

Abstract
Ultraviolet (UV) light is a primary environmental risk factor for melanoma, a deadly form of skin cancer derived from the pigmented cells called melanocytes. UVB irradiation causes DNA damage, mainly in the form of pyrimidine dimers (cis-syn cyclobutane pyrimidine dimers and pyrimidine (6-4) pyrimidone photoproducts), and organisms have developed complex multiprotein repair processes to cope with the DNA damage. Zebrafish is becoming an important model system to study the effects of UV light in animals, in part because the embryos are easily treated with UV irradiation, and the DNA damage repair pathways appear to be conserved in zebrafish and mammals. We are interested in exploring the effects of UV irradiation in young adult zebrafish, so that we can apply them to the study of gene–environment interactions in models of skin cancer. Using the Xiphophorus UV melanoma model as a starting point, we have developed a UV irradiation treatment chamber, and established UV treatment conditions at different ages of development. By translating the Xiphophorus UV treatment methodology to the zebrafish system, we show that the adult zebrafish skin is competent for nucleotide excision DNA damage repair, and that like in mammalian cells, UV treatment promotes phosphorylation of H2AX and a p53-dependent response. These studies provide the groundwork for exploring the role of UV light in melanoma development in zebrafish.
Introduction
The incidence of cutaneous melanoma is rapidly increasing worldwide, most commonly among the Caucasian population, with the highest incidence rates in Australia and the United States.1,2 Geographical and epidemiological studies have established a strong correlation between solar ultraviolet (UV) radiation, skin color, and incidence of melanoma.3 For example, among the Caucasian population in the Queensland region of Australia, melanoma incidence is the highest in the world, with 82.1 male and 55.3 female patients per 100,000 residents.1 In the United Kingdom, high rates of melanoma in Scotland are seen in men and women, with the trunk as the commonest primary site in men, and the lower limb the primary site in women.4 In young English women, overseas holiday sun exposure is responsible for increased nevus count, and an increased risk of melanoma.5 With changing lifestyles, melanoma has also increased in countries where this disease was traditionally rare, such as in the City of Beijing, China, where the incidence rate of malignant melanoma has increased from 0.2 per 100,000 inhabitants in the year 2000, to 1 per 100,000 inhabitants in 2004.6
Melanin, the pigment produced in the melanosome of melanocytes and transferred to the keratinocytes, plays a critical role in protecting melanocytes from transformation. Functioning as a natural sunscreen, melanin protects the melanocytes and surrounding keratinocytes from UV light–induced DNA damage. In people, melanocytes can produce two types of melanins, red/yellow pheomelanin and brown/black eumelanin, and the levels and types of melanin determine the range of skin colors in the human population.7 Genetic mutations in the enzymes that control the biogenesis of melanin can affect the quantity and type of pigmentation in mammals and fish. For example, in people, mutations in pigmentation enzymes, such as tyrosinase, lead to reduced melanin synthesis and albinism, and can also confer an increased risk for melanoma.8 In humans and zebrafish, mutations in SLC24A5 cause a reduction in the quantity of melanin in melanosomes, resulting in a golden (pale) phenotype in zebrafish, and contributing to lighter skin pigmentation some human populations.9 MC1R functions as a membrane receptor of melanocytes for the α-melanocyte-stimulating hormone, a primary regulator of eumelanin synthesis. In people, there are more than 70 allelic variants in the MC1R gene, and its extensive polymorphism indicates that it is a major contributor to the diversity of human pigmentation. Some MC1R variants contribute to the production of phenomelanin, with reduced eumelanin production, leading to red hair and increased skin burning, rather than tanning, after sunlight exposure.10
UV radiation can be divided into three wavelength ranges according to their photochemistry: UVA (320–400 nm), UVB (290–320 nm), and UVC (240–290 nm). The stratospheric ozone absorbs much of the UVC radiation before it reaches the earth's surface. UV light can cause damage by direct absorption by DNA and proteins, and by the indirect generation of reactive oxygen species.11,12 The most common DNA lesions induced by UVB radiation are the cis–syn cyclobutane pyrimidine dimers (CPDs) and the pyrimidine (6-4) pyrimidone photoproducts [(6-4)PDs]. UVA is absorbed about 10-fold less efficiently into DNA; however, given the increased abundance in sunlight and penetration in the skin, coupled with the generation of UVA induced reactive oxygen species, UVA is an important mutagen in human skin.13 Organisms have evolved effective photoprotective and DNA repair mechanisms to remove these lesions: the CPD and (6-4)PDs are removed from DNA by either photoenzymatic repair (PER) or nucleotide excision repair (NER). Absent in humans, the PER is a light-dependent process that reverses the lesions by an enzyme-catalyzed reaction using energy absorbed from the visible light. For example, PER in Xiphophorus skin can efficiently remove most UV-induced CPDs within 15 min, and (6-4)PDs within 60 min.14 By contrast, in Xiphophorus, NER is a less efficient, light-independent process involving the removal of damaged DNA and the replacement by DNA polymerases.15
Our understanding of DNA damage repair and the role of UV in melanomagenesis has been enhanced by animal models, including genetically engineered mice, the Xiphophorus hybrid fish, the South American opossum, and human skin xenografts. One of the oldest is the Xiphophorus hybrid model (platyfishes and swordtails) that has been used to study spontaneous melanoma for over 80 years.16 Small, internally fertilizing, and live bearing, they are native to Central America and can be adapted to the laboratory environment.17 By treating young fry, Setlow et al. developed the first Xiphophorus hybrid UV-induced melanoma model,18 associated by genetic linkage with a CDKN2-like gene.19 Using a unique radioimmunoassay (RIA) to quantify photoproducts,20 Mitchell et al. have shown that in Xiphophorus hybrids that develop melanoma after UVB irradiation there is a decreased NER capacity of (6-4)PD photoproduts,14 giving insight into the mechanism of UV-induced damage.
Despite the utility of Xiphophorus as a model for cancer biology, the zebrafish and medaka model systems offer additional advantages in understanding the genetics of melanoma development.21 Zebrafish and medaka fishes are born ex utero, and their embryos can be easily studied for melanocyte development, and manipulated by microinjection and/or chemical treatment.22–26 In contrast, Xiphophorus are internally fertilized, and live bearing, making embryo manipulation impractical. The zebrafish and medaka genomic resources, coupled with the range of genetic tools and ease of handling, make these model systems a tractable and practical alternative to other fish systems.27,28 Medaka and zebrafish develop cancer, and the zebrafish has especially emerged as an excellent animal model for cancer research.21 Zebrafish develop a wide tumor spectrum that can resemble human malignancies both by histopathology and at the molecular level. Zebrafish cancers can be induced by chemical mutagens (such as 7,12-dimethylbenz(a)anthracene [DMBA]), specific genetic mutations, or oncogene transgenesis.21 For example, expression of the BRAFV600E mutation, the most common mutation in human nevi and melanoma, is sufficient to induce ectopic nevi in zebrafish, and can collaborate with p53 mutations to promote melanoma.29,30 As UV light is the primary environmental risk factor for melanoma, and zebrafish can develop melanoma, and UV light can promote melanoma in Xiphophorus, we reasoned that we could develop a UV light-induced model of melanoma in zebrafish. This model would be important for future studies to identify melanoma susceptibility loci, the role of pigment in melanoma protection and development, and UV light DNA damage mutations in cancer genes. The first step toward this aim is to establish a UV light treatment methodology, and translate the UV light protocols from the Xiphophorus to the zebrafish system. In this study, we test the suitability and parameters of zebrafish for photocarcinogenesis, including the dose response, the pathologic consequence of zebrafish to UV irradiation, and the DNA repair capacity.
Materials and Methods
Zebrafish husbandry
Adult and zebrafish embryos were raised and maintained at 28.5°C. Embryos were staged according to Kimmel et al.31 The p53 mutant fish carry a point mutation (M214K) in the DNA-binding domain of p53 protein.32
UVB irradiation
An irradiation chamber similar to the one used for the Xiphophorus was built for this study. The UVB source is four UVB lamps emitting 311–312 nm light (TL 20W/01; Philips, Eindhoven, The Netherlands). UVB was quantified with a UVB detector (SEL005/WBS320/TD; International Light, Peabody, MA) coupled to a radiometer (ILT1400A; International Light). Fish were housed in a UVB transparent irradiation box filled with fish water for UVB irradiation, with a UVB dose rate from both sides at 12 J/m2/s, and the fish swam freely during the course of exposure. Twenty-four hours before irradiation, fish were moved to a dark room to prevent exposure to visible light. After UVB exposure, fish were sacrificed immediately, or returned to fish tanks and kept in the dark for the first 24 h to avoid light-dependent PER.
Acridine orange assay
After irradiation, 24 hpf embryos were kept in the dark for 6 h. Embryos were then incubated with 5 μg/mL of acridine orange (Sigma, St. Louis, MO) in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4) at 28.5°C for 30 min, and washed with E3 medium twice. Embryos were observed under a fluorescent stereo-microscope (Leica macroFluo™, Wetzlar, Germany), and photographs were taken using a monochrome camera (Qimaging, Surrey, BC, Canada) for fluorescent imaging.
DNA extraction
Fish were sacrificed, skin removed, and frozen in liquid nitrogen before isolating the genomic DNA using phenol/chloroform extraction adopted for the use in zebrafish.33 Briefly, 400 μL of DNA extraction buffer (10 mM Tris pH 8, 100 mM ethylenediaminetetraacetic acid pH 8, and 0.5% sodium dodecyl sulfate) was added to each sample, and the skin tissues were homogenized using plastic pestles, and treated with 10 μg/mL RNase A at 37°C for 1 h. Then, 10 μL of proteinase K (10 mg/mL) was added to each sample, and the lysate was incubated at 37°C overnight. Samples were sequentially extracted with equal volumes of phenol, phenol/chloroform (1:1), and chloroform/isoamyl alcohol (24:1), and DNA was precipitated by adding 40 μL of 3 M sodium acetate (pH 5.2) and 800 μL of 100% ethanol. The mixture stood at room temperature for 30 min and was centrifuged at 12,000 rpm for 10 min, rinsed with 70% ethanol, air-dried, and dissolved in 200 μL of 100 mM Tris-HCl, pH 7.5 and 10 mM EDTA, pH 8.0 (TE) buffer. The DNA was quantified using a spectrum meter (NanoDrop–Thermo Fisher Scientific, Wilmington, DE). If the value of A260/280 is less than 1.8 or that of A260/230 is less than 2.0, DNA samples were further purified.
Reverse transcription polymerase chain reaction
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) following the manufacture's recommendations. First-strand cDNA was synthesized from 2 μg of total RNA using a Superscript first-strand synthesis kit (Invitrogen). cDNA was then amplified by polymerase chain reaction (PCR) using gene-specific primers. PCR was carried out according to the following protocol: initial denaturation at 95°C for 2 min, followed by variable number of cycles of denaturation at 95°C for 15 s, annealing at variable temperature for 30 s, extension at 72°C for 60 s, and a final extension period of 5 min at 72°C. PCR products were observed on 0.8% agarose gel. Primers and gene-specific PCR conditions used in this study are shown in Table 1.
Table 1.
Polymerase Chain Reaction Primers and Conditions
| |
|
|
PCR conditions |
|
|
|---|---|---|---|---|---|
| Gene | Mechanism | NCBI accession number | Annealing temperature (°C) | Cycles | Primers (5′–3′) |
| β-Actin | Cytoskeleton | AF057040 | 58 | 28 | F: TGCCATGTATGTGGCCATCCA |
| R: ACCTCCAGACAGCACTGTGT | |||||
| Caspase3a | Apoptosis | NM131877 | 57 | 31 | F: TGTGTTGCTCAGTCACGGC |
| R: GGCATGTTGGAGGTGGACTC | |||||
| DDB2 | DNA repair (NER) | NM001083061 | 57 | 38 | F: AAAAGACCGAATGAAGAAACTCC |
| R: TAGTAAGCAACTTGGTGCTGTCA | |||||
| Ku80 | DNA repair (NHEJ) | AY877316 | 57 | 31 | F: TGGAGGAGATTGAGAGAGAACTG |
| R: GTTTCATCATCATCGTTCAGACA | |||||
PCR, polymerase chain reaction; DDB, damaged DNA binding proteins; NER, nucleotide excision repair; NHEJ, nonhomologous end joining; NCBI, National Center for Biotechnology Information.
Radioimmunoassay
RIA was used to quantify UVB photoproducts and was carried out as described by Mitchell.34 Briefly, 2–5 μg DNA of heat-denatured sample DNA was incubated with 5–10 pg of poly(2′-deoxyadenosine)-poly(2′-deoxythymidine) (labeled to ∼5 × 108 cpm/μg by nick translation with 32P-deoxythymidine triphosphate) in a total volume of 1 mL 10 mM Tris (pH 8.0), 1 mM ethylenediaminetetraacetic acid, 150 mM NaCl, and 0.2% gelatin. Antiserum was added at a dilution that yielded optimal binding to labeled ligand. After 3 h incubation at 37°C, the immune pellet was precipitated for 2 days at 4°C with goat anti-rabbit immunoglobulin (Calbiochem, San Diego, CA) and normal rabbit serum (UTMDACC; Science Park/Veterinary Division, Bastrop, TX). The immune complex was centrifuged at 3700 rpm for 45 min at 10°C and the supernatant discarded. The pellet was dissolved in 100 μL tissue solubilizer (NCS; Amersham, Piscataway, NJ), mixed with 6 mL ScintiSafe (Fisher, Pittsburgh, PA) containing 0.1% glacial acetic acid, and quantified using LSC (Packard Instruments, Meriden, CT).
Immunochemistry
After exposure to 2.16 kJ/m2 UVB light (3 min), fish were kept in the dark, sacrificed 12 h after treatment, and fixed overnight in 4% paraformaldehyde in phosphate-buffered saline (PBS) at 4°C, followed by washing in PBS and preservation in 70% ethanol. Samples were then dehydrated in graded alcohol solutions, cleared in xylene, and embedded in paraffin. Seven-micrometer sections were cut and processed for antibody staining as described.29 Sections were immersed twice in xylene for 5 min to dewax, and then were rehydrated through graded alcohol solutions (100%, 90%, 70%, 50%, and 30%; 3 min each) and stopped in water. Slides were boiled in 0.01 M citrate buffer (2 mM citrate acid and 8 mM Sodium citrate, pH 6.0) in a pressure cooker for 5 min to retrieve the antigen. To quench the endogenous peroxide, slides were then immersed in 3% H2O2 for 10 min. After washing, sections were incubated with DAKO block solution (Dako, Glostrup, Denmark) for 30 min at room temperature, and then incubated overnight at 4°C with a rabbit anti-zebrafish anti-phospho-H2AX primary antibody diluted 1:1000 in DAKO antibody dilutent. The phospho H2AX antibody was made against a phospho-peptide corresponding to residues 131–142 of zebrafish Histone H2AX:SGKKGSSQ[S]QEY, where the phospho-serine is in brackets. Antiserum was subtracted against the unphosphorylated peptide and then affinity purified against the phospho-peptide. The immunization was done at Open Biosystems. Horseradish peroxidase–conjugated secondary antibody and reagents for detection of signals are included in the DAKO REALTMEnVisionTM detection system, and detection was performed according to the manual.
Statistics
The survival rate was corrected after applying the modified Schneider–Orelli's formula35 if death was found in untreated control group. The formula is % S = 1 − (T − C/100 − C), where % S = percent survival, T = percent mortality in the treatment, and C = percent mortality in the control. The mean and standard deviation of fish survival from replicates were calculated using Excel (Microsoft, Redmond, WA). Regression analysis was used to estimate the LD50.
Results
Construction of a UVB irradiation chamber
To apply the protocols for UVB induction of melanoma in Xiphophorus to zebrafish, we constructed a UVB irradiation chamber adapted from Mitchell et al.36 with minor alterations (Fig. 1). Banks of two Philips TL20W/01 UVB lamps were installed on each side. To keep the temperature constant in the chamber over the course of experiments, a fan was installed at one end of the chamber, and a vent at the other end. Two rails between the two banks of lamps held a UVB transparent irradiation box, which can house up to 5 adults, 10 juveniles, and 50 larvae. Two light controls were installed to adjust the emission of UVB light from each bank of lamps, with a UVB light emission range of 5.1–15.4 J/m2/s. This feature is helpful when an adjustable UV light is needed, for example, in the case of a low irradiation rate and long duration. The lamps and fan are controlled by two individual switches, as well as one main switch, and for safety reasons, the lamps can only be turned on when the lid is closed.
FIG. 1.
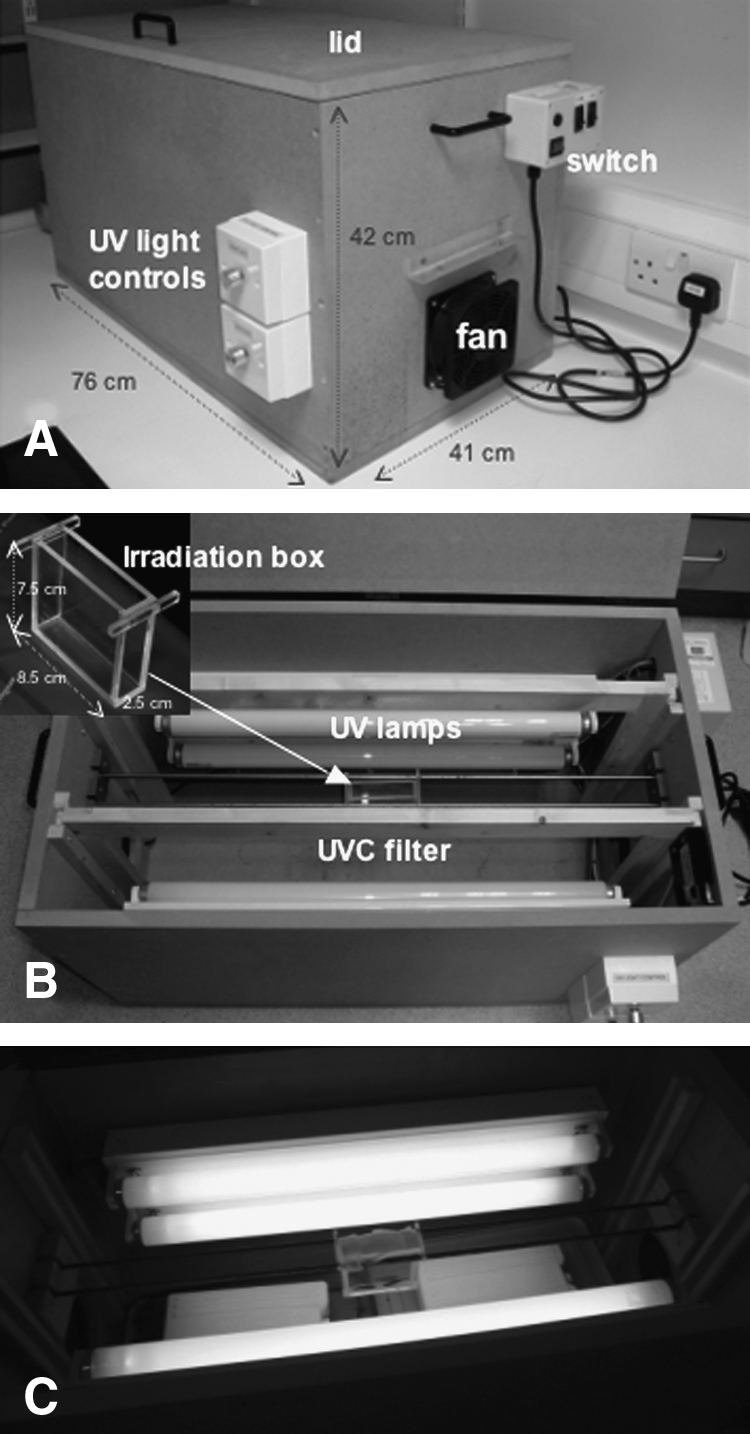
The UV irradiation chamber. (A) The closed chamber showing switches, fan, and UV light control. The overall outer dimensions of the chamber are 76.0 cm long, 41.0 cm wide, and 42.0 cm tall. (B) The opened chamber showing two banks of UVB lamps, irradiation box (inset), and rails holding the irradiation box. The irradiation box is 8.5 cm long, 2.5 cm wide, and 7.5 cm tall. A removable UVC filter is also included. Any additional details required regarding the construction of the chamber will be readily given by the authors upon request. (C) Adult zebrafish are being irradiated in the chamber. UV, ultraviolet.
UVB-induced p53-dependent cell death and DNA repair gene expression in zebrafish embryos
After UV DNA damage, the tumor suppressor p53 plays a critical role in halting the cell cycle to allow for repair of the damage, or initiating cell death.37 Loss of p53 in zebrafish by morpholino oligonucleotide knockdown,38 or the p53M214K mutant line32 results in reduced levels of DNA-damage-induced apoptosis in response to gamma-irradiation, UV treatment, or chemical DNA damage agents. The sensitivity of zebrafish embryos to UVB treatment can vary depending on specific embryonic stages.39 To test the cellular response of zebrafish embryos to UVB irradiation in our UV chamber, we exposed 24 hpf embryos to a sublethal dose of UVB (1.08 kJ/m2), kept the fish in the dark to prevent PER, and stained with acridine orange dye to observe cell death in living embryos at 6 h after treatment (Fig. 2A–D). Wild-type zebrafish embryos revealed cell death 6 h after UVB treatment. In contrast, the p53 mutant zebrafish did not show cell death, and appeared similar to the untreated control embryos. This sublethal dose of UVB irradiation did not cause significant death in the embryos, as determined 3 days after treatment (n = 100/genotype; experiment repeated three times). When 24 hpf embryos were treated with a higher UVB irradiation dose (4.32 kJ/m2) and observed 2 days posttreatment, significant death and morphological phenotypes were observed (93.7% in UV-treated p53-deficient embryos [n = 348]; 92.2% in UV-treated wild-type embryos [n = 90]; and 0% in untreated p53-deficient embryos [n = 76] and wild-type embryos [n = 32]).
FIG. 2.


UVB-induced apoptosis and expression of DNA damage repair–related genes. (A–D) Twenty-four hpf wt and p53 mutant embryos were irradiated with a sublethal dose of UVB (1.08 kJ/m2). After incubated in dark for 6 h, embryos were stained with acridine orange solution to detect cell death induced by UVB treatment in living embryos. (E) Total RNA isolated and reverse transcription polymerase chain reaction analysis was performed to examine the expression of DNA damage repair–related genes after UVB exposure. wt, wild type.
To establish the DNA damage repair pathways stimulated after UV treatment, we examined the expression of several DNA-repair-related genes in wild-type and p53 mutant embryos at 6 h after sublethal UV exposure (1.08 kJ/m2) (Fig. 2E). Damaged DNA binding proteins (DDB) initiate the recognition of DNA lesions, the first step of NER.40 We find that expression of DDB2 increased in UV-exposed wild-type and p53 mutant embryos, with a greater increase in wild-type embryos. In contrast, KU80 involved in nonhomologous end joining pathway41 did not change after UV treatment, consistent with the finding in zebrafish hepatocytes, suggesting that nonhomologous end joining is not the main mechanism of DNA repair in zebrafish after UVB irradiation.42 Light-induced apoptosis can also cause an increase in expression of the pro-apoptotic caspase3 gene that correlates with Caspase 3 activity.43 We also found that caspase3 RNA was upregulated in the wild-type embryos, but not the p53 mutant embryos after UVB treatment. These initial experiments show that the UV treatment chamber can cause a p53-dependent, PER-independent DNA damage response in the developing zebrafish embryos.
Sensitivity of larvae and young adult zebrafish to UV light
Sunburn in early life has been implicated as an important risk factor for melanoma.3 Having determined that our UV chamber effectively causes UV-induced cell death in early embryos, we performed dose–response experiments to determine the level of UV treatment that was required to cause DNA damage in the skin in a wild-type and p53-deficient background. In the Xiphophorus model, melanoma is effectively induced when 5-day-old Xiphophorus fry are exposed to UV irradiation. As embryonic development is within the mother Xiphophorus, and their development is not directly comparable to zebrafish development, we chose five to 6-week-old zebrafish that were approximately 1 cm in length, thereby being approximately the same size and stage in development as Xiphophorus 5 days after birth. For comparison, we also examined the effects of UV treatment on 6-day-old embryos. During these experiments we found that the animals were sensitive to the UV doses used, and in the 6-day-old zebrafish, the survival of the p53 mutant lines was not significantly different from the response of wild-type fish (Fig. 3A). Notably, the UV sensitivity was higher in the 6-day-old animals than in the 24 hpf animals; when we applied the same UV treatment conditions used on the 24 hpf embryos (1.08 kJ/m2) to 6-day-old zebrafish, all animals died. This may reflect differences in UV tolerance through development, as previously described.39 Linear regression lines show a dose-dependent response to UVB irradiation in wild-type and p53 mutant fish (R2(wt) = 0.977 and R2(p53) = 0.935), allowing us to calculate the LD50 to be 0.60 kJ/m2 for both wild-type and p53 mutant 6-day-old zebrafish (Fig. 3C). In the 5–6-week-old fish, higher UV doses were used, and the survival curves of the p53 mutant lines showed an enhanced overall sensitivity compared to the wild-type fish (Fig. 3B). Polynomial regression lines show a dose-dependent response to UVB irradiation in wild-type and p53-deficient young adult fish (R2(wt) = 0.951 and R2(p53) = 0.975), allowing us to calculate the LD50 to be 2.86 and 2.65 kJ/m2 for young adult wild-type and p53 zebrafish, respectively (Fig. 3D). We noted that the common abnormalities caused by UVB irradiation included an enhanced curve to the body of the fish, and the fish could sometimes recover from this phenotype (Fig. 3E, F).
FIG. 3.

p53 Mutant zebrafish are sensitive to UVB irradiation. (A) Comparison of sensitivity of 6-day-old wt and p53 mutant fish at various UVB doses. Ten fish were used at each dose in each experiment. Experiments had three replicates. Means and standard deviations of the three experiments are presented. (B) Comparison of sensitivity in 5–6-week-old wt and p53 fish at various UVB doses. (C) The data from 6-day-old wt and p53 fish fit to linear regression lines (R2 = 0.977 and 0.9352 for wt and p53 fish, respectively; R2 = 1 when curves best fit the regression line). The calculated LD50s (where survival is 50%) are 0.6 kJ/m2 for both wt and p53 fish. (D) The data from 5–6-week-old wt and p53 young adults fit to polynomial regression lines (R2 = 0.9512 and 0.9747 for wt and p53 fish, respectively). (E, F) The typical morphology of UVB-exposed zebrafish. Curved spinal is the common abnormalities in both juvenile and young adult fish, also in wt and p53 mutant fish.
Histone H2AX is phosphorylated after UVB treatment in zebrafish skin
To determine if the lower UVB treatment doses (2.16 kJ/m2) were able to promote DNA damage in adult zebrafish skin, we used an antibody that recognizes the zebrafish phospho-H2AX histone variant. The histone variant H2AX is phosphorylated along tracks of chromatin at double-strand breaks after ionizing radiation.44 UV treatment also induces H2AX phosphorylation in human cell lines, in a pan-nuclear staining that is highest in S-phase, and in contrast to the characteristic discrete nuclear foci after ionizing radiation.45 We observed the DNA damage sites in the tissues of UVB irradiated adult fish by staining sections with a zebrafish phospho-H2AX antibody. Twelve hours after UVB exposure (and being kept in the dark), pan-nuclear staining of phospho-H2AX was detected in the skin and fin tissues of exposed wild-type and p53 mutant fish (Fig. 4). No positive signals in the fin were detected in unexposed fish or fish killed shortly after exposure. In both wild-type and p53-deficient fish, the H2AX staining pattern was more intense in the fin than in the skin, consistent with findings in Xiphophorus that the photoproduct level in the fin is two- to threefold higher than in the skin,46 suggesting that the scales may be natural shields of UVB irradiation. Interestingly, the phospho-H2AX staining in the fin was localized to the outermost side of the fin, presumably reflecting that the inside of the fin was protected from UV damage (Fig. 4C). Thus, using phospho-H2AX as a marker for DNA damage, we find that UVB treatment is able to promote DNA damage in adult skin.
FIG. 4.
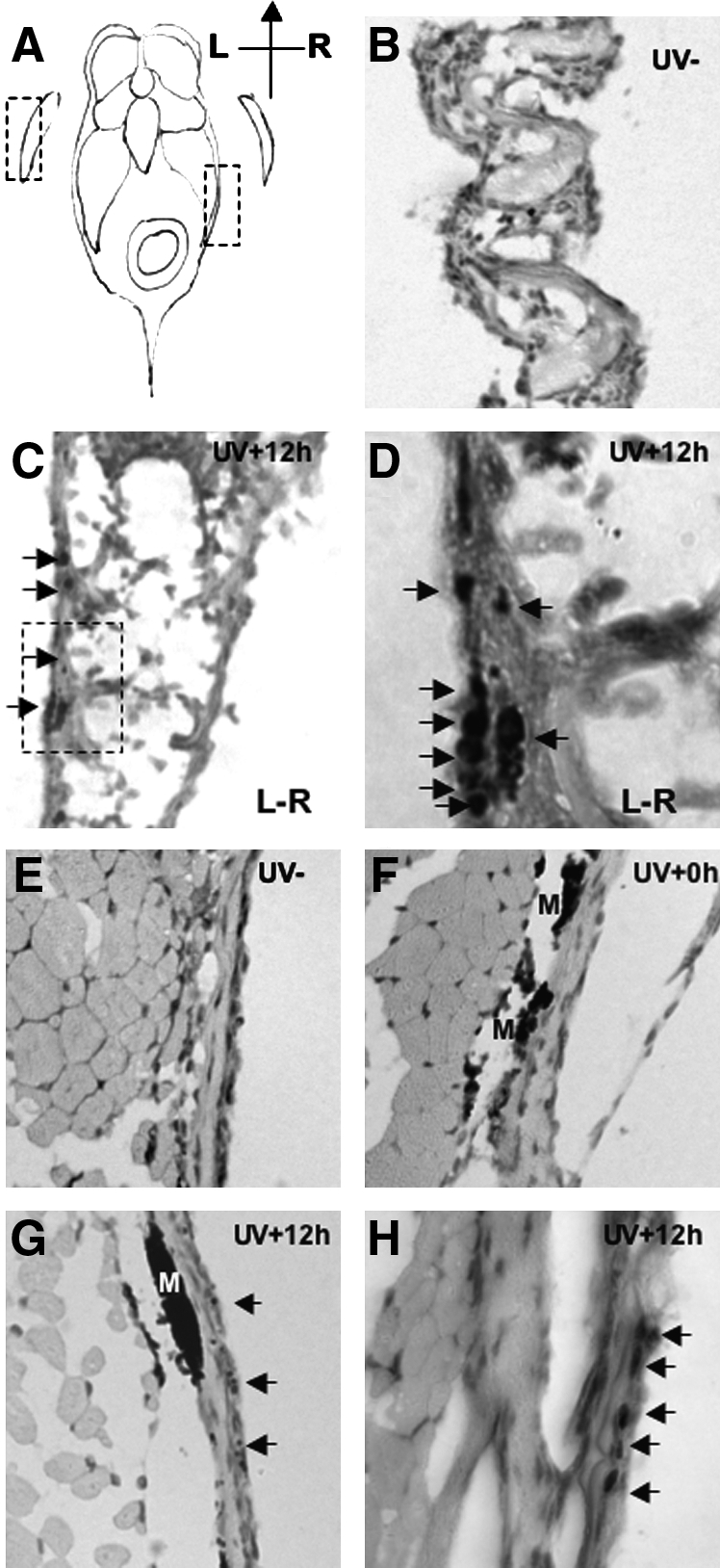
Observation of DNA damage sites in zebrafish fin (B–D) and skin (E–H). Histological sections of adult zebrafish 12 hours after of UVB (2.16 kJ/m2) exposure, stained with an antibody to detect phospho-histone H2AX (phospho-H2AX). (A) Schematic diagram of zebrafish section; dotted boxes denote the examined fin and skin tissues in (B) and (E), respectively. (B) Section of pectoral fin of untreated wt fish (hematoxylin and eosin stain; magnification 200×). (C) Section of pectoral fin of UVB exposed wt fish. Phospho-H2AX signals are located in the epidermis on the most outer side of fin (arrows); magnification 200×. Dotted box indicates region examined in (D). (D) High magnification (630×) view of the phospho-H2AX positive cells in the fin. (E) No phospho-H2AX signal is detected in the skin of untreated fish; magnification 200×. (F) Skin of fish sacrificed immediately after UVB exposure reveals no staining for phospho-H2AX; magnification 200×. (G) Skin at 12 hours postirradiation reveals specific phospho-H2AX staining; magnification 200×. (H) High magnification (630×) view of the phospho-H2AX positive cells in the skin. Phospho-H2AX positive nuclei are indicated with arrows; M denotes melanin.
UVB DNA damage repair is dependent on p53 in adult zebrafish
Exposure to UVB wavelengths results in pyrimidine dimers (covalent adducts between adjacent pyrimidines) called CPDs and (6-4)PDs. Decreased NER ability is correlated with the inducibility of melanoma in Xiphophorus F1 hybrids.14 We wanted to ask if adult zebrafish also had active repair systems to remove photoproducts and repair the DNA damage, like the Xiphophorus species, and if we could detect differences in the p53 mutant line. Using RIA, we measured the CPDs and (6-4)PDs levels in 6-month-old wild-type and p53 mutant zebrafish at 0, 3, 6, and 24 h after exposure to 2.16 kJ/m2 UVB light, the same dose used to study phospho-H2AX levels in the skin. Low background DNA damage frequencies were detected in wild-type and p53 fish (Table 2 and Fig. 5A, B), and significant levels of CPDs and (6-4)PDs were induced in zebrafish skin by the challenge dose. Consistent with the photochemistry, considerably more CPDs were induced compared to (6-4)PDs (i.e., ∼fivefold).47 Apparent increases in DNA damage immediately after irradiation (3 and 6 h) are typical observations associated with interindividual variation in photoproduct measurements derived from RIA as well as other techniques,48 and there is essentially no repair at these early time points. We also measured the rate of photoproduct repair over 24 h postirradiation (Fig. 5A, B). Keeping fish in the dark to limit repair to NER pathways, we found that wild-type zebrafish skin had repaired most photoproducts by 24 h, with only 23% CPDs and 12% (6-4)PDs remaining. By contrast, in the p53 mutant zebrafish, most CPDs (74%) and (6-4)PDs (64%) remained in the skin. This result shows that p53 mutant zebrafish are impaired in the NER of DNA damage in the skin caused by UVB exposure. Wild-type zebrafish showed a similar NER capacity compared with three Xiphophorus species (Fig. 5C).
Table 2.
Nucleotide Excision Repair in Zebrafish Skin
| |
|
CPDs/mb DNA |
(6-4)PDs/mb DNA |
||||
|---|---|---|---|---|---|---|---|
| Time of repair | Mean (n = 5) | SD | Remaining% | Mean (n = 5) | SD | Remaining% | |
| wt | UV | 98.6 | 6.9 | 8.7 | 2.5 | ||
| 0 | 567.2 | 106.7 | 100 | 92.2 | 26.4 | 100 | |
| 3 | 766.4 | 100.9 | 135 | 115.1 | 22.6 | 125 | |
| 6 | 692.3 | 109.3 | 122 | 89.7 | 21.9 | 97 | |
| 24 | 133.0 | 18.4 | 23 | 10.8 | 2.9 | 12 | |
| p53 | UV | 99.6 | 16.4 | 8.8 | 3.0 | ||
| 0 | 767.6 | 206.6 | 100 | 130.3 | 33.6 | 100 | |
| 3 | 783.7 | 127.1 | 102 | 123.2 | 18.4 | 95 | |
| 6 | 709.5 | 141.1 | 92 | 105.4 | 24.6 | 81 | |
| 24 | 570.3 | 130.8 | 74 | 83.0 | 22.4 | 64 | |
CPD, cyclobutane pyrimidine dimers; SD, standard deviation; UV, ultraviolet; wt, wild type.
FIG. 5.


DNA damage repair in the skin of adult zebrafish. Fish were exposed to UVB (2.16 kJ/m2) light; photoproducts frequencies at 0, 3, 6, and 24 h postirradiation were quantified using radioimmunoassay and normalized to the amount of damage present immediately after UVB exposure in wt zebrafish (100%). DNA repair in wt and p53 mutant fish was determined and compared for (A) the repair of (6-4)PDs and (B) the repair of cyclobutane pyrimidine dimers. (C) Comparison of cyclobutane pyrimidine dimers repair rate at 24 h postirradiation in wt zebrafish and three Xiphophorus species: X. couchianus, X. maculates, and X. andersi.
Discussion
While UV exposure is a clear risk factor for melanoma, the molecular mechanisms affected by UV, and how these contribute to melanoma progression in humans are not yet understood. In mice, neonatal UV exposure can initiate melanoma, supporting a role for early UV exposure and melanoma risk in people.49,50 In the mice models, UVB contributes to melanoma initiation,51 while in the Xiphophorus both UVA and UVB can initiate melanoma.18 In Xiphophorus, melanoma is correlated both to UV exposure, as well as the capacity to repair lesions, providing important evidence that genetic background can play a key role in UV-induced melanoma susceptibility. As zebrafish is a tractable genetic system, and can be engineered to develop melanoma, the zebrafish provides a unique opportunity in which to explore the genetic relationship between UV exposure and melanoma development.
Previous studies have already demonstrated that the zebrafish system can be an important tool to investigate the biological effects of UV light in cells and development. Like the Xiphophorus species, zebrafish have a competent antioxidant response and photorepair system to repair UV-induced DNA damage.39,52,53 DNA repair appears to vary at specific stages in development, with 12 hpf embryos showing the greatest sensitivity to UVA and UVB treatment.39 In cultured zebrafish hepatocytes, the early response to UVB irradiation involves DNA repair genes such as XPC and DDB2, and the late response includes upregulation of p53 and cell cycle arrest.42 In this study, we describe a methodology for treating larval and young adult zebrafish with UV. Based on the established Xiphophorus models of UV-induced melanoma, we have designed a UV treatment chamber that can be used to provide accurate UV treatment to zebrafish without the additional stress of adding anesthetics or removing the fish from the water (Fig. 1). In the Xiphophorus model, 5-day postbirth larvae are exposed to UV exposure.54 As Xiphophorus are live born, this corresponds to approximately 5–6 weeks of development and size for zebrafish, with some differences, for example, in the development of the immune system.55,56 We show that using this chamber to administer UV treatment can cause a DNA damage response in zebrafish embryos (Fig. 2), larvae, and adults (Figs. 3–5). Importantly, we show that UVB treatment can induce phospho-H2AX staining in the skin of young adult zebrafish, indicating DNA damage and showing an important example of UV-induced phospho-H2AX staining in an animal. Phospho-H2AX staining in zebrafish skin appears to be throughout the nucleus rather than in discrete foci, supporting the notion that UVB treatment is promoting single, but not double strand breaks in the skin.45 Thus, we have described a simple methodology for promoting a UV-induced NER DNA damage response in young adult zebrafish.
Unlike most cancers, p53 mutations are surprisingly rare in melanoma. This may be because of frequent mutations in the CDKN2A locus that encodes the tumor suppressor proteins, p16INK4a and p19ARF, and regulate the Rb and p53 pathways, respectively.57 Loss of ARF expression, or changes in MDM2 levels may inactivate the p53 pathway in human melanoma development.58–61 In mice, HRAS mutations in a p53-null background cause melanoma,62 and in zebrafish, p53 mutations can contribute to the promotion of BRAFV600E melanocytic nevi to malignant melanoma.29 In addition, compelling evidence from genome wide expression analysis reveals that the p53 pathway is impaired in human melanomas, and that dysfunction of the p53 pathway contributes to the transition from nevi to melanoma.63
While evidence for a role for p53 in human melanoma progression is accumulating, the relationship between UV exposure and p53 in melanoma is not yet clear. Strong evidence supports a role for p53 in pigmentation after UV exposure,64 but signature mutations in p53 are not a consistent molecular feature of human melanomas. Recently, it has been reported that p53 appears to contribute to distinct melanocyte cell death pathways after either UVA or UVB exposure.65 In zebrafish embryos, we see an enhanced rate of cell death throughout the developing embryo after UV treatment, which is absent in the p53-deficient embryos and is consistent with the previous reports showing p53-dependent cell death in UV and ionizing radiation conditions.32,38,66 Our data also show that the p53 mutant zebrafish are deficient in NER in the adult skin of treated fish (Fig. 5A, B). This correlates with a significant reduced survival rate of p53-deficient young adult lines after UV treatment (Fig. 2B). Of interest, we find that young larval forms deficient for p53 do not have an enhanced sensitivity to UV compared with wild-type fish at 6 days of development (Fig. 2A). Strong genetic evidence in mouse and zebrafish indicates that keeping p53 at low levels during embryogenesis is critical to protect normal development.38,67–71 In zebrafish embryos, an ionizing radiation p53-independent DNA damage response has recently been identified.72 While speculative, our observation may point to an early developmental period when the UV DNA damage response is p53 independent, perhaps because of a developmental requirement to suppress p53 activity. Evidence from medaka shows p53 gene expression is developmentally regulated, with less p53 gene expression during the first few days of embryogenesis.73 Unlike mammalian cells, in response to UV light medaka fry do not show an increase in p53 gene expression.73 However, p53 most likely still plays an important role in tumor suppression in medaka, as loss of p53 has been shown to collaborate with oncogenic Xmrk to direct pigment cell tumor spectrum and pathology.74
While our studies test the role of p53-dependent NER in the skin of UV-treated fish, it will be of great interest to specifically examine the UV response and p53-dependent NER within zebrafish melanocytes, and to relate these to melanoma development. For example, in melanocyte cell culture, reduced levels of Mitf, a critical gene in melanocyte development and melanoma, causes enhanced sensitivity to UV-induced apoptosis.75 As well, loss-of-function mutations in the MC1R gene sensitize human melanocytes to the DNA damaging effects of UV radiation.76 With genetic control of these melanocyte genes,26,77 and other genes that control pigmentation (including golden9), coupled with the increasing genetic resources for cancer biology (including pten mutations and BRAFV600E transgenic lines21), the zebrafish is well poised to make a significant contribution to the gene–environment interactions that contribute to melanoma development. This work contributes to this aim by providing a methodology for administering consistent UV to zebrafish at all ages, and showing that adult zebrafish skin has a competent p53-dependent NER pathway.
Acknowledgments
We gratefully acknowledge Dr. James Amatruda (UT Southwestern Medical Center, Dallas, TX) for the H2AX antibody, and we would like to thank Dr. Karthika Paranthaman for excellent management of the Zebrafish Facility. We would like to thank Professor Ian Jackson for critical reading of the article. We are grateful for funding of this work from Medical Research Council and the Association of International Cancer Research 07-0421 to E.E.P., a British Council Award to Z.Z., and an NCI Grant CA113671 and NIEHS Center Grant ES07784 to D.L.M.
Disclosure Statement
No competing financial interests exist.
References
- 1.Coory M. Baade P. Aitken J, et al. Trends for in situ and invasive melanoma in Queensland, Australia, 1982–2002. Cancer Causes Control. 2006;7:21–27. doi: 10.1007/s10552-005-3637-4. [DOI] [PubMed] [Google Scholar]
- 2.Linos E. Swetter SM. Cockburn MG, et al. Increasing burden of melanoma in the United States. J Invest Dermatol. 2009;129:1666–1674. doi: 10.1038/jid.2008.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tran TT. Schulman J. Fisher DE. UV and pigmentation: molecular mechanisms and social controversies. Pigment Cell Melanoma Res. 2008;21:509–516. doi: 10.1111/j.1755-148X.2008.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacKie RM. Bray C. Vestey J, et al. Melanoma incidence and mortality in Scotland 1979–2003. Br J Cancer. 2007;96:1772–1777. doi: 10.1038/sj.bjc.6603801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silva Idos S. Higgins CD. Abramsky T, et al. Overseas sun exposure, nevus counts, and premature skin aging in young English women: a population-based survey. J Invest Dermatol. 2009;129:50–59. doi: 10.1038/jid.2008.190. [DOI] [PubMed] [Google Scholar]
- 6.Guidelines for Diagnosis and Treatment of Melanoma in China. first. Beijing, China: CSCO office; 2008. Chinese Society for Clinical Oncology (CSCO) p. 2. [Google Scholar]
- 7.Sturm RA. Molecular genetics of human pigmentation diversity. Hum Mol Genet. 2009;18(Review Issue 1):R9–R17. doi: 10.1093/hmg/ddp003. [DOI] [PubMed] [Google Scholar]
- 8.Pharoah PD. Shedding light on skin cancer. Nat Genet. 2008;40:817–818. doi: 10.1038/ng0708-817. [DOI] [PubMed] [Google Scholar]
- 9.Lamason RL. Mohideen MA. Mest JR, et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310:1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- 10.Valverde P. Healy E. Jackson I, et al. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet. 1995;11:328–330. doi: 10.1038/ng1195-328. [DOI] [PubMed] [Google Scholar]
- 11.Halliday GM. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat Res. 2005;571:107–120. doi: 10.1016/j.mrfmmm.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 12.Phillipson RP. Tobi SE. Morris JA. McMillan TJ. UV-A induces persistent genomic instability in human keratinocytes through an oxidative stress mechanism. Free Radic Biol Med. 2002;32:474–480. doi: 10.1016/s0891-5849(01)00829-2. [DOI] [PubMed] [Google Scholar]
- 13.Bennett DC. Ultraviolet wavebands and melanoma initiation. Pigment Cell Melanoma Res. 2008;21:520–524. doi: 10.1111/j.1755-148X.2008.00500.x. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell DL. Nairn RS. Johnston DA, et al. Decreased levels of (6-4) photoproduct excision repair in hybrid fish of the genus Xiphophorus. Photochem Photobiol. 2004;79:447–452. doi: 10.1562/ca-03-14.1. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell DL. Meador JA. Byrom M. Walter RB. Resolution of UV-induced DNA damage in Xiphophorus fishes. Mar Biotechnol (NY) 2001;3(Suppl 1):S61–S71. doi: 10.1007/s101260000000. [DOI] [PubMed] [Google Scholar]
- 16.Meierjohann S. Schartl M. From Mendelian to molecular genetics: the Xiphophorus melanoma model. Trends Genet. 2006;22:654–661. doi: 10.1016/j.tig.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell DL. Nairn RS. Photocarcinogenesis in Xiphophorus hybrid models. Zebrafish. 2006;3:311–321. doi: 10.1089/zeb.2006.3.311. [DOI] [PubMed] [Google Scholar]
- 18.Setlow RB. Woodhead AD. Grist E. Animal model for ultraviolet radiation-induced melanoma: platyfish-swordtail hybrid. Proc Natl Acad Sci U S A. 1989;86:8922–8926. doi: 10.1073/pnas.86.22.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nairn RS. Kazianis S. McEntire BB, et al. A CDKN2-like polymorphism in Xiphophorus LG V is associated with UV-B-induced melanoma formation in platyfish-swordtail hybrids. Proc Natl Acad Sci U S A. 1996;93:13042–13047. doi: 10.1073/pnas.93.23.13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell DL. Haipek CA. Clarkson JM. Further characterization of a polyclonal antiserum for DNA photoproducts: the use of different labeled antigens to control its specificity. Mutat Res. 1985;146:129–133. doi: 10.1016/0167-8817(85)90002-1. [DOI] [PubMed] [Google Scholar]
- 21.Amatruda JF. Patton EE. Genetic models of cancer in zebrafish. Int Rev Cell Mol Biol. 2008;271:1–34. doi: 10.1016/S1937-6448(08)01201-X. [DOI] [PubMed] [Google Scholar]
- 22.Lynn Lamoreux M. Kelsh RN. Wakamatsu Y, et al. Pigment pattern formation in the medaka embryo. Pigment Cell Res. 2005;18:64–73. doi: 10.1111/j.1600-0749.2005.00216.x. [DOI] [PubMed] [Google Scholar]
- 23.Kelsh RN. Genetics and evolution of pigment patterns in fish. Pigment Cell Res. 2004;17:326–336. doi: 10.1111/j.1600-0749.2004.00174.x. [DOI] [PubMed] [Google Scholar]
- 24.Rawls JF. Mellgren EM. Johnson SL. How the zebrafish gets its stripes. Dev Biol. 2001;240:301–314. doi: 10.1006/dbio.2001.0418. [DOI] [PubMed] [Google Scholar]
- 25.White RM. Zon LI. Melanocytes in development, regeneration, and cancer. Cell Stem Cell. 2008;3:242–252. doi: 10.1016/j.stem.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Richardson J. Lundegaard PR. Reynolds NL, et al. mc1r pathway regulation of zebrafish melanosome dispersion. Zebrafish. 2008;5:289–295. doi: 10.1089/zeb.2008.0541. [DOI] [PubMed] [Google Scholar]
- 27.Ekker SC. Stemple DL. Clark M, et al. Zebrafish genome project: bringing new biology to the vertebrate genome field. Zebrafish. 2007;4:239–251. doi: 10.1089/zeb.2007.9979. [DOI] [PubMed] [Google Scholar]
- 28.Mitani H. Kamei Y. Fukamachi S, et al. The medaka genome: why we need multiple fish models in vertebrate functional genomics. Genome Dyn. 2006;2:165–182. doi: 10.1159/000095103. [DOI] [PubMed] [Google Scholar]
- 29.Patton EE. Widlund HR. Kutok JL, et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249–254. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 30.Patton EE. Zon LI. Taking human cancer genes to the fish: a transgenic model of melanoma in zebrafish. Zebrafish. 2005;1:363–368. doi: 10.1089/zeb.2005.1.363. [DOI] [PubMed] [Google Scholar]
- 31.Kimmel CB. Ballard WW. Kimmel SR, et al. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 32.Berghmans S. Murphey RD. Wienholds E, et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci U S A. 2005;102:407–412. doi: 10.1073/pnas.0406252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westerfield M. Eugene, OR: University of Oregon Press; 1995. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio), edition 3. [Google Scholar]
- 34.Mitchell DL. Quantification of DNA photoproducts in mammalian cell DNA using radioimmunoassay. In: Henderson DS, editor. Methods in Molecular BiologyDNA Repair Protocols. 2nd. Totowa, NJ: The Humana Press Inc; 2006. pp. 239–249. [Google Scholar]
- 35.Duffield S. Jordan S. Evaluation of insecticides for the control of Helicoverpa armigera (Hübner) and Helicoverpa punctigera (Wallengren) (Lepidoptera: Noctuidae) on soybean, and the implications for field adoption. Aust J Entomol. 2001;39:322–327. [Google Scholar]
- 36.Mitchell DL. Paniker L. Douki T. DNA damage, repair and photoadaptation in a Xiphophorus fish hybrid. Photochem Photobiol. 2009;85:1384–1390. doi: 10.1111/j.1751-1097.2009.00591.x. [DOI] [PubMed] [Google Scholar]
- 37.Vazquez A. Bond EE. Levine AJ. Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 38.Langheinrich U. Hennen E. Stott G. Vacun G. Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr Biol. 2002;12:2023–2028. doi: 10.1016/s0960-9822(02)01319-2. [DOI] [PubMed] [Google Scholar]
- 39.Dong Q. Svoboda K. Tiersch TR. Monroe WT. Photobiological effects of UVA and UVB light in zebrafish embryos: evidence for a competent photorepair system. J Photochem Photobiol B. 2007;88:137–146. doi: 10.1016/j.jphotobiol.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scrima A. Konícková R. Czyzewski BK, et al. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bladen CL. Lam WK. Dynan WS. Kozlowski DJ. DNA damage response and Ku80 function in the vertebrate embryo. Nucleic Acids Res. 2005;33:3002–3010. doi: 10.1093/nar/gki613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandrini JZ. Trindade GS. Nery LE. Marins LF. Time-course expression of DNA repair-related genes in hepatocytes of zebrafish (Danio rerio) after UV-B exposure. Photochem Photobiol. 2009;85:220–226. doi: 10.1111/j.1751-1097.2008.00422.x. [DOI] [PubMed] [Google Scholar]
- 43.Wu J. Gorman A. Zhou X, et al. Involvement of caspase-3 in photoreceptor cell apoptosis induced by in vivo blue light exposure. Invest Ophthalmol Vis Sci. 2002;43:3349–3354. [PubMed] [Google Scholar]
- 44.Rogakou EP. Pilch DR. Orr AH. Ivanova VS. Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 45.Marti TM. Hefner E. Feeney L, et al. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meador JA. Walter RB. Mitchell DL. Induction, distribution and repair of UV photodamage in the platyfish, Xiphophorus signum. Photochem Photobiol. 2000;72:260–266. doi: 10.1562/0031-8655(2000)072<0260:idarou>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 47.Mitchell DL. Nairn RS. The biology of the (6-4) photoproduct. Annu Rev Photochem Photobiol. 1989;49:805–819. doi: 10.1111/j.1751-1097.1989.tb05578.x. [DOI] [PubMed] [Google Scholar]
- 48.Wheeler DL. Martin KE. Ness KJ, et al. Protein kinase C epsilon is an endogenous photosensitizer that enhances ultraviolet radiation-induced cutaneous damage and development of squamous cell carcinomas. Cancer Res. 2004;64:7756–7765. doi: 10.1158/0008-5472.CAN-04-1881. [DOI] [PubMed] [Google Scholar]
- 49.Noonan FP. Dudek J. Merlino G. De Fabo EC. Animal models of melanoma: an HGF/SF transgenic mouse model may facilitate experimental access to UV initiating events. Pigment Cell Res. 2003;16:16–25. doi: 10.1034/j.1600-0749.2003.00014.x. [DOI] [PubMed] [Google Scholar]
- 50.Hacker E. Muller HK. Irwin N, et al. Spontaneous and UV radiation-induced multiple metastatic melanomas in dk4R24C/R24C/TPras mice. Cancer Res. 2006;66:2946–2952. doi: 10.1158/0008-5472.CAN-05-3196. [DOI] [PubMed] [Google Scholar]
- 51.De Fabo EC. Noonan FP. Fears T. Merlino G. Ultraviolet B but not ultraviolet A radiation initiates melanoma. Cancer Res. 2004;64:6372–6376. doi: 10.1158/0008-5472.CAN-04-1454. [DOI] [PubMed] [Google Scholar]
- 52.Charron RA. Fenwick JC. Lean DR. Moon TW. Ultraviolet-B radiation effects on antioxidant status and survival in the zebrafish, Brachydanio rerio. Photochem Photobiol. 2000;72:327–333. doi: 10.1562/0031-8655(2000)072<0327:ubreoa>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 53.Dong Q. Todd Monroe W. Tiersch TR. Svoboda KR. UVA-induced photo recovery during early zebrafish embryogenesis. J Photochem Photobiol B. 2008;93:162–171. doi: 10.1016/j.jphotobiol.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Setlow RB. Woodhead AD. Temporal changes in the incidence of malignant melanoma: explanation from action spectra. Mutat Res. 1994;307:365–374. doi: 10.1016/0027-5107(94)90310-7. [DOI] [PubMed] [Google Scholar]
- 55.Zapata A. Diez B. Cejalvo T. Gutiérrez-de Frías C. Cortés A. Ontogeny of the immune system of fish. Fish Shellfish Immunol. 2006;20:126–136. doi: 10.1016/j.fsi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Leknes IL. Uptake of foreign ferritin in platy Xiphophorus maculatus (Poeciliidae: Teleostei) Dis Aquat Organ. 2002;51:233–237. doi: 10.3354/dao051233. [DOI] [PubMed] [Google Scholar]
- 57.Lowe SW. Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 58.Sharpless E. Chin L. The INK4a/ARF locus and melanoma. Oncogene. 2003;22:3092–3098. doi: 10.1038/sj.onc.1206461. [DOI] [PubMed] [Google Scholar]
- 59.Gluck I. Simon AJ. Catane R. Germline analysis of thymidine/guanidine polymorphism at position 309 of the Mdm2 promoter in malignant melanoma patients. Melanoma Res. 2009 doi: 10.1097/CMR.0b013e32832ccd27. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 60.de Sá BC. Fugimori ML. Ribeiro Kde C, et al. Proteins involved in pRb and p53 pathways are differentially expressed in thin and thick superficial spreading melanomas. Melanoma Res. 2009;19:135–141. doi: 10.1097/CMR.0b013e32831993f3. [DOI] [PubMed] [Google Scholar]
- 61.Firoz EF. Warycha M. Zakrzewski J, et al. Association of MDM2 SNP309, age of onset, and gender in cutaneous melanoma. Clin Cancer Res. 2009;15:2573–2580. doi: 10.1158/1078-0432.CCR-08-2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bardeesy N. Bastian BC. Hezel A, et al. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol. 2001;21:2144–2153. doi: 10.1128/MCB.21.6.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu H. McDaid R. Lee J, et al. The role of BRAF mutation and p53 inactivation during transformation of a subpopulation of primary human melanocytes. Am J Pathol. 2009;174:2367–2377. doi: 10.2353/ajpath.2009.081057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cui R. Widlund HR. Feige E, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128:853–864. doi: 10.1016/j.cell.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 65.Wäster PK. Ollinger KM. Redox-dependent translocation of p53 to mitochondria or nucleus in human melanocytes after UVA- and UVB-induced apoptosis. J Invest Dermatol. 2009;129:1769–1781. doi: 10.1038/jid.2008.421. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H. p53 plays a central role in UVA and UVB induced cell damage and apoptosis in melanoma cells. Cancer Lett. 2006;244:229–238. doi: 10.1016/j.canlet.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 67.Jones SN. Roe AE. Donehower LA. Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 68.Montes de Oca Luna R. Wagner DS. Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 69.Lee H. Kimelman D. A dominant-negative form of p63 is required for epidermal proliferation in zebrafish. Dev Cell. 2002;2:607–616. doi: 10.1016/s1534-5807(02)00166-1. [DOI] [PubMed] [Google Scholar]
- 70.Chen J. Ng SM. Chang C, et al. p53 isoform Δ113p53 is a p53 target gene that antagonizes p53 apoptotic activity via Bcl-xL activation in zebrafish. Genes Dev. 2009;23:278–290. doi: 10.1101/gad.1761609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vousden KH. Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 72.Sidi S. Sanda T. Kennedy RD, et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell. 2008;133:864–877. doi: 10.1016/j.cell.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen S. Hong Y. Scherer SJ. Schartl M. Lack of ultraviolet-light inducibility of the medakafish (Oryzias latipes) tumor suppressor gene p53. Gene. 2001;264:197–203. doi: 10.1016/s0378-1119(01)00340-7. [DOI] [PubMed] [Google Scholar]
- 74.Schartl M. Wilde B. Laisney JA et al. A mutated EGFR is sufficient to induce malignant melanoma with genetic background-dependent histopathologies. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.213. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 75.Hornyak TJ. Jiang S. Guzmán EA, et al. Mitf dosage as a primary determinant of melanocyte survival after ultraviolet irradiation. Pigment Cell Melanoma Res. 2009;22:307–318. doi: 10.1111/j.1755-148X.2009.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abdel-Malek Z. Knittel J. Kadekaro AL, et al. The melanocortin 1 receptor and the UV response of human melanocytes—a shift in paradigm. Photochem Photobiol. 2008;84:501–508. doi: 10.1111/j.1751-1097.2008.00294.x. [DOI] [PubMed] [Google Scholar]
- 77.Lister JA. Robertson CP. Lepage T, et al. Nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development. 1999;126:3757–3767. doi: 10.1242/dev.126.17.3757. [DOI] [PubMed] [Google Scholar]