

Abstract
A family of IGF-binding proteins (IGFBP) exerts biological actions both dependent on and independent of IGF-I. A major effector of the insulin/IGF-I signaling pathway, the serine/threonine protein kinase Akt, mediates cellular processes such as glucose uptake, protein synthesis, cell survival, and growth. IGF-I is required for normal organismal growth, and in the pancreatic β-cell, the insulin/IGF-I signaling pathway is critical for normal and adaptive maintenance of β-cell mass. Expression of myrAkt1, an activated form of Akt, in the endocrine pancreas drives β-cell expansion through dramatic increases in both islet and β-cell size and number. Herein we present a comparative expression profiling of myrAkt1 transgenic islets that demonstrates the increased abundance of transcripts encoding proteins associated with growth, suppression of apoptosis, RNA processing, and metabolism. Although IGFBP5 is identified as a gene induced by Akt1 activation in the β-cell, Igfbp5 expression is not necessary for myrAkt1 to augment β-cell size or mass in vivo. However, in the absence of Igfbp5, mice demonstrate an increase in size and mild glucose intolerance. This is accentuated during diet-induced obesity, when Igfbp5-deficient mice have increased adiposity compared with wild-type mice on the same diet. These studies reveal a novel role for Igfbp5 in the control of growth and metabolism.
IGFBP5 is not required for enhanced beta cell growth in response to active Akt, but is important to normal organismal growth and glucose metabolism.
The insulin signaling pathway is responsible for regulation of glucose homeostasis via stimulation of glucose uptake and storage in peripheral tissues and by inhibition of glucose production in the liver, whereas IGF-I is a major regulator of organ and organismal growth. In addition to the classical insulin target tissues, insulin and IGF-I also act on the β-cell and are critical for the regulation of insulin secretory function and maintenance of normal and adaptive β-cell mass (1). For example, conditional deletion of either the insulin receptor (IR) (2) or the IGF-I receptor (3) in the β-cell impairs β-cell glucose-stimulated insulin secretion with only minor or no effect, respectively, on maintenance of β-cell mass. However, combined deletion of both receptors leads to severe hyperglycemia due to both β-cell functional defects and an inability to maintain normal β-cell mass (4). In wild-type animals and humans, when insulin demand is increased during physiological or pathological states such as pregnancy, obesity, or insulin resistance, insulin output must increase to maintain glucose homeostasis. This compensatory adaptation involves β-cell hyperplasia, hypertrophy, suppression of apoptosis, and enhanced insulin secretion. It is now generally accepted that inadequate β-cell compensation is the critical factor in the progression from insulin resistance to type 2 diabetes (5).
The canonical insulin/IGF-I signaling pathway involves activation of receptor tyrosine kinase activity and recruitment of the scaffolding proteins, IR substrates 1 (IRS1) and 2 (IRS2) (6). Receptor phosphorylation of the IRS proteins then leads to activation of phosphatidylinositol-3 kinase (PI3K) and increased levels of phosphoinositide-3,4,5-phosphate (PIP3) at the plasma membrane. A major target of PI3K activation is the serine/threonine kinase Akt (6). Previously, mice with β-cell-specific expression of an activated form of Akt1, myrAkt1, were generated to investigate the function of Akt1 in the β-cell (7,8). These mice display an approximately 8-fold increase in β-cell mass, which is due to an increase in the number and size of islets and a doubling in β-cell size. In addition, the proliferation rate is increased, suggesting enhanced cell division contributes to the augmentation of β-cell mass (7). Consistent with the increase in β-cell mass, the myrAkt1 transgenic (TG) mice are hyperinsulinemic and hypoglycemic and exhibit improved glucose tolerance. Thus, Akt1 positively affects β-cell expansion via stimulation of β-cell growth (size), proliferation, and potentially neogenesis.
The ability of Akt to mediate the effects of growth factors and nutrients on cell growth, proliferation, and survival is well documented in various cell types. These studies have also led to the identification of several targets of Akt, both direct and indirect, that are candidates for transmitting the growth and survival signals (9). For instance, in addition to its role in regulation of glycogen breakdown, the Akt substrate glycogen synthase kinase 3 (GSK3) is also involved in targeting various substrates for ubiquitin-mediated degradation. Many of the proteins that GSK3 phosphorylates are regulators of cell growth, such as c-Myc, β-catenin, and cyclin D1 and D2 (9). Akt also regulates the cell cycle inhibitors p21CIP and p27KIP1 by phosphorylation and sequestration in the cytosol (10,11). In addition, Akt influences transcription of cell cycle regulators such as p27KIP1 via phosphorylation and inhibition of the Foxo family of forkhead transcription factors (12).
Specific to pancreatic β-cells, the G1 cell cycle proteins cyclin-dependent kinase 4 (CDK4) and the cyclins D1 and D2 are involved in postnatal β-cell proliferation and maintenance of β-cell mass (13,14). Recently, Fatrai et al. (15) demonstrated that CDK4 activity is required for the enhanced β-cell proliferation observed in mice with β-cell-specific overexpression of myrAkt1. In these mice, activation of Akt in the β-cell is associated with increased phosphorylation of GSK3β, increased protein expression of cyclins D1 and D2, and CDK4 activation. Although this study suggests one potential mechanism for the effect of Akt activation on β-cell proliferation, enhanced proliferation is only one component contributing to the overall increase in β-cell mass. Thus, the precise mechanism by which myrAkt1 elicits its TG phenotype, particularly the increase in cell size, remains uncertain (16). For example, haploinsufficiency of FOXO1, a transcription factor negatively regulated by Akt, restores PDX-1 expression and β-cell mass in IRS2 knockout mice; this suggests a potential link between Akt activation and PDX-1 expression (17). PDX-1 is an important transcription factor for both pancreas and islet development and normal β-cell function.
In this study, we have used comparative expression profiling of myrAkt1 TG islet mRNA to identify potential targets and gene networks that might mediate the effect of Akt1 activation on pancreatic β-cell mass expansion. To narrow the list of candidates, we have compared the transcriptional profile secondary to expression of active Akt1 with a physiological condition that causes enhanced growth, i.e. pregnancy. Analysis of differentially regulated genes provides insight into specific cellular functions such as growth, suppression of apoptosis, RNA processing, and metabolism that are influenced by Akt1 activation in the β-cell. Furthermore, we have identified the IGF-binding protein 5 (IGFBP5) as a gene highly up-regulated by myrAkt1 in the β-cell and have investigated its role in mediating the effect of myrAkt1 on augmentation of β-cell mass. In the course of these studies, we have serendipitously identified an unexpected role for Igfbp5 in the regulation of growth and metabolism.
Results
Effect of Akt1 activation on gene expression in pancreatic β-cells
To identify genes that are differentially regulated by Akt1 activation in the β-cell, we performed large-scale expression profiling on pancreatic islets isolated from 4- to 6-wk-old RIP-myrAkt1 TG mice. This microarray expression analysis identified 131 genes of 6299 distinct genes spotted on the PancChip 4.0 as differentially regulated (2.1%) (supplemental Fig. 1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). Of the 131 genes, 68 were significantly (>1.4-fold) up-regulated and 63 significantly down-regulated in the RIP-myrAkt1 TG islets; genes encoding multiple classes of proteins were represented (supplemental Fig. 2).
To elucidate the physiological processes in the β-cell that are influenced by Akt1 activation, functional annotation analysis was performed to determine gene ontology functions and to identify functional groups that are overrepresented within the set of differentially expressed genes (Table 1). As expected, genes affecting both cell growth and cell death were overrepresented among the genes differentially regulated by myrAkt1 in TG islets. Importantly, the thymoma viral protooncogene protein 1 gene, encoding Akt1, was up-regulated 1.53-fold. The increased Akt1 expression is a good positive control and lends confidence to the validity of results from this analysis. Energy availability and the capacity to enhance protein synthesis are requirements for cellular growth. Therefore, it is interesting that several genes involved in the regulation of carbohydrate metabolism and RNA processing were differentially regulated in the myrAkt1 TG islets (Table 1).
Table 1.
Classified list of known genes identified by PancChip 4.0 analysis as significantly differentially expressed (>1.4 and 20% false discovery rate) in myrAkt1 TG islets compared with NTG islets
| Entrez ID | Gene name | Fold change |
|---|---|---|
| Cell adhesion/extracellular matrix | ||
| 20750 | Secreted phosphoprotein 1 (Spp1) | 3.13 |
| 13602 | SPARC-like 1 (Sparcl1) | 2.10 |
| 20733 | Serine protease inhibitor, Kunitz type 2 (Spint2) | 1.66 |
| 22329 | Vascular cell adhesion molecule 1 (Vcam1) | 1.58 |
| 53378 | Syndecan binding protein (Sdcbp) | 1.43 |
| 15894 | Intercellular adhesion molecule (Icam1) | −1.45 |
| Membrane- and organelle-associated proteins | ||
| 12653 | Chromogranin B (Chgb) | 2.94 |
| 12505 | CD44 antigen (CD44) | 2.27 |
| 17748 | Metallothionein 1 (Mt1) | 2.14 |
| 16783 | Lysosomal membrane glycoprotein 1 (Lamp1) | 1.52 |
| 12010 | β2-Microglobulin (B2m) | 1.42 |
| 67182 | Membrane-associated protein 17 (Pdzk1ip1) | −1.47 |
| 68585 | Reticulon 4 (Rtn4; Nogo) | −1.59 |
| 80876 | Interferon-induced transmembrane protein 2 (Ifitm2) | −1.60 |
| 216350 | Transmembrane 4 superfamily member 3 (Tspan8) | −2.20 |
| Receptors | ||
| 67168 | Purinergic receptor (family A group 5) (Plac8) | 1.67 |
| 16004 | IGF-II receptor (Igfr2) | −1.50 |
| 23920 | Insulin receptor-related receptor (Insrr) | −1.89 |
| 77773 | IGF-I receptor 1 (Igf1r) | −1.93 |
| Glucose-dependent insulinotropic polypeptide receptor (Gipr) | −2.30 | |
| Transporters | ||
| 74413 | Membrane-targeting (tandem) C2 domain containing 1 (Mtac2d1) | 1.93 |
| 11927 | ATX1 (antioxidant protein 1) homolog 1 (yeast) (Atox1) | 1.89 |
| 69150 | Sorting nexin 4 (Snx4) | 1.60 |
| 66868 | Major facilitator superfamily domain containing 1 (Mfsd1) | 1.51 |
| 20515 | Solute carrier family 20, member 1 (Slc20a1) | 1.41 |
| 69354 | Solute carrier family 38, member 4 (Slc38a4) | −1.59 |
| 18111 | Neuronatin (Nnat) | −1.63 |
| 22139 | Transthyretin (Ttr) | −1.89 |
| Signal transduction | ||
| 83965 | Ectonucleotide pyrophosphatase/phosphodiesterase 5 (Enpp5) | 1.57 |
| 106572 | RIKEN cDNA 1700093E07 gene (Rab31) | 1.52 |
| 19344 | RAB5B, member RAS oncogene family | 1.41 |
| 57436 | γ-Aminobutyric acid (GABA(A)) receptor-associated protein-like 1 (Gabarapl1) | −1.40 |
| 66120 | FK506 binding protein 11 (Fkbp11) | −1.98 |
| Cytokines and chemokines | ||
| 17319 | Macrophage migration inhibitory factor (Mif) | 1.57 |
| 67213 | Chemokine-like factor super family 6 (Cmtm6) | 1.42 |
| Metabolism | ||
| Glycolysis | ||
| 18746 | Pyruvate kinase, muscle (Pkm2) | 1.51 |
| 13806 | Enolase 1, α nonneuron (Eno1) | 1.45 |
| 14121 | Fructose bisphosphatase 1 (Fbp1) | −1.56 |
| TCA cycle | ||
| 17449 | Malate dehydrogenase 1, NAD (soluble) (Mdh1) | 1.41 |
| Oxidative phosphorylation | ||
| 11951 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit c (subunit 9), isoform 1 (Atp5g1) | 1.64 |
| 228033 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit c (subunit 9), isoform 3 (Atp5g3) | 1.43 |
| 11973 | ATPase, H+ transporting, V1 subunit isoform 1 (Atp6v1e1)E | 1.41 |
| Lipid | ||
| 26971 | Phospholipase A2, group IIF (Pla2g2f) | 1.50 |
| 16956 | Lipoprotein lipase (Lpl) | −3.61 |
| (Continued) | ||
Table 1A.
Continued
| Entrez ID | Gene name | Fold change |
|---|---|---|
| Amino acid | ||
| 67092 | l-Arginine:glycine amidinotransferase (Gatm) | 2.86 |
| 14719 | Glutamate oxaloacetate transaminase 2, mitochondrial (Got2) | 1.79 |
| 12411 | Cystathionine β-synthase (Cbs) | −1.50 |
| Vitamin | ||
| 27973 | Vitamin K epoxide reductase complex, subunit 1 (Vkorc1) | −1.43 |
| Coenzyme | ||
| 14871 | Glutathione S-transferase, θ1 (Gstt1) | 1.41 |
| Other | ||
| 68133 | Glycine cleavage system protein H (aminomethyl carrier) (Gcsh) | 1.51 |
| 432720 | Similar to 3(20)α-hydroxysteroid/dihydrodiol/indanol dehydrogenase (Akr1c19) | −1.76 |
| RNA-protein complex | ||
| 76936 | Heterogeneous nuclear ribonucleoprotein M (Hnrpm) | 3.39 |
| 20813 | Signal recognition particle 14 (Srp14) | 1.75 |
| 54208 | ADP-ribosylation factor-like 6 interacting protein 1 (Arl6ip1) | 1.55 |
| 12972 | Crystallin, ζ(Cryz) | 1.45 |
| 11837 | Acidic ribosomal phosphoprotein P0 (Arbp) | −1.40 |
| 67891 | Ribosomal protein L4 (Rpl14) | −1.41 |
| 17938 | Nascent polypeptide-associated complex-α polypeptide (Naca) | −1.41 |
| 12696 | Cold inducible RNA-binding protein (Cirbp) | −1.57 |
| 19981 | Ribosomal protein L37a (Rpl37a) | −1.62 |
| 13627 | Eukaryotic translation elongation factor 1 α 1 (Eef1a1) | −1.66 |
| 20102 | Ribosomal protein S4, X-linked (Rps4x) | −1.67 |
| 319195 | Ribosomal protein L17 (Rpl17) | −1.85 |
| Nuclear and DNA-binding proteins | ||
| 21888 | Transducin-like enhancer of split 4, homolog of Drosophila E(spl) (Tle4) | 1.64 |
| 68024 | Histone 1, H2bc (Hist1h2bc) | 1.50 |
| 17536 | Myeloid ecotropic viral integration site-related gene 1 (Mrg1) | −1.42 |
| 18124 | Nuclear receptor subfamily 4, group A, member 3 (Nr4a3) | −1.42 |
| 18129 | Notch gene homolog 2 (Drosophila) (Notch2) | −1.45 |
| 50496 | E2F transcription factor 6 (E2f6) | −1.46 |
| 15331 | High-mobility group nucleosomal binding domain 2 (Hmgn2) | −1.48 |
| 18033 | Nuclear factor of κ−light chain gene enhancer in B cells 1, p105 (Nfkb1) | −1.65 |
| 15081 | H3 histone, family 3B (H3f3b) | −1.76 |
| 13170 | D site albumin promoter binding protein (Dbp) | −1.79 |
| 14009 | Ets variant gene 1 (Etv1) | −1.98 |
| Cell growth and death | ||
| 16011 | IGFBP5 (Igfbp5) | 4.30 |
| 77579 | Myosin, heavy polypeptide 10, nonmuscle (Myh10) | 2.26 |
| 21987 | Tumor protein D52-like 1 (Tpd52l1) | 2.16 |
| 22350 | Villin 2 (Vil2) | 1.85 |
| 12700 | Cytokine inducible SH2-containing protein (Cish) | 1.75 |
| 12759 | Clusterin (Clu) | 1.58 |
| 11651 | Thymoma viral protooncogene 1 (Akt1) | 1.53 |
| 22195 | Ubiquitin-conjugating enzyme E2 liter 3 (Ube213) | 1.50 |
| 13643 | Ephrin B3 (Efnb3) | −1.41 |
| 75723 | Angiomotin-like 1 (Amotl1) | −1.54 |
| 21761 | Mortality factor 4 like 1 (Morfl1) | −1.55 |
| 11746 | Annexin A4 (Anxa4) | −1.75 |
| 18616 | Paternally expressed 3 (Peg3) | −2.63 |
| Cytoarchitecture | ||
| 18040 | Neurofilament 3, medium (Nefm) | 2.72 |
| 11867 | Actin-related protein 2/3 complex, subunit 1B (Arpc1b) | 1.63 |
| 22145 | Tubulin, α 4 (Tuba4) | 1.53 |
| 19241 | Thymosin, β 4, X chromosome (Tmsb4x) | −1.49 |
| Hormones | ||
| 16334 | Insulin II (Ins2) | −1.73 |
| 14526 | Glucagon (Gcg) | −2.08 |
| 20604 | Somatostatin (Sst) | −2.33 |
| 56373 | Carboxypeptidase B2 (plasma) (Cpb2) | 4.64 |
| (Continued) | ||
Table 1B.
Continued
| Entrez ID | Gene name | Fold change |
|---|---|---|
| 94284 | UDP glycosyltransferase 1 family polypeptide A6 (Ugt1a6a), | 1.99 |
| 26879 | UDP-Gal:βGlcNAc β1,3-galactosyltransferase, polypeptide 3 (B3galnt1) | 1.87 |
| 13039 | Cathepsin L (Cts1) | 1.79 |
| 11883 | Arylsulfatase A (Arsa) | −1.45 |
| 12876 | Carboxypeptidase E (Cpe) | −1.56 |
| 76485 | Glycosyltransferase 8 domain containing 1 (Glt8d1) | −1.62 |
| Miscellaneous | ||
| 103537 | Mbt domain containing 1 (Mbtd1) | 2.03 |
| 66442 | Spindle pole body component 25 homolog (S. cerevisiae) (Spbc25) | 2.00 |
| 100604 | RIKEN cDNA E430036I04 gene (Lrrc8c) | 1.98 |
| 55951 | Brain protein 44-like (Brp441) | 1.87 |
| 225131 | WW domain containing adaptor with coiled-coil (Wac) | 1.83 |
| 20363 | Selenoprotein P, plasma, 1 (Sepp1) | 1.79 |
| 16061 | Ig heavy chain (J558 family) (Igf-VJ558) | 1.53 |
| 67702 | Ring finger protein 149 (Rng149) | 1.44 |
| 81018 | Zinc finger protein 313 (Zfp313) | −1.43 |
| 27886 | Expressed sequence 2 embryonic lethal | −1.43 |
| 406217 | RIKEN cDNA 2410004M13 gene (Bex4) | −1.48 |
| 373070 | miRNA-containing gene (Mirg) | −1.98 |
Pregnancy is a condition of increased body mass and metabolic stress that stimulates increases in β-cell mass (18). Because overexpression of activated Akt is a very artificial means to increase growth, we compared the myrAkt1 microarray data set with an array comparing differential gene expression in islets experiencing a physiological form of enhanced growth, i.e. pregnancy. Interestingly, we found that mRNA encoding Akt1 was increased in the islets from pregnant mice and that many genes are differentially regulated and change in a similar direction in both paradigms. This analysis is most informative when the Ingenuity metabolic/cell-to-cell signaling network for each array is compared (supplemental Fig. 3). In addition to Akt1 induction, IGFBP5, cytokine-inducible Src homology-2-containing protein (CISH), leucine-rich repeat containing 8C (LRRC8C), and villin (Vil2) are up-regulated in both experimental systems. The genes encoding lipoprotein lipase (Lpl), insulin 2 (Ins), preproglucagon (GCG), and somatostatin (SST) are similarly decreased in both expression profiles. This analysis demonstrates that Akt1 expression is induced in pregnancy, a condition during which β-cell mass expands and islet function is optimized. Comparison of both gene expression profiles points to novel Akt targets, such as IGFBP5 and lipoprotein lipase, that might contribute to the effect of Akt1 on β-cell growth and function. Furthermore, the fact that these genes are similarly regulated in two situations in which β-cell mass is increased provides additional support for an important role downstream of Akt1 activation.
As described above, Igfbp5 was identified both as a gene induced in the islet during pregnancy and as one of the most highly up-regulated genes in the myrAkt1 TG islet array. IGFBP5 is a member of the IGF-I-binding protein family that bind with high affinity to both IGF-I and IGF-II (19). Igfbp5 mRNA expression is induced by treatment with IGF-I in both aortic and vascular smooth muscle cells in a wortmannin- and rapamycin-sensitive manner, suggesting that induction of Igfbp5 is downstream of the PI3K/mTOR signaling pathway (20,21). Considering the numerous studies implicating IGFBP5 as a regulator of cellular growth and differentiation (22,23), we selected Igfbp5 as a plausible candidate to mediate the effects of myrAkt1 in the β-cell.
In vivo and in vitro confirmation of myrAkt1-induced increase in Igfbp5 expression
Igfbp5 mRNA and protein expression was assessed in islets isolated from non-TG (NTG) or myrAkt1 TG mice (Fig. 1). Igfbp5 mRNA expression was increased 7-fold in myrAkt1 TG islets compared with NTG islets, in agreement with the microarray analysis (Fig. 1A). Western blot analysis also showed a dramatic increase in IGFBP5 protein expression in the myrAkt1 TG islets (Fig. 1B). Additionally, Igfbp5 mRNA expression was examined in the MIN6 β-cell line, in which the myrAkt1 transgene was stably expressed. Figure 2A shows confirmation of myrAkt1 protein expression by Western blot analysis. The myrAkt1 transgene migrates faster than the endogenous protein due to deletion of the pleckstrin homology domain. Increased Akt1 phosphorylation and phosphorylation of the downstream target GSK indicates activation of the Akt1 signaling pathway compared with cells expressing the empty vector (pLNCX). Analysis of Igfbp5 expression by quantitative PCR demonstrated an increase of approximately 2-fold in the myrAkt1-expressing cells compared with controls (Fig. 2B). The relatively lesser induction of Igfbp5 mRNA in the myrAkt1 stable MIN6 cells compared with the myrAkt1 TG islets might be due to differences in the level of expression of the transgene or a higher basal Igfbp5 expression in the immortalized β-cell line. To ask whether regulation of Igfbp5 by Akt is a generalized phenomenon, we examined the effect of acute Akt1 activation on induction of Igfbp5 expression in primary mouse embryonic fibroblasts (MEFs). Wild-type MEFs were infected with either adenovirus encoding green fluorescent protein (ad-GFP) or myrAkt1 (ad-myrAkt1). The myrAkt1 cDNA expressed by this virus contains the full-length Akt1 sequence. As shown in Fig. 3A, ad-myrAkt1 infection increased total and phosphorylated Akt1 expression compared with ad-GFP-infected MEFs. Adenoviral overexpression of myrAkt1 induced a dramatic 50-fold increase in Igfbp5 mRNA expression compared with ad-GFP-infected cells (Fig. 3B). Together, these data confirm that Akt1 activation increases expression of Igfbp5 in the β-cell.
Figure 1.

Igfbp5 mRNA and protein is up-regulated in the myrAkt1 TG islets, and expression is absent in Igfbp5−/− islets. A, Islets from 4- to 6-wk-old mice were isolated and total RNA was extracted and assessed by quantitative PCR for Igfbp5. Igfbp5 mRNA was corrected for Gapdh expression (n = 3 per genotype). Error bars represent sd. *, P < 0.01. B, Islets were isolated from 12- to 14-wk-old mice, and protein lysates were prepared from a total of 30 islets per genotype and processed for immunoblotting as described in Materials and Methods. WT, Wild-type; P-Akt, phospho-Akt.
Figure 2.

Igfbp5 expression is increased in MIN6 cells by stable expression of myrAkt1. MIN6-myrAkt1 cells were plated into six-well plates and harvested at approximately 80% confluency after an overnight culture in DMEM containing 3 mm glucose. A, Cell extracts were processed for immunoblotting as described in Materials and Methods. B, Total RNA was extracted, and Igfbp5 expression was assessed by quantitative PCR. Igfbp5 mRNA level was corrected for TBP expression. The graph reflects a representative experiment from a total of three separate experiments performed in triplicate. Error bars represent sd. *, P < 0.01. P-GSK, Phospho-GSK.
Figure 3.
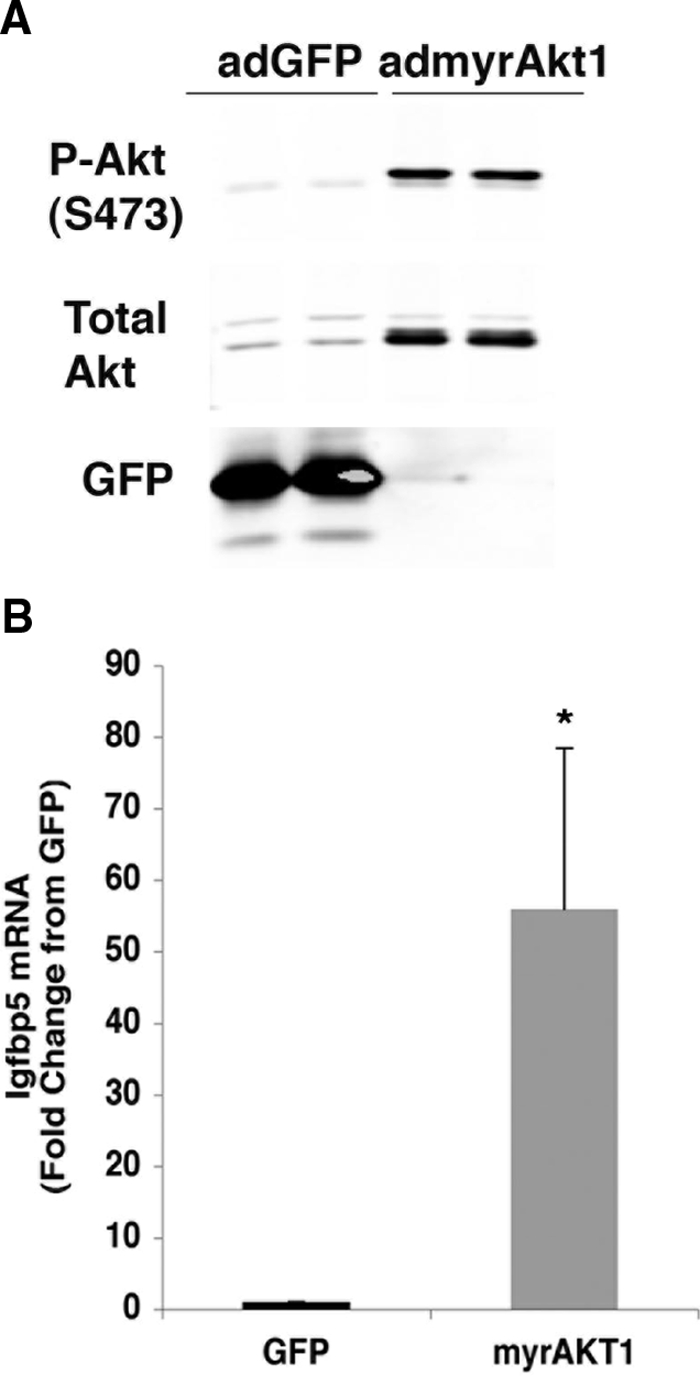
MyrAkt1 induces Igfbp5 mRNA expression in primary MEFs. Primary MEFs were infected with either ad-GFP or myrAkt1 and then subcultured for 48 h before harvesting. A, Cells were lysed and processed for immunoblotting as described in Materials and Methods. B, Igfbp5 expression was assessed by quantitative PCR. Igfbp5 mRNA levels were normalized to TBP expression. The graph represents the average of four separate experiments. The error bars indicate sd. *, P < 0.001. P-Akt, Phospho-Akt.
Derivation and analysis of myrAkt1 TG mice deficient for Igfbp5
We next sought to determine whether the induction of Igfbp5 is required for increased β-cell size or islet number stimulated by myrAkt1. The most convincing strategy to determine whether IGFBP5 is required for changes in β-cell mass in the myrAkt1 TG islets is to examine, in vivo, the consequences of myrAkt1 expression in Igfbp5-deficient mice. To this end, we intercrossed the myrAkt1 TG mice with mice deficient for Igfbp5 (Igfbp5−/−;myrAkt1 TG). The Igfbp5-null mice have been characterized in regard to the effect of Igfbp5 on whole-body and organ growth as well as mammary gland development and involution after lactation (24). Development and growth is normal in the absence of Igfbp5, but there is a small delay in mammary gland involution. The effect of Igfbp5 deletion on regulation of glucose homeostasis has not been examined previously (24).
Deletion of Igfbp5 was confirmed in the Igfbp5−/− mice by examining Igfbp5 protein expression in both kidney and islet tissue. Igfbp5 expression is abundant in the kidney and was detected by Western blot in Igfbp5 wild-type samples. No Igfbp5 was detected in tissues taken from the Igfbp5−/− mice (data not shown). Although Igfbp5 levels were below the detection limit in wild-type islets, myrAkt1 greatly increased IGFBP5 protein expression. However, in the Igfbp5−/−;myrAkt1 TG islets, IGFBP5 protein expression was absent, confirming deletion of Igfbp5 in these mice (Fig. 1B).
Analysis of β-cell mass and size in Igfbp5−/−;myrAkt1 TG mice
To determine whether β-cell mass was augmented in Igfbp5−/−;myrAkt1 TG mice, pancreas sections were stained with insulin and glucagon (Fig. 4). Igfbp5−/−;myrAkt1 TG mice displayed a similar expansion in islet size and number as the Igfbp5+/+;myrAkt1 TG. Of note, although the Igfbp5−/− mice exhibited impaired glucose tolerance (see below), islet size and number did not appear different from the Igfbp5+/+ mice.
Figure 4.

Islet size and β-cell mass are similarly augmented in Igfbp5−/−;myrAkt1 TG mice. Indirect immunofluorescence of pancreas sections from 9- to 10-wk-old male mice of the indicated genotypes. Insulin is green, glucagon is red, and Hoechst 33358 (nuclei) is blue.
To determine whether β-cell size was altered in either the Igfbp5−/−;NTG mice or the Igfbp5−/−;myrAkt1 TG compared with Igfbp5+/+ mice, we stained pancreas sections with β-catenin to highlight the cell periphery and hemagglutinin (HA) to confirm transgene expression (Fig. 5). β-Cell size appeared similar in Igfbp5+/+;NTG and Igfbp5−/−;NTG pancreata. As expected, expression of the myrAkt1 transgene led to a dramatic increase in β-cell size, but there was no difference between cells expressing the myrAkt1 transgene in the presence or absence of Igfbp5. Together, these results demonstrate that deletion of Igfbp5 does not impair the ability of myrAkt1 to augment both β-cell size and mass in vivo.
Figure 5.

β-Cell size is similarly increased in Igfbp5−/−;myrAkt1 TG mice as in Igfbp5+/+;myrAkt1 TG mice. Indirect immunofluorescence of pancreatic sections from male 9- to 10-wk-old mice. A and C, Igfbp5+/+;NTG; B and D, Igfbp5−/−;NTG; E and G, Igfbp5+/+;myrAkt1 TG; F and H, Igfbp5−/−;myrAkt1 TG. Cells were stained for HA (green) to detect expression of the myrAkt1 transgene and for β-catenin (red) and with Hoechst 33358 (nuclei, blue). A, B, E, and F show β-catenin staining only.
Because the β-cell expansion in myrAkt1 TG mice leads to metabolic abnormalities such as hyperinsulinemia, hypoglycemia, and improved glucose tolerance, we assessed glucose homeostasis in myrAkt1 mice deficient for Igfbp5 (7,8). As demonstrated previously, mice wild-type for Igfbp5 and expressing the myrAkt1 transgene (Igfbp5+/+;myrAkt1 TG) exhibit fasting hypoglycemia. Igfbp5−/− mice expressing the myrAkt1 transgene (Igfbp5−/−;myrAkt1 TG) did not exhibit a significant decrease in fasting blood glucose level, although there was a trend in this direction (Fig. 6A). Random fed blood glucose levels were not significantly different regardless of myrAkt1 transgene expression and/or the absence of Igfbp5 (Fig. 6B). As expected, glucose tolerance in the Igfbp5+/+;myrAkt1 TG mice was significantly improved compared with the Igfbp5−/−;NTG mice. Likewise, the Igfbp5−/−;myrAkt1 TG mice displayed improved glucose tolerance compared with either the Igfbp5+/+;NTG mice or Igfbp5−/−;NTG mice, such that the glucose tolerance curves in myrAkt1 TG mice were indistinguishable whether or not they expressed Igfbp5. Surprisingly, Igfbp5−/−;NTG mice were significantly glucose intolerant (Fig. 6C). As measured using the euglycemic-hyperinsulinemic clamp technique, Igfbp5−/− mice showed a trend toward reduced glucose disposal and enhanced hepatic glucose output in the presence of insulin that did not achieve statistical significance (data not shown). Serum insulin levels after an overnight fast or during the glucose tolerance test were increased in the Igfbp5+/+;myrAkt1 TG mice compared with Igfbp5+/+;NTG mice; however, this increase did not reach significance, possibly due to the small number of mice. In contrast, the serum insulin levels in the Igfbp5−/−;myrAkt1 TG mice were significantly elevated at both fasting and 15 min after glucose injection compared with Igfbp5−/−;NTG mice (Fig. 6D). The improved insulin secretion observed in the latter mice suggests that Igfbp5 is not required for the effects of myrAkt1 on the β-cell.
Figure 6.

Regulation of glucose metabolism in male Igfbp5−/−;myrAkt1 TG mice. Glucose values were obtained through tail bleeds from either 14- to16-h fasted (n = 9–24) (A) or random fed (n = 5–12) (B) mice. *, P < 0.001 for NTG vs. myrAkt1 TG. C, Intraperitoneal glucose tolerance tests were performed on 14- to 16-h fasted male mice (8–10 wk old) of the indicated genotypes (n = 9–24). Error bars indicate se. *, P < 0.05; **, P < 0.01 vs. NTG values. D, Serum insulin levels in blood obtained by tail bleeds were ascertained on animals immediately before and 15 min after glucose injection (n = 8–19). Error bars represent se. *, P < 0.01 for 0 vs. 15 min; **, P < 0.01 for Igfbp5−/−;NTG vs. Igfbp5−/−;myrAkt1 TG mice.
Both the Igfbp5−/− and Igfbp5−/−;myrAkt1 TG mice exhibited a small but significant increase in body weight (8.5%) at 8–10 wk of age compared with wild-type mice (Fig. 7A). To determine whether the increased body weight is present at birth or develops postnatally, we weighed pups daily from d 1–7, once per week for 12 wk, and then every 2 wk. The weights of the pups were not significantly different during the first 7 d after birth (data not shown). Male Igfbp5−/− mice did not show a significant difference in body weight until 6 wk of age with the increase reaching 11% by 12 wk. In contrast, female Igfbp5−/− mice exhibited significantly enhanced weight gain by the end of the first week (Fig. 7C). At 5 wk of age, there was an approximately 12% increase in body weight in the Igfbp5−/− mice compared with wild type; this difference grew to about 19% by 7 wk of age. At 20 wk of age, male Igfbp5−/− mice exhibit increased length (from rump to snout) and a significant increase in lean mass as determined by dual-energy x-ray absorptiometry (DEXA) analysis (Figs. 7D and 10E). The increased weight in the knockout mice can be specifically attributed to the changes in lean mass, because fat mass and bone mineral composition and density were the same in both groups (data not shown). We also monitored energy expenditure in these mice to examine whether changes in thermogenesis, respiration, or activity attributed to the enhanced growth phenotype. As shown in Table 2, there were no differences between genotypes in respiration, heat generation, or food intake. However, the Igfbp5−/− mice exhibited decreased total activity compared with wild type during the 24 h they were monitored.
Figure 7.

Igfbp5−/− mice exhibit increased postnatal body weight. A, Body weights were measured on 14- to 16-h fasted male mice between 8 and 10 wk of age (n = 9–24). Error bars indicate se. *, P < 0.01 for Igfbp5−/− and Igfbp5−/−;myrAkt1 TG compared with NTG or myrAkt1 TG mice. Growth curves for male (B) and female (C) mice. Weights were monitored on random fed mice beginning 1 wk after birth and once per week thereafter for 12 wk and then every 2 wk until 14 wk (males, n = 10–12; females, n = 7–9). Error bars indicate se. *, P < 0.01 for Igfbp5+/+ vs. Igfbp5−/− mice. D, DEXA analysis; E, body length.
Figure 10.

Igfbp5−/− mice have significantly elevated fasting blood glucose and are more insulin resistant than Igfbp5+/+ mice after 9 wk on a high-fat diet. A, Glucose levels were determined from blood obtained through tail bleeds on 5- to 6-h fasted mice (Igfbp5+/+ n = 5; Igfbp5−/− n = 4). *, P < 0.05. B, Plasma insulin levels in blood obtained from tail bleeds were ascertained on 5- to 6-h fasted mice (Igfbp5+/+ n = 5; Igfbp5−/− n = 4). *, P < 0.04. C, Blood glucose levels after a single injection of insulin (Igfbp5+/+ n = 5; Igfbp5−/− n = 4). Error bars indicate se. *, P < 0.04.
Table 2.
Energetics in Igfbp5-deficient mice
| Genotype | Igfbp5+/+ perbody mass | Igfbp5−/− perbody mass | Igfbp5+/+ perlean mass | Igfbp5−/− perlean mass |
|---|---|---|---|---|
| VO2 (ml/kg · h) | 125,336 ± 6,066.5 | 117,029 ± 4,046.3 | 146,931 ± 6,441.1 | 131,370 ± 3,909.7 |
| VCO2 (ml/kg · h) | 103,830 ± 7,434.3 | 101,193 ± 3,251.8 | 114,251 ± 5,008.4 | 113,507 ± 7,009.6 |
| RER | 46.1 ± 1.18 | 48.3 ± 1.24 | NA | NA |
| Heat (kcal/h · kg) | 659.8 ± 25.08 | 596.2 ± 23.01 | 802.8 ± 27.86 | 731.0 ± 22.14 |
| Food intake (g) | 2.40 ± 0.375 | 3.00 ± 0.368 | NA | NA |
| Normalized food intake (g/kg) | 89.0 ± 16.06 | 92.5 ± 14.26 | 106.7 ± 17.68 | 112.5 ± 15.06 |
| Total activity | 71,405 ± 13,914.6 | 31,886 ± 5,758.0a | NA | NA |
Wild-type (n = 4) and Igfbp5−/− (n = 4) male, approximately 20-wk-old mice were used to assess food intake and respiratory exchange ratio (RER) under conditions of free access to food and water using the Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH) for 24 h. Oxygen consumption (VO2) and carbon dioxide production (VCO2) were measured in CLAMS and used to calculate respiratory quotient/RER. NA, Not appropriate; RER, respiratory exchange ratio.
P < 0.05, wild type vs. Igfbp5−/−.
To elicit a more dramatic metabolic phenotype, wild-type and Igfbp5−/− mice were placed on a high-fat diet. Igfbp5−/− gained significantly more weight than wild-type mice and demonstrated greater linear growth (Fig. 8). During diet-induced obesity, Igfbp5−/− had a different body composition compared with wild-type mice, with a significant increase in adiposity (Fig. 9, A and B). This was evident by DEXA scanning or by assessing the weight of the fat pad relative to body weight (Fig. 9, B and C). As expected, associated with the enhanced obesity, Igfbp5−/− mice had elevated serum glucose and insulin concentrations compared with wild-type mice on the same diet (Fig. 10). Moreover, they were insulin resistant as assessed by an insulin tolerance test. Thus, Igfbp5 is required for maintenance of normal body size and insulin sensitivity, particularly when challenged by a high-fat diet.
Figure 8.

Igfbp5−/− (BP5−/−) mice gain more weight on a high-fat diet compared with Igfbp5+/+ (BP5+/+) mice. A, Mice were fasted overnight, and weight gain was calculated by subtracting the weight at the indicated time from weight at the initiation of high-fat diet feeding. *, P < 0.01. B, Body weight expressed as percentage of weight at initiation of diet (n = 5 per genotype, except at 4 wk, Igfbp5+/+ n = 2 and Igfbp5−/− n = 3). *, P < 0.04. C, Igfbp5+/+ and Igfbp5−/− mice at 16 wk on high-fat diet. D, Body length of mice at 16 wk on HFD (n = 5 per genotype). E, Body weight at 16 wk on high-fat diet (random fed value) (n =5 per genotype). Error bars indicate se. *, P < 0.05.
Figure 9.

Body composition analysis of Igfbp5−/− mice after 16 wk on a high-fat diet. A, Total fat and lean mass (n =5 per genotype); B, fat and lean mass corrected for total mass; C, isolated liver and epididymal fat pad weights; D, liver and epididymal fat pad weights corrected for body weight (n = 5 per genotype). Error bars represent se. *, P < 0.01.
Discussion
Regulation of glucose homeostasis throughout life requires dynamic fluctuations in pancreatic β-cell mass. The endocrine organ achieves this plasticity through a careful balance of β-cell renewal and β-cell loss. The serine/threonine kinase Akt has been implicated as a critical factor linking diverse nutrient, growth factor, and incretin stimuli with processes related to β-cell mass expansion such as proliferation, hypertrophy, and cell survival. Importantly, the factors downstream of Akt1 that mediate these growth-related effects have not been defined completely. In this study, by assessing the gene expression profile of RIP-myrAkt1 TG islets, we identify potential targets and/or gene networks that might contribute to the effect of Akt1 activation on enhancing β-cell mass and/or function. We demonstrate that Akt1 activation induces gene networks with functions related to cell growth, inhibition of apoptosis, RNA processing, and metabolism. From this analysis, we also identify the IGFBP5 as a gene induced by Akt1 in the β-cell. We find that Igfbp5 deficiency does not prevent the effect of myrAkt1 on expansion of β-cell mass in vivo. However, IGFBP5 is important for regulation of postnatal growth and possibly glucose homeostasis.
Gene expression analysis identified genes with interesting functions that might contribute to the myrAkt1 TG phenotype. We decided to focus our efforts on IGFBP5, based on its induction in the array analyses and its purported effects on growth. IGFBP5 is a member of a family of six IGFBPs that bind to and modulate the activity of IGF-I and IGF-II (19). Both IGF-I and IGF-II are potent stimuli for pancreatic β-cell growth (25). In β-cell tissue culture lines, IGF-I potentiates the mitogenic effect of glucose (26). Overexpression of IGF-I in the β-cell protects them from streptozotocin-induced β-cell death (27). Furthermore, IGF-I mRNA increases rapidly in the area of the regenerating pancreas after 90% pancreatectomy (28). Lastly, Devedjian et al. (29) demonstrated that overexpression of IGF-II in the β-cell leads to a 3-fold increase in β-cell mass. Together, these data indicate that IGF-I can positively influence β-cell growth and development under various conditions. The IGFBPs have been postulated to modulate IGF activity by various mechanisms, including protecting IGFs from proteolytic degradation, targeting IGFs in serum to specific tissues, and regulating local IGF availability to receptors by sequestration in extracellular storage pools (19). There has been limited investigation into the effects of IGFBP on β-cell growth and/or function.
Many studies have demonstrated a role for IGFBP5 in promoting aspects of growth such as differentiation and proliferation in non-β-cell lines and tissues. For example, Yin et al. (30) report that endogenous IGFBP5 is important for maintaining bone cell survival and differentiation in human osteoblastic cells. Igfbp5 has also been identified by serial analysis of gene expression (SAGE) as an abundant transcript during cardiac development (31). The ability of IGFBP5 to regulate growth has been divided into both IGF-dependent and -independent effects. IGFBP5 stimulates proliferation of murine calvarial cells, a primary osteoblast cell line, independently of IGF-I (32). IGFBP5 expression is also up-regulated in various tumor sections, and cell lines such as pancreatic islet adenocarcinomas, thyroid carcinoma, and prostate and breast cancers (33,34,35,36).
IGF-I induces IGFBP5 expression in aortic and vascular smooth muscle cells. This induction is blocked by both LY294002 and rapamycin, suggesting that the PI3K/mTOR signaling pathway is involved in stimulation of IGFBP5 expression (20,21,37). IGFBP5 has also been identified as a gene induced by TG overexpression of myrAkt1 in the heart, a model of cardiac hypertrophy (38,39). Furthermore, acute overexpression of myrAkt1 by adenovirus infection induces IGFBP5 expression in neonatal rat ventricular cardiomyocytes (38). However, the role of IGFBP5 in regulating cardiac hypertrophy in these models has not been assessed.
Igfbp5 mRNA and protein are expressed in islets and ductal cells in the developing and adult mouse pancreas (40). Interestingly, Igfbp5 is recognized as a transcript enriched in the developing pancreatic endoderm (41). However, the role of IGFBP5 in β-cell growth and/or function has not been examined previously. In this study, we present data that IGFBP5 is induced by overexpression of myrAkt1 in the β-cell and during pregnancy, both conditions of augmented β-cell mass. However, even in mice deficient for IGFBP5, myrAkt1 overexpression still augmented β-cell mass. The lack of IGFBP5 does not compromise β-cell function because the Igfbp5−/−;myrAkt TG mice exhibit fasting hyperinsulinemia and improved glucose tolerance. These experiments demonstrate that Igfbp5 is not necessary for the effect of myrAkt1 on expansion of β-cell mass in vivo. However, it is possible that other members of the Igfbp family could have compensated for the absence of Igfbp5, because IGFBPs 1–5 are expressed in the β-cell and surrounding islet cell types (40).
Ubiquitous overexpression of IGFBP5 inhibits growth and development in vivo, suggesting that IGFBP5 might function primarily as an inhibitor of IGF action (42). A requirement for IGFBP5 in normal growth was not detected in several previous studies (24,43). The most likely explanation for the different findings reported herein are the additional crosses of Igfbp5−/− alleles onto a C57BL/6 genetic background. In this study, we have demonstrated that Igfbp5 is required for normal growth only during the postnatal period. Of note, overexpression of IGFBP5 results in decreased prenatal growth and maximal growth retardation that occurred by about 2–3 wk after birth, suggesting that IGFBP5 regulates growth before the onset of GH-stimulated IGF synthesis (42). From this analysis, the authors surmised that the effect of IGFBP5 on growth was either through regulation of IGF-II action or through IGF-independent means. However, our results show that in the absence of IGFBP5, enhanced growth does not develop until after GH-stimulated IGF synthesis has begun. In contrast to the IGFBP5 overexpression study, the Igfbp5−/− phenotype presents the antithesis of Igf1-null mice, which exhibit progressively increased growth retardation after about 3 wk postnatal development (44). Thus, although artificially high levels of IGFBP5 lead to IGF-I-independent inhibition of the early stages of growth, we suggest that IGFBP5 influences growth during postweaning development most likely through IGF-I.
Interestingly, during this study, we observed that IGFBP5-deficient mice exhibit impaired glucose tolerance. Given this, it was surprising that the euglycemic hyperinsulinemic clamp showed no abnormalities. Perhaps this simply reflects the subtlety of the defect; alternatively, the resistance might have been related to a change in a circulating factor, for example glucose, that is held constant in the clamp but not the glucose tolerance test. In any case, the insulin-resistance phenotype was greatly enhanced when the mice were placed on a high-fat diet. In addition to its well-known anabolic effects, IGF-I also influences carbohydrate metabolism. For example, mice with either liver- or skeletal muscle-specific deletion of IGF-I exhibited impaired glucose intolerance and insulin action (45,46). Intraperitoneal injection of IGF-I induces a pronounced decrease in blood glucose level in IR-deficient mice (47). IGFBP proteins influence glucose homeostasis as well. For instance, hyperglycemia and glucose intolerance occur as a result of global overexpression of either IGFBP1 or IGFBP3 (48,49). Our findings demonstrate that in addition to effects on growth, IGFBP5 is also important for normal glucose homeostasis.
Materials and Methods
Materials
DMEM, fetal bovine serum (FBS), penicillin/streptomycin solution, and sodium pyruvate solution were obtained from Invitrogen (Carlsbad, CA). Fugene transfection reagents were from Roche (Mannheim, Germany). The Igfbp5 antibodies were from R&D Systems (Minneapolis, MN) and Santa Cruz Biotechnology (Santa Cruz, CA). The GFP antibody was from BD Biosciences (Palo Alto, CA), and the actin antibody was from Abcam (Cambridge, MA). Horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology. Secondary antibodies for use with the Odyssey infrared imaging system (LICOR Biosciences, Lincoln, NE) were purchased from Rockland Inc. (Gilbertsville, PA). All other antibodies were from Cell Signaling Technology, Inc. (Beverly, MA).
Animals and genotype analysis
The derivation of both the RIP-myrAkt1 TG mice (8) and the Igfbp5 knockout mice (24) have been reported previously. To generate Igfbp5−/−;myrAkt TG mice, Igfbp5−/− mice were mated to wild-type C57BL/6 females (The Jackson Laboratory, Bar Harbor, ME). Igfbp5+/− progeny were then mated inter se to generate Igfbp5−/− mice. The Igfbp5+/− or Igfbp5−/− mice were mated among each other to propagate the Igfbp5 line. Next, either Igfbp5+/− or Igfbp5−/− mice were intercrossed to RIP-myrAkt1 TG mice (>10 generations on C57bL/6 background). The resulting Igfbp5+/−;myrAkt1 TG mice were mated to either Igfbp5+/− or Igfbp5−/− mice to generate mice of the appropriate genotypes. Heterozygous Igfbp5 mice were bred together to generate cohorts for growth-curve analysis. Weights for the growth-curve analysis were obtained on normally fed mice from 1400–1600 h. Genotyping was performed by PCR analysis using genomic DNA isolated from tail snips of newborn mice. All procedures involving mice were conducted in accordance with approved Institutional Animal Care Use Committee protocols.
Islet isolation and Western blot
Pancreatic islets of Langerhans were isolated from 4- to 6-wk-old NTG or RIP-myrAkt1 TG mice by collagenase (Crescent Chemicals, Hauppauge, NY) digestion followed by purification through a Ficoll gradient as described previously (50). Purified islets were handpicked using a dissecting microscope. For Western blot analysis, islets were washed once in ice-cold 1× PBS and then lysed in ice-cold buffer containing 140 mm NaCl, 10 mm Tris (pH 7.4), 200 mm NaF, 10% glycerol, 1% Nonidet P-40, 1× Complete protease inhibitor cocktail (Roche) and 1× phosphatase inhibitor cocktail 1 and 2 (Sigma Chemical Co., St. Louis, MO). The islet lysates were homogenized by freezing-thawing three times in liquid nitrogen followed by a 37 C water bath. Insoluble material was pelleted by centrifugation at 10,000 rpm at 4 C for 10 min. The supernatant was collected and protein was measured by bicinchoninic acid assay (Pierce, Rockland, IL).
RNA isolation and real-time PCR analysis
Total RNA from islets was isolated in Trizol (Invitrogen) according to the manufacturer’s instructions. Total RNA from MIN6 cells or MEFs was isolated using the RNeasy Plus kit (Ambion, Austin, TX). RNA was reverse transcribed using random decamer primers, Retroscript II reverse transcriptase, and the accompanying reagents (Ambion) following the manufacturer’s directions. PCR mixes were assembled using the Brilliant SYBR Green QPCR Master Mix (Stratagene, La Jolla, CA). Reactions were performed using the SYBR Green (with dissociation curve) program on the Mx3000 quantitative PCR system (Stratagene). All reactions were performed in triplicate with the reference dye normalization, and median cycle threshold values were used for analysis. The primer sequences were as follows: Igfbp5 forward, 5′-GCAAAGCGTTGGAATAGAGG-3′; Igfbp5 reverse, 5′-AGATTCAGCCTTGGTTGTGG-3′; Gapdh forward, 5′-GGCAAAGTGGAGATTGTTGC-3′; Gapdh reverse, 5′-TGACAAGCTTCCCATTCTCG-3′; TATA-binding protein (TBP) forward, 5′-CCCCTTGTACCCTTCACCAAT-3′; TBP reverse, 5′-GAAGCTCGGTACAATTCCAG-3′.
Microarray expression profiling, data analysis, and gene ontology analysis
Total RNA (100 ng) for each sample (n = 3 per genotype) was amplified using the MessageAmp aRNA kit (Ambion). The concentration and integrity of each sample was determined both before and after amplification using an Agilent Bioanalyzer Lab-ON-A-Chip Nano 6000 chip. Microarray expression profiling, data, and gene ontology analysis were described previously (51). Fluorescently labeled (CyDye; Amersham Pharmacia Biotech Ltd., Piscataway, NJ) cDNA was hybridized to the PancChip version 4.0. This chip contains a set of 13,824 cDNAs including 3450 IMAGE clones chosen for their expression in pancreas and liver, 300 samples from the University of Pennsylvania’s Functional Genomics Core in-house collection, and 7226 nonredundant clones collected from pancreas libraries and control genes. The data were normalized using the print-tip lowess method using the SMA (Statistical Microarray Analysis) package in R (52). For statistical analysis, genes were called differentially expressed using the latest version of Significance Analysis of Microarrays (SAM) with a false discovery rate of 20% (53).
Cell culture and Western blot analysis
MIN6 cells (kindly provided by Prof. Jun-Ichi Miyazaki, Osaka University, Osaka, Japan) were used between passages 28 and 40 at approximately 80% confluence. MIN6 cells were grown in DMEM containing 25 mm glucose supplemented with 15% FBS, 1 mm sodium pyruvate, 50 μm β-mercaptoethanol, 100 μg/ml streptomycin, and 100 U/ml penicillin, equilibrated with 5% CO2, 95% air at 37 C. MIN6 myrAkt1 stable cells were generated by retroviral infection followed by antibiotic selection with neomycin. The myrAkt1 construct was generated by inserting cDNA for human Akt1 (containing a deletion of the pleckstrin homology domain (amino acids 4–129) and addition of a 14-amino-acid myristoylation sequence (8) into the retroviral vector pLNCX1. The generation of retrovirus was as described previously with slight modifications (54). Briefly, ecotropic BOSC cells (a gift from Warren S. Pear, University of Pennsylvania, Philadelphia, PA) were transiently transfected with pVSV G and pCgP pantropic retroviral packaging constructs and retroviral vector (pLNCX1-HAmyrAkt1). Cell-free viral supernatants were harvested at both 24 and 48 h and used to infect MIN6 cells (passage 29). Primary wild-type MEFs were established from embryonic d-13.5 embryos as described previously (55). MEFs were cultured in DMEM containing 25 mm glucose supplemented with 10% FBS and 100 μg/ml streptomycin, 100 U/ml penicillin. For western blot analysis, cells were washed twice in ice-cold 1× PBS and then lysed in ice-cold buffer (see islet isolations above for recipe). 5 μl of lysate was removed for analysis of total protein by BCA assay (Pierce, Rockland, IL).
Adenovirus infection
The myrAkt1 adenoviral constructs have been described previously (56). The CMV-eGFP adenovirus was obtained from the University of Pennsylvania Viral Vector Core. For adenovirus infection of MEFs, the infection was optimized by preincubation of the virus in DMEM containing 0.5% BSA (Sigma) and 0.5 μg/ml poly-l-lysine (Sigma) for 1.5 h at room temperature. Wild-type primary MEFs were washed once with unsupplemented DMEM before addition of the adenovirus mixture. MEFs were incubated with adenovirus overnight and then washed twice in DMEM plus 10% FBS and subcultured an additional 48 h before harvesting for preparation of protein and total RNA.
Analytical techniques
Blood glucose levels were measured with a Bayer Glucometer Elite. Insulin levels on serum obtained from tail bleeds were determined by ultrasensitive ELISA (Crystal Chem, Downers Grove, IL). Intraperitoneal glucose tolerance tests were performed on 14- to 16-h fasted mice at a glucose dose of 1 g/kg body weight. Insulin tolerance tests were performed on 5- to 6-h fasted mice. Animals were injected with 1 U/kg body weight with Humulin-R (Eli Lilly, Indianapolis, IN). For the high-fat diet study, male mice were placed on a diet containing 58% of energy in kcal as fat from coconut oil and high sucrose (catalog number D12331; Research Diets, Inc., New Brunswick, NJ) at 5 wk of age. The hyperinsulinemic euglycemic clamp studies, DEXA, and energy expenditure analysis were performed by the Institute for Diabetes, Obesity, and Metabolism Mouse Phenotyping and Physiology Core facility.
Histological analyses
For all histological studies, pancreata were dissected and fixed in 4% paraformaldehyde for 24 h and then laid flat for paraffin embedding and sectioning by the University of Pennsylvania Morphology Core. For immunofluorescence microscopy, after deparaffinization, all samples were exposed to boiling 10 mm citric acid buffer (pH 6.0) for 6 min. Slides were then blocked in protein blocking solution reagent 1481 for 10 min at room temperature. Primary antibodies were diluted in PBS containing 0.1% BSA and 0.2% Triton X-100 and incubated overnight at 4 C. The appropriate secondary antisera were also diluted in PBS containing 0.1% BSA and 0.2% Triton X-100 and added to slides for 2 h at room temperature. The following antisera were used: insulin (Linco, St. Charles, MO), glucagon (Biodesign, Saco, ME), HA (Santa Cruz Biotechnology), β-catenin (BD Biosciences). The secondary antibodies Cy2-conjugated donkey anti-guinea pig IgG, Cy3-conjugated donkey antirabbit IgG, and Cy3-conjugated donkey antimouse IgG were from Jackson ImmunoResearch, West Grove, PA.
Statistical analysis
Data are expressed as means ± sd or ± sem as indicated in the figure legends. Statistically significant differences between groups were analyzed using ANOVA (JMP Start Statistics). P < 0.05 was considered statistically significant.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health Grant P01 DK49210 to M.J.B. and the cores of the Penn Diabetes and Endocrinology Research Center, P30 DK019525, and National Institutes of Health Grants T32-GM07229.
Disclosure Summary: The authors of this manuscript have nothing to declare.
First Published Online November 6, 2009
Abbreviations: ad-GFP, Adenovirus encoding green fluorescent protein; CDK4, cyclin-dependent kinase 4; DEXA, dual-energy x-ray absorptiometry; GSK3, glycogen synthase kinase; HA, hemagglutinin; IGFBP5, IGF-binding protein 5; IR, insulin receptor; IRS1, IR substrate 1; MEF, mouse embryonic fibroblast; NTG, nontransgenic; TBP, TATA-binding protein; TG, transgenic.
References
- Lingohr MK, Buettner R, Rhodes CJ 2002 Pancreatic β-cell growth and survival: a role in obesity-linked type 2 diabetes? Trends Mol Med 8:375–384 [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR 1999 Tissue-specific knockout of the insulin receptor in pancreatic β-cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–339 [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Holzenberger M, Shih DQ, Ozcan U, Stoffel M, Magnuson MA, Kahn CR 2002 β-Cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter β-cell mass. Nat Genet 31:111–115 [DOI] [PubMed] [Google Scholar]
- Ueki K, Okada T, Hu J, Liew CW, Assmann A, Dahlgren GM, Peters JL, Shackman JG, Zhang M, Artner I, Satin LS, Stein R, Holzenberger M, Kennedy RT, Kahn CR, Kulkarni RN 2006 Total insulin and IGF-I resistance in pancreatic β-cells causes overt diabetes. Nat Genet 38:583–588 [DOI] [PubMed] [Google Scholar]
- Prentki M, Nolan CJ 2006 Islet β-cell failure in type 2 diabetes. J Clin Invest 116:1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR 2006 Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96 [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA 2001 Islet β-cell expression of constitutively active Akt1/PKBα induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 108:1631–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, Polonsky KS, Naji A, Birnbaum MJ 2001 Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat Med 7:1133–1137 [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC 2007 AKT/PKB signaling: navigating downstream. Cell 129:1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM 2002 PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 8:1153–1160 [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC 2001 Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 3:245–252 [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM 2000 AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404:782–787 [DOI] [PubMed] [Google Scholar]
- Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF 2005 Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Mol Cell Biol 25:3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M 1999 Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nat Genet 22:44–52 [DOI] [PubMed] [Google Scholar]
- Fatrai S, Elghazi L, Balcazar N, Cras-Méneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E 2006 Akt induces β-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes 55:318–325 [DOI] [PubMed] [Google Scholar]
- Elghazi L, Balcazar N, Bernal-Mizrachi E 2006 Emerging role of protein kinase B/Akt signaling in pancreatic β-cell mass and function. Int J Biochem Cell Biol 38:157–163 [DOI] [PubMed] [Google Scholar]
- Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs 3rd WH, Wright CV, White MF, Arden KC, Accili D 2002 The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β-cell growth. J Clin Invest 110:1839–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson RL, Brelje TC 1997 Adaptation of islets of Langerhans to pregnancy: β-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 29:301–307 [DOI] [PubMed] [Google Scholar]
- Firth SM, Baxter RC 2002 Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev 23:824–854 [DOI] [PubMed] [Google Scholar]
- Duan C, Hawes SB, Prevette T, Clemmons DR 1996 Insulin-like growth factor-I (IGF-I) regulates IGF-binding protein-5 synthesis through transcriptional activation of the gene in aortic smooth muscle cells. J Biol Chem 271:4280–4288 [DOI] [PubMed] [Google Scholar]
- Duan C, Liimatta MB, Bottum OL 1999 Insulin-like growth factor (IGF)-I regulates IGF-binding protein-5 gene expression through the phosphatidylinositol 3-kinase, protein kinase B/Akt, and p70 S6 kinase signaling pathway. J Biol Chem 274:37147–37153 [DOI] [PubMed] [Google Scholar]
- Duan C, Xu Q 2005 Roles of insulin-like growth factor (IGF) binding proteins in regulating IGF actions. Gen Comp Endocrinol 142:44–52 [DOI] [PubMed] [Google Scholar]
- Schneider MR, Wolf E, Hoeflich A, Lahm H 2002 IGF-binding protein-5: flexible player in the IGF system and effector on its own. J Endocrinol 172:423–440 [DOI] [PubMed] [Google Scholar]
- Ning Y, Hoang B, Schuller AG, Cominski TP, Hsu MS, Wood TL, Pintar JE 2007 Delayed mammary gland involution in mice with mutation of the insulin-like growth factor binding protein 5 gene. Endocrinology 148:2138–2147 [DOI] [PubMed] [Google Scholar]
- Swenne I 1992 Pancreatic β-cell growth and diabetes mellitus. Diabetologia 35:193–201 [DOI] [PubMed] [Google Scholar]
- Hügl SR, White MF, Rhodes CJ 1998 Insulin-like growth factor I (IGF-I)-stimulated pancreatic β-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem 273:17771–17779 [DOI] [PubMed] [Google Scholar]
- George M, Ayuso E, Casellas A, Costa C, Devedjian JC, Bosch F 2002 β-Cell expression of IGF-I leads to recovery from type 1 diabetes. J Clin Invest 109:1153–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FE, Rosen KM, Villa-Komaroff L, Weir GC, Bonner-Weir S 1991 Enhanced insulin-like growth factor I gene expression in regenerating rat pancreas. Proc Natl Acad Sci USA 88:6152–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devedjian JC, George M, Casellas A, Pujol A, Visa J, Pelegrín M, Gros L, Bosch F 2000 Transgenic mice overexpressing insulin-like growth factor-II in β-cells develop type 2 diabetes. J Clin Invest 105:731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Xu Q, Duan C 2004 Paradoxical actions of endogenous and exogenous insulin-like growth factor-binding protein-5 revealed by RNA interference analysis. J Biol Chem 279:32660–32666 [DOI] [PubMed] [Google Scholar]
- Anisimov SV, Tarasov KV, Riordon D, Wobus AM, Boheler KR 2002 SAGE identification of differentiation responsive genes in P19 embryonic cells induced to form cardiomyocytes in vitro. Mech Dev 117:25–74 [DOI] [PubMed] [Google Scholar]
- Miyakoshi N, Richman C, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S 2001 Evidence that IGF-binding protein-5 functions as a growth factor. J Clin Invest 107:73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SK, Dennis RA, Barone GW, Lamps LW, Haun RS 2006 Differential expression of insulin-like growth factor binding protein-5 in pancreatic adenocarcinomas: identification using DNA microarray. Mol Carcinog 45:814–827 [DOI] [PubMed] [Google Scholar]
- Miyake H, Nelson C, Rennie PS, Gleave ME 2000 Overexpression of insulin-like growth factor binding protein-5 helps accelerate progression to androgen-independence in the human prostate LNCaP tumor model through activation of phosphatidylinositol 3′-kinase pathway. Endocrinology 141:2257–2265 [DOI] [PubMed] [Google Scholar]
- Sheikh MS, Shao ZM, Hussain A, Clemmons DR, Chen JC, Roberts Jr CT, LeRoith D, Fontana JA 1993 Regulation of insulin-like growth factor-binding-protein-1, 2, 3, 4, 5, and 6: synthesis, secretion, and gene expression in estrogen receptor-negative human breast carcinoma cells. J Cell Physiol 155:556–567 [DOI] [PubMed] [Google Scholar]
- Stolf BS, Carvalho AF, Martins WK, Runza FB, Brun M, Hirata Jr R, Jordão Neves E, Soares FA, Postigo-Dias J, Kowalski LP, Reis LF 2003 Differential expression of IGFBP-5 and two human ESTs in thyroid glands with goiter, adenoma and papillary or follicular carcinomas. Cancer Lett 191:193–202 [DOI] [PubMed] [Google Scholar]
- Kiepe D, Ciarmatori S, Hoeflich A, Wolf E, Tönshoff B 2005 Insulin-like growth factor (IGF)-I stimulates cell proliferation and induces IGF binding protein (IGFBP)-3 and IGFBP-5 gene expression in cultured growth plate chondrocytes via distinct signaling pathways. Endocrinology 146:3096–3104 [DOI] [PubMed] [Google Scholar]
- Cook SA, Matsui T, Li L, Rosenzweig A 2002 Transcriptional effects of chronic Akt activation in the heart. J Biol Chem 277:22528–22533 [DOI] [PubMed] [Google Scholar]
- Schiekofer S, Shiojima I, Sato K, Galasso G, Oshima Y, Walsh K 2006 Microarray analysis of Akt1 activation in transgenic mouse hearts reveals transcript expression profiles associated with compensatory hypertrophy and failure. Physiol Genomics 27:156–170 [DOI] [PubMed] [Google Scholar]
- Hill DJ, Hogg J, Petrik J, Arany E, Han VK 1999 Cellular distribution and ontogeny of insulin-like growth factors (IGFs) and IGF binding protein messenger RNAs and peptides in developing rat pancreas. J Endocrinol 160:305–317 [DOI] [PubMed] [Google Scholar]
- Gu G, Wells JM, Dombkowski D, Preffer F, Aronow B, Melton DA 2004 Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development 131:165–179 [DOI] [PubMed] [Google Scholar]
- Salih DA, Tripathi G, Holding C, Szestak TA, Gonzalez MI, Carter EJ, Cobb LJ, Eisemann JE, Pell JM 2004 Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. Proc Natl Acad Sci USA 101:4314–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Y, Schuller AG, Bradshaw S, Rotwein P, Ludwig T, Frystyk J, Pintar JE 2006 Diminished growth and enhanced glucose metabolism in triple knockout mice containing mutations of insulin-like growth factor binding protein-3, -4, and -5. Mol Endocrinol 20:2173–2186 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhou J, Powell-Braxton L, Bondy C 1999 Effects of Igf1 gene deletion on postnatal growth patterns. Endocrinology 140:3391–3394 [DOI] [PubMed] [Google Scholar]
- Fernández AM, Kim JK, Yakar S, Dupont J, Hernandez-Sanchez C, Castle AL, Filmore J, Shulman GI, Le Roith D 2001 Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev 15:1926–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjögren K, Wallenius K, Liu JL, Bohlooly-Y M, Pacini G, Svensson L, Törnell J, Isaksson OG, Ahrén B, Jansson JO, Ohlsson C 2001 Liver-derived IGF-I is of importance for normal carbohydrate and lipid metabolism. Diabetes 50:1539–1545 [DOI] [PubMed] [Google Scholar]
- Di Cola G, Cool MH, Accili D 1997 Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J Clin Invest 99:2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar K, Krsek M, Dheen ST, Murphy LJ 1996 Impaired glucose homeostasis in insulin-like growth factor binding protein-1 transgenic mice. J Clin Invest 98:1818–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silha JV, Gui Y, Murphy LJ 2002 Impaired glucose homeostasis in insulin-like growth factor-binding protein-3-transgenic mice. Am J Physiol Endocrinol Metab 283:E937–E945 [DOI] [PubMed] [Google Scholar]
- Scharp DW, Kemp CB, Knight MJ, Ballinger WF, Lacy PE 1973 The use of ficoll in the preparation of viable islets of langerhans from the rat pancreas. Transplantation 16:686–689 [DOI] [PubMed] [Google Scholar]
- Gupta RK, Gao N, Gorski RK, White P, Hardy OT, Rafiq K, Brestelli JE, Chen G, Stoeckert Jr CJ, Kaestner KH 2007 Expansion of adult β-cell mass in response to increased metabolic demand is dependent on HNF-4α. Genes Dev 21:756–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP 2002 Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G 2001 Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98:5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JT, Birnbaum MJ 2001 ADP-ribosylation factor 6 delineates separate pathways used by endothelin 1 and insulin for stimulating glucose uptake in 3T3-L1 adipocytes. Mol Cell Biol 21:5276–5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SS, Cho H, Mu J, Birnbaum MJ 2003 Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem 278:49530–49536 [DOI] [PubMed] [Google Scholar]
- Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K 2000 Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101:660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.