encapsulation systems using the thermally activated ammonium persulfate-tetramethylethylenediamine (APS/TEMED) initiation.9–11 Additionally, utilizing ascorbic acid in combination with persulfate ions, Mikos and coworkers described the significant advantages of redox initiation pathways for generating hydrogels and the importance of the redox initiator components on cellular environments.12 Another redox initiation system involving Fenton’s reagent,13,14 comprising a ferrous salt (Fe+2) and hydrogen peroxide (H2O2), was successfully employed for the polymerization of poly(vinyl alcohol) (PVA) hydrogels showing unique and advantageous drug release profiles compared with UV cured PVA hydrogels.15 However, this rapid Fenton system also demonstrates unique challenges in finely controlling cure times and avoiding the formation of microgel domains.16
Other light-independent initiation systems include enzymes that catalyze electron transfer reactions and subsequently generate products capable of initiating chain polymerization of water-soluble vinyl monomers; these include certain oxidoreductase catalytic mechanisms which directly form a primary radical appropriate for initiating the vinyl polymerization reaction. An eminent example involves the horseradish peroxidase (HRP) enzyme that catalyzes the oxidation of 2,4-pentanedione by H2O2 generating a radical initiating species on the acetylacetone molecule.17 Although this HRP ternary system demonstrates successful polymerization of monomers including acrylamide and hydroxyethylmethacrylate,18,19 the scheme also shows extended and irregular inhibitory periods of between 45 and100 minutes.20 Another ternary system consisting of manganese peroxidase, H2O2 and 2,4-pentanedione initiated the polymerization of acrylamide but required 12 hours under a nitrogen atmosphere.21 Such extended polymerization times do not facilitate the rapid formation of hydrogels as needed in cellular encapsulation. To date, no enzyme-mediated chain initiation systems have been employed for cellular encapsulation into hydrogel scaffolds.
Enzyme-mediated initiation systems involving oxidase enzymes that produce H2O2, including the glucose oxidase (GOX) enzyme, offers a beneficial approach for rapidly polymerizing hydrogels. In the GOX initiation system, the enzyme binds to the glucose substrate generating gluconolactone and subsequently regenerates the flavin adenine dinucleotide (FAD) cofactor by binding oxygen and producing H2O2. By coupling ferrous ions to this enzymatic process, hydroxyl radical species are produced. Notably, a unique aspect of this system involves the utilization and elimination of oxygen, a powerful inhibitor of radical chain polymerization, by the GOX enzyme during the initiation process. This feature offers unique advantages, including the ability to reduce initiator quantities and eliminate the requirement of performing reactions under inert atmospheric conditions which precludes the ability to encapsulate cells. For example, the GOX initiation system was actually completely suppressed under a nitrogen atmosphere until the introduction of oxygen whereupon polymerization quickly commenced.22 This compatibility with oxygen further prompted the use of this GOX system for the polymerization of detachable balloon catheters used in endovascular surgery where oxygen flow to blood and tissue is critical.23 The tolerance of the GOX system to oxygen and the elimination of an extraneous energy source for initiation, such as light or heat, prompted our investigations to understand further the polymerization kinetics and employ this system for cellular encapsulation.
Herein, we detail the chain polymerization kinetic reactions and mechanism of the GOX mediated initiation system for forming polymers with biomaterials applications. The outlined kinetic parameters provide a simple means to control the final vinyl conversion and tailor the polymerization rates. The understanding of these kinetic reactions facilitated the encapsulation of mammalian cells with high viability.
Experimental Section
Polymerization Kinetic Studies
The polymerization reactions were monitored using Fourier transform infrared (FTIR) spectroscopy with a Nicolet 750 Magna FTIR instrument. The acrylate double bond conversion was followed in real time using the near-IR absorption peak between 6212–6150 cm−1 corresponding to the C-H stretch of the acrylate functional group. The polymerization reaction commenced with the addition of GOX from Aspergillus niger (Sigma-Aldrich) to a mixing solution of all other reaction components comprising iron (II) sulfate (Fe+2) (Sigma-Aldrich), glucose (Sigma-Aldrich), 2-hydroxyethyl acrylate (HEA), poly(ethylene glycol) diacrylate (Mn~575 Da) (Sigma-Aldrich) and 2-(N-morpholino)ethanesulfonic acid (MES) buffer stabilized at pH=4.5 (Teknova). The MES buffer was chosen for these kinetic studies to maintain slightly acidic conditions favorable for the GOX enzyme. A 10% (w/v) glucose stock solution was stabilized to ensure mutarotation of the sugar. After mixing, the reactions were immediately transferred to a 1mm thick glass sample compartment and placed in the FTIR instrument that utilized a horizontal transmission apparatus.24 All the reactions were performed sealed from the open atmosphere at ambient temperature without a solution purge of oxygen. Negative controls were performed by eliminating either GOX, glucose or Fe+2 from the reaction mixture and each resulted in no polymerization after 30 minutes. A slight induction period was partly attributed to the presence of the hydroquinone monomethylether (MEHQ) inhibitor in the unpurified commercial monomers. This induction period permitted the acquisition of a reliable zero conversion baseline utilized in the data analysis. The initial polymerization rates (Rp) were obtained by determining the time required to react from 15% to 30% double bond conversion and each experimental condition was performed three times. Rheological measurements using an ARES rheometer (TA Instruments) were employed to verify the polymerization of the poly(ethylene glycol) tetra-acrylate (Mn~20000 Da). For rheological measurements, the reaction materials were mixed in the same manner used for the near-IR experiments and promptly sandwiched between 20mm plates in parallel configuration.
Cell Culture
The NIH3T3 fibroblast cells were cultured using Dulbecco’s modified eagle medium (DMEM) containing 25mM glucose (Gibco) and further supplemented with 10% fetal bovine serum (FBS), 1µg/mL amphotericin, 50U/mL penicillin, 50ug/mL streptomycin and 20ug/mL gentamicin. The cells were cultured under standard conditions (37°C and 5% CO2) both prior to and following encapsulation
Fibroblast encapsulation
The poly(ethylene glycol) tetra-acrylate (Mn~20000) (PEGTA20000) was synthesized according to previously published protocols.25 The characterization of the PEGTA20000 product with H-NMR confirmed 95% acrylation. A peptide comprising CRGDS (cysteine, arginine, glycine, aspartic acid, serine) was synthesized using a 433A peptide synthesizer (Applied Biosystems) and purified by reverse phase high performance liquid chromatography. The identity of the peptide was verified by MALDI-MS. An Ellman’s colorimetric assay26 was also used to verify and quantify the presence of reduced thiols on cysteine residues, which permit the facile incorporation of the peptide within the hydrogel network.27 Briefly, the cysteine residues present on the CRGDS peptide are incorporated into the hydrogel by chain transfer during the homopolymerization of the acrylate groups. Inclusion of this peptide in the hydrogel facilitates cellular adhesion and survival in synthetic hydrogels.28
Fibroblasts were encapsulated into hydrogels at a density of 30 × 106 cells/mL, by gently suspending the cells in a PEGTA20000 monomer formulation to obtain a final concentration of 2.5 × 10−5 M GOX, 1.25mM Fe+2, 4mM glucose, 15wt% PEGTA20000, and 1mM of CRGDS in a 1X Dulbecco’s Phosphate Buffered Saline (PBS) solution (pH=7.2–7.4). The mixture was added to a cylindrical mold (4mm diameter, 1.5mm height) and permitted to polymerize for approximately 5 minutes. The gels were incubated for 30 minutes in 1X PBS (pH=7.2–7.4) at 37°C before transferring the gels to the appropriate media or cell viability solution. For experiments involving the catalase enzyme, the gels were incubated under standard conditions in DMEM cell culture media containing a final catalase concentration of 2.0 × 10−6 M for 24 hours. The catalase enzyme catalyzes the degradation of H2O2 to water and O2, both products well tolerated by cells. Cell viability was determined using the Live/Dead cellular stain (Invitrogen), a membrane integrity assay that stains living cells green and dead cells red. The fluorescence images were obtained using confocal microscopy, 3 images taken at random positions for each gel examined. Live, green cells were counted using MetaMorph software; dead, red cells were counted manually.
Results and Discussion
Enzymatic H2O2 generation and Initial Polymerization Rates
The GOX mediated radical generation process has been shown to be comprised of four significant reaction steps (Equations 1–4).23
| (1) |
| 29 | (2) |
| 29 | (3) |
| 30 | (4) |
The GOX enzyme catalyzes the oxidation of glucose to gluconolactone and subsequently regenerates the FAD cofactor by reducing O2 to H2O2 (Equation 1). By combining the enzymatically produced H2O2 with ferrous ions, a hydroxyl radical initiator is generated (Equation 2) which may further react with vinyl monomer [M] to produce a chain initiating species (Equation 4) or react with additional ferrous ion to inhibit polymerization (Equation 3). Overall, the present studies found that this GOX-mediated initiation system promotes rapid polymerization rates reaching high acrylic double bond conversion, usually within about three minutes under current conditions at ambient temperature and atmosphere. Moreover, the polymerization rates were easily tailored using a HEA-PEGDA575 monomer formulation by changing the initiator component concentrations (i.e., glucose, GOX enzyme, Fe+2). Detailed investigations of polymerization kinetics were required to understand further this initiation system for use in cellular encapsulation studies.
Studies were performed to investigate the effects of H2O2 generation (Equation 1) on the polymerization kinetics, including the effects of glucose and GOX concentrations on polymerization rates. First, the effects of glucose on polymerization rates were evaluated by monitoring ambient temperature polymerization reactions at differing glucose concentrations using near-IR spectroscopy. Representative acrylate conversion profiles for various glucose concentrations using 6.25 × 10−7 M GOX enzyme and 2.5 × 10−4 M Fe+2 (Figure 1A) display the general trend that the increase in glucose concentrations results in an increase in polymerization rates when all other reaction components are maintained constant. This trend would be expected since increasing the glucose substrate results in the increase in the H2O2 generation rate by the GOX enzyme.
Figure 1.

(A) A representative double bond conversion profile of the GOX-mediated initiation system with varying concentrations of glucose and a fixed Fe+2 concentration of 2.5 × 10−4 M. The conversion plot displays that the rate of polymerization increases with increasing glucose concentrations. (B) The dependence of initial polymerization rates on glucose concentrations for (○) 2.5 × 10−4 M, (■) 1.25 × 10−4 M, and (▲) 6.25 × 10−5 M of Fe+2. All reactions were performed with 6.25 × 10−7 M GOX, 10mM MES pH= 4.5, 20wt% HEA and 15wt% PEGDA575 with ambient temperature and oxygen.
These acrylate conversion profiles were further used to determine scaling relationships between the initial polymerization rates (Rp) and glucose concentrations at different fixed ferrous values (Figure 1B). Since the polymerization rate scales with initiation rate31, direct monitoring of the polymerization reaction (i.e., double bond conversion) permits a further understanding of the initiation reaction and ultimately its mechanisms. Classically, assuming pseudosteady state and chain length independent termination, the radical chain polymerization rate is proportional to the square root of the initiation rate (Ri) indicating bimolecular termination (Rp ∞ Riα, where α = 0.5).31,32 A divergence of the scaling exponent (α) from 0.5 is attributed to alterations in the typical bimolecular termination mode, including α < 0.5 for systems involving chain length dependent termination33 and α > 0.5 for systems encountering inhibitory or unimolecular termination.34 Here, the dependence of polymerization rates on glucose concentrations displayed a dynamic variation of scaling exponents changing from α = 1, at lower glucose values, to α = 0.5 at increased glucose values. At α = 1, a first order dependence of polymerization rate on glucose concentration indicates a deviation from bimolecular termination likely associated with an inhibitory termination reaction. Evidence for such an inhibitory reaction is also suggested by the conversion profiles in Figure 1A where at lower glucose value (i.e., below 1.75 × 10−4 M of glucose), the reactions do not achieve complete double bond conversion. This inhibitory reaction is further evaluated and discussed in the following section concerning iron and polymerization.
Interestingly, at higher glucose concentrations, approximately 1.0 × 10−3 M and above, the initial polymerization rates do not change as the glucose concentration changes and ultimately maintain a zero order dependence with respect to glucose. This behavior implies that as the polymerization rate saturates, another component of the initiation system becomes limiting (i.e., Fe+2, GOX, O2). Further tests were performed by monitoring polymerization reactions at various GOX enzyme concentrations (Figure 2). In Figure 2, all reactions contain identical [Fe+2] at 2.0 × 10−4M, whereas each line represents a unique and fixed glucose value. Each line displays a near square root dependence of the polymerization rate on the enzyme concentration until a saturation point is achieved, after which the polymerization rate maintains a zero order dependence on GOX. Notably, the saturation point is reached at, and above, 6.25 × 10−7 M of GOX enzyme, the same concentration of enzyme employed in Figure 1. This result indicates that a point is reached whereupon additional glucose or GOX enzyme does not increase the polymerization rate.
Figure 2.
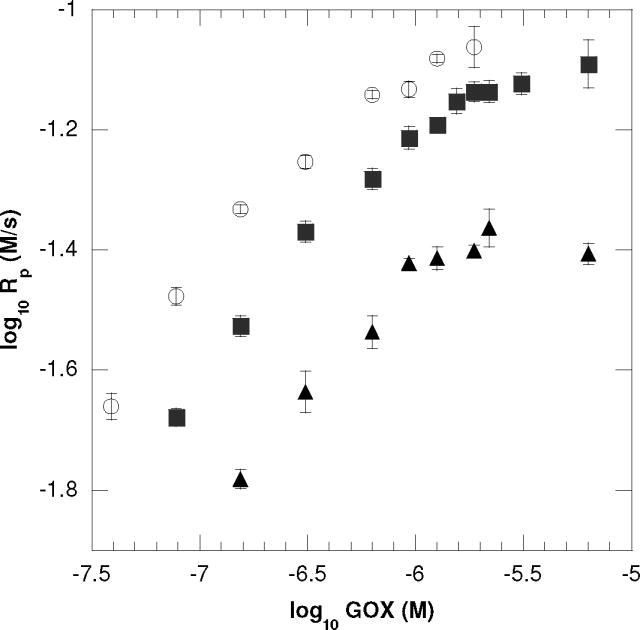
The dependence of initial polymerization rates on GOX concentrations for (○) 1.0 × 10−3 M, (■) 5.0 × 10−4 M and (▲) 2.5 × 10−4 M of glucose. These reactions exhibit a near square root dependence of the polymerization rate on the enzyme concentration (i.e., 0.42 ±0.04, 0.45 ±0.04, 0.41 ±0.04 for 1.0 × 10−3 M, 5.0 × 10−4 M and 2.5 × 10−4 M of glucose, respectively) until a saturation point is achieved at, and above, 6.25 × 10−7 M of GOX. All reactions were performed with ambient oxygen and temperature with 2.0 × 10−4 M Fe+2, 10mM MES pH=4.5, 20wt% HEA and 15wt% PEGDA575.
Taken together, the saturation rate behavior achieved at high concentrations of glucose (Figure 1B) and high concentrations of GOX (Figure 2) is partly explained by understanding the enzymatic H2O2 production mechanism. In this mechanism, increased glucose utilization is accompanied by increased oxygen consumption due to the enzymatic regeneration of the FAD cofactor. Before the polymerization reaction begins, the total dissolved oxygen concentration in these GOX reactions should approximate that of the dissolved oxygen found in a typical acrylate formulation (~1.0 × 10−3M) since the reactions were sealed without an initial solution purge of oxygen.34 The zero order dependence of polymerization rates implies that enzymatic consumption of oxygen contributes to the polymerization rate plateau observed at these higher glucose and GOX levels. Additionally, although the iron species is catalytic in this system (see below discussion concerning iron), the consumption of Fe+2 ferrous may also contribute to this rate plateau behavior at high glucose and GOX concentrations.
Effects of Iron Concentration on Polymerization
The quantity of iron present in a GOX-mediated initiation reaction is crucial since Fe+2 can either promote polymerization by forming the hydroxyl primary radical (Equation 2) or hinder polymerization by destroying the same hydroxyl radical, thus limiting the chain initiation events (Equation 3). Understanding the effects of iron on final double bond conversion and polymerization rates is essential for using the GOX initiation system for biomedical applications such as cellular encapsulation. Incomplete functional group conversion could inhibit the formation of mechanically stable and fully crosslinked hydrogel scaffolds or could result in cell viability loss due to the presence of unreacted monomer. Moreover, understanding the kinetic effects of iron is essential for tuning cure times.
The effect of the [Fe+2] on polymerization kinetics was monitored using near-IR spectroscopy by varying the ferrous concentrations while maintaining identical concentrations of all remaining initiation components (i.e., glucose, GOX, monomer). An increase of initial Rp values with an increase in Fe+2 concentration was expected for a certain range of iron and confirmed as shown in Figure 3. The results in Figure 3 also illustrate that an increase in fixed glucose concentrations (each line represents a unique and fixed glucose value) increases the overall Rp values while maintaining the same scaling exponents (α) values at 0.5 (Table 1). This polymerization rate dependence on the square root of the ferrous ion concentration indicates typical bimolecular termination with negligible inhibitory reactions within this range of Fe+2 values. Moreover, when varying Fe+2 concentrations, the polymerization rates overlap for 1.0 × 10−3 M and 1.5 × 10−3M of fixed glucose amounts (Figure 3). This behavior is consistent with the zero order dependence on glucose observed above 1.0 × 10−3 M as discussed in the previous section. However, at lower glucose and Fe+2 values, the Rp rapidly and significantly drops as shown for 3.0 × 10−4 M and 1.75 × 10−4 M of glucose. This drop in Rp is attributed to the low concentration of hydroxyl initiating species which are unable to overcome termination reactions.
Figure 3.

The dependence of initial polymerization rates on Fe+2 concentrations for (●) 1.5 × 10−3 M, (□)1.0 × 10−3 M, (△) 5.0 × 10−4 M, (◇) 3.0 × 10−4 M, and (■)1.75 × 10−4 M of glucose. This plot displays that the Rp is dependent upon the square root of the Fe+2 concentrations indicating a typical biomolecular termination mode for this range of iron values. Additionally, at (●) 1.5 × 10−3 M and (□)1.0 × 10−3 M of glucose, the Rp values are near identical with increasing Fe+2. This is consistent with zero order dependence of glucose above 1.0 × 10−3 M. All reactions were performed with ambient oxygen and temperature with 6.25 × 10−7 M GOX, 10mM MES pH=4.5, 20wt% HEA and 15wt% PEGDA575.
Table 1.
Initiation rate scaling exponents for the GOX mediated reactions when [Fe+2] was varied between 1 × 10−4 M and 5 × 10−4 M at different fixed glucose concentrations. All reactions contained 6.25 × 10−7 M GOX, 20wt% HEA, 15wt% PEGDA575, 10mM MES pH=4.5
| Glucose (M) | Scaling exponent (α) |
|---|---|
| 1.5 × 10−3 | 0.47 ± 0.02 |
| 1.0 × 10−3 | 0.48 ± 0.02 |
| 5.0 × 10−4 | 0.47 ± 0.02 |
| 3.0 × 10−4 | 0.49 ± 0.02 |
| 1.75 × 10−4 | 0.53 ± 0.09 |
To understand the termination mechanism further, the final double bond conversions were examined. As shown in Figure 4A, a considerable decrease in final conversion occurs with an increase in [Fe+2], suggesting an inhibitory reaction. The increase in Fe+2 accompanies the radical wasting reaction (Equation 3) and likely promotes the decrease in double bond conversion as previously reported.23 However, in addition to this wasting reaction, certain metal salts, including Fe+3, may terminate propagating radical chains by oxidizing the growing polymer chain radical (Mn•) through an electron transfer process as shown in Equation 5.35
Figure 4.

(A) The change in final double bond conversion as a result of differing Fe+2 concentrations is shown for (○) 1.5 × 10−3 M, (□) 1.0 × 10−3 M, (▲) 5.0 × 10−4 M, (◇) 3.0 × 10−4 M, and (■) 1.75 × 10−4 M of glucose. (B) The change in final double bond conversion as a result of differing Fe+2 concentrations with a fixed [glucose] of 1.0 × 10−3 M for all lines is shown. Each line displays the drop in conversion when reactions were supplemented with (□) 0 M, (●) 5.0 × 10−4 M, (▼;) 7.5 × 10−4 M and (◆) 1.0 × 10−3 M of Fe+3 ions. This indicates that the polymerization is susceptible to ferric termination. All reactions were performed with ambient oxygen and temperature with 6.25 × 10−7 M GOX, 10mM MES pH=4.5, 20wt% HEA and 15wt% PEGDA575.
| (5) |
Since ferric ions are generated during the initiation reaction (Equations 2 and 3), it was anticipated that the conversion decrease was partially attributed to the accumulation of Fe+3. However, all monomers are not similarly affected by Fe+3 as shown by the resistance of poly(methyl methacrylate) radicals to ferric termination attributed to steric influence.35 To test the susceptibility of the current monomer formulation to ferric termination, the GOX mediated polymerization reaction with the HEA-PEGDA575 formulation and 1.0 × 10−3 M glucose was performed with supplemented ferric ions (Figure 4B). Significant drops in conversion occurred with increasing Fe+3, indicating that these inhibitors cause premature chain termination in this system. For example, an average decrease in final conversion of 9%(± 3%), 30%(± 5%) and 59%(± 4%) occurred with the addition of 5.0 × 10−4 M, 7.5 × 10−4 M and 1.0 × 10−3 M of Fe+3, respectively.
Importantly, Figure 4A also displays that the reactions become more susceptible to iron inhibition as the glucose concentration used to initiate the reaction is reduced. For example, 95% (± 0.3%) conversion was achieved with reactions containing 5 × 10−4 M Fe+2 and 1.0 × 10−3 M glucose whereas only 74% (± 2%) conversion was reached with reactions containing 5 × 10−4 M Fe+2 and 1.75 × 10−4 M glucose. This result would indicate that, in general, maintaining a high glucose:iron concentration ratio may largely eliminate iron’s inhibitory reactions and promote polymerization. Interestingly, the results in Figure 4A nearly collapse onto a single line when re-plotted as the mole ratio of (Fe+2)2/(glucose) versus final double bond conversion (Figure 5). This plot displays the trend that as the mole ratio of (Fe+2)2/(glucose) becomes smaller (i.e., reaching 10−7), the final double bond conversion approaches 100%. This again emphasizes the benefits of maintaining a high glucose: iron ratio.
Figure 5.

The change in final double bond conversion versus the mole ratio of (Fe+2)2/(glucose) is shown for (○) 1.5 × 10−3 M, (□) 1.0 × 10−3 M, (▲) 5.0 × 10−4 M, (◇) 3.0 × 10−4 M, and (■) 1.75 × 10−4 M of glucose. This result further emphasizes the importance of maintaining a high mole ratio of glucose: iron to prevent excessive termination reactions. All reactions were performed with ambient oxygen and temperature with 6.25 × 10−7 M GOX, 10mM MES pH=4.5, 20wt% HEA and 15wt% PEGDA575.
The capacity for high conversion with trace ferrous iron is explained by understanding the catalytic nature of iron in this initiation system. As shown in Equation 5, the termination of propagating chains by Fe+3 regenerates Fe+2 which can subsequently interact with another H2O2 molecule to initiate polymerization. Since the generation of H2O2 scales with the quantity of glucose present in the GOX system, providing excess glucose promotes the formation of the hydroxyl radical from the catalytic iron. These findings are in agreement with the importance of the stoichiometric relationship between the components of Fenton’s reagent, namely the [Fe+2], [Fe+3] and [H2O2].36 For example, an optimized molar ratio of [H2O2]/[Fe+2] = 10 was found when using only Fenton’s reagent for the production of poly(N-vinyl-2-pyrrolidone).14
GOX Mediated Initiation System for Cellular Encapsulation
Cellular encapsulation in hydrogels requires biocompatible, crosslinked and high molecular weight macromers (e.g., acrylated PEGs), typically with a molecular weight greater than 3kDa, to ensure diffusion of bioactive molecules between the cellular hydrogel and the surrounding media.37 Here, we employed PEGTA20000 (Figure 6A) to encapsulate fibroblasts into a hydrogel using the GOX-mediated polymerization reaction. Monitoring the polymerization kinetics of PEGTA20000 with IR spectroscopy proved infeasible due to the relatively insignificant absorption of the carbon-carbon double bond of the acrylate group. However, the characteristics of the GOX-mediated initiation reaction, as revealed by the HEA-PEGDA575 polymerization studies, should remain unaffected despite the slightly different monomer formulation (i.e., PEGTA20000). For the encapsulation reactions during the polymerization of PEGTA20000, the glucose concentration was maintained in excess of the ferrous ion concentrations, consistent with Figure 4, to eliminate excessive radical termination reactions with excess iron that would potentially result in incomplete acrylate conversion. Moreover, since the polymerization rates are also dependent on monomer concentration,31 tailoring the GOX initiation reaction for the polymerization using the 15wt% PEGTA20000 monomer formulation (that contains a lower concentration of acrylate groups and a neutral pH) required employing an overall higher concentration of glucose and iron to achieve gelation within an appropriate time period. The polymerization kinetics are not expected to be greatly affected from the CRGDS peptide (present at 1mM) as the propagation reaction is dominated by the chain growth mechanism associated with the excess of PEGTA20000 acrylate (~30mM acrylate concentration). The polymerization of a 15wt% PEGTA20000 monomer solution using 1mM CRGDS peptide, 4.0 × 10−3 M glucose, 1.25 × 10−3 M Fe+2 and 2.5 × 10−5 M of GOX enzyme was monitored using rheometry, obtaining a final modulus of 10kPA within three minutes.
Figure 6.

(A) Chemical structure of the PEGTA20000 macromer used to encapsulation fibroblast cells using the GOX-mediated initiation system. (B) Fibroblast (NIH3T3) cells were encapsulated into a 15wt% PEGTA20000 using the GOX-mediated initiation system with 4.0 × 10−3 M glucose, 1.25 × 10−3 M Fe+2, 2.5 × 10−5 M GOX, 1mM CRGDS and 1X PBS. The cell-laden gels were incubated in DMEM glucose media without catalase for either 0, 1 or 24 hours or in DMEM glucose media supplemented with 2.0 × 10−6 M of catalase enzyme for 24 hours. The 24 hour incubations with, and without, catalase were significantly different (α = 0.01). The cell viability was quantified using a LIVE/DEAD membrane integrity assay.
For the cellular encapsulation reactions, immediately upon combining all GOX initiation components, the cells were gently mixed with this precursor solution, transferred to cylindrical molds and permitted to polymerize. The GOX initiation system rapidly cured (~ 3 minutes) into a cylindrical shape hydrogel (4mm diameter × 1.5mm height) using a 1X solution of PBS containing 1mM CRGDS peptide, 4.0 × 10−3 M glucose, 1.25 × 10−3 M Fe+2, 15wt% PEDTA20000, and 2.5 × 10−5 M GOX. It would be expected that post-polymerization hydrogels will contain residual initiator components including the GOX enzyme as the diffusion of proteins out of high molecular weight PEG gels requires days.38
To investigate the effects of the remaining encapsulated initiator constituents on cell viability, gels were incubated in cell media for 0, 1 and 24 hours post-polymerization. The hydrogels were swollen in DMEM media containing 25mM of glucose. Thus, the cells are subject to H2O2 production from any remaining active GOX enzyme. However, cell viability was 98% (±1%) for incubation in media not containing catalase for one hour and 96% (±3%) for incubation in media supplemented with 2.0 × 10−6 M of catalase for 24 hours (Figure 6B). Since the catalase enzyme catalyzes the decomposition of H2O2 to water and oxygen, it would be expected that the presence of catalase post-polymerization would assist in eliminating potential H2O2 generated from residual and active GOX enzyme. The gels incubated in media lacking catalase displayed lower viability (87% ±3%), suggesting that the catalase enzyme may provide protection for cells encapsulated using GOX-mediated initiation by eliminating excess H2O2 generated by the GOX enzyme. Overall, the cells tolerate the GOX-mediated initiation reaction for cellular encapsulation (independent of catalase) as demonstrated by the high viability after one hour. Furthermore, the cellular viability is enhanced at 24 hours post-encapsulation by including the catalase enzyme in the cell media.
Due to the recognized cytotoxicity of H2O2, the high viability of cells encapsulated with the GOX mediated system was unanticipated. However, this result does agree with prior studies demonstrating that the presence of poly(vinyl alcohol) (PVA) polymer significantly decreases the cytotoxicity of murine fibroblasts exposed to H2O2.16 In this study with PVA, the presence of the hydrogel material was suggested to provide cellular protection against the hydroxyl radical. An additional report using oxidative polymerization of phenol by the horseradish peroxidase enzyme that exposed cells to H2O2 during cellular encapsulation into alginate-phenol gels also demonstrated high cellular viability.39 Altogether, these investigations imply that polymeric hydrogel materials may provide protection against the H2O2 during initiation and against potential residual oxidant species generated from any remaining active GOX enzyme within the hydrogel scaffold. Additional investigations would be necessary to understand further the potential protective qualities of the hydrogel material with respect to the GOX initiation system.
Overall, these results demonstrate the first use of the GOX-mediated radical initiation system for the generation of biocompatible hydrogels used to encapsulate fibroblasts with high cellular viability. In these studies, the GOX-mediated cellular encapsulation was performed at ambient temperature. However, this reaction would also be expected to perform well, and with potentially increased polymerization rates, at physiological temperature (i.e., 37°C) as GOX remains stable and active at this temperature. It is also anticipated that the GOX system would not encounter iron inhibitory termination reactions (as discussed in Figure 4) from endogenous iron in biological samples such as serum. Under normal physiological conditions, extracellular iron pools are sequestered by iron binding proteins (e.g., ferritin) or chelating agents and are present at lower concentrations than the current Fe+2 initiation component. As with any new material, this initiating system would require further understanding molecular release from hydrogels (e.g., the GOX enzyme) and general biocompatibility for future in vivo applications. However, the utilization of the GOX-enzyme in implantable devices such as the commercially available, subcutaneous glucose sensor40 is encouraging for the overall biological tolerance of GOX-containing materials.
Given the rapid and predictable nature of the initiation, the GOX system may offer benefits for additional biomedical applications. For instance, GOX immobilization in hydrogels is highly significant for producing glucose responsive hydrogel biosensors to mimic islet cell activity for the treatment and monitoring of diabetes.41 Current techniques for GOX immobilization in a hydrogel involve employing other radical chain initiators in the presence of GOX,42,43 or swelling hydrogels in a GOX solution.44 In the current work, the GOX enzyme is encapsulated within the hydrogel during the initiation process and, in effect, immobilizes itself into the polymer matrix. By using GOX to generate the H2O2 initiator, a facile approach for fabricating GOX immobilized glucose hydrogels for sensor applications may be achieved.
Conclusion
The benefits and mechanisms of the GOX-mediated initiation reaction are demonstrated by monitoring a series of polymerization reactions using near-IR spectroscopy with a HEA-PEGDA575 monomer formulation. Utilizing this formulation, we demonstrated that the GOX-mediated initiation system provides a rapid and tunable approach for generating hydrogels. The polymerization occurs within minutes at ambient temperature without requiring any oxygen purge and using low concentrations of initiation components. Moreover, this system uniquely overcomes potential oxygen inhibition by using the GOX enzyme to locally consume oxygen and subsequently generate the H2O2 initiator. The polymerization rates increase with increasing glucose until, ultimately, achieving zero order dependence above 1 × 10−3 M glucose. Additionally, the initiation reaction shows susceptibility to inhibitory reactions from an excess of iron and this behavior is avoided by using a high ratio of glucose: iron.
The GOX initiation system was employed for cellular encapsulation of a fibroblast cell line (NIH3T3s) into a 15wt% PEGTA20000 hydrogel scaffold. The decrease in acrylate concentration using this PEGTA20000 monomer formulation contributed to the requirement to increase the initiation components while still maintaining a high glucose: iron mole ratio. At 24 hours post-encapsulation, the cells demonstrate high viability indicating that residual initiation components have minimal deleterious effects. This system is the first demonstration of using an enzyme-mediated chain initiation reaction for cellular encapsulation and offers an alternative light-independent approach for generating hydrogel scaffolds to support cellular growth. This elimination of light dependence may provide additional benefits including the capability to cure intricate shapes without concerns associated with shadowing effects and the potential capacity to perform in clinical applications that are limited by light penetration.
Supplementary Material
Acknowledgments
This work has been supported by the State of Colorado and the University of Colorado Technology Transfer Office, by National Institutes of Health Grant No. R21 CA 127884, and by NSF grant CBET 0626023. L.M.J. and B.D.F. acknowledge the Graduate Assistantship in Areas of National Need Fellowship from the U.S. Department of Education.
Footnotes
Supporting Information Available. The supplemental data includes fluorescent images of cell-laden gels post-encapsulation with the GOX-mediated initiation system. These images display cells after incubation in glucose containing media for differing time points. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv Mater. 2006;18:1345–1360. [Google Scholar]
- 2.Cushing MC, Anseth KS. Science. 2007;316:1133–1134. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- 3.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 4.Bryant SJ, Bender RJ, Durand KL, Anseth KS. Biotechnol Bioeng. 2004;86:747–755. doi: 10.1002/bit.20160. [DOI] [PubMed] [Google Scholar]
- 5.Bryant SJ, Nuttelman CR, Anseth KSJ. Biomater Sci Polymer Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 6.Sntjens SHN, Nettles DL, Carnahan MA, Setton LA, Grinstaff MW. Biomacromolecules. 2006;7:310–316. doi: 10.1021/bm050663e. [DOI] [PubMed] [Google Scholar]
- 7.Bryant SJ, Anseth KS. J Biomed Mater Res, Part A. 2003;64:70–79. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 8.Rice MA, Anseth KS. Tissue Eng. 2007;13:683–691. doi: 10.1089/ten.2006.0142. [DOI] [PubMed] [Google Scholar]
- 9.Hong Y, Song H, Gong Y, Mao Z, Gao C, Shen J. Acta Biomater. 2007;3:23–31. doi: 10.1016/j.actbio.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Temenoff JS, Park H, Jabbari E, Conway DE, Sheffield TL, Ambrose CG, Mikos AG. Biomacromolecules. 2004;5:5–10. doi: 10.1021/bm030067p. [DOI] [PubMed] [Google Scholar]
- 11.Betz MW, Modi PC, Caccamese JF, Coletti DP, Sauk JJ, Fisher JP. J Biomed Mater Res, Part A. 2008;86:662–670. doi: 10.1002/jbm.a.31640. [DOI] [PubMed] [Google Scholar]
- 12.Temenoff JS, Shin H, Conway DE, Engel PS, Mikos AG. Biomacromolecules. 2003;4:1605–1613. doi: 10.1021/bm030056w. [DOI] [PubMed] [Google Scholar]
- 13.Baxendale JH, Evans MG, Park CS. Trans Faraday Soc. 1946;42:155–169. [Google Scholar]
- 14.Barros JAG, Fechine GJM, Alcantara MR, Catalani LH. Polymer. 2006;47:8414–8419. [Google Scholar]
- 15.Mawad D, Odell R, Poole-Warren LA. Int J Pharm. 2009;366:31–37. doi: 10.1016/j.ijpharm.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 16.Mawad D, Martens PJ, Odell RA, Poole-Warren LA. Biomaterials. 2007;28:947–955. doi: 10.1016/j.biomaterials.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Durand A, Lalot T, Brigodiot M, Marechal E. Polymer. 2000;41:8183–8192. [Google Scholar]
- 18.Durand A, Lalot T, Brigodiot M, Marechal E. Polymer. 2001;42:5515–5521. [Google Scholar]
- 19.Derango RA, Chiang L, Dowbenko R, Lasch JG. BioTechniques. 1992;6:523–526. [Google Scholar]
- 20.Lalot T, Brigodiot M, Marechal E. Polym Int. 1999;48:288–292. [Google Scholar]
- 21.Iwahara K, Hirata M, Honda Y, Watanabe T, Kuwahara M. Biotechnol Lett. 2000;22:1355–1361. [Google Scholar]
- 22.Iwata H, Hata Y, Matsuda T, Ikada Y. J Polym Sci, Part A. 1991;29:1217–1218. [Google Scholar]
- 23.Iwata H, Hata Y, Matsuda T, Taki W, Yonekawa Y, Ikada Y. Biomaterials. 1992;13:891–896. doi: 10.1016/0142-9612(92)90111-z. [DOI] [PubMed] [Google Scholar]
- 24.Berchtold KA, Hacioglu B, Lovell L, Nie J, Bowman CN. Macromolecules. 2001;34:5103–5111. [Google Scholar]
- 25.Cruise GM, Scharp DS, Hubbell JA. Biomaterials. 1998;19:1287–1294. doi: 10.1016/s0142-9612(98)00025-8. [DOI] [PubMed] [Google Scholar]
- 26.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego, CA: 1995. pp. 88–90. [Google Scholar]
- 27.Salinas CN, Anseth KS. Macromolecules. 2008;41:6019–6026. [Google Scholar]
- 28.Hersel U, Dahmen C, Kessler H. Biomaterials. 2003;24:4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 29.Walling C. Acc Chem Res. 1975;8:125–131. [Google Scholar]
- 30.Biro A, Takacs E, Wojnarovits L. Macromol Rapid Commun. 1996;17:353–357. [Google Scholar]
- 31.Odian G. Principles of Polymerization. 4. John Wiley & Sons, Inc; Hoboken, NJ: 2004. pp. 212–214. [Google Scholar]
- 32.Bowman CN, Kloxin CJ. AIChE J. 2008;54:2775–2795. [Google Scholar]
- 33.Lovestead TM, Berchtold KA, Bowman CN. Macromolecules. 2005;38:6374–6381. [Google Scholar]
- 34.Cramer NB, O’Brien CP, Bowman CN. Polymer. 2008;49:4756–4761. [Google Scholar]
- 35.Dainton FS, Seaman PH. J Polym Sci. 1959;39:279–297. [Google Scholar]
- 36.Neyens E, Baeyens J. J Hazard Mater. 2003;B98:33–50. doi: 10.1016/s0304-3894(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 37.Nicodemus GD, Bryant SJ. Tissue Eng Part B. 2008;14:149–165. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West JL, Hubbell JA. Reactive Polymers. 1995;25:139–147. [Google Scholar]
- 39.Sakai S, Hashimoto I, Ogushi Y, Kawakami K. Biomacromolecules. 2007;8:2622–2626. doi: 10.1021/bm070300+. [DOI] [PubMed] [Google Scholar]
- 40.Tubiana-Rufi N, Riveline JP, Dardari D. Diabetes Metab. 2007;33:415–420. doi: 10.1016/j.diabet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Ravaine V, Ancla C, Catargi B. J Controlled Release. 2008;132:2–11. doi: 10.1016/j.jconrel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 42.Podual K, Doyle FJ, III, Peppas NA. J Controlled Release. 2000;67:9–17. doi: 10.1016/s0168-3659(00)00195-4. [DOI] [PubMed] [Google Scholar]
- 43.Kang SI, Bae YH. J Controlled Release. 2003;86:115–121. doi: 10.1016/s0168-3659(02)00409-1. [DOI] [PubMed] [Google Scholar]
- 44.Godjevargova T, Dayal R, Turmanova S. Macromol Biosci. 2004;4:950–956. doi: 10.1002/mabi.200400058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.