

Abstract
The usurping of translational control by sustained activation of translation initiation factors is oncogenic. Here we show that the primary negative regulators of these oncogenic initiation factors - the 4E-BP protein family - operate as guardians of a translational control checkpoint in lung tumor defense. When challenged with the tobacco carcinogen NNK, 4ebp1−/−/4ebp2−/− mice showed increased sensitivity to tumorigenesis compared to their wild type counterparts. The 4E-BP deficient state per se creates pro-oncogenic, genome-wide skewing of the molecular landscape - with translational activation of genes governing angiogenesis, growth and proliferation; and translational activation of the precise cytochrome p450 enzyme isoform (CYP2A5) that bioactivates NNK into mutagenic metabolites. Our study provides in vivo proof for a translational control checkpoint in lung tumor defense.
Introduction
Biomedical scientists have recognized for more than a decade that components of the cap-dependent translation initiation machinery are more abundant and active in cancer than in their non-malignant counterparts; an adjustment previously assumed to serve the increased metabolic needs of cancer. For example, increased expression of eukaryotic translation initiation factor 4E (eIF4E) is one of the early events in breast tumorigenesis (1) where it serves as an independent prognostic factor (2, 3). In lung neoplasia, the level of eIF4E increases with morphological aberrancy ranging from low levels in atypical adenomatous hyperplasia to high levels in invasive papillary adenocarcinoma (4). Nonetheless, it came as somewhat of a surprise when studies of hematological malignancies, breast cancer and lung cancer all documented that cancer is subject to translational control (5–8). In fact, more than three quarters of the approximately 300 cancer-related genes recognized to date activate the eukaryotic translational initiation complex 4F (eIF4F) directly or indirectly (9, 10).
If sustained activation of cap-dependent translation is oncogenic, then it is reasonable to posit the existence of a translational control checkpoint in tumor defense. The logical guardians of this checkpoint would be the primary negative regulators of eIF4F, the eukaryotic translation initiation factor 4E binding protein (4E-BP) family of proteins which negatively regulate translation initiation by sequestering the rate-limiting component of the eIF4F complex, eIF4E. Under physiological circumstances, a wide variety of regulatory cues govern 4E-BP activity by phosphorylation through the Ras/PI3K/AKT/mTOR kinase cascade (11). Hyperphosphorylation decreases the affinity of 4E-BPs for eIF4E, permitting eIF4E to participate in eIF4F assembly. However, the most ubiquitous translational repressor, 4E-BP1, displays attributes of a tumor suppressor: (a) it is functionally inactivated in aggressive breast carcinomas by hyperphosphorylation (7); and (b) when ectopically expressed in breast and lung carcinoma cells, 4E-BP1 suppresses tumorigenicity (7, 8). These findings support the idea that the 4E-BP family of translational repressors might not only function in their canonical role as global regulators of mRNA recruitment to the ribosome, but also in tumor surveillance at a translational control checkpoint.
To test whether this checkpoint exists, we used mice genetically engineered to lack two of the three members of the 4E-BP family (4ebp1−/−/4ebp2−/−) – and selected the widely used mouse model of lung tumorigenesis induced by the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). The advantages of this model are well chronicled: i) NNK is an abundant and potent tobacco carcinogen; ii) the molecular mechanism of its bioactivation by a specific cytochrome p450 enzyme isoform (CYP2A5) and its mechanism of mutagenicity are well established; iii) it produces lung tumors that progress from hyperplasia to adenocarcinoma in a manner very similar to the human disease; iv) tobacco induced adenocarcinoma of the lung is currently the most frequent form of lung cancer, and lung cancer is the leading cause of cancer mortality in the world (12, 13). Here we provide the first in vivo proof for the existence of a translational control checkpoint in lung tumor defense, whose integrity depends on the 4E-BP family of proteins. We further show that the 4E-BP deficient state per se – a condition now known to prevail in many human cancers – leads to a genome-wide skewing of the molecular landscape as reflected by translational activation of genes for angiogenesis, growth and proliferation.
Materials and Methods
NNK model of lung carcinogenesis
Protocol for NNK administration was approved by University of Minnesota IACUC. Balb/c mice doubly knocked out for Eif4ebp1 and Eif4ebp2 were generously provided by Dr. Sonenberg (14). Wild type Balb/c mice were purchased from Charles River Laboratories (Wilmington, MA) and the A/J mice were purchased from Jackson Laboratory (Bar Harber, ME). At 5 weeks age, mice were started on AIN-93M purified diet (Research Diets, New Brunswick, NJ) until the end of the experiment. At 6 weeks age, mice were injected i.p. with NNK (Toronto Research Chemicals Inc, Ontario, Canada) at doses of 0 (vehicle only), 0.5, 1, 2, and 4 mmol/kg body weight. At 28 weeks post NNK injection, all mice were sacrificed for analysis.
Histological analysis
Lungs were formalin fixed, paraffin embedded, and sectioned for histological analysis. The University of Minnesota Cancer Center Histology Core performed H&E for histological grade distribution; and the Anatomic Pathology Core performed Ki-67 (Lab Vision, Fremont, CA) for proliferation, activated caspase-3 (Calbiochem, San Diego, CA) for apoptosis and CD31 (Biocare Medical, Concord, CA) for microvessel quantification.
Western blot analysis
Lung tissue was flushed with PBS and homogenized before cells were lysed with RIPA buffer (150mM NaCl, 6mM NaHPO4, 4mM NaH2PO4, 2mM EDTA, 1% NaDOC, 1% NP40, 0.1% SDS) supplemented with Protease Inhibitor Cocktail Tablets (Roche, Switzerland), phosphatase inhibitors Na3OV4 (1mM) and beta-glycerophosphate (50mM). The protein content of lysates was quantified and equal amounts resolved by 15% SDS-PAGE. Proteins were transferred onto nitrocellulose and analyzed for eIF4E, eIF4G, 4E-BP1, 4E-BP2, and 4E-BP3. Antibodies for eIF4E (BD Biosciences, San Diego, CA) and 4E-BP1 (Abcam, Cambridge, MA) were purchased, and antibodies for 4E-BP2 and 4E-BP3 were provided by Dr. Sonenberg.
NNK metabolism by lung microsomes
Mouse lung microsomes (MLM) were prepared according to a previously published protocol (15). 20 μM [5-3H]NNK was incubated with MLM (100 μg protein) for 30 min at 37 °C in a reaction mixture containing 50 mM Tris buffer, pH 7.4, and a NADPH generating system (GS) (0.4 mM NADP+, 100 mM glucose-6-phosphate and 0.4 units of glucose-6-phosphate dehydrogenase). Reactions were terminated by the addition of 20 μl each of 0.3 M zinc sulfate and saturated barium hydroxide. The samples were analyzed by reverse-phase HPLC with radioflow detection. A Phenomenex Gemini C18 column (3.9 × 300 mm; 10 μm; Torrance, CA) was used to separate the NNK metabolites. The analytes were eluted by a linear gradient from 100% A (20 mM sodium phosphate, pH 7.0) to 70% A and 30% B (methanol) over 60 min and then to 50% B over 10 min at a flow rate of 1 ml/min. The coumarin 7-hydroxylation activity in the MLM was analyzed according to a previously published method (16). 20 μg MLM was incubated with 20 μM coumarin for 20 minutes at 37 °C in the presence of GS. The reaction was terminated with 15% TCA and the formation of 7- hydroxycoumarin measured by reverse phase HPLC with fluorescence detection.
DNA isolation from lungs for adduct analysis
5 week old wild type and 4ebp1−/−/4ebp2−/− mice were started on AIN-93M diet. At 6 weeks of age, mice were injected i.p with NNK (2 mmol/kg body weight). Mice were sacrificed at 5 time points post injection: 1h, 8h, 24h, 4 days and 9 days. Lungs were isolated and protein digested with Proteinase K overnight. RNA, DNA and protein were separated using TRIZOL (Invitrogen, Carlsbad, California). The RNA layer was removed and DNA precipitated using ethanol.
Pyridyloxobutylation Adduct Analysis
Pyridyloxobutylation adduct analysis was performed as previously described with minor modifications (17). Genomic DNA (150 μg) was dissolved in buffer (10mM Sodium Succinate 5mM CaCl2, pH 7.0) and subjected to neutral thermal hydrolysis at 100 °C for 30 min. Micrococcal nuclease phosphodiesterase II (Worthington Biochemical, New Jersey) and alkaline phosphatase (Roche, Switzerland) was used to digest DNA to individual nucleosides. Samples were loaded onto Strata-X cartridges (Phenomenex, California) for solid phase extraction. LC-ESI-MS/MS analysis was carried out with an Agilent 1100 capillary flow HPLC (Agilent Technologies, Palo Alto, CA) equipped with a Luna 5 μm C18 column (Phenomenex) and coupled to a Discovery Max (ThermoElectron, San Jose, CA) triple quadrupole mass spectrometer. The solvent elution program was a gradient from 5 to 65% CH3OH in 15 mM NH4OAc buffer in 30 min at a flow rate of 10 μL/min at 30 °C.
O6-methylguanine Adduct Analysis
Genomic DNA (100 μg) was dissolved in 10 mM sodium phosphate (NaPO4,pH = 7) and heated at 95 °C for 30 min. Internal standard, d3-O6-methylguanine (300 fmol), was added. A 10% volume of 1N HCl was added and acid hydrolysis performed at 80°C for 1 hour. Samples were filtered through Ultracel YM-10 filters (Millipore, Massachusetts) after neutralization with an equal volume of 0.2N ammonium hydroxide. Samples were dried and re-suspended in 16 μl 25 mM ammonium acetate buffer. Half the sample volume was used for capillary HPLC LC/MS/MS analysis. The analysis was performed on an Agilent Technologies 1100 series capillary HPLC system interfaced with a Finnigan Quantum Discovery triple quadrupole mass spectrometer. HPLC separation was achieved using a Phenomenex Synergy 4μ Hydro-RP column (250 × 0.5 mm, 4 micron) with a flow rate of 12 μl/min and a column temperature of 40 °C. The mobile phase consisted of 25 mM ammonium acetate (A) and methanol (B). O6-methylguanine was eluted using a gradient from 5% B to 27% B in 13 min then to 40% B in 1 minute with a hold of 3 minutes before going back to initial conditions. The mass spectrometer was operated in positive ESI mode using selective ion monitoring as previously described (18).
Polyribosome-microarray analysis
Mouse lungs were removed and processed for total and polyribosome bound RNA as previously described (19) (n=6). Polyribosome microarray analysis was performed as previously described (20). The polyribosome stratified RNA was fractionated and ten, 0.5 ml fractions were collected into tubes containing 50 μl 10% SDS. RNA in the fractions was purified using Tri-reagent (Sigma) and fractions 7 to10 were combined into the “heavy” fraction (3 or more ribosomes per transcript). 100 ng of sample RNA was labeled utilizing the Affymetrix Two-Cycle Target Labeling and Control Reagent Kit. Samples were hybridized onto Affymetrix Murine 430A 2.0 arrays using the FS450 Fluidics Work Station and were scanned using the Gene Chip Scanner 3000 by the University of Minnesota Biomedical Genomics Center.
Data analysis
All data analysis was performed in the statistical environment “R”1 or the programming language “Perl” using the packages indicated. To assure data quality, we performed several levels of quality control. First we assessed technical parameters including RNA integrity and scaling factor (21) using the Simpleaffy package (22). At this step, one sample was removed due to reduced RNA integrity. The remaining samples were normalized with GCRMA using updated probe set definitions (23) “RefSeq version 10” (24). To test how well the biological conditions were replicated, we used Principal Components Analysis and found that the samples clustered according to biological origin within the two first components, indicating high data set quality. We identified genes as differentially expressed using Significance Analysis of microarrays (25). We performed a non-paired analysis using a fixed random seed (1, 2, 3, 4, 5, 6, 7, 8, 9) and a large delta table (400) using the package Samr v1.25. Three data sets were assessed for differential expression between the 4ebp1−/−/4ebp2−/− and wild type mice: transcriptional data, translational data and translational data that had been corrected for transcription. The correction was performed by subtracting the log2 mean signal derived from the transcriptional sample from each translational sample according to sample class. To identify genes that were statistically differentially translated, we filtered for genes that showed differential expression (FDR<5%) both at the translational level and in the transcriptionally corrected translational data set. To identify pathways that were enriched either among translationally activated or inactivated genes, we used the GO::Termfinder v0.72 module (26). The annotation used by GO::Termfinder from the Gene Ontology consortium (27) had been modified to also include miRNA target sites as described previously (20). Categories that showed a FDR<5% were considered significant. The data set has been deposited at Gene Expression Omnibus accession GSE17406.
RT-qPCR
We followed established procedures we have previously described (28). Real-time PCR was performed using the Roche Light-Cycler with SYBR Green (Roche). Primer sequences specific to Cytochrome C, CYP2A5, and VEGFa were as follows: Cytochrome C Forward: ACCTGGTGGGAGTGTGCTAC Reverse: CATCCTGGACCTCCACCTC. CYP2A5 Forward: CACTGCTTCGAATGATGCTG, Reverse: GAGTCGATGAAGTCCCTTGG. VEGFa Forward: GATCATGCGGATCAAACCTC, Reverse: TTTCTGGCTTTGTTCTGTCTTTC. For all RT-qPCR, an annealing temperature of 60°C was used.
Statistical Analysis
For statistical analyses, ANOVA, Wilcoxon rank sum or the student’s t-test was used with 2-tails and unequal variance expressed as mean ± s.e.m. unless otherwise stated.
Results
4ebp1−/−/4ebp2−/− mice manifest increased sensitivity to NNK-induced lung tumorigenesis
To determine whether the 4E-BPs function at a translational checkpoint in lung tumor defense, we performed a dose ranging study in Balb/c mice genetically engineered to be null for Eif4ebp1 and Eif4ebp2 (14). At baseline, these mice have normal levels of eIF4E, eIF4E binding partner eukaryotic translational initiation factor 4G (eIF4G), and the remaining 4E-BP family member, 4E-BP3 (Fig. 1A). At the highest dose of NNK examined (4 mmol NNK/kg body weight), both wild type and null mice developed typical milky white lung surface tumors (Fig. 1B). These lesions were indistinguishable from tumors produced in the more commonly used A/J strain of mice (12) which served as a positive control (not shown). However, when the dose-response characteristics of 4ebp1−/−/4ebp2−/− mice were compared to wild type, 4ebp1−/−/4ebp2−/− mice manifested a significantly increased sensitivity to the tumorigenic effects of NNK (Fig. 1C). For Balb/c mice, independent of genotype, lung tumors were infrequently seen up to 1 mmol NNK/kg body weight. However, a 5-fold increase in tumor frequency was apparent in 4ebp1−/−/4ebp2−/− mice at 2 mmol NNK/kg body weight (P=0.00018) with convergence of tumor frequencies at the highest dose examined. These data indicate that the lack of 4E-BP1 and 4E-BP2 sensitizes mice to the tumorigenic effects of NNK.
Figure 1.

NNK induced lung tumorigenesis. A. Steady state levels of eIF4G, eIF4E, 4E-BP1, 4E-BP2, 4E-BP3 and actin in lung extracts from 4ebp1−/−/4ebp2−/− (left) and wild type (right) mice. B. Representative surface tumors in lungs from NNK treated 4ebp1−/−/4ebp2−/− (left) and wild type (right) mice. C. Lung surface tumor frequency as a function of NNK dose (n=15, p=0.00018, student’s t-test).
Tumors from 4ebp1−/−/4ebp2−/− mice have increased microvessel density
4ebp1−/−/4ebp2−/− and wild type tumors were analyzed to determine if their morphological pattern or cytokinetic properties differed. In accord with the published literature (13), histological analysis showed that all tumors were of the adenocarcinoma subtype, with all stages of tumor progression - hyperplasia, adenoma, and adenocarcinoma – represented. The distribution ratio of these lesions in wild type and 4ebp1−/−/4ebp2−/− mice did not differ (Fig. 2A). To define the cytokinetic properties of tumors, we carried out immunohistochemical analysis to quantify the frequency of proliferating (Ki-67) and apoptotic (caspase-3) cells. Analysis of proliferating cells showed no difference between 4ebp1−/−/4ebp2−/− and wild type tumors (Fig. 2B). The frequency of apoptotic cells was negligible in all tumors (not shown). We also performed an immunohistochemial quantification of tumor vascularity using the endothelial determinant CD31 which showed that microvessel density was increased approximately 5-fold in 4ebp1−/−/4ebp2−/− tumors (Fig. 2C). Given the increased microvessel density in 4ebp1−/−/4ebp2−/− mice, we analyzed baseline (i.e. pre-NNK treatment) levels of VEGF; a potent angiogenic factor that is tightly controlled at the translational level (29). Quantitative PCR analysis showed that although the total abundance of the Vegf transcript did not differ between 4ebp1−/−/4ebp2−/− and wild type mice, there was a 3-fold increase in the amount of Vegf transcript bound to polyribosomes (≥ 3 bound ribosomes) in the 4ebp1−/−/4ebp2−/− mice (Fig. 2D). This indicated translational activation of VEGF in the 4ebp1−/−/4ebp2−/− mice at baseline before NNK exposure, providing an explanation for their propensity to develop tumors with a higher density of microvessels.
Figure 2.

Histology, microvessel density and cytokinetic properties of lung tumors. A. Lung lesions were analyzed for the frequency of hyperplasia, adenoma, and adenocarcinoma. B. Proliferation. Adenomas were analyzed for Ki-67 positive nuclei. C. Microvessel density. Shown is the number of CD31 positive pixels per tumor (resolved at 400x; n = 7, p=0.046, student’s t-test) in adenomas. D. Quantitative PCR analysis of VEGF in mouse lung tissue at baseline before NNK treatment. Shown is the quantity of total cellular and polyribosome bound VEGF RNA in arbitrary fluorescence units (n = 4, p= 0.0065, Wilcoxon rank sum test)
The mutagenic potency of NNK is increased in 4ebp1−/−/4ebp2−/− mice
Target tissue metabolism of NNK by members of the cytochrome P450 superfamily of enzymes converts it into active metabolites that form adducts with nucleotide bases in DNA (30). As a consequence, in addition to losing the ability to restrain eIF4F under oncogenic stress, 4ebp1−/−/4ebp2−/− mice might also have baseline changes in the translational machinery that make them more efficient at catalyzing NNK into carcinogens. To address this possibility, we measured the extent of NNK α-hydroxylation in wild type and 4ebp1−/−/4ebp2−/− lung microsomes. NNK α-hydoxylation ultimately results in the formation of methane diazohydroxid and 4-(3-pyridyl)-4-oxobutane-1-diazohydroxide which reacts with DNA leading to mutations (Fig. 3A). The diazohydroxides are reactive and cannot be directly measured. Instead we measured the formation of ketoaldehyde (4-hydroxy-1-(3-pyridyl)-1-butanone, OPB) and keto alcohol (4-oxo-1-(3-pyridyl)-1-butanone, HPB), which are formed in a 1:1 ratio with the diazohydroxides (13). 4ebp1−/−/4ebp2−/− mice had a nearly 2-fold increase in OPB and HPB compared to wild type mice, whereas non-P450 mediated carbonyl reduction of NNK to NNAL was unchanged (Fig. 3B). Enzymes from the P450 2A subfamily, in particular CYP2A5, are the most efficient catalysts of NNK α-hydroxylation in mouse lung (13). To determine if the α-hydroxylation activity was an isolated phenomenon or a reflection of a generalized increase in P450 2A activity, we examined the production of 7-hydroxy coumarin from coumarin, a CYP2A5 specific reaction. In accord with the data for NNK metabolism, coumarin 7-hydroxylation was also increased 2-fold in 4ebp1−/−/4ebp2−/− mice. These findings indicate that the increased sensitivity of 4ebp1−/−/4ebp2−/− mice to NNK carcinogenesis might not only result from a biological defect in tumor defense, but also from metabolic alterations leading to a higher effective dose of electrophiles from NNK.
Figure 3.
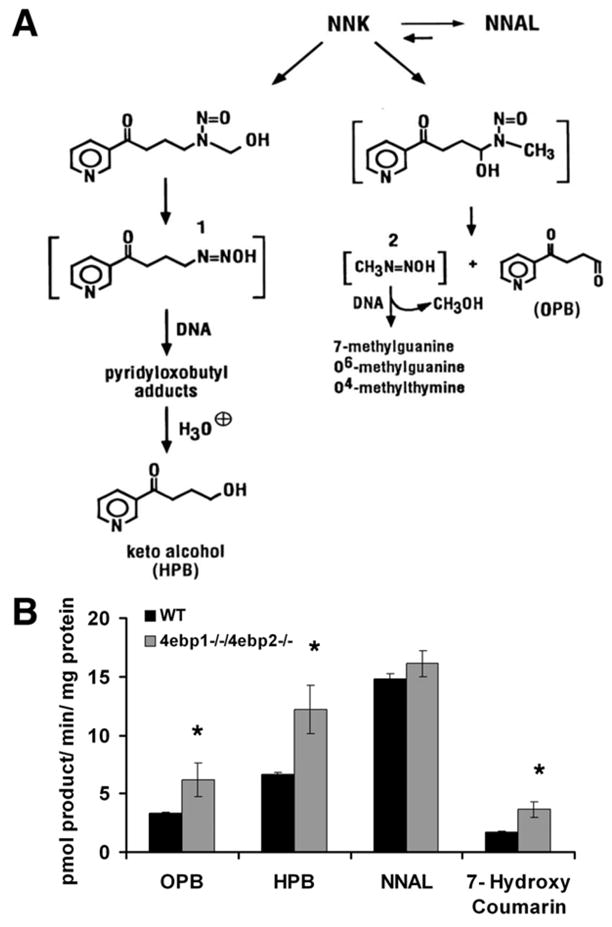
NNK metabolism. A. NNK Metabolism. Depicted are the pathways showing the conversion of NNK to 1. 4-(3-pyridyl)-4-oxobutane-1-diazohydroxide and 2. methane diazohydroxide. B. Quantification of NNK and coumarin metabolism. Shown are the end products of NNK metabolism (OPB, HPB, and NNAL) and the CYP2A5 specific metabolism of coumarin to 7-hydroxy coumarin. (n=3, * designates p < 0.05, student’s t- test)
As a direct measure of NNK mutagenicity, we next quantified 2 major classes of DNA adducts methylation (O6-methylguanine) and pyridyloxobutylation (POB: O2-POB-thymidine and 7-POB-guanine) (31, 32) - in the lungs of mice for up to 9 days after administration of 2 mmol NNK/kg body weight (the dose generating the largest difference in tumor count between wild type and 4ebp1−/−/4ebp2−/−). While the pattern of O2-POB-thymidine and 7-POB-guanine adducts was independent of genotype, O6-methylguanine adducts were more abundant in the 4ebp1−/−/4ebp2−/− mice - reaching 3-fold higher levels at 96 hours (p=0.012) after NNK injection, before declining to the wild type levels at 9 days (Fig. 4; 2-way ANOVA for pattern comparison, student’s t-test to compare the wild type and null value at each time point). This result indicates that the potency of NNK as a mutagen was altered by genotype, and that the increased tumor frequency seen in 4ebp1−/−/4ebp2−/− mice might not only result from an impaired ability to negatively regulate eIF4F during an oncogenic stress, but also from an enhanced vulnerability to NNK mutagenicity due to baseline changes in the translational machinery.
Figure 4.
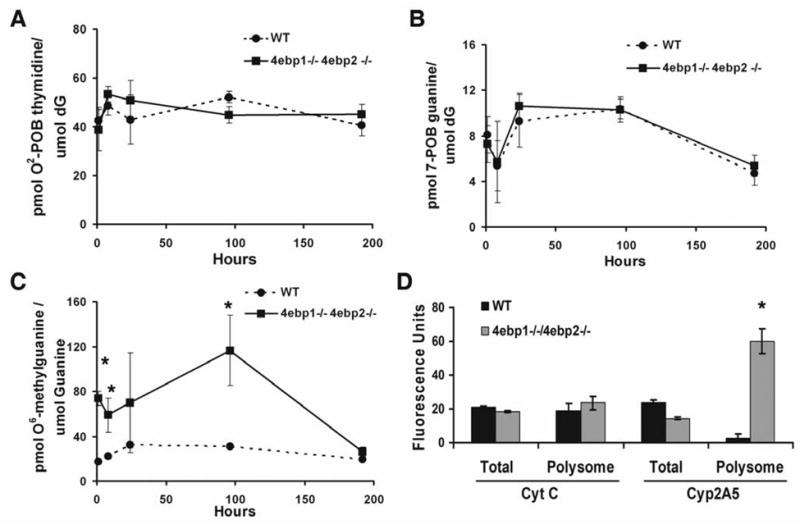
DNA adduct formation. A. O2-POB-thymidine adducts; B. 7-POB-guanine adducts; C. O6-methylguanine adducts (n=4, p < 0.0001, 2-way ANOVA; student’s t-test for individual time points denoted by asterisk). D. Quantitative PCR analysis of cytochrome P450 2A5 in mouse lung tissue at baseline before NNK treatment. Shown is the quantity of total and polyribosome bound CYP2A5 RNA in arbitrary fluorescence units (n = 4, p = 0.034, Wilcoxon rank sum test)
To explore this possibility, we analyzed baseline, pre-NNK treatment expression of CYP2A5 in the mouse lung. Since available immunological reagents do not distinguish between CYP2A4 and CYP2A5 (CYP2A4 is a poor bioactivater of NNK), we quantified Cyp2a5 transcript abundance and ribosome binding using RT-qPCR. Steady state levels of Cyp2a5 transcript did not differ between wild type and 4ebp1−/−/4ebp2−/− mice; however, we observed a striking, 40-fold increase in the quantity of Cyp2a5 transcript present in the polyribosome bound pool (Fig. 4D). This dramatic translational activation of CYP2A5 at baseline provides a molecular mechanism for the increased NNK bioactivation and adduct formation we observed in the 4ebp1−/−/4ebp2−/− mice.
Translationally activated genes in 4ebp1−/−/4ebp2−/− mice are skewed towards neoplastic functions
The 4E-BPs function by competing with eIF4G for eIF4E binding (33). In principle, this implies that the pattern of ribosome recruitment to all transcripts in the cell could be altered by knockout of 4E-BP1 and 4E-BP2. However, recent genome-wide studies of ribosome recruitment in vitro identify key potentially oncogenic transcripts as being disproportionately affected when eIF4E is ectopically over expressed (20, 28, 34); therefore we reasoned the same might be true when 4E-BP inhibition of eIF4E was diminished.
To define the pattern of ribosome recruitment, we performed combined polyribosome-microarray analysis of RNA (both total RNA and actively translating RNA) obtained from the lungs of wild type and 4ebp1−/−/4ebp2−/− mice in the basal state, before NNK administration. To perform a pathway-oriented analysis, we examined the genome-wide dataset at three levels: transcriptional (total RNA), translational (polyribosome RNA) and the translational level after correction for transcription (an in silico correction of the data from polyribosome RNA). We identified those genes that were differentially expressed at both the translational level and the translational level corrected for transcription. This analysis yielded a set of genes which are regulated more at the translational level compared to the transcriptional level. We separated the genes into those that were activated at the translational level and those that were inactivated and looked for enrichment of any pathway defined by the Gene Ontology consortium. This analysis determines if a pathway is under active translational control, and not if the activity is driven by positive or negative regulators of the pathway’s function. In addition, this analysis does not determine which changes in ribosome recruitment represent primary effects of the 4ebp1−/−/4ebp2−/− genotype, and which represents compensatory feed-back or feed-forward effects.
As might be anticipated, we found 35 translationally activated pathways and only 2 inactivated pathways in 4ebp1−/−/4ebp2−/− mice (Table 1). Strikingly, the majority of translationally activated genes belonged to pathways in only 2 categories: growth and proliferation – both cancer-defining properties. Some of the genes belonging to the proliferation pathways were genes promoting proliferation (e.g. Cyclin D1), while others were genes restraining proliferation (e.g. Rb). The remaining genes belonged to the transcription category; a category that - while not cancer-specific - reveals how alterations in translational control can directly influence gene expression at the transcriptional level. Of note, the 2 translationally down regulated pathways appeared to be non-specific (cellular component and nuclear-ER network). These data disclose a significant skewing of baseline ribosome recruitment to cancer-related transcripts in 4ebp1−/−/4ebp2−/− mice.
Table 1.
Gene Ontology Analysis of all genes enriched in the polyribosome bound pool of 4ebp1−/−/4ebp2−/− mice (FDR < 5%)
| Function | FDR | P - VALUE | ENRICHMENT |
|---|---|---|---|
| Growth and Metobolism | |||
| biopolymer metabolic process | 0.00% | 1.2E-07 | 1.51 |
| nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 0.00% | 2.0E-06 | 1.57 |
| RNA metabolic process | 0.40% | 1.7E-05 | 1.59 |
| mRNA metabolic process | 0.80% | 1.3E-04 | 3.03 |
| regulation of macromolecule metabolic process | 1.09% | 1.4E-04 | 1.54 |
| regulation of cellular metabolic process | 1.85% | 2.2E-04 | 1.51 |
| regulation of metabolic process | 1.89% | 2.9E-04 | 1.50 |
| regulation of nucleoside, nucleotide and nucleic acid metabolic process | 2.78% | 5.1E-04 | 1.51 |
| macromolecule biosynthetic process | 0.50% | 5.4E-05 | 1.50 |
| biopolymer biosynthetic process | 0.67% | 3.7E-05 | 1.58 |
| regulation of biosynthetic process | 1.79% | 3.0E-04 | 1.52 |
| regulation of macromolecule biosynthetic process | 2.13% | 2.5E-04 | 1.53 |
| RNA biosynthetic process | 2.53% | 6.8E-04 | 1.50 |
| Proliferation | |||
| cell division | 0.67% | 7.2E-06 | 3.50 |
| M phase of mitotic cell cycle | 2.50% | 7.7E-04 | 3.36 |
| mitosis | 2.55% | 6.6E-04 | 3.43 |
| mitotic cell cycle | 4.39% | 1.3E-03 | 2.79 |
| cell cycle | 4.51% | 1.2E-03 | 1.89 |
| chromosome condensation | 4.54% | 1.3E-03 | 12.73 |
| Transcription | |||
| nucleic acid binding | 0.00% | 1.6E-06 | 1.60 |
| nucleus | 0.00% | 1.6E-13 | 1.75 |
| zinc ion binding | 0.00% | 2.9E-06 | 1.76 |
| gene expression | 0.50% | 1.1E-05 | 1.55 |
| regulation of gene expression | 1.80% | 4.3E-04 | 1.50 |
| transcription | 1.88% | 2.5E-04 | 1.53 |
| transcription, DNA-dependent | 2.64% | 6.4E-04 | 1.51 |
| regulation of transcription | 2.74% | 5.9E-04 | 1.50 |
| mRNA processing | 0.44% | 9.5E-05 | 3.28 |
| RNA splicing | 2.45% | 7.1E-04 | 3.39 |
| nuclear mRNA splicing, via spliceosome | 2.46% | 5.1E-04 | 3.54 |
| RNA splicing, via transesterification reactions | 2.67% | 5.1E-04 | 3.54 |
| RNA processing | 2.71% | 8.6E-04 | 2.37 |
| RNA binding | 4.00% | 1.1E-03 | 2.06 |
| Others | |||
| ribonucleoprotein complex assembly | 2.00% | 4.7E-04 | 4.95 |
| protein prenyltransferase activity | 4.11% | 1.0E-03 | 8.49 |
Discussion
The concept that sustained activation of cap-dependent translation initiation is oncogenic is now firmly established (5–7). Unproved was its logical corollary - that a translational control checkpoint exists in the tumor defense system, and that the 4E-BP protein family functions at this checkpoint. Using mice genetically engineered to lack two of the three 4E-BP family members; and a high fidelity tobacco carcinogen mouse model of lung cancer, here we report that 4E-BP deficiency sensitizes mice to NNK-induced lung tumorigenesis, establishing a role for the 4E-BPs at a translational control checkpoint in lung tumor defense.
Evidence for the role of eIF4F activation in cancer has been accumulating for more than a decade (35, 36). Since the 4E-BPs are the primary negative regulators of eIF4F function, our hypothesis that 4ebp1−/−/4ebp2−/− mice would be sensitized to NNK-induced lung tumorigenesis had strong antecedent experimental grounding (7, 8). Indeed, as we predicted, our study shows an increase in the sensitivity of 4ebp1−/−/4ebp2−/− mice to NNK, establishing a role for the 4E-BP family of cap-dependent translation repressors in tumor defense.
Morphological analysis of tumors revealed a striking increase in tumor microvessel density in 4ebp1−/−/4ebp2−/− mice; a result identifying a role for the 4E-BPs in restraint of tumor neovascularization. Activation of eIF4F is known to selectively activate translation of the angiogenic protein VEGF (29). At baseline before NNK treatment, we found increased recruitment of ribosomes to Vegf mRNA in 4ebp1−/−/4ebp2−/− mice; creating a primed state for increased neovascularization of NNK-triggered tumors. However, we note that translational control of angiogenesis in not simply unidirectional. In breast carcinoma, high level over expression of 4E-BP1 promotes angiogenesis by triggering a switch from cap-dependent to internal ribosome entry site (IRES) mediated translation (37). This leads to translational activation of VEGF which can be translated in a cap-independent as well as a cap-dependent manner – giving it a selective advantage to recruit ribosomes when cap-dependent translation is repressed. These data highlight the importance of translational control in governing the propensity of tumors to progress by recruiting a circulation, and provide a molecular mechanism for the increased vascularity we observed in the lung tumors from 4ebp1−/−/4ebp2−/− mice.
To elucidate the molecular mechanism by which 4E-BP deficiency sensitizes mice to NNK, we analyzed the baseline lung RNA ribosome recruitment pattern genome-wide. There was a marked skewing of the 4ebp1−/−/4ebp2−/− molecular landscape towards translational activation of genes governing growth and proliferation; as well as for genes related to transcription. These data are in accord with genome-wide in vitro studies that reveal selective translation of genes belonging to proliferation pathways - and those governing gene expression itself - when the translation initiation machinery is activated (20, 38). Our findings reveal what may be considered a cancer-primed state in 4ebp1−/−/4ebp2−/− mice, since unstressed mice do not show a cancer phenotype, but do have a translational profile for key cancer related functions that is significantly different from normal; and display a lower NNK-threshold for tumorigenesis. Based on our findings, it is tempting to speculate that a similar shift in the global translational profile towards cancer may be recapitulated in the smoker’s lung; a possibility in accord with the clinical finding that hyperphosphorylated 4E-BP1 (i.e. the inactive form) is a negative prognostic factor in breast, ovarian, and prostate cancer (39).
Our most unexpected finding was that 4ebp1−/−/4ebp2−/− mice were more efficient at catalyzing NNK into carcinogenic metabolites, and that this resulted in increased methylated DNA adducts. The mechanism involved translational activation of CYP2A5, the primary enzyme catalyzing NNK into its carcinogenic metabolites in the lung (40). Thus we have identified a novel role for the 4E-BPs in regulating the cytochrome P450 enzyme system – a finding with important clinical implications. Combining the published literature with our current findings, we may have unveiled a pernicious feed-forward loop that can operate in human smokers (Fig. 5A). When smoking commences, physiological levels of CYP2A5 in the lung catalyze NNK into active carcinogens forming DNA adducts, preferentially creating codon 12 gain-of-function mutations in RAS (41). Oncogenic Ras activates the PI3K/Akt/mTOR pathway which in turn phosphorylates the 4E-BPs (42), i.e. a 4ebp1−/−/4ebp2−/− phenocopy. We show here that CYP2A5 is translationally activated in the 4E-BP deficient state, increasing the catalysis of NNK into carcinogenic metabolites, resulting in accelerated accumulation of DNA adducts (Fig. 5B). We speculate that such a feed-forward loop in human smokers may help explain the carcinogenic potency of cigarette smoke and the exponential relationship between lung cancer incidence and smoking duration (43, 44).
Figure 5.

Hypothetical model for how the translational control checkpoint is disabled in human lung cancer. A. Mouse: Knockout of 4E-BP1 and 4E-BP2 leads to steady state translational activation of genes governing growth, proliferation, angiogenesis and CYP2A5, the enzyme bioactivating NNK. B. Human smokers: Exposure to NNK causes mutations leading to Ras/PI3K/Akt/mTOR pathway activation. This inactivates the 4E-BP system and creates a 4E-BP null phenocopy. A potential feed-forward loop is set up in which the genome-wide translational profile is shifted to activate genes driving growth, proliferation, angiogenesis and NNK bioactivation; so that the target cell is pushed further along the cancer pathway and NNK is a more potent mutagen.
Although our study does not definitively establish a role for the 4E-BP family of proteins in defense against malignancies other than adenocarcinoma of the lung; together with the published literature, it strongly suggests a more general role in cancer defense. Our findings, therefore, provide impetus for current efforts to develop new classes of cancer therapeutics aimed at normalizing translational control and reestablishing integrity of the translational control checkpoint. These efforts include mTOR inhibitors designed to restore 4E-BP function and compounds targeting the integrity or activity of the eIF4F complex (45–47). Our data also support the potential utility of array-based translational profiling to begin identifying and stratifying at-risk states for the development of lung and other malignancies.
Acknowledgments
This work was supported by the Knut and Alice Wallenberg foundation (O. L.), NIH 5P50DA013333 (Transdisciplinary Tobacco Use Research Center; we would like to thank Dorothy Hatsukami, PI for one of the pilot funding awards to P.B.B.), and NIH T32 HL07741 (Y.Y.K). We would like to thank Dr. Sonenberg for his generous donation of the 4ebp1−/−/4ebp2−/− mice and the 4E-BP2 and 4E-BP3 antibodies. We would also like to thank the University of Minnesota Cancer Center Pathology Core for the histological analyses; and the Minnesota Super Computer Institute.
Footnotes
References
- 1.Nathan CA, Carter P, Liu L, et al. Elevated expression of eIF4E and FGF-2 isoforms during vascularization of breast carcinomas. Oncogene. 1997;15:1087–94. doi: 10.1038/sj.onc.1201272. [DOI] [PubMed] [Google Scholar]
- 2.Li BD, Liu L, Dawson M, De Benedetti A. Overexpression of eukaryotic initiation factor 4E (eIF4E) in breast carcinoma. Cancer. 1997;79:2385–90. [PubMed] [Google Scholar]
- 3.Coleman LJ, Peter MB, Teall TJ, et al. Combined analysis of eIF4E and 4E-binding protein expression predicts breast cancer survival and estimates eIF4E activity. Br J Cancer. 2009;100:1393–9. doi: 10.1038/sj.bjc.6605044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seki N, Takasu T, Mandai K, et al. Expression of eukaryotic initiation factor 4E in atypical adenomatous hyperplasia and adenocarcinoma of the human peripheral lung. Clin Cancer Res. 2002;8:3046–53. [PubMed] [Google Scholar]
- 5.Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 6.Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 7.Avdulov S, Li S, Michalek V, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell. 2004;5:553–63. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 8.Jacobson BA, Alter MD, Kratzke MG, et al. Repression of cap-dependent translation attenuates the transformed phenotype in non-small cell lung cancer both in vitro and in vivo. Cancer Res. 2006;66:4256–62. doi: 10.1158/0008-5472.CAN-05-2879. [DOI] [PubMed] [Google Scholar]
- 9.De Benedetti A, Harris AL. eIF4E expression in tumors: its possible role in progression of malignancies. Int J Biochem Cell Biol. 1999;31:59–72. doi: 10.1016/s1357-2725(98)00132-0. [DOI] [PubMed] [Google Scholar]
- 10.Zimmer SG, DeBenedetti A, Graff JR. Translational control of malignancy: the mRNA cap-binding protein, eIF-4E, as a central regulator of tumor formation, growth, invasion and metastasis. Anticancer Res. 2000;20:1343–51. [PubMed] [Google Scholar]
- 11.Raught B, Gingras AC. eIF4E activity is regulated at multiple levels. Int J Biochem Cell Biol. 1999;31:43–57. doi: 10.1016/s1357-2725(98)00131-9. [DOI] [PubMed] [Google Scholar]
- 12.Belinsky SA, Devereux TR, Foley JF, Maronpot RR, Anderson MW. Role of the alveolar type II cell in the development and progression of pulmonary tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the A/J mouse. Cancer Res. 1992;52:3164–73. [PubMed] [Google Scholar]
- 13.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 14.Le Bacquer O, Petroulakis E, Paglialunga S, et al. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest. 2007;117:387–96. doi: 10.1172/JCI29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, D’Agostino J, Wu H, et al. CYP2A13: variable expression and role in human lung microsomal metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. J Pharmacol Exp Ther. 2007;323:570–8. doi: 10.1124/jpet.107.127068. [DOI] [PubMed] [Google Scholar]
- 16.von Weymarn LB, Felicia ND, Ding X, Murphy SE. N-Nitrosobenzylmethylamine hydroxylation and coumarin 7-hydroxylation: catalysis by rat esophageal microsomes and cytochrome P450 2A3 and 2A6 enzymes. Chem Res Toxicol. 1999;12:1254–61. doi: 10.1021/tx990128y. [DOI] [PubMed] [Google Scholar]
- 17.Lao Y, Villalta PW, Sturla SJ, Wang M, Hecht SS. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem Res Toxicol. 2006;19:674–82. doi: 10.1021/tx050351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajesh M, Wang G, Jones R, Tretyakova N. Stable isotope labeling-mass spectrometry analysis of methyl- and pyridyloxobutyl-guanine adducts of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in p53-derived DNA sequences. Biochemistry. 2005;44:2197–207. doi: 10.1021/bi0480032. [DOI] [PubMed] [Google Scholar]
- 19.Mullany LK, Nelsen CJ, Hanse EA, et al. Akt-mediated liver growth promotes induction of cyclin E through a novel translational mechanism and a p21-mediated cell cycle arrest. J Biol Chem. 2007;282:21244–52. doi: 10.1074/jbc.M702110200. [DOI] [PubMed] [Google Scholar]
- 20.Larsson O, Li S, Issaenko OA, et al. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814–24. doi: 10.1158/0008-5472.CAN-07-0752. [DOI] [PubMed] [Google Scholar]
- 21.Larsson O, Sandberg R. Lack of correct data format and comparability limits future integrative microarray research. Nat Biotechnol. 2006;24:1322–3. doi: 10.1038/nbt1106-1322. [DOI] [PubMed] [Google Scholar]
- 22.Wilson CL, Miller CJ. Simpleaffy: a BioConductor package for Affymetrix Quality Control and data analysis. Bioinformatics. 2005;21:3683–5. doi: 10.1093/bioinformatics/bti605. [DOI] [PubMed] [Google Scholar]
- 23.Dai M, Wang P, Boyd AD, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandberg R, Larsson O. Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinformatics. 2007;8:48. doi: 10.1186/1471-2105-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyle EI, Weng S, Gollub J, et al. GO::TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–5. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Gene Ontology project in 2008. Nucleic Acids Res. 2008;36:D440–4. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsson O, Perlman DM, Fan D, et al. Apoptosis resistance downstream of eIF4E: posttranscriptional activation of an anti-apoptotic transcript carrying a consensus hairpin structure. Nucleic Acids Res. 2006;34:4375–86. doi: 10.1093/nar/gkl558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung J, Bachelder RE, Lipscomb EA, Shaw LM, Mercurio AM. Integrin (alpha 6 beta 4) regulation of eIF-4E activity and VEGF translation: a survival mechanism for carcinoma cells. J Cell Biol. 2002;158:165–74. doi: 10.1083/jcb.200112015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crespi CL, Penman BW, Gelboin HV, Gonzalez FJ. A tobacco smoke-derived nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, is activated by multiple human cytochrome P450s including the polymorphic human cytochrome P4502D6. Carcinogenesis. 1991;12:1197–201. doi: 10.1093/carcin/12.7.1197. [DOI] [PubMed] [Google Scholar]
- 31.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 32.Peterson LA, Hecht SS. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991;51:5557–64. [PubMed] [Google Scholar]
- 33.Mader S, Lee H, Pause A, Sonenberg N. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4 gamma and the translational repressors 4E-binding proteins. Mol Cell Biol. 1995;15:4990–7. doi: 10.1128/mcb.15.9.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mamane Y, Petroulakis E, Martineau Y, et al. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS ONE. 2007;2:e242. doi: 10.1371/journal.pone.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonenberg N. eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. Biochem Cell Biol. 2008;86:178–83. doi: 10.1139/O08-034. [DOI] [PubMed] [Google Scholar]
- 36.Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008;68:631–4. doi: 10.1158/0008-5472.CAN-07-5635. [DOI] [PubMed] [Google Scholar]
- 37.Braunstein S, Karpisheva K, Pola C, et al. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell. 2007;28:501–12. doi: 10.1016/j.molcel.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 38.Provenzani A, Fronza R, Loreni F, Pascale A, Amadio M, Quattrone A. Global alterations in mRNA polysomal recruitment in a cell model of colorectal cancer progression to metastasis. Carcinogenesis. 2006;27:1323–33. doi: 10.1093/carcin/bgi377. [DOI] [PubMed] [Google Scholar]
- 39.Castellvi J, Garcia A, Rojo F, et al. Phosphorylated 4E binding protein 1: a hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer. 2006;107:1801–11. doi: 10.1002/cncr.22195. [DOI] [PubMed] [Google Scholar]
- 40.Felicia ND, Rekha GK, Murphy SE. Characterization of cytochrome P450 2A4 and 2A5-catalyzed 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) metabolism. Arch Biochem Biophys. 2000;384:418–24. doi: 10.1006/abbi.2000.2128. [DOI] [PubMed] [Google Scholar]
- 41.Ronai ZA, Gradia S, Peterson LA, Hecht SS. G to A transitions and G to T transversions in codon 12 of the Ki-ras oncogene isolated from mouse lung tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and related DNA methylating and pyridyloxobutylating agents. Carcinogenesis. 1993;14:2419–22. doi: 10.1093/carcin/14.11.2419. [DOI] [PubMed] [Google Scholar]
- 42.Patel MR, Jacobson BA, De A, et al. Ras pathway activation in malignant mesothelioma. J Thorac Oncol. 2007;2:789–95. doi: 10.1097/JTO.0b013e31811f3aab. [DOI] [PubMed] [Google Scholar]
- 43.Flanders WD, Lally CA, Zhu BP, Henley SJ, Thun MJ. Lung cancer mortality in relation to age, duration of smoking, and daily cigarette consumption: results from Cancer Prevention Study II. Cancer Res. 2003;63:6556–62. [PubMed] [Google Scholar]
- 44.Doll R, Peto R. Cigarette smoking and bronchial carcinoma: dose and time relationships among regular smokers and lifelong non-smokers. J Epidemiol Community Health. 1978;32:303–13. doi: 10.1136/jech.32.4.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lane HA, Wood JM, McSheehy PM, et al. mTOR Inhibitor RAD001 (Everolimus) Has Antiangiogenic/Vascular Properties Distinct from a VEGFR Tyrosine Kinase Inhibitor. Clin Cancer Res. 2009;15:1612–22. doi: 10.1158/1078-0432.CCR-08-2057. [DOI] [PubMed] [Google Scholar]
- 46.Ghosh P, Park C, Peterson MS, Bitterman PB, Polunovsky VA, Wagner CR. Synthesis and evaluation of potential inhibitors of eIF4E cap binding to 7-methyl GTP. Bioorg Med Chem Lett. 2005;15:2177–80. doi: 10.1016/j.bmcl.2005.01.080. [DOI] [PubMed] [Google Scholar]
- 47.Moerke NJ, Aktas H, Chen H, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–67. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]