

Abstract
We mathematically modeled the receptor-dependent mitogen-activated protein kinase (MAPK) signaling by incorporating the regulation through cellular phosphatases. Activation induced the alignment of a phosphatase cascade in parallel with the MAPK pathway. A novel regulatory motif was, thus, generated, providing for the combinatorial control of each MAPK intermediate. This ensured a non-linear mode of signal transmission with the output being shaped by the balance between the strength of input signal and the activity gradient along the phosphatase axis. Shifts in this balance yielded modulations in topology of the motif, thereby expanding the repertoire of output responses. Thus, we identify an added dimension to signal processing wherein the output response to an external stimulus is additionally filtered through indicators that define the phenotypic status of the cell.
Keywords: Receptors/Leukocyte/Lymphocyte, Signal Transduction/Phosphoprotein Phosphatases/Dual Specificity Phosphatase Cascade, MAPK, MKP3, BCR
Introduction
The prototypic mitogen-activated protein kinase (MAPK)4 pathway consists of a three-tiered module composed of a MAPK (ERK1/2), which is activated via phosphorylation by a MAPK kinase (MEK1/2), which in turn is phosphorylated by a MAPK kinase kinase (Raf). This pathway exists in all eukaryotic organisms and controls fundamental cellular processes such as proliferation, differentiation, migration, survival, and apoptosis (1–3). Importantly, the cascade arrangement of this module permits integration of a wide range of conserved cellular process, thereby enabling a precise control of the amplitude, kinetics, and duration of ERK1/2 (ERK) activation. Several studies have documented that it is the ability to modulate these individual parameters of ERK activation that confers signaling specificity to this pathway in terms of regulating the cellular response (4–7).
The contrast between the apparent biochemical simplicity of this pathway and the range of complex cellular functions that it controls has prompted extensive investigations on the structure and regulation of this pathway. One approach toward this goal has been through computational modeling of this pathway. The earliest model by Huang and Ferrell (8) demonstrated that ERK activation exhibited ultrasensitivity and that this was primarily due to the distributive mechanism involved in the dual phosphorylation of ERK (9). Subsequent to these pioneering findings, an increasing number of models that have gained both in size and complexity have been developed over the years. The various aspects examined by these models include feedback cooperativity and inhibition, hysteresis, oscillations, Ras activation, scaffolding proteins, signal specificity, and robustness among others (10–13). From the standpoint of input/output relationships, these models have provided important new insights on feedback, sequestration, and scaffolding influences on the ERK response (14–17).
A less explored aspect of MAPK regulation, however, has been the role of phosphatases in regulating signal specificity (18). An earlier study by Bhalla et al. (19) identified MAPK phosphatase (MKP) as the locus of flexibility that regulated between monostable and bistable regimes of operation. More recently, the protein tyrosine phosphates SHP-1 was shown to be critical for defining the ligand discrimination threshold of the ERK response in T lymphocytes (20). Nonetheless, in view of the multiple phosphatases known to be associated with components of the MAPK pathway, a more integrated view of how such phosphatases regulate input/output relationships is presently lacking (21).
Our earlier study in murine B lymphoma A20 cells had demonstrated that the B cell antigen receptor (BCR)-dependent phosphorylation profiles of all the three constituents of the MAPK module (i.e. Raf MEK1/2 and ERK1/2) were profoundly influenced when cells were depleted of a range of cellular phosphatases by siRNA (22). Interestingly, depending upon the phosphatase depleted, these effects were either positive or negative, suggesting diverse modes of regulation. Therefore, in the present study we integrated these experimental data with existing literature to generate a mathematical model for ERK phosphorylation. The resulting model revealed an additional level of regulation of the MAPK pathway that was enforced through the parallel alignment of a cascade of phosphatases. Importantly, the cross-talk between the MAPK and the phosphatases cascade led to the assembly of a novel regulatory motif that enabled independent calibration of signal at each successive node. As a result, signal transmission through the MAPK pathway was nonlinear, leading to a combinatorial expansion of the landscape of potential output responses. Significantly, in addition to parameters such as signal strength and duration, the bounds of this landscape also incorporated variations in the relative levels (or activities) of the associated phosphatases. Thus, our studies identify a novel processing principle, the result of which the signal output represents an integrated expression of the properties of the stimulus and the phenotypic status of the cell.
EXPERIMENTAL PROCEDURES
Model Building and Numerical Simulations
A complete description of model building and details of the reaction scheme and parameters including rate equations for each of the reactions is provided in supplemental data S1A (description of model) and S1B (model content, parameters, reaction, and their rates, initial conditions). All reactions were converted into molecule-molecule interactions defining either binding interactions or catalytic reactions. We have used Simbiology2.2 (Mathworks) on MATLAB7.5 as a platform for implementing the model and for carrying out numerical simulations. All calculations except for that of restimulation experiments were done using ode15s, which is a solver for stiff differential equations and differential algebraic equations. It uses the variable order method based on numerical differentiation formula for stiff differential equations. Restimulation experiments were done using Sundials solver CVODE provided with Simbiology. Parameters were either taken from literature or estimated. Estimation of unknown parameters was done either by iteratively fitting to the experimental constraints or by using local optimization with lsqnonlin and lsqcurvefit (wherever experimental data were available for immediate readout). Sensitivity analysis of the complete model was done by using SBML_SAT (23), a freely available SBML-based MATLAB toolbox.
Stimulation of Cells and Detection of Phosphoproteins
A20 cells (1 × 107/ml) were stimulated with the F(ab)2 fragment of goat anti-mouse IgG at a final concentration of 25 μg/ml in RPMI for a period of up to 30 min (24). At appropriate times aliquots of cells were collected and centrifuged, and the cell pellets were stored in liquid nitrogen. When required, cells were lysed, the detergent-soluble proteins were resolved by SDS-PAGE, and specific proteins and phosphoproteins were detected (and quantified) by Western blot using the procedure and antibodies as previously described (25).
Co-immunoprecipitation
Lysates were prepared from 2 × 107 cells/group/time point in a buffer containing a mixture of protease and phosphatase inhibitors as previously described (24). Lysates were precleared with protein A-agarose and then incubated with appropriate antibody (0.6–1 mg at 4 °C for 2 h), after which the immune complexes were precipitated with protein A-Sepharose. The immunoprecipitates were identified by immunoblotting with specific antibodies (Santa Cruz Biotechnology).
Overexpression of MKP3 in A20 Cells
MKP3 was overexpressed as a Strep-tag II fusion protein in A20 cells (26). For this the cDNA was cloned into the pEXPR-IBA103 vector and then transfected into cells by electroporation. Subsequent selection for neomycin resistance over a 4-week period yielded stably transfected cells. A Western blot analysis revealed that these cells expressed about 2-fold higher levels of MKP3 when compared with untransfected A20 cells.
Confocal Microscopy; Staining and Image Analysis
Immunofluorescence staining was performed following the standardized protocol reported previously (Kumar et al. (22)). Stained cells were observed with a Nikon TE 2000E microscope equipped with 60×/1.4NA PlanApochromat differential interference contrast objective lens. Secondary antibodies tagged either with Alexa 488 or Alexa 568 were excited at 488 and 543 nm with an argon ion and a helium-neon laser, respectively. The emissions were recorded through emission filters set at 515/30 and 605/75. Serial confocal sections (0.5 μm thick) within a z-stack spanning a total thickness of 10–12 μm were taken in individual channels green and red using the motor drive focusing system. The transmission and detector gains were set to achieve the best signal-to-noise ratios, and the laser powers were tuned to limit bleaching fluorescence. The refractive index of the immersion oil used was 1.515 (Nikon). All settings were rigorously maintained for all the experiments. All images were quantified using Image-Pro Plus Version 6.0, a commercially available software package from Media Cybernetics.
The merged confocal images were subjected to co-localization analysis to determine the “Overlap Coefficient” proposed by Manders et al. (27).
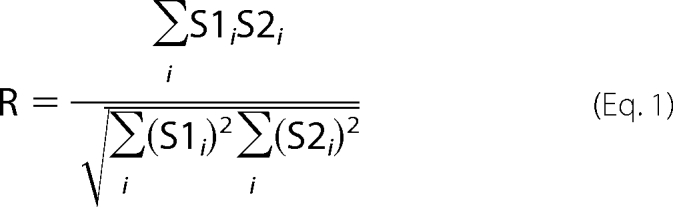 |
where S1 is signal intensity of pixels in the first channel, and S2 is signal intensity of pixels in the second channel.
At least 18–20 cells were analyzed in 3 sets of experiments for the co-localization studies. All the images are in the Tiff RGB 24 format, and phycoerythrin fluorescence has been given a green pseudocolor. To reduce the unwanted background noise generated by the photomultiplier signal amplification, all the image stacks were treated with two-dimensional filters (Gaussian and sharpening filtering).
RESULTS
Modeling BCR-dependent Activation of the MAPK Network
The BCR is a hetero-oligomeric transmembrane protein in which the surface immunoglobulin constitutes the ligand binding unit, whereas the signaling function is encoded within the non-covalently associated heterodimer of CD79a and CD79b (28). Activation of BCR-dependent signaling is mediated by three distinct cytoplasmic protein tyrosine kinases, which are Lyn, Syk, and Btk. Activation of Lyn serves as the critical first step, which then further phosphorylates the immunoreceptor tyrosine-based activation motifs encoded within the CD79a-CD79b component of the BCR complex. The activated BCR subsequently recruits multiple downstream pathways, some of which include those dependent upon phospholipase Cγ, the Rho family of GTPases, and the RAS/phosphatidylinositol 3-kinase (for review, see Ref. 29).
Our previous experimental studies of the plasticity of the BCR signaling network and its response to specific perturbations (22, 24, 25) provided us with an internally normalized dataset for modeling salient features of the signaling network. We, therefore, combined these results with data from the literature to develop a block diagram for the BCR-activated MAPK signaling network. Here we specifically included the results from our most recent experiments probing the consequences of siRNA-mediated depletion of various cellular phosphatases on BCR-dependent phosphorylation of the constituents of the MAPK pathway (22). Data generated from the depletion of five of the tested phosphatases were taken for this study, and these five phosphatases were PP1, PP2A, MKP1, MKP2, and MKP3. The selection of these phosphatases was guided by the fact that MKPs are known regulators of phosphorylation of the intermediates in the MAPK pathway (19, 30, 31). Furthermore, Raf, MEK, MKP1, and MKP3 are all regulated through phosphorylation at Ser/Thr residues (30, 32), thereby implicating a possible role for PP1 and PP2A.
A detailed description of the model development, parameter estimation, and sensitivity and robustness analyses is provided in supplemental data S1, A and B, whereas a “line diagram” of this model is depicted in Fig. 1A. Briefly, however, our approach was divided into two stages. The first involved compilation of BCR-dependent signaling events through data-mining for processes that included immunoreceptor tyrosine-based activation motif phosphorylation and recruitment of early signaling intermediates such as Lyn, Syk, B cell linker protein, phospholipase Cγ2, Btk, and phosphatidylinositol 3-kinase up to activation of the MAPK pathway. In our model we summarized phosphorylation of the BCR-associated immunoreceptor tyrosine-based activation motif as phosphorylated BCR, whereas the receptor-dependent activation of Lyn and Syk was modeled according to the reported positive feedback system at BCR (33). Furthermore, we also incorporated the role of the protein-tyrosine phosphatase SHP1, which has previously been shown to function as a negative regulator of Lyn and BCR (24). The phosphatidylinositol 3-kinase and inositol signaling system was modeled by including both B cell adaptor protein- and CD19-mediated recruitment of the phosphatidylinositol 3-kinase subunits. This has been shown to induce both phosphatidylinositol 3-kinase activation and phosphatidylinositol 3-mediated Btk and Akt recruitment to the membrane, where Akt is phosphorylated by phosphoinositide-dependent protein kinase (29, 34). On the other hand, at least in B cells, phospholipase Cγ and RasGTP accumulation is mediated through the scaffolding protein B cell linker protein and was incorporated in our model accordingly (34–37). Consistent with the literature, we distinguished between the pool of B cell linker protein that was bound to Grb2/Sos and that bound to Btk/phospholipase Cγ at the level of activation of the downstream pathways. Phospholipase Cγ-dependent hydrolysis of phosphatidylinositol 2 into diacylglycerol and phosphatidylinositol 1,4,5-trisphosphate results in the activation of protein kinase C, which along with the RasGTP produced by the B cell linker protein·Grb2·SoS complex, has been shown to constitute an “AND” gate input function for activation of the MAPK cascade at the level of Raf (MAPK kinase kinase) (34).
FIGURE 1.

Signaling through the Lymphocyte receptor induces a novel regulatory motif for MAP kinase activation. A schematic overview of the reactions for BCR dependent activation of MAP kinase ERK1/2 is shown in panel A. The highlighted area represents the novel signaling motif identified and this is expanded in panel B. IP3, phosphatidylinositol 1,4,5-trisphosphate; PIP2, phosphatidylinositol 2; PI3K, phosphatidylinositol 3-kinase; DAG, diacylglycerol; PKC, protein kinase C; BLNK, B cell linker protein; BCAP, B cell adaptor protein.
The reaction schemes for recruitment and activation of the signaling intermediates were strictly in accordance with previously published models. In addition to the general schematic of BCR signal transduction leading to ERK, however, we also included regulation of the core MAPK module by cellular phosphatases. Here, care was taken to incorporate these phosphatases in the network in a manner that was consistent with the known literature information. In instances where adequate information was not available, assumptions were made on the basis of our own experimental data. For instance, although previous reports suggested that MKP1 was primarily localized within the nucleus of cells (38–40), we found that, at least in A20 cells, this phosphatase was distributed between both the cytoplasmic and the nuclear compartments (see supplemental data S1, A and B). Accordingly, a cytoplasmic pool of MKP1 was also taken into consideration in our scheme.
The second stage of our model-building exercise involved several rounds of iterations to ensure that the model accurately reproduced the experimental dataset. Model parameters such as initial concentrations, Km and Kd values, and other rate parameters were either taken from literature or, when not available, were optimized for producing the best experimental fit. The model was constructed by employing generic rate laws such as mass action and Michaelis-Menten reaction schemes to represent the interactions among the various species. The system was taken as appropriately modeled when it could fit the experimental data, which also included our previous findings on the effects of siRNA-mediated depletion of cellular phosphatases on BCR-dependent phosphorylation of ERK (22). Recapitulation of these latter findings, however, required us to incorporate two novel interactions for MKP3, wherein in addition to ERK it was also involved in the dephosphorylation of both MEK and MKP1. Furthermore, the functional properties of MKP3 could only be accounted for by postulating that it was the phosphorylation state of this protein that dictated its relative substrate specificity for ERK on the one hand and MEK and MKP1 on the other. Although these aspects are discussed in more detail later, inclusion of these links provided (over multiple iterations) the unique solution wherein the simulated results matched well with the experimental data.
Although the resulting model was fairly complex, it nonetheless encompassed the span of regulatory events emanating from the receptor and terminating at ERK (Fig. 1, A and B). To test the model we performed local and global sensitivity analyses using several independent methods to evaluate the parameters for their robustness against fluctuations that are normally inherent to noisy biological conditions. We found that parameters to which the model was most sensitive were those that are already known to be critical for shaping the BCR-specific signaling response (see supplemental data S1, A and B for details). In other words, our model indeed reflected the true kinetic properties of BCR-dependent signaling. Parameters specific for the MAPK-phosphatase module (see Fig. 1B) were robust in terms of the output behavior and could tolerate a broad range of perturbations against the ERK activation response. Furthermore, we also verified that the robustness of our model compared well with that of other prominent models described for the MAPK pathway (see supplemental data S1, A and B). At the experimental level, the model also successfully recapitulated results available in the literature, which included our own earlier experimental data on BCR-dependent signaling in A20 cells (supplemental Figs. S2 and S3).
Analysis of the Model
The model described in Fig. 1A exhibited several intriguing topological and dynamical features. This is especially evident from Fig. 1B, which highlights the co-alignment of a phosphatase cascade alongside the MAPK pathway, leading to several cross-regulatory interactions between the two. Notably, these interactions also included two novel links for MKP3. That is, in addition to ERK, our model described MKP3 to also interact with (and negatively regulate) MEK and MKP1 (Fig. 1B). Furthermore, another related prediction by the model was that MKP3-dependent regulation was tightly controlled through a modulation of its substrate bias. In other words, whereas phosphorylated MKP3 was described to exhibit a preference for MEK and MKP1, its non-phosphorylated form was predicted to be more selective for ERK (see supplemental data S1). As a result, shifts in the distribution of MKP3 between the phosphorylated and non-phosphorylated pools potentially served as a mechanism for diversifying the influence of MKP3 on MAPK signaling.
An in silico prediction of the time-dependent profile of ERK phosphorylation was consistent with the experimentally obtained results (Fig. 2A). Furthermore, a similar in silico analysis of ERK phosphorylation in response to varying ligand concentrations yielded an incremental dose-response response profile (Fig. 2B). This prediction of a graded ERK output could also be experimentally confirmed by stimulating A20 cells with increasing concentrations of anti-IgG (supplemental Fig. S4). Importantly, ERK response to ligand dose was found to be graded regardless of whether it was monitored at the level of the cell population by Western blot analysis (Fig. 2C) or at the level of single cells through intracellular staining for phospho-ERK and detection by flow cytometry (Fig. 2D).
FIGURE 2.

The ERK phosphorylation response is proportional to the stimulus strength. Panel A shows the concordance obtained between experiment (red diamonds, values are the mean ± S.D., n = 3) and simulation (black line) examining the time course of ERK phosphorylation obtained upon stimulation of A20 cells with a saturating (25 μg/ml) concentration of anti-IgG. Panel B shows the results of an in silico analysis estimating the magnitude of ERK phosphorylation (ppERK) obtained after stimulation of cells with varying anti-IgG concentrations. Panel C gives the corresponding results of an experiment where A20 cells were stimulated for 10 min, with the indicated doses of anti-IgG. ERK phosphorylation was then determined in lysates by Western blot analysis (see supplemental Fig. S4). Values are the mean (±S.D.) of three independent experiments and are shown here as normalized raw signal intensity (pSignal intensity). A semi-log plot of these results yielded a Hill coefficient of 0.6 and 0.56 for simulated and the experimental results, respectively. Stimulated cells were also subjected to staining for intracellular phospho-ERK using antibodies specific for double-phosphorylated ERK followed by fluorescein isothiocyanate-labeled secondary antibodies. Stained cells were then analyzed by flow cytometry, and the results are shown in panel D. Depicted here are the profiles obtained for cells stimulated with either 0.1 (black line), 0.5 (green line), 5 (pink line), or 25 μg/ml (blue line) of anti-IgG. The profile for unstimulated cells overlapped with that for cells stimulated with 0.1 μg/ml of ligand. For the negative control, cells were stained with rabbit IgG instead of the anti-phospho-ERK antibody.
Experimental Verification of the Model
Although our model successfully described the activation profile of several of the incorporated intermediates in a manner that was consistent with experimental observations, we, however, wanted to further verify some of its novel topological features. Of particular interest in this connection was the identification of two new links for MKP3. Although the interaction between MKP3 and ERK is well known (41–43), our model also predicted the existence of interactions of MKP3 with both MEK and MKP1 (Fig. 1B). It was this ability to regulate function of multiple intermediates that then highlighted MKP3 as an important regulator of MAPK signaling. For this we first performed co-immunoprecipitation experiments from lysates of A20 cells using antibodies specific either for MEK, MKP1, or MKP3. As shown in Fig. 3A, immunoprecipitation of either MEK or MKP1 also led to the simultaneous enrichment of MKP3, whereas immunoprecipitates of MKP3 were enriched for both MEK and MKP1. Although immunoprecipitation results do not permit quantification, they nonetheless support at least the existence of MKP3 in a simultaneous complex with MKP1 and MEK, thereby confirming the interactions depicted in Fig. 1B. In subsequent experiments we observed that the siRNA-mediated depletion of MKP3 resulted in a significant (∼4-fold) increase in the basal levels of MKP1 (Fig. 3B and supplemental Fig. S4). These levels were further increased upon stimulation of cells through the BCR (Fig. 3B). Importantly, this effect of MKP3 could not be inhibited by the inclusion of cycloheximide in the cultures (supplemental Fig. S4), suggesting that it was the stability rather than the rate or extent of synthesis of the MKP1 protein that was affected by MKP3-silencing. The stability of intracellular MKP1 is dictated by its phosphorylation status. Although the non-phosphorylated form exhibits a short half-life, phosphorylation enhances its stability (30). Thus, the observed inverse relationship between MKP3 and MKP1 levels in Fig. 3B provides additional support for the functional nature of the link between these two phosphatases. In this connection it is pertinent to note that under conditions of MKP3 silencing, the level of both phosphorylated ERK and p38 are significantly reduced after receptor stimulation (22). Thus, the increased stability of MKP1 observed here after MKP3 silencing is more likely due to reduced dephosphorylation of MKP1 by MKP3.
FIGURE 3.

Verification of novel interactions incorporated in the model. In panel A, lysates from stimulated or unstimulated A20 cells were immunoprecipitated with antibodies specific either for MKP3, MEK, or MKP1 (“Experimental Procedures”) (left column, IP). Immunoprecipitates were then subjected to a Western blot analysis using the antibodies indicated in the right column (WB). As a negative control (ctrl), parallel immunoprecipitates with rabbit IgG were also probed with the corresponding antibodies. Panel B shows the results of Western blot analyses for MKP1 in A20 cells treated either with MKP3-specific siRNA (red line) or with non-silencing (i.e. GFP-specific) siRNA (blue line). Stimulation times with anti-IgG (25 μg/ml) are indicated, and values are the mean ± S.D. of three separate experiments. We have previously demonstrated that siRNA-mediated silencing of MKP3 results in a >70% reduction of this phosphatase protein in A20 cells (22). In C, the left panel shows the profiles obtained from an in silico analysis in the presence (pink) and absence (blue) of DRB. The right panel shows the corresponding results obtained experimentally, where cells were treated with or without DRB (20 μm) before stimulation with anti-IgG. ppERK, ERK phosphorylation.
We also verified whether the phosphorylation status of MKP3 indeed influenced its substrate specificity. For this we stimulated A20 cells with anti-IgG in the presence of the CK2α inhibitor, DRB (44). As shown in Fig. 1B, MKP3 is primarily phosphorylated by CK2α. Inhibition of this kinase, therefore, leads to a concomitant inhibition of MKP3 phosphorylation (32). According to our model then, this should also result in an increased efficiency of MKP3-mediated dephosphorylation of ERK. As expected, stimulation of DRB-treated cells led to inhibition of BCR-dependent phosphorylation of ERK (Fig. 3C and supplemental Fig. S4). These results are also consistent with previous findings that CK2α-dependent phosphorylation of MKP3 attenuates its inhibitory effect on ERK phosphorylation (32).
The results in Fig. 3C, therefore, provide experimental support for our proposal that MKP3 displays differential substrate specificity depending upon its phosphorylation status. To further establish this, however, we also employed an alternate and more direct approach. Here, A20 cells were stimulated either in the presence or absence of the CK2α inhibitor, DRB. Subsequently, these cells were analyzed by confocal microscopy to determine the effects of CK2α inhibition on the co-localization of MKP3 with ERK, MEK, and MKP1. Fig. 4 reveals that in stimulated cells inhibition of CK2α led to a substantial increase in the extent of co-localization between MKP3 and ERK. In contrast, co-localization of MKP3 with both MEK and MKP1 was significantly reduced (Fig. 4). Essentially similar results were obtained in experiments where, instead of inhibition, CK2α was also specifically silenced through targeted siRNA (not shown). At one level these results provide direct evidence for the novel links between MKP3 and MEK and between MKP3 and MKP1 identified by our model. In addition to this, however, the observed alterations in the relative extents of co-localization of MKP3 with ERK, MEK, and MKP1, induced by the inhibition of CK2α-dependent phosphorylation of MKP3, also provide experimental confirmation for our proposal that the substrate specificity of MKP3 is influenced by its phosphorylation status. Collectively, therefore, the consistency between predicted and experimentally obtained phosphorylation profiles of the various signaling intermediates described above as well as the experimental confirmation of the new MKP3-dependent regulatory links proposed provide experimental support for the model depicted in Fig. 1A.
FIGURE 4.

Verifying the MKP3 substrate bias in response to CK2 inhibition. For the experiment, A20 cells were stimulated for 10 min with anti-IgG in the presence or absence of casein kinase inhibitor DRB (20 μm). Cells were then fixed and stained for ERK1/2 and MKP3 (top panel), MEK1/2 and MKP3 (middle panel), or MKP1 and MKP3 (bottom panel) and observed under laser scanning confocal microscope (“Experimental Procedures”). Merged images for co-localization between green (ERK1/2, MEK1/2, or MKP1) and red (MKP3) are shown in the figure. The quantitative differences between DRB-treated and -untreated cells for co-localization with MKP3-MEK1/2, MKP3-ERK1/2, and MKP3-MKP1 upon anti-IgG treatment are shown on the right-hand side. Values are the average (±S.E.) of more than at least 40 cells (***, p < 0.0005; **, p < 0.005).
Signal-dependent Assembly of the MAPK Cascade into a Unique Regulatory Module
Our model described that each intermediate in the MAPK pathway was under the regulatory control of both the co-aligned and its immediately upstream phosphatase. Thus, MEK was regulated by both PP2A and MKP3, whereas ERK was regulated by MKP3 and MKP1 (Fig. 1B). The MAPK module contained links that collectively belonged to all of the four possible categories; activation of activator (protein kinase C to Raf and Raf to MEK), inhibition of activator (PP2A to Raf and MKP3 to MEK), activation of inhibitor (protein kinase C to MKP3 and ERK to MKP1), and inhibition of inhibitor (PP2A to MKP3 and MKP3 to MKP1/2) (Fig. 1, A and B).
A significant aspect here was that co-alignment of the two cascades resulted in the generation of a novel regulatory motif that was composed of a contiguous set of two square units. The first of these consisted of Raf, MEK, PP2A, and MKP3 as the vertices, whereas the second unit involved MEK, ERK, MKP3, and MKP1 (Fig. 1B). Furthermore, the linkage relationships between the corresponding vertices were also replicated (Fig. 1B). Interestingly, a closer inspection of this motif revealed that it was in fact composed of four smaller regulatory units (each triangle in the motif) that represented alternately placed type-2 coherent and incoherent feed-forward loops (defined as in Ref. 45). A more detailed analysis of these substructures and their individual contributions to the overall properties of the parent motif is currently under way as a separate study. In this report we focused solely on the role of the larger motif in regulating signal processing by the MAPK cascade.
Assembly of this regulatory motif was dependent upon two separate input signals originating from the BCR. The first of these was a protein kinase C- and RasGTP-dependent AND gate that mediated Raf activation in a signal-dependent manner (19, 46). The second input signal was defined by the CK2α-dependent phosphorylation of MKP3 (Fig. 1B). It has previously been demonstrated that MKP3 and CK2α form a protein complex wherein CK2α specifically phosphorylates MKP3, with the corresponding increase in ERK2 phosphorylation (32). As discussed later, the regulation of MKP3 between its phosphorylated and non-phosphorylated states has significant implications for signal processing by the MAPK pathway. Finally, phosphorylation of MKP1 by ERK was also a prerequisite for the assembly of this motif, signifying the importance of cross-talk between these two cascades in this process.
Phosphatase-mediated Regulation of the ERK Response
We first examined how the connected network of phosphatases influenced signal output from the MAPK module. This was achieved through in silico experiments examining the ligand dose dependence of ERK phosphorylation under conditions where the individual phosphatases were depleted one at a time. Fig. 5 shows the results of these experiments where Normal depicts the profile obtained in the unperturbed condition. It is evident that, with the exception of PP1, no other phosphatase depletion had any significant effect on the proportional nature of ERK activation (Fig. 5). The observed effect of PP1 depletion is consistent with our earlier experimental findings involving depletion of this enzyme by siRNA. In these experiments a marked decrease in the magnitude of ERK activation was obtained at the later time points (22). Thus, these results suggest that PP1 may play an important role in buffering the inactive pool of hyperphosphorylated Raf, which is generated by activated ERK through a positive feedback loop, and Akt (19, 47, 48).
FIGURE 5.

Phosphatase-mediated regulation of the MAPK signaling response. The influence of individual phosphatases in sensitizing the ERK output was monitored in silico by analyzing ligand dose versus peak phospho-ERK (ppERK) levels within the first 30 min of activation. This analysis was performed either in normal cells (Normal) or in cells where the indicated phosphatase was depleted from the system. In each case, the -fold change in ligand concentration required to increase ERK phosphorylation from 10 to 90% of its maximal value is also given (F). These values confirm that the ERK response remains proportional under all of these conditions.
Depletion of any of the remaining phosphatases resulted only in a modulation of the amplitude of ERK phosphorylation, with MKP1 depletion yielding the most pronounced effect (Fig. 5). Notable, however, were the qualitatively distinct effects seen upon depletion of the individual, MAPK-associated, phosphatases. Suppression of PP2A expression yielded a marginal effect on the shape of the dose-response curve (Fig. 5), whereas that of MKP3 resulted in a diminished activation window for ERK due to a simultaneous increase in sensitivity to lower ligand concentrations and a decrease in the peak level that was achieved (Fig. 5). In contrast, removal of MKP2 induced a comparable increase in both the upper and lower bounds of ERK activity (Fig. 5). Thus, individual phosphatases comprising the MAPK module exert characteristic influences on the ligand-dependent dose-response profile of ERK activation.
Perturbation in Phosphatase Concentrations Modulates the MAPK Response
The output of a signaling network is primarily determined by the topology of the network and the concentrations of its nodal constituents. Furthermore, Fig. 5 also confirmed that the topological allocation had a significant bearing in terms of defining the extent of regulation that each phosphatase exerted on the MAPK signaling pathway. Therefore, we next examined how variations in the concentration of these phosphatases would impact on BCR-initiated MAPK signaling. This involved in silico experiments in which the concentration of the individual phosphatase was varied from one that was 10-fold lower to one that was 10-fold higher than its constitutive level in A20 cells. The phosphorylation of both MEK1/2 and ERK were examined under these conditions.
Although similar experiments were also carried out for PP1 and MKP2 (see supplemental Fig. S5), only the results obtained for the three phosphatases that are incorporated in the “two-square” structural motif are shown in Fig. 6. The figure describes the effect of varying the concentration of these individual phosphatases on the peak levels of MEK and ERK phosphorylation. Of particular note here was the fact that changing PP2A concentrations exerted dissimilar effects on MEK and ERK. A bimodal effect was observed for MEK, and this feature was retained at all strengths of signal (i.e. ligand doses) employed (Fig. 6). Contrary to this, ERK yielded a PP2A-dependent bimodal profile only at low ligand concentrations, with the response to this phosphatase becoming linear as the signal strength was increased (Fig. 6). Thus, PP2A-mediated regulation yielded non-identical effects on these two consecutively positioned, signaling intermediates of the MAPK pathway. Furthermore, the extent of deviation in behavior between these two intermediates was also dependent upon the strength of the BCR stimulus. Increasing PP2A concentrations also led to a progressive reduction in the amplitude of both MEK and ERK responses, although the effect on the latter molecule was more pronounced (Fig. 6). This aspect could be experimentally verified by stimulating A20 cells in the presence of the PP2A inhibitor okadaic acid. Although okadaic acid is a known inhibitor of both PP2A and PP1, the 100-fold lower IC50 value for PP2A allowed us to use a concentration range that was specific for PP2A.
FIGURE 6.

Modulation in phosphatase concentrations further shape ligand sensitivity of the MAPK pathway. Peak phosphorylation of ERK (ppERK) and MEK (ppMEK) was monitored in response to variations in both ligand and individual phosphatase concentrations. Three separate sets of simulation within a defined concentration window were used to reduce the computing time. For each phosphatase, 100 different concentrations of phosphatase were used varying from 10× lower to 10× higher (PP2A, 0.0007–0.07 μm; MKP3, 0.00015–0.015 μm; MKP1, 0.00002–0.002 μm) than its constituent concentration level in A20 cells. The combined data are plotted here. Panel A profiles peak phospho-MEK levels as a combined function of both varying ligand doses and varying concentrations of each of the three phosphatases. Here, the top panel shows the three-dimensional plot, whereas the lower panel depicts the same results in the form of a heat map. Similarly, panel B shows peak phospho-ERK levels as a combined function of both ligand dose and varying concentrations of the three phosphatases.
As shown in supplemental Fig. S6, whereas inhibition of PP2A activity led to a linear increase in ERK phosphorylation, that of MEK displayed a bimodal response to PP2A (see also Fig. 8A). The sensitivity of ERK to PP2A activity likely derives from the concomitant decrease in the size of the pool of non-phosphorylated MKP3 as a result of decreasing PP2A levels. As discussed earlier, this would decrease the efficiency of MKP3-dependent dephosphorylation of ERK. In addition to this, however, a diminished pool of phospho-MKP3 would also imply a consequent reduction in the negative regulation of MKP1 and, thereby, an increase in the MKP1-dependent inactivation of ERK.
FIGURE 8.

Experimental confirmation of the systems properties of the MAPK-associated regulatory module. Panel A depicts peak MEK and ERK phosphorylation (10 min of stimulation with anti-IgG) levels obtained as a function of changes either in MKP3 or PP2A concentrations as described in the text. For the profiles obtained in silico (Predicted), the concentration ranges utilized is described in supplemental Fig. S8, whereas the extent of variation in phosphatase concentration/activity obtained experimentally (Experimental) is described under “Experimental Support for Signal Processing Function of Phosphatase Cascade.” The anti-IgG concentrations employed for the experiment in high ligand dose (upper panel) were 10 (blue line) and 25 (red line) μg/ml, whereas for low ligand dose (lower panel), it was 0.05 (blue line) and 0.5 (red line) μg/ml. OA, okadaic acid. Panel B describes the effects of combined variations in levels/activities of both MKP3 and PP2A on the magnitude of ERK phosphorylation. Here again, a comparison between the in silico (Predicted) and the experimentally (Experimental) obtained results is shown. Although a similarity in profiles between the two groups is clearly evident, the absence of a higher degree of concordance was primarily due to the limited number of data points in the experimental group. As described in the text, MKP3 levels were varied in the experimental sets either through specific depletion by siRNA (KD) or by overexpression (OE).
In similar experiments where MKP3 concentrations were varied, we observed an inverse relationship between the amplitude of MEK phosphorylation and MKP3 concentrations. In contrast to this, ERK activation displayed a bell-shaped profile (Fig. 6). The curvature of the bell was, however, more pronounced at higher ligand concentrations, supporting that ERK activation remained responsive to the strength of the stimulus. The bimodal effect of MKP3 on ERK activation again probably reflects the consequence of concentration-dependent shifts in the distribution of MKP3 between its phosphorylated and non-phosphorylated states. At low concentrations, the non-phosphorylated form of MKP3 would predominate due to the dominant action of PP2A. The increased specificity of this form for ERK can then explain the reduced levels of peak ERK phosphorylation. As MKP3 levels increase, however, the proportion of its phosphorylated form is also expected to increase. The consequences of this on ERK activation would then be defined by the balance of the effects of MKP3 on ERK and that of phospho-MKP3 on MEK and MKP1. At one level, MKP3 would negatively regulate ERK activity by ensuring its dephosphorylation. As opposed to this, the phosphorylated subset of MKP3 (phospho-MKP3) would preferentially target MEK and MKP1 but with opposing consequences. Phospho-MKP3-mediated inactivation of MEK would also inhibit transmission of signal to ERK. On the other hand, the phospho-MKP3 dependent dephosphorylation and resultant destabilization of MKP1 would lead to attenuation in the negative regulation of ERK by this enzyme. Consequently, it is the quantitative shifts in balance between these distinct activities that would then shape the ERK response to variations in MKP3 concentrations.
Consistent with expectations, increasing MKP1 levels led to a steep decline in peak levels of ERK phosphorylation, whereas the effect on MEK was only marginal (Fig. 6). Thus, the cumulative results in Fig. 6 emphasize that signal-dependent formation of the two-square motif enables tight regulation of the MAPK signaling by the associated phosphatases. Importantly, both quantitative and qualitative aspects of signal transmission between two consecutive nodes were selectively influenced, thereby providing a mechanism by which signal transmission at the MEK/ERK interface could be further calibrated.
The Phosphatase Axis Orients Signal Processing by the MAPK Module
Concentrations of phosphatases frequently vary from one cell type to another. Furthermore, even within a given cell type, either differences in environmental conditions (histories of stimulus) and/or developmental stages can lead to significant changes in intracellular phosphatase concentrations (31, 49, 50). This is particularly true for MKPs in lymphocytes, where changes either in the activation or differentiation state of the cell can lead to marked alteration in the relative levels of this class of phosphatases (31). Thus, given the high connectivity between the MAPK and the phosphatase cascade, it was reasonable to suspect that variations in the relative concentration or activity of the MAPK-associated phosphatases would have a significant impact on signal processing. To explore this we examined the consequences of simultaneously varying the concentration of any two of the three MAPK-associated phosphatases while keeping the third at its constitutive level. The effects on the magnitude of peak phosphorylation of both MEK and ERK at a fixed ligand dose were monitored. All the three possible phosphatase combinations were tested over a range of 10,000 different pairs (in a 100 × 100 matrix) of their respective concentrations. The range of phosphatase concentrations employed here was identical to that described for Fig. 6.
Fig. 7A depicts the results of such an exercise in which PP2A and MKP3 concentrations were varied while keeping MKP1 at a fixed level. As shown, MEK activity yields a bell-shaped curve in the direction of increasing PP2A levels, although this profile tends to progressively “flatten” as MKP3 concentrations also increase. Although the inhibitory effect of MKP3 on MEK activation was monotonic, it was, however, buffered at high PP2A concentrations (Fig. 7A). In other words, the relative concentrations of PP2A and MKP3 have a significant role to play in defining the sensitivity of MEK phosphorylation to ligand-induced stimulation of cells. This was also true in the case of ERK, although here a bimodal activation profile was obtained in the directions of both increasing PP2A and MKP3 concentrations. This latter observation suggests the existence of compensatory effects between these two phosphatases whereby multiple combinations of their respective concentrations yield the same level of peak ERK activity.
FIGURE 7.

Inter-phosphatase cross-talk regulates signal transmission through the MAPK pathway. The effects of simultaneous variations in the concentration SAM-associated phosphatases on peak phospho-MEK (ppMEK) and peak phospho-ERK (ppERK) levels within a 30-min activation window are shown here. A total of 100 different concentrations were employed for each phosphatase. These varied from 10× lower to 10× higher than the constituent concentration in A20 cells (PP2A, 0.0007–0.07 μm; MKP3, 0.00015–0.015 μm; MKP1, 0.00002–0.002 μm). The three combinations shown here are MKP3-PP2A (A), MKP3-MKP1 (B), and PP2A-MKP1 (C). In each case the top panel shows peak phospho-MEK levels, whereas the lower panel shows peak phospho-ERK responses. The left and the right panels depict the three-dimensional landscape and the corresponding heat map representation, respectively. The corresponding profiles for the remaining components of the module that are also regulated by phosphorylation (i.e. Raf, MKP1, and MKP3) are shown in supplemental Fig. S9.
Fig. 7B shows the results of a similar experiment where the concentrations of MKP3 and MKP1 were varied under conditions where PP2A levels were kept constant. As may be expected, variations in MKP1 concentrations exerted little or no effect on peak MEK activity, and the resulting landscape was heavily biased in favor of the concentration-dependent inhibitory effects of MKP3. In contrast, a bimodal activation curve was again observed for ERK along the MKP3 axis. The curvature of this profile, however, was stringently controlled by MKP1, with ERK activation completely inhibited even at moderately higher concentrations of this phosphatase (Fig. 7B).
Similar to the results in Fig. 7A, varying combinations of PP2A and MKP1 also yielded a bimodal MEK profile in the direction of PP2A but with a significant reduction in the phosphatase concentration window where at least detectable MEK activation was allowed (Fig. 7C). With the exception of attenuating the amplitude of MEK activation, varying MKP1 levels had little effect on the profile. Qualitatively similar effects were also noted for ERK where increasing MKP1 levels served to further exacerbate the inhibition of ERK activation that was observed as a function of increasing PP2A levels. However, here the inhibitory effect of MKP1 was far more pronounced (Fig. 7C). Thus, although the MAPK module functions as a proportional response system, these results emphasize that the net output is further specified by the relative concentrations of the individual MAPK-associated phosphatases that are present. In this connection the assembly of these phosphatases into a regulatory axis is particularly relevant. As a result, variations in the concentration gradient along this axis exercise a combinatorial influence on signal processing by the MAPK pathway.
Finally, another intriguing feature revealed from a cursory examination of Fig. 7 is the fact that the range of allowed concentration regimens of phosphatases was significantly more restricted for ERK than it was for MEK. This was generally true for all three pairs of phosphatase combinations studied where the activation landscape for MEK was always larger in area than that obtained for ERK (Fig. 7). These observations suggest that the control exerted by the co-aligned phosphatase axis is further accentuated during transmission of signal from MEK to ERK, the effector molecule of the MAPK pathway.
Experimental Support for Signal Processing Function of Phosphatase Cascade
To confirm the above findings, we examined as a representative case the consequences of experimentally modulating MKP3 levels in conjunction with variations in the concentration of active PP2A. MKP3 levels in A20 cells were altered either through the use of specific siRNA, which reduces the amount of the protein by >70% (25), or by stably overexpressing it in these cells to achieve a net 2-fold increase in its concentration (“Experimental Procedures”). To modulate the concentration of active PP2A, we again employed its inhibitor okadaic acid. Thus, cells expressing either constitutive, reduced (through siRNA), or increased (through over-expression) levels of MKP3 were individually treated with varying okadaic acid concentrations, and each of these groups was then stimulated with a range of ligand (i.e. anti-IgG) doses. Peak phosphorylation levels of both MEK and ERK were determined in each case, and the results obtained are presented in supplemental Figs. S6 and S7.
We first compared these results with the predicted effects on peak phosphorylation of MEK and ERK of varying levels of the individual phosphatases shown in Fig. 6. The panel a in Fig. 8A shows an expanded region of these predicted profiles obtained at the higher range of ligand doses and over the estimated range of either MKP3 concentrations or PP2A activities employed in the present set of experiments (see supplemental Fig. S8). Panel b of Fig. 8A depicts the experimentally obtained results under these conditions. A high degree of correspondence between the simulated and the experimentally derived profiles is clearly evident here. This was also equally true for peak MEK and ERK phosphorylation profiles in cells stimulated with suboptimal doses of the ligand. In addition to this, the data obtained in supplemental Fig. S6 also served to substantiate the results of our in silico experiments describing the effects of simultaneously varying MKP3 and PP2A levels on the magnitude of ERK phosphorylation (see Fig. 7A). A comparison of results from the two approaches is shown in Fig. 8B. Here, panel a shows the results of our in silico analysis performed by employing either a high or a suboptimal concentration of ligand for cell stimulation. The corresponding profiles derived from our experimental data (supplemental Fig. S7) are shown in panels b of these figures. A good correlation between results from the two approaches is again clearly evident here. This was despite the limited range of concentrations that could feasibly be tested in the experimental groups. A similar degree of correspondence was also obtained when experimental and predicted results for MEK phosphorylation were similarly compared in terms of a heat map (data not shown).
Thus, by verifying the accuracy of the resulting predictions, the data in Fig. 8 support the fidelity of the model described in Fig. 1A. In addition, these findings also firmly establish the role played by the co-associated phosphatase axis in terms of calibrating the signal processing capabilities of the MAPK pathway.
DISCUSSION
The remarkable versatility of the MAPK module is evident from the fact that ERK activation can display a wide range of activity profiles depending on the interfaces that regulate signal transmission through this module. Thus, depending on the cell type, ERK activation can be ultrasensitive, converting graded inputs into a digital output response (10). Embedding this pathway within a positive feedback loop can lead to a dramatic amplification of signal and also endow it with both bistability and hysteresis (19). In contrast, a negative feedback loop combined with intrinsic ultrasensitivity brings about sustained oscillations in ERK phosphorylation (12, 14). The sensitivity of the MAPK pathway to the cellular milieu is further underscored by the fact that under certain conditions it can also function as a monostable system yielding a proportional output response at the level of ERK phosphorylation (51–53). At least some of the determinants of the nature of input/output relationships identified so far are the concentration of MKPs (19), the concentration of the scaffolding proteins involved (15), and the extent of sequestration of ERK in a complex with its kinase, MEK (16). In addition to defining the threshold between a switch-like versus a proportional response, these parameters also play a critical role in modulating the amplitude and kinetics of the output signal.
In the present case the proportional nature of the input/output relationship, despite incorporating a distributive mechanism for the dual phosphorylation of ERK, appears to be combinatorially defined through the relative concentrations/activities of the MAPK pathway-associated phosphatases. Thus, in silico exercises involving select modulations in phosphatase concentrations yielded an enhanced slope for ERK activation under conditions where either PP1 or MKP3 levels were significantly increased (supplemental Fig. S10). Alterations in levels of the MAPK pathway components had no such effect. Consistent with this is our experimental demonstration that siRNA-mediated silencing of these individual phosphatases had a significant effect on the amplitude, the stability, the kinetics, and the base-line levels of ERK phosphorylation. A role for filtering spurious noise by MAPK-associated phosphatases has also been recently discussed (17).
Our finding that phosphatase-dependent regulation of the MAPK pathway involves organization of the phosphatases into a co-aligned cascade was especially interesting. Traditionally, negative regulation of intracellular signal transduction is conceived to occur as a discrete set of localized interactions between a phosphatase and its target molecule, and a cascade organization for phosphatases has not been considered so far (31, 54). Importantly, this co-alignment resulted in the formation of a novel regulatory module that was composed of a contiguous set of two square units, with the MAPK constituents and the phosphatases PP2A, MKP3, and MKP1 constituting the vertices of these units. We have termed this novel signaling motif, induced upon BCR-dependent stimulation of cells, as the signal-activated motif (SAM).
Assembly of the SAM involved two distinct input signals that, in turn, separately activated the functionally distinct components of the module. The first of these involved the combined action of protein kinase C and RasGTP to activate Raf, thereby initiating signal flow through the MAPK cascade. The second signal, on the other hand, involved the CK2α-dependent phosphorylation of MKP3. This event was critical to the regulatory function of the phosphatase axis. Thus, whereas BCR activation activated signal flow through the MAPK pathway on the one hand, it also simultaneously embedded this pathway within a regulatory module so that the net signal output could be carefully regulated.
Central to the functioning of the SAM was the distribution of MKP3 between its phosphorylated and non-phosphorylated states where the balance between these two forms was defined by the reciprocal actions of the constitutively active PP2A and the signal- activated CK2α. This segregation of input signals to activate both the positive and negative regulatory components of the SAM motif provided the mechanism by which filtration and appropriate calibration of the signal output could be achieved. Key to this was the differential substrate bias displayed by phosphorylated and non-phosphorylated MKP3, with the relative proportion of these two subsets being governed by the extent to which the input signal (i.e. activated CK2α) could buffer the action of PP2A. The conditional shifts in the equilibrium between these two pools played a critical role in defining the signal output in terms of the ERK activation profile. In other words this ability to modulate substrate preferences enabled MKP3 to function as an additional response element for signal discrimination whereby both signal amplitude and sensitivity to ligand concentration could be further tuned.
Our subsequent analysis of how output responses were shaped by alterations in phosphatase concentrations also provided interesting new insights into the properties of the SAM. Although MKP3 levels were important in defining the amplitude of ERK activation, it was the MKP1 concentrations that strictly defined the window of ERK activation. That output regulation involved the coordinated action of associated phosphatases was also emphasized in our studies examining the effect of simultaneously varying concentrations of two phosphatases. For example, the available levels of the remaining two phosphatases further modulated the effects of varying MKP3 concentrations. This was equally true for PP2A and MKP1, thereby reaffirming the signal-dependent assembly of these phosphatases into a functional axis that intimately regulates signal processing.
The net outcome of the coordinated action of phosphatases was that information flow through the MAPK pathway did not occur in a linear fashion. Rather, signal was differentially processed at each intermediate in the pathway. Consequently, in addition to the strength of the stimulus, transmission of signal between successive nodes was also rendered sensitive to the relative concentrations of the associated phosphatases. Thus, for example, conditions yielding a potent activation of MEK did not necessarily translate into a similar profile for ERK. Instead, signal transmission from MEK to ERK was additionally regulated by the activity and/or concentration of the two remaining phosphatase components of the SAM. The resultant combinatorial effects then ranged from a marked amplification to a complete suppression of ERK activation. Indeed even the limited investigations performed here reveal how the intricate coupling of a kinase with a phosphatase axis can provide for flexible regimes of regulation where the activity and/or concentrations of the participating phosphatases make key contributions to calibration of the signal output. A direct consequence of this was an expanded landscape of potential ERK responses where a given output characterized not only the strength of activating stimulus but also the phenotypic state of the cell, at least when characterized in terms of the MAPK-associated phosphatase milieu.
Differences in the tissue-specific origin of cells are also often reflected at the level of differences in concentration of the individual MKPs. Similarly, variable MKP levels frequently characterize the individual stages of lineage commitment in lymphocytes (55). Furthermore, even within a given cell type, activation of ERK leads to the up-regulation of both MKP1 and MKP3 (31), with the consequent de-sensitization of ERK to any further stimulation of the cell. Thus, the composition of at least MKPs within a cell may well provide a phenotypic description of its activation and/or differentiation status, which then is also incorporated when defining the ERK output response.
Thus, in summary, our present delineation of an emergent motif that endows the MAPK pathway with flexible regimes for modulating signal output provides additional insights into mechanisms that facilitate information processing by the signaling machinery. At one level, these findings reveal a novel organizational principle wherein a kinase cascade is intricately coupled to a regulatory cascade of phosphatases. The resulting structural motif that links each kinase intermediate to both the iso-stage and the upstream phosphatase ensures that signal transmission is subjected to tight scrutiny at every step of the pathway. This allows the “strength” of the links between the phosphates axis and the kinase cascade to also function as determinants of signal processing. As a result, even a simple proportional response system becomes endowed with the ability to generate an output that, in addition to signal duration and strength, is also sensitized to variations in phosphatase concentrations and activities. As shown here, for instance, the landscape of ERK activation responses that can potentially be generated as a result of the SAM can fully account for the diversity of cellular responses that this pathway is known to generate. Furthermore, as noted earlier, differences in the phenotypic status of cells either at the level of cell type or differentiation state or with a history of prior stimulations can also be integrated when defining the ERK output response. It, however, remains to be seen whether such a regulatory module is also associated with the other pathways of the signaling network.
Supplementary Material
Acknowledgment
We thank Ubaid Ullah for help with the flow cytometry experiments.
This work was supported by a grant from Department of Biotechnology, Government of India (to K. V. S. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental data S1 and Figs. S2–S10.
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- BCR
- B cell antigen receptor
- SAM
- signal-activated motif
- MEK
- MAPK kinase
- MKP
- MAPK phosphatase
- siRNA
- small interfering RNA
- DRB
- 5,6-dichloro-1-,8-d-ribofuranosylbenzimidazole.
REFERENCES
- 1.Cobb M. H. (1999) Prog. Biophys. Mol. Biol. 71, 479–500 [DOI] [PubMed] [Google Scholar]
- 2.Widmann C., Gibson S., Jarpe M. B., Johnson G. L. (1999) Physiol. Rev. 79, 143–180 [DOI] [PubMed] [Google Scholar]
- 3.Chang L., Karin M. (2001) Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 4.Tan P. B., Kim S. K. (1999) Trends Genet 15, 145–149 [DOI] [PubMed] [Google Scholar]
- 5.Ebisuya M., Kondoh K., Nishida E. (2005) J. Cell Sci. 118, 2997–3002 [DOI] [PubMed] [Google Scholar]
- 6.Kolch W., Calder M., Gilbert D. (2005) FEBS Lett. 579, 1891–1895 [DOI] [PubMed] [Google Scholar]
- 7.Friedman A., Perrimon N. (2006) Nature 444, 230–234 [DOI] [PubMed] [Google Scholar]
- 8.Huang C. Y., Ferrell J. E., Jr. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10078–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrell J. E., Jr., Bhatt R. R. (1997) J. Biol. Chem. 272, 19008–19016 [DOI] [PubMed] [Google Scholar]
- 10.Orton R. J., Sturm O. E., Vyshemirsky V., Calder M., Gilbert D. R., Kolch W. (2005) Biochem. J. 392, 249–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eungdamrong N. J., Iyengar R. (2007) Biophys. J. 92, 808–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiao L., Nachbar R. B., Kevrekidis I. G., Shvartsman S. Y. (2007) PLoS Comput. Biol. 3, 1819–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das J., Ho M., Zikherman J., Govern C., Yang M., Weiss A., Chakraborty A. K., Roose J. P. (2009) Cell 136, 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kholodenko B. N. (2000) Eur. J. Biochem. 267, 1583–1588 [DOI] [PubMed] [Google Scholar]
- 15.Levchenko A., Bruck J., Sternberg P. W. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5818–5823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blüthgen N., Bruggeman F. J., Legewie S., Herzel H., Westerhoff H. V., Kholodenko B. N. (2006) FEBS J. 273, 895–906 [DOI] [PubMed] [Google Scholar]
- 17.Locasale J. W., Shaw A. S., Chakraborty A. K. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13307–13312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raman M., Chen W., Cobb M. H. (2007) Oncogene 26, 3100–3112 [DOI] [PubMed] [Google Scholar]
- 19.Bhalla U. S., Ram P. T., Iyengar R. (2002) Science 297, 1018–1023 [DOI] [PubMed] [Google Scholar]
- 20.Altan-Bonnet G., Germain R. N. (2005) PLoS Biol. 3, e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinrich R., Neel B. G., Rapoport T. A. (2002) Mol. Cell 9, 957–970 [DOI] [PubMed] [Google Scholar]
- 22.Kumar D., Dua R., Srikanth R., Jayaswal S., Siddiqui Z., Rao K. V. (2008) BMC Res. Notes 1, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zi Z., Zheng Y., Rundell A. E., Klipp E. (2008) BMC Bioinformatics 9, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh D. K., Kumar D., Siddiqui Z., Basu S. K., Kumar V., Rao K. V. (2005) Cell 121, 281–293 [DOI] [PubMed] [Google Scholar]
- 25.Kumar D., Srikanth R., Ahlfors H., Lahesmaa R., Rao K. V. (2007) Mol. Syst. Biol 3, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt T. G., Skerra A. (2007) Nat. Protoc. 2, 1528–1535 [DOI] [PubMed] [Google Scholar]
- 27.Manders E. M., Verbeek F. J., Aten J. A. (1993) J. Microsc. 169, 375–382 [DOI] [PubMed] [Google Scholar]
- 28.Reth M. (1992) Annu. Rev. Immunol. 10, 97–121 [DOI] [PubMed] [Google Scholar]
- 29.Kurosaki T. (2000) Curr. Opin. Immunol. 12, 276–281 [DOI] [PubMed] [Google Scholar]
- 30.Brondello J. M., Pouysségur J., McKenzie F. R. (1999) Science 286, 2514–2517 [DOI] [PubMed] [Google Scholar]
- 31.Dickinson R. J., Keyse S. M. (2006) J. Cell Sci. 119, 4607–4615 [DOI] [PubMed] [Google Scholar]
- 32.Castelli M., Camps M., Gillieron C., Leroy D., Arkinstall S., Rommel C., Nichols A. (2004) J. Biol. Chem. 279, 44731–44739 [DOI] [PubMed] [Google Scholar]
- 33.Rolli V., Gallwitz M., Wossning T., Flemming A., Schamel W. W., Zürn C., Reth M. (2002) Mol. Cell 10, 1057–1069 [DOI] [PubMed] [Google Scholar]
- 34.Kurosaki T. (1999) Annu Rev. Immunol. 17, 555–592 [DOI] [PubMed] [Google Scholar]
- 35.Kurosaki T., Tsukada S. (2000) Immunity 12, 1–5 [DOI] [PubMed] [Google Scholar]
- 36.Kurosaki T., Maeda A., Ishiai M., Hashimoto A., Inabe K., Takata M. (2000) Immunol. Rev. 176, 19–29 [DOI] [PubMed] [Google Scholar]
- 37.Kurosaki T. (2002) Nat. Rev. Immunol. 2, 354–363 [DOI] [PubMed] [Google Scholar]
- 38.Camps M., Nichols A., Arkinstall S. (2000) FASEB J. 14, 6–16 [PubMed] [Google Scholar]
- 39.Bollen M., Beullens M. (2002) Trends Cell Biol. 12, 138–145 [DOI] [PubMed] [Google Scholar]
- 40.Cyert M. S. (2001) J. Biol. Chem. 276, 20805–20808 [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y., Zhang Z. Y. (2001) J. Biol. Chem. 276, 32382–32391 [DOI] [PubMed] [Google Scholar]
- 42.Karlsson M., Mathers J., Dickinson R. J., Mandl M., Keyse S. M. (2004) J. Biol. Chem. 279, 41882–41891 [DOI] [PubMed] [Google Scholar]
- 43.Muda M., Boschert U., Dickinson R., Martinou J. C., Martinou I., Camps M., Schlegel W., Arkinstall S. (1996) J. Biol. Chem. 271, 4319–4326 [DOI] [PubMed] [Google Scholar]
- 44.Zandomeni R., Zandomeni M. C., Shugar D., Weinmann R. (1986) J. Biol. Chem. 261, 3414–3419 [PubMed] [Google Scholar]
- 45.Alon U. (2007) Nat. Rev. Genet. 8, 450–461 [DOI] [PubMed] [Google Scholar]
- 46.Kolch W. (2000) Biochem. J. 351, 289–305 [PMC free article] [PubMed] [Google Scholar]
- 47.Hatakeyama M., Kimura S., Naka T., Kawasaki T., Yumoto N., Ichikawa M., Kim J. H., Saito K., Saeki M., Shirouzu M., Yokoyama S., Konagaya A. (2003) Biochem. J. 373, 451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ueki K., Matsuda S., Tobe K., Gotoh Y., Tamemoto H., Yachi M., Akanuma Y., Yazaki Y., Nishida E., Kadowaki T. (1994) J. Biol. Chem. 269, 15756–15761 [PubMed] [Google Scholar]
- 49.Echevarria D., Martinez S., Marques S., Lucas-Teixeira V., Belo J. A. (2005) Dev. Biol. 277, 114–128 [DOI] [PubMed] [Google Scholar]
- 50.Dickinson R. J., Eblaghie M. C., Keyse S. M., Morriss-Kay G. M. (2002) Mech. Dev. 113, 193–196 [DOI] [PubMed] [Google Scholar]
- 51.Birtwistle M. R., Hatakeyama M., Yumoto N., Ogunnaike B. A., Hoek J. B., Kholodenko B. N. (2007) Mol. Syst. Biol. 3, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santos S. D., Verveer P. J., Bastiaens P. I. (2007) Nat. Cell Biol. 9, 324–330 [DOI] [PubMed] [Google Scholar]
- 53.Tian T., Harding A., Inder K., Plowman S., Parton R. G., Hancock J. F. (2007) Nat. Cell Biol. 9, 905–914 [DOI] [PubMed] [Google Scholar]
- 54.Keyse S. M. (2000) Curr. Opin. Cell Biol. 12, 186–192 [DOI] [PubMed] [Google Scholar]
- 55.Bettini M. L., Kersh G. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16257–16262 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.