

Abstract
We introduce the Gaussian-mixture umbrella sampling method (GAMUS), a biased molecular dynamics technique based on adaptive umbrella sampling that efficiently escapes free energy minima in multi-dimensional problems. The prior simulation data are reweighted with a maximum likelihood formulation, and the new approximate probability density is fit to a Gaussian-mixture model, augmented by information about the unsampled areas. The method can be used to identify free energy minima in multi-dimensional reaction coordinates. To illustrate GAMUS, we apply it to the alanine dipeptide (2D reaction coordinate) and tripeptide (4D reaction coordinate).
INTRODUCTION
The free energy surfaces of biomolecular systems are characterized by a large number of local minima (basins) [1-7]. Consequently, approaches to sampling the full surface with simulation times accessible to present-day molecular dynamics (MD) require special methods to escape from the free energy basins. A frequently used approach augments the physical potential by a biasing potential. If the biasing potential is adjusted during the simulation, the probability to explore unvisited areas increases [8-14].
Many biasing methods are based on umbrella sampling [15]. In umbrella sampling, specific values of one or more progress variables, the “reaction coordinate”, are made more probable by a local biasing potential. In a widely used extension of this approach, called adaptive umbrella sampling, the biasing potential is updated during multiple simulations, until the potential is sufficiently uniform so that the system visits all values of the reaction coordinate(s) with equal probability [10, 11]. Modern implementations of the umbrella sampling concept include coordinate flooding [8], hyperdynamics [16] and its variant, accelerated MD [12], and metadynamics [9, 17]. All of these methods focus on escaping from free energy minima. In the coordinate flooding method the potential is augmented by a Gaussian term that moves the trajectory out of the current basin. This flooding potential acts on the lowest frequency principal modes. The biasing potential in metadynamics consists of a history-dependent sum of Gaussians that are centered on previously visited conformations. In hyperdynamics and accelerated MD, the biasing potential is based solely on information from the underlying potential energy surface; in accelerated MD, values of the potential energy below a preset threshold are augmented by a “boost” energy [12]. Related approaches for sampling free energy surfaces can be regarded as outgrowths of thermodynamic integration [18]. Such methods often sample a predetermined, possibly vectorial, reaction coordinate, as in adaptive biasing force sampling [19], adiabatic molecular dynamics [20], and blue moon sampling [21, 22]. Alternatively, an optimal reaction coordinate is determined during the sampling, as in the finite temperature string method [23-26]. We compare and contrast GAMUS with conformational flooding and metadynamics, both of which also introduce Gaussians, in Appendix A.
To obtain physical (“unbiased”) properties from one or more such biased simulations, the statistics need to be reweighted [15, 27, 28]. In adaptive umbrella sampling, the reweighting is now most commonly performed with the weighted histogram analysis method (WHAM) [27, 29-31]. In the WHAM, the probability distributions are described by histograms of a predetermined bin size. The unbiased probability distribution is obtained by minimizing the variance in each of the bins. In practice, both the memory and the sampling requirements associated with binning limit the WHAM to low-dimensional (usually one or two) progress variables. This is due to the fact that larger grids require more memory, and also more sampling for statistical accuracy (each bin needs at least one sample). These limitations restrict the use of biasing methods to low-dimensional systems if thermodynamic properties are desired, or if the biasing depends on the reweighted statistics from previous biasing simulations (which is the case in adaptive umbrella sampling, for example). We note that low dimensional sampling might be enough, in principle, since for diffusive dynamics there exists a “perfect” one-dimensional reaction coordinate that can reproduce the kinetics [32-35]. However, since it is unclear if in practice one can always find such a coordinate, higher dimensional sampling remains important. The results of the latter can also be useful in obtaining the reduction to a one dimensional description of the free energy surface.
We propose a new adaptive umbrella sampling method, the Gaussian-mixture umbrella sampling method (GAMUS), that significantly increases the dimensionality of systems that can be studied. The outline of the algorithm is shown in Figure 1. The method employs a Gaussian-mixture model to fit the probability distribution by multivariate Gaussians, and bypasses the use of bins for the collection and reweighting of the statistics. The method avoids areas of low probability, and will be shown to map efficiently free energy basins as a function of progress variables involving up to four degrees of freedom. This is demonstrated by the application of the method to the alanine dipeptide (CH3CO-Ala-NHCH3) and the alanine tripeptide (CH3CO-Ala2-NHCH3). Although much smaller than proteins, these peptides are good systems for test purposes, since their free energy surface as a function of the backbone dihedral angles can be calculated directly [11, 36]. Both peptides have recently been used to test a method for the calculation of low free energy pathways between two known free energy basins [37]. The goal of the present method is not to connect known free energy basins, but rather to escape from free energy minima. Our test calculations show that GAMUS efficiently explores the conformational space. During the relatively short simulations (4 ns total for each system), many free energy basins were identified for the alanine dipeptide and the alanine tripeptide. For the alanine dipeptide the relative free energies of these basins were correct; no independent free energy data are available for the alanine tripeptide.
Figure 1.
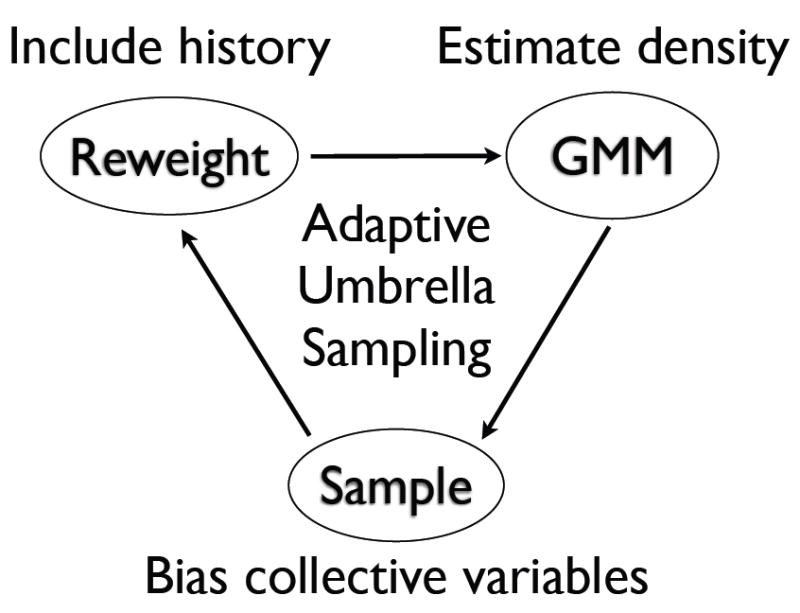
The diagram outlines the adaptive umbrella sampling scheme of GAMUS. The Gaussian mixture model (GMM; top right) receives weighted samples of a set of collective variables and estimates the probability density of those variables. The umbrella sampling (bottom) uses the estimate of this density to bias the sampling of unexplored regions. The reweighting method (top left) combines the most recent samples from the umbrella sampling with samples from previous iterations of GAMUS and updates the weight of each sample.
We stress that the method is designed to explore free energy basins and is less appropriate to describe the location and free energy of the barriers. In GAMUS, the probability density, not the free energy, is fit by the Gaussian-mixture model. We focus on the probability density, since there exists a very efficient algorithm, the expectation-maximization algorithm [38, 39], that can be used for the fit. This algorithm determines the weight, the covariance, and the location of all Gaussians used in the fit in an iterative manner. Fitting the probability density means that the free energy surface, which is obtained from the logarithm of the probability density surface, will be less accurate (will have higher statististical errors) for regions of high free energy. Therefore, we expect the method to be most useful for the identification of free energy basins, and less so for barriers on high dimensional free energy surfaces. Also, while GAMUS quickly locates basins, the convergence of the free energy of these basins takes longer. Once the basins have been identified with GAMUS, accurate free energy differences, and pathways with free energy profiles connecting the basins can be obtained by more specialized methods [24-26, 35, 37, 40, 41], which require a priori knowledge of the basins.
The paper is organized as follows. The Theory section discusses the adaptive umbrella sampling method used here, the reweighting of statistics from the biased simulations, and the construction of the biasing potential from multi-variate Gaussian functions. Applications of the method to the alanine dipeptide and the alanine tripeptide are presented in the Results section, and the conclusions are summarized in the final section. Technical details on the multi-state acceptance ratio method used in the reweighting of the data, and the expectation-maximization method used for the multi-variate Gaussian fit are described in the Appendices.
THEORY
Terminology and definitions
The Hamiltonian H0(X) describes a system with coordinates X. The symbol q is used to designate a vector of collective variables (like the φ and ψ protein dihedral angles); the dimension of q (the number of collective variables) is D. During the GAMUS procedure we collect samples of q and estimate their weight in the canonical ensemble of H0 at temperature T. We reweight (and subsequently fit) N samples from previous iterations of GAMUS; we index these samples with n, so that the n-th sample of q is qn with weight wn. We follow the standard notation and terminology for probabilities in the Bayesian statistics literature [42] and do not discriminate between probabilities and probability densities when such distinctions are clear from the context; we denote all probabilities with the symbol P. The probability density of q for the Hamiltonian H0(X) at temperature T is P(q); the estimate of that same density after the i-th iteration of GAMUS is P(i)(q) — the parenthesis in the subscript serves as a reminder that P(i)(q) is an estimate of P(q) using all available data up to and including iteration i. The number of Gaussians used in the mixture model is M. The m-th Gaussian, centered at μm with covariance matrix Σm, is g(q|μm, Σm); its weight in the mixture model is πm. During the i-th iteration of GAMUS the umbrella sampling proceeds with the Hamiltonian Hi(X), the result of adding to H0(X) the biasing potential Vb,i(q); the canonical partition function corresponding to Hi(X) at temperature T is Zi.
Adaptive umbrella sampling [10, 11]
During the i + 1 iteration of the sampling part of the GAMUS algorithm, the system is propagated by constant-temperature molecular dynamics (or Monte Carlo) subject to the Hamiltonian Hi+1(X):
| (1) |
with Vb,i+1 the biasing potential in the (i + 1)th simulation. This biasing potential is set equal to minus the estimated free energy (or potential of mean force) F(i)(q) from the previous simulation [10, 11]:
| (2) |
For a canonical ensemble, the estimated free energy follows from:
| (3) |
where k is the Boltzmann constant, T the absolute temperature, and Pi(q) the estimate of the probability distribution of q calculated after the ith simulation (Eq. 7) [18, 28]. Thus, the biasing potential enhances the sampling of q by increasing the energy of the visited regions (which, due to Eq. 3, have a low free energy), encouraging the system to explore unvisited areas. In the limit that the estimated free energy equals the true free energy, all possible values of q are visited with equal probability during the sampling.
Reweighting the measured statistics
In the biasing simulations, the history of the configurations q that was visited is recorded. To account for the use of the biasing potential, the estimate of the probability distribution P(i)(q) is obtained by reweighting of the biased statistics from one or several previous simulations (simulations i, i−1, etc.) Formally, the weight wn of each sample qn coming from simulation i with biasing potential Vb,i in an unbiased simulation of the same length is [15, 28]:
| (4) |
Zi is the partition function of simulation i:
| (5) |
and Z0 is the partition function of the system of interest:
| (6) |
Similar expressions are obtained for simulations i − 1, etc.
In standard adaptive umbrella sampling [11] the ratios Zi/Z0, Zi−1/Z0, etc., that appear in Eq. 4, (or, equivalently, Zi/Zi−1, etc.) are solved simultaneously by the weighted histogram analysis method (WHAM) [27] in a manner that optimizes the estimates of the histograms of the configurations. WHAM usually uses a grid to store the statistics for each configuration (each value of the reaction coordinate). The memory limitations due to the use of large grids limit the method to low dimensional systems (~ 1 – 3 dimensions). Several empirical observations as well as recent theoretical analysis of the equations leading to WHAM [31, 43, 44] suggest that one could avoid the need for a grid and use only the statistics of the energy to obtain accurate estimates of the ratios of the partition functions. Although such an approach should be viable within GAMUS, we have not tested this method in the present work. Instead, we use the multi-state acceptance ratio method [45] to calculate the ratios of the partition functions. The multi-state acceptance ratio method does not store the statistics on a grid, so that the reweighting of data is not limited to low dimensions. The multi-state acceptance ratio method can also use non-equilibrium work data as input, but in our current application we do not take advantage of this feature. For a summary of this method, see Appendix B.
Construction of the biasing potential
In standard adaptive umbrella sampling [11] the probability P(i)(q) (Eq. 3) is stored on a grid, and the biasing potential is obtained from the interpolation of the associated free energies on the grid (Eq. 2). For the interpolation a Fourier series [11] or splines [37] can be used, for example. The use of a grid again limits the method to low (≈ 1 – 3) dimensions due to memory constraints. To circumvent these limitations, we obtain the estimate for the probability distribution P(i)(q) from a Gaussian-mixture model fit [39, 46] using the reweighted statistics from several previous simulations. In this fit P(i)(q) is described by a sum of M Gaussians:
| (7) |
where πm,i are positive constant weights. The gm,i(q) are multivariate Gaussian probability distribution functions, centered at μm,i:
| (8) |
with Σm,i the symmetric real positive definite variance-covariance matrix of gm,i with determinant |Σm,i|, and D the dimension of q. The periodic components of q (for example, the φ or ψ backbone dihedral angles) can be treated with periodic generalizations of Gaussian distributions, such as the von Mises, and the Fisher-von Mises distributions [47]. If the periodic components of the distribution are localized within their period, the resulting periodic generalization of Eq. 8 is equivalent to shifting the difference q − μm,i to the minimal image.
The Gaussian-mixture model fit is performed with multiple tries of the memory-efficient expectation-maximization (EM) algorithm [38, 39] (see Appendix C). The EM algorithm returns a locally optimal solution to the fit problem and may thus be trapped at unsuitable stationary points (for a discussion, see, e.g., Ref. [48]). The quality of the fit can be improved by selecting the highest likelihood solution of an ensemble of EM fits to the same data set (where each ensemble member is a fit starting from a different initial condition).
Typically, not the entire domain of q is visited in a simulation (especially not in the first few simulations). To avoid extrapolating the biasing potential outside the area visited in q, the biasing potential is localized to the visited space. This localization can be achieved by using a generalization of Eq. 7:
| (9) |
with γi(q) an appropriate function to describe our prior knowledge (or lack thereof) of the possible values of q outside the already visited region of space [42, 49]. Such priors can make use of the symmetry, if any, of the collective variables [50] or any other prior information about those variables; e.g. for angular coordinates like the backbone dihedral angles φ and ψ one could use a uniform flat prior, or the average Ramachandran plot based on the PDB. In the applications presented here, we use a constant γi, independent of q.
In the biasing potential used for the first simulation (i = 0) all πm,0 except π0,0 are set to zero. In subsequent simulations, the value of π0,i is chosen to localize the probability distribution on the sampled points and the remaining πm,i are renormalized. In the applications presented here, we either match the lowest probability sampled data of simulation i to π0,i or we set π0,i to a predefined (small) value.
Using the Gaussian-mixture model fit to describe the measured probability distribution, the force corresponding to the biasing potential to be used in Eq. 1 for simulation i is given by:
| (10) |
The transform of the forces back to Cartesian coordinates follows from:
| (11) |
with qa the components of the vector q.
SIMULATION SETUP
The simulations of the alanine dipeptide (CH3CO-Ala-NHCH3) and the alanine tripeptide (CH3CO-Ala2-NHCH3) were performed with the CHARMM polar hydrogen parameter set param19 [51, 52], and the ACE I implicit solvent model [53] as implemented in the CHARMM program [51]. ACE I [53] was used with a smoothing parameter of 1.6 and a solvent dielectric constant of 78.5 [54]. It has been shown previously that these ACE I parameters give good agreement with results from explicit solvent simulations for the alanine dipeptide [54]. In all cases, Langevin dynamics [55] were used with a friction coefficient of 6 ps−1 and a time step of 1 fs; SHAKE [56] was not used in the simulations.
The free energy surface of the alanine dipeptide as a function of the φ and ψ dihedral angles was obtained by adaptive umbrella sampling using a grid spacing of 2° [11, 37]. The free energy surface of the alanine tripeptide as a function of the main chain φ1, ψ1, φ2 and ψ2 dihedral angles was calculated from an aggregate 301 μs replica exchange simulations [57-59] using 7 replicas at 300, 348, 406, 475, 558, 657, 777, and 920 K. A total of 1.5·109 frames from these simulations were used for the calculation of the free energy surface; the frames were 0.2 ps apart. The surface was calculated on a four dimensional grid with a grid spacing of 10°. The WHAM method [27] was used to reweight the statistics of the higher temperature replica simulations to 300 K, and error estimates were obtained from a block analysis [28]. The basins were identified by the method described in Ref. [37]. Briefly, the free energy was monitored along 100 random vectors centered at a local free energy minimum. To minimize the effect of noise due to the discretization from the use of a grid, insufficient sampling, and inherent roughness in the actual surface, this monitoring was performed on a coarse-grained grid coalescing 4×4×4×4 original grid points (corresponding to a grid with a grid spacing of 40°), with the free energy of each coarse-grained grid point set equal to the minimum free energy of its associated 256 original grid points. If the free energy at the center is a local minimum for each random vector a basin has been identified; the minimum of this basin is at the center of the random vectors. We repeated this method for each local free energy minimum in the original 10° grid.
In the Gaussian-mixture fit umbrella sampling (GAMUS) simulations, the probabilities as a function of the φ and ψ coordinates (for the dipeptide) or the φ1, ψ1, φ2, and ψ2 coordinates (for the tripeptide) were fit to the Gaussian-mixture model (Eq. 9) after each MD run of 100,000 steps. For the fit, a total of 4 + i Gaussians were used (where i is the index of the simulation); the fits were initialized by centering the Gaussians at randomly selected sampled data points and by setting all variance-covariance matrices to a diagonal matrix that corresponds to a width of 10° in each dimension. The data points from all previous simulations were reweighted (Eq. 4) and included in the fits; as described in the Theory section, the multi-state acceptance ratio method [45] was used to determine the ratio of partition functions needed for the weights of each sampled point. The data points used for the fits were 100 fs apart, giving a total of 1000 points for each simulation. The fits were repeated twenty times using different random seeds, and the fit with the highest likelihood was selected for the next biasing potential. This procedure, consisting of a simulation followed by the fits, was repeated 40 times. This corresponds to a total simulation time of 4 ns, and results in a total of 44 Gaussians for the final fit. The first simulation was started in the C7eq (dipeptide) and (C7eq, C7eq) (tripeptide) conformation; the subsequent simulations were restarted using the velocities and coordinates from the last frame of the previous simulation.
To test the convergence of the method as a function of the number of Gaussians used for the fits, we also performed simulations of the alanine dipeptide in which the maximum number of Gaussians were set to 11 and 22. As before, the number of Gaussians was given by 4 + i (where i is the index of the simulation), but no Gaussians were added after the 6th or 18th simulation. All Gaussians were fully optimized after each run, however. In the dipeptide simulations, the prior γ(q) (Eq. 9) was set equal to the lowest sampled probability; the actual value of γ(q) had no influence on the results. We found empirically that the prior became more important for the tripeptide. This is due to the fact that the volume of the 4D space (tripeptide) is much larger than the volume of the 2D space (dipeptide); hence, extrapolation effects are exascerbated. We minimized these effects by capping the log γ(q) to a small value (between -30 to -50); the actual value did not have a significant impact on the results. For the reported results, min(log γ(q)) = −30. GAMUS simulations without such caps typically had smaller values of γ(q) and sometimes showed deep artificial minima in later runs (n > 20), and large ratios Zn/Zn−1 between the runs.
Free energy basins from the GAMUS simulations were identified with the method described in Ref. [37]. The free energy surfaces were calculated on a 10° grid, using the Gaussian mixture fits (Eq. 9). We note that this procedure can be easily extended to higher dimensions due to the sparseness of the visited space [37]; grid points are only stored for the sampled space, rather than for the entire space. The minima of the basins obtained in this way were used as the starting points for a Monte Carlo search of the GAMUS free energy minima. Since the Gaussian mixture fit yields an analytical expression for the free energy surface, these minima are not necessarily at the centers of the grid points used in the procedure of Ref. [37].
RESULTS
Alanine dipeptide
Fig. 2 shows the free energy surface of the alanine dipeptide as a function of the φ and ψ dihedral angles, as obtained by adaptive umbrella sampling using a grid spacing of 2° [11]. Five basins are observed: C7eq at (φ, ψ) = (−79, 139) with F = 0.0 kcal/mol, αR at (-79, -39) with F = 1.0 kcal/mol, αL at (55, 49) with F = 4.6 kcal/mol, C7ax at (61, -73) with F = 3.8 kcal/mol, and h at (63, -179) with F = 5.1 kcal/mol (angles in degrees, and all energies relative to the C7eq basin). The location and free energy of the saddle points S1 – S9 that separate these basins is given in the caption of Fig. 2.
Figure 2.
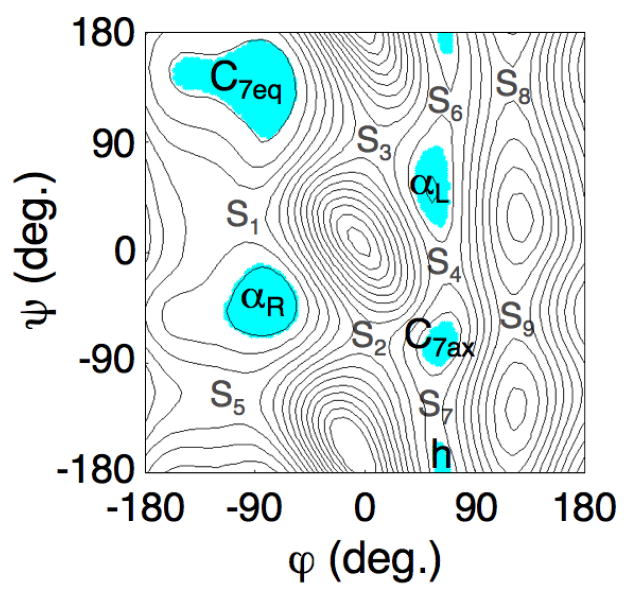
Converged free energy surface of the alanine dipeptide as a function of the φ and ψ dihedral angles. The surface was obtained by exhaustive adaptive umbrella sampling [11, 37]. Each isocontour is separated by 1 kcal/mol; the minimum value of the surface is set to zero. The 5 free energy basins are highlighted. The location (φ, ψ) and free energy of the minima of the basins is C7eq (-79, 139) 0.0, αR (-79, -39) 1.0, αL (55, 49) 4.6, C7ax (61, -73) 3.8, and h (63, -179) 5.1 kcal/mol. The location and energies of the saddle points are: S1 (-105, 35) 3.4, S2 (5, -71) 7.0, S3 (9, 91) 7.2, S4 (65, -7) 5.9, S5 (-103, -119) 4.3, S6 (67, 123) 5.7, S7 (55, -133) 5.5, S8 (121, 139) 9.3, and S9 (121, -51) 10.0.
Fig. 3A shows the sampled configurations of the first four simulations (4 × 100 ps) of a typical GAMUS run, superposed on the converged surface (Fig. 2). The effective potential of the dipeptide for these runs, given by the difference between the converged free energy (Fig. 2) and the GAMUS biasing potential, are given in Fig. 3B–D for the 2nd–4th biased simulation, respectively. Since no biasing was used for the first simulation, the effective potential for the first simulation equals the converged free energy surface (Fig. 2). In the first simulation the system explored only the free energy minimum of the starting C7eq conformation (Fig. 3A, blue). Since the C7eq basin is well visited in the first run, the effective potential for the second simulation is flattened around the C7eq basin (Fig. 3B). Due to this flattening, the S1 barrier has effectively disappeared, and the second simulation escaped along the S1 saddle point region to the αR basin (Fig. 3A, green). Since the C7eq and the αR basins are well visited in the first two simulations, the effective potential for the third simulation is flattened around these basins and the S2 and S3 barrier regions are lowered (Fig. 3C). Therefore, in the third simulation the system iterated between the first two basins and escaped to the C7ax basin via the S2 region (Fig. 3A, red). The regions around the C7eq, αR, and C7ax basins are flattened in the effective potential for the fourth simulation, and the S4, S6 and S7 barrier regions are also lowered (Fig. 3D), allowing the system to explore the region north of the αL basin around (φ, ψ) = (42, 88) in the fourth simulation (Fig. 3A, black). This region corresponds to an artificial minimum in the effective potential.
Figure 3. Gaussian-mixture umbrella sampling of the alanine dipeptide.
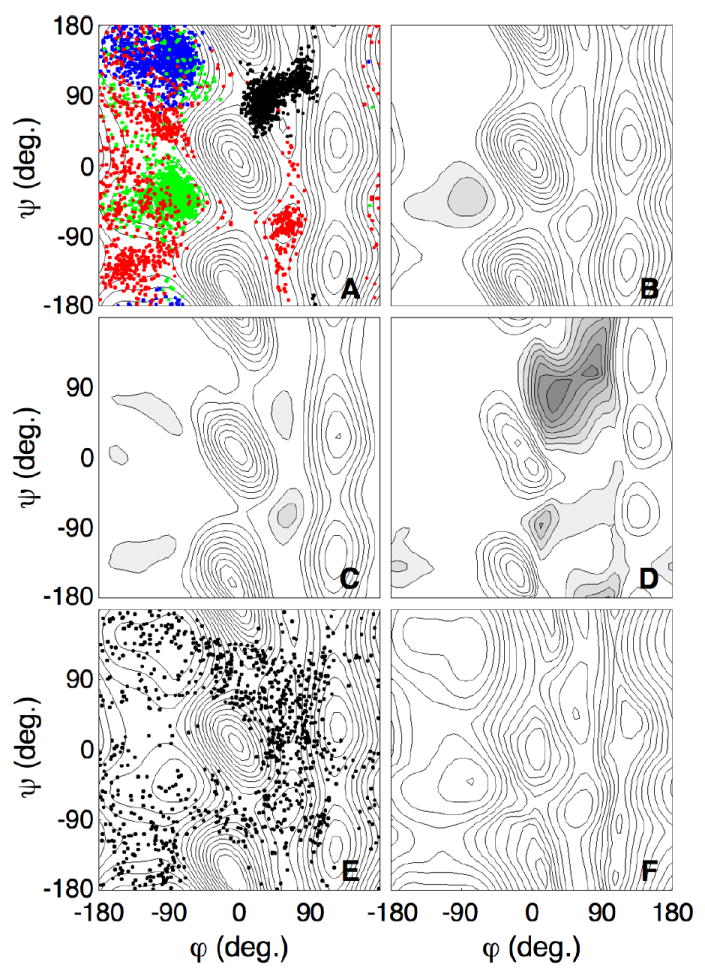
A. The samples of the first four simulations of the Gaussian-mixture umbrella sampling overlayed on the converged free energy surface of Fig. 2. The blue, green, red, and black dots denote the samples from simulations 1 – 4 respectively.
B-D. The effective potential on the dipeptide for simulations 2 (B), 3 (C) and 4 (D), given by the difference between the converged free energy and the biasing potential.
E. The samples from simulation 40 overlayed on the converged free energy surface of Fig. 2.
F. Free energy surface from the Gaussian-mixture fit after 40 simulations. This fit used a total of 44 Gaussians and all data points sampled in the 40 × 100 ps simulations. Each isocontour is separated by 1 kcal/mol; the minimum value of the surface is set to zero.
Analogous to other adaptive umbrella potential methods [11], the Gaussian-mixture fit may introduce artificial minima and maxima in the effective potential due to extrapolations: the Gaussian functions also extend into the areas that were not visited in any of the simulations. These artifacts were observed in a number of the simulations (see also the grey areas in Fig. 4F, for example). Although the trajectory may spend considerable time in such artificial minima, we found that in practice, these minima only have short-lasting effects. Generally, the trajectory escaped from the artificial minima within a single simulation. However, the artificial minima affect the efficiency of the calculation (by spending more than the expected time in areas that are energetically unimportant). This averse effect can be minimized by monitoring the simulation and restarting from a different configuration if desired, or by controlling the value of the prior parameter γ(q), to restrict the sampling within a desired band of free energies.
Figure 4. Gaussian-mixture umbrella sampling of the alanine dipeptide using 44, 22, and 11 Gaussian functions.
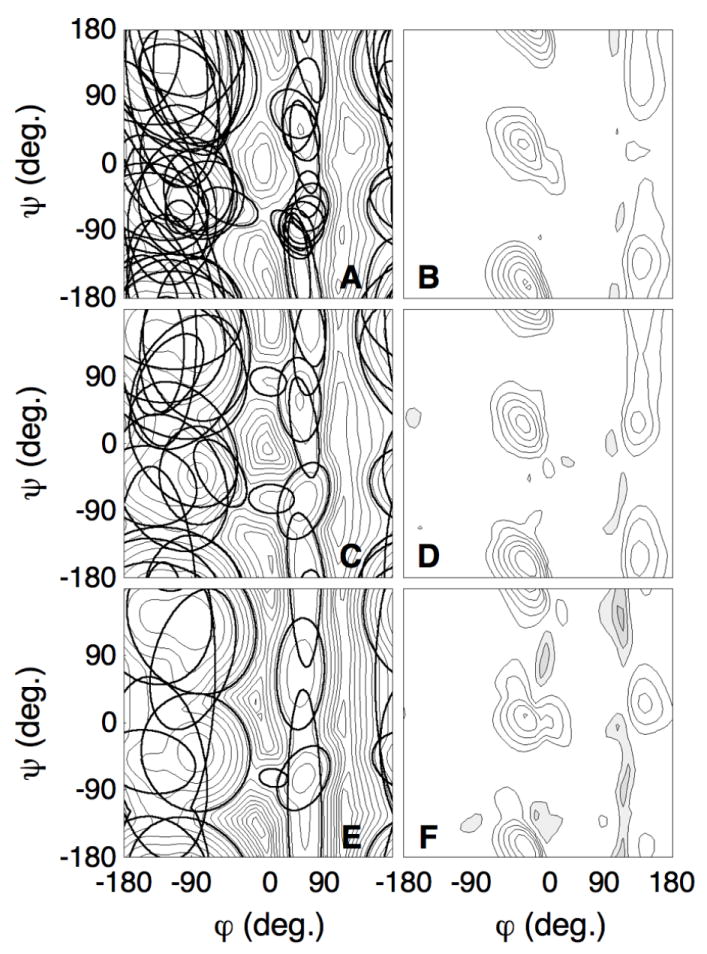
A, C, E. Isocontours of all Gaussians of the mixture-fit obtained after 40 simulations, over-layed on the free energy surface. The isocoutours of the Gaussian gi with mixing coefficient πi are plotted such that πigi(φ, ψ) = exp(−20). A. 44, C. 22, E. 11 Gaussians.
B, D, F. Free energy difference between the converged free energy surface (Fig. 3A) and the fitted surface. Each isocontour is separated by 1 kcal/mol. The filled gray areas show negative values of the plot, the white areas show positive values. The fitted free energy surfaces are within 1 kcal/mol in the areas of the dominant minima and are lower than the converged free energy surface on the barriers. B. 44, D. 22, F. 11 Gaussians.
After 40 simulations the system freely diffused between the various minima of the underlying free energy surface (Fig. 3E). Fig. 3F shows the fitted free energy surface of the alanine dipeptide after 40 fits. Comparison with the converged free energy surface (Fig. 2) showed that the shapes of the five basins are well described by the fit. Moreover, the artificial minima have disappeared.
Table I lists the location and the minimum of the free energy of the basins as identified by the Gaussian-mixture fit. The C7eq basin was identified after the first simulation, the αR basin after the second, the C7ax basin after the third, the αL basin after the fifth, and the h basin after the eigth simulation. Once a basin had been identified, the location of the minimum of the basin normally did not change much during subsequent fits; however, there are exceptions (e.g. αL changed from (59,61) to (56, 46) degrees, with the latter in significantly better agreement with the converged results of (55, 49) degrees (Table I)). All final Gaussian-mixture fit minima differ by a few degrees from the converged minima, with the largest deviations of 12° and 20° occurring for the C7eq and the h basin, respectively. The deviations are due to the relative flatness of the basins near the minima. In all cases, the free energy difference on the converged surface between the converged minimum structure and the GAMUS minimum structure is less than 0.1 kcal/mol. The relative free energies of the basins converged more slowly than their locations. After 8 fits, when all the basins had been identified for the first time, the maximum and the average unsigned errors were 1.2 kcal/mol and 0.52 kcal/mol, after 10 fits 0.7 kcal/mol and 0.3 kcal/mol, and after 40 fits 0.2 kcal/mol and 0.06 kcal/mol, respectively. In addition to the free energy estimates at specific angles shown in Table I, we can define the free energy of a state by integrating the normalized probability over the shaded area in Fig. 2. The free energy differences (in kcal/mol) of these states between the GAMUS and the converged free energy for iteration 10 of GAMUS are: C7eq: 0.04; αR: 0.1; C7ax: 0.7; αL: 0.4; h: 0.7, and for iteration 40 of GAMUS: C7eq: 0.05; αR: -0.03; C7ax: 0.09; αL: 0.09; h: 0.2.
TABLE I.
Location and free energy of the free energy basin minima of the alanine dipeptide. Shown are the (φ, ψ) dihedral angles (in degrees) and the free energy estimate for those specific angles relative to the free energy at the minimum of the C7eq basin (in kcal/mol).
| Simulation | C7eq | αR | C7ax | αL | h |
|---|---|---|---|---|---|
| 0a | (-79, 139) 0.0 | (-79, -39) 1.0 | (61, -73) 3.8 | (55, 49) 4.6 | (63, -179) 5.1 |
| 1 | (-81, 138) 0.0 | ||||
| 2 | (-83, 135) 0.0 | (-78, -40) 1.8 | |||
| 3 | (-82, 134) 0.0 | (-85, -43) 1.6 | (60, -75) 5.5 | ||
| 4 | (-85, 133) 0.0 | (-87, -45) 1.5 | (61, -55) 5.6 | ||
| 5 | (-87, 133) 0.0 | (-84, -43) 1.6 | (60, -91) 5.5 | (59, 61) 5.1 | |
| 6 | (-85, 132) 0.0 | (-99, -45) 1.4 | (59, -75) 4.7 | (59, 51) 5.5 | |
| 7 | (-84, 132) 0.0 | (-89, -45) 1.3 | (59, -78) 4.8 | (57, 47) 4.9 | (64, 140) 5.8 |
| 8 | (-83, 130) 0.0 | (-90, -44) 1.2 | (59, -77) 5.0 | (60, 39) 4.9 | (61, -170) 6.0 |
| 9 | (-84, 131) 0.0 | (-86, -42) 1.1 | (60, -76) 4.8 | (62, 39) 4.7 | (62, -166) 5.7 |
| 10 | (-83, 127) 0.0 | (-88, -44) 1.1 | (59, -77) 4.5 | (57, 49) 4.9 | (59, -151) 5.6 |
| 39 | (-82, 127) 0.0 | (-77, -40) 0.9 | (59, -81) 3.7 | (57, 45) 4.6 | (62, -178) 5.2 |
| 40 | (-81, 130) 0.0 | (-79, -40) 0.9 | (59, -80) 3.8 | (56, 46) 4.6 | (60, -163) 5.3 |
From the converged free energy surface as obtained from adaptive umbrella sampling using a 2 degree grid (Fig. 3A).
To test the convergence of the method as a function of the number of Gaussians functions used for the fits, we repeated the analysis for simulations in which the maximum number of Gaussians were set to 11 and 22. As before, the number of Gaussians was given by 4 + i (where i is the index of the simulation); in the tests, no Gaussians were added after the 6th or 18th simulation, but all Gaussians were fully optimized after each run. Again, we used a total of 40 simulations of 100 ps each; previously, this resulted in a total of 44 Gaussians for the last fit. Fig. 4A,C,E show the isocontours of all Gaussians used in the last fits. These contours are projected onto the free energy surface as obtained from the last simulation; they show where the Gaussian functions tend to localize. Fig. 4B,D,F show the differences between these calculated free energy surfaces and the converged free energy surface; the top graphs (Fig. 4A,B) are for the fit with 44 Gaussians, the middle (Fig. 4C,D) for the fit with 22 Gaussians, the bottom graphs (Fig. 4E,F) for the fit with 11 Gaussians.
Fig. 4 shows that the differences between the fit and the converged free energy surface were mainly located in the areas of high free energy (≳ 10 kcal/mol): in the fitted surface the free energy of these regions was underestimated. This means that the effective potential of the dipeptide, given by the difference between the converged free energy and the biasing potential, was flat around the basins but remained high around the true high barriers (Fig. 4B,D,F). Thus, the trajectories were diffusive around the basins but avoided the high barrier areas (see also Fig. 3E), explaining the efficiency of the method in locating the free energy basins (no time was wasted in the areas of high free energy). The errors in the high barrier regions were large (several kcal/mol). This is becase we fit the probability density, rather than the free energy, so that we can use the very efficient expectation-maximization algorithm [38, 39] for the fit. Since the free energy is obtained from the logarithm of the probability density, large statistical errors will occur for areas of low probability (high free energy). As expected, the performance of the method increased with the number of Gaussians used for the fit: the quality of the free energy surface and the free energy difference map is better for the fit with 44 Gaussians than for the fits with 22 or 11 Gaussians. The general location of the Gaussians is similar for the three fits (Fig. 4A,C,E). A comparison of Fig. 4A,C,E shows that the additional Gaussians are mostly used to fine tune the geometrical shape of the basins.
We also calculated the free energy profiles along the minimum free energy pathways (MFEPs) for the C7eq ↔ C7ax transition obtained on he converged surface. Here, the MFEPs are defined as the steepest descent pathways on the free energy surface [37]. There are four MFEPs connecting these basins [37]:
| (12) |
| (13) |
| (14) |
| (15) |
other paths have higher energies (see the caption of Fig. 2 for the location of the basins and saddle points). In Fig. 5 the free energy profiles along these pathways are shown for the final fit using 11 Gaussians (light grey), 22 Gaussians (grey), 44 Gaussians (dark grey) and for the converged free energy surface (black); in each case, the profiles were calculated along the same pathways (the MFEPs on the converged surface). Fig. 5 shows that the fits identify the position of the basins, and their relative free energies (especially when more Gaussians are used). For the barrier region, however, larger deviations occur between 0.5-1 kcal/mol (for S1 − S2 and S4 − S7) and 2 kcal/mol (S3 using 11 Gaussians).
Figure 5.

Free energy profiles along the minimum free energy pathways for the alanine dipeptide. The shape of the MFEPs correspond to those on the fully converged free energy surface (Eq. 12-15)[37]; the free energy profiles on this surface are shown in black. The profiles along these pathways after 40 GAMUS simulations are shown in light grey (using 11 Gaussians), grey (using 22 Gaussians) and dark grey (using 44 Gaussians for the fit).
A. MFEP 1.
B. MFEP 2.
C. MFEP 3.
D. MFEP 4.
Alanine tripeptide
Table II presents the minima of the free energy basins of the alanine tripeptide as a function of the main chain φ1, ψ1, φ2, and ψ2 dihedral angles, as obtained from replica exchange simulations of aggregate duration 301 μs. The energies are relative to the global free energy minimum of the alanine tripeptide, which is the (C7eq,C7eq) conformation at (φ1, ψ1, φ2, ψ2) = (− 75, 135, −75, 135) (the apparent mismatch with the C7eq conformation of the alanine dipeptide is due to the different grid spacing used in determining the free energy surface). As observed before [37], the (φ1, ψ1) angles are mostly independent of the (φ2, ψ2) angles of the alanine tripeptide: the relative free energies are generally a sum of the energies of the corresponding basins in the alanine dipeptide. It is interesting to note that the observed independence gives support to the use of “build-up” procedures, in which the accessible conformational space is built from the conformational space of small fragments [60]. In total, 25 basins were identified; it is possible that some very high (≳ 10 kcal/mol) free energy basins were missed due to insufficient sampling.
TABLE II.
Location and free energy of the free energy basin minima of the alanine tripeptide. Shown are the (φ1, ψ1, φ2, ψ2) dihedral angles (in degrees) and the free energy (F) for those specific angles relative to the free energy at the minimum of the (C7eq, C7eq) basin (in kcal/mol). Distances between the basins identified by GAMUS and the replica exchange simulations are given in degrees.
| Basin | Simulation | (φ1, ψ1, φ2, ψ2), F | Distancea |
|---|---|---|---|
| (C7eq, C7eq) | REXb | (-75, 135, -75, 135), 0.0±0.0 | |
| 10 | (-89, 158, -76, 129), 0.0 | 28 | |
| 20 | (-90, 140, -87, 147), 0.0 | 23 | |
| 30 | (-93, 151, -84, 145), 0.0 | 28 | |
| 40 | (-99, 144, -86, 148), 0.0 | 31 | |
| (C7eq, αR) | REXb | (-75, 135, -75, -45), 1.1±0.0 | |
| 10 | (-81, 151, -77, -47), 0.4 | 17 | |
| 20 | (-92, 148, -67, -55), 2.8 | 25 | |
| 30 | (-83, 144, -65, -57), 2.3 | 20 | |
| 40 | (-84, 138, -80, -52), 1.8 | 13 | |
| (C7eq, αL) | REXb | (-65, 125, 55, 35), 4.3±0.0 | |
| 40 | (-74, 125, 57, 38), 7.0 | 10 | |
| (C7eq, C7ax) | REXb | (-75, 135, 65, -75), 3.7±0.0 | |
| 20 | (-78, 126, 63, -68), 5.9 | 12 | |
| 30 | (-100, 134, 61, -48), 7.4 | 37 | |
| 40 | (-101, 136, 62, -71), 6.7 | 26 | |
| (C7eq, h) | REXb | (-155, 145, 65, 175), 5.9±0.1 | |
| (αR, C7eq) | REXb | (-75, -45, -85, 135), 1.2±0.0 | |
| 10 | (-106, -57, -94, 148), -2.3 | 37 | |
| 20 | (-105, -54, -95, 149), -1.4 | 36 | |
| 30 | (-103, -52, -96, 150), 0.2 | 34 | |
| 40 | (-84, -46, -120, 147), 1.2 | 38 | |
| (αR, αR) | REXb | (-75, -45, -75, -45), 2.5±0.0 | |
| 10 | (-89, -31, -84, -42), 3.9 | 22 | |
| 20 | (-82, -30, -73, -47), 3.2 | 17 | |
| 30 | (-85, -30, -75, -47), 4.8 | 18 | |
| 40 | (-83, -29, -72, -45), 2.2 | 18 | |
| (αR, αL) | REXb | (-75, -45, 55, 45), 5.6±0.0 | |
| (αR, C7ax) | REXb | (-75, -45, 65, -75), 4.7±0.0 | |
| (αR, h) | REXb | (-75, -45, 65, 175), 6.1±0.1 | |
| (αL, C7eq) | REXb | (55, 45, -85, 135), 4.7±0.0 | |
| 40 | (58, 33, -105, 134), 8.4 | 24 | |
| (αL, αR) | REXb | (55, 55, -75, -35), 5.8±0.1 | |
| (αL, αL) | REXb | (55, 45, 55, 25), 7.9±0.2 | |
| (αL, C7ax) | REXb | (55, 75, 65, -85), 7.8±0.2 | |
| (αL, h) | REXb | (55, 55, 55, 155), 9.6±0.5 | |
| (C7ax, C7eq) | REXb | (65, -65, -75, 135), 3.9±0.0 | |
| 20 | (63, -68, -86, 127), 7.2 | 14 | |
| 30 | (63, -69, -98, 125), 7.9 | 25 | |
| 40 | (64, -67, -83, 129), 7.1 | 10 | |
| (C7ax, αR) | REXb | (55, -85, -75, -35), 3.6±0.0 | |
| 20 | (55, -79, -92, -35), 7.0 | 18 | |
| 30 | (55, -79, -82, -35), 7.4 | 9 | |
| 40 | (57, -77, -89, -33), 7.5 | 16 | |
| (C7ax, αL) | REXb | (55, -65, 65, 25), 7.4±0.1 | |
| (C7ax, C7ax) | REXb | (55, -75, 65, -65), 7.3±0.1 | |
| (C7ax, h) | REXb | (45, -65, 65, 115), 7.8±0.2 | |
| (h, C7eq) | REXb | (65, 175, -85, 135), 5.3±0.0 | |
| 20 | (61, 140, -86, 128), 8.4 | 36 | |
| (h, αR) | REXb | (65, -165, -75, -45), 6.1±0.1 | |
| (h, αL) | REXb | (75, -175, 55, 55), 8.5±0.3 | |
| (h, C7ax) | REXb | (55, 175, 55, -65), 8.1±0.2 | |
| (h, h) | REXb | (75, 145, 55, -155), 8.9±0.3 | |
| GAMUS Artificial Minima: | |||
| (αR, ?) | 20 | (-83, -48, -125, 150), 1.0 | |
| (αR, ?) | 30 | (-87, -46, -131, 148), 0.8 | |
| (C7eq, ?) | 30 | (-92, 124, -125, 145), 0.7 | |
| (?, ?) | 30 | (-120, 152, -119, 124), 2.8 | |
| (αR, ?) | 40 | (-75, -50, -149, 162), 1.4 | |
| (C7eq, ?) | 40 | (-87, 138, -131, 149), 1.0 | |
Since the bin size in the replica exchange simulations was 10° (see text), basins overlap perfectly for distances less than degrees.
From the replica exchange simulations (in bold).
Table II shows the location and free energy of the minima of the basins as identified in a typical sequence of GAMUS simulations. Four basins were identified after 10 simulations (1 ns), and 8 after 40 simulations (4 ns). The identified basins are the basins with the lowest free energy, and the location of the basins closely corresponds to those identified by the replica exchange simulations. Since the replica exchange simulations used a grid spacing of 10°, basins overlap perfectly for distances less than . Although the GAMUS simulations correctly identified the location of the lowest free energy basins, the relative free energies of the basins generally differed from those of the replica exchange simulations. For the lowest basins ((C7eq,αR), (αR,C7eq), and (αR,αR)) these deviations were less than 1 kcal/mol after 40 simulations; for the highest basins ((C7eq,C7ax) and (αL,C7eq)) the deviations were several kcal/mol after 40 simulations. In addition, GAMUS yields 2 artificial minima after 40 simulations due to the extrapolation in the fitting procedure (Table II).
Since the GAMUS simulations depend on random numbers (in the Langevin dynamics and in the fitting procedure), different basins are found in different runs. We performed two additional sets of 40 GAMUS simulations starting in the (C7eq, C7eq) configuration but with different random seeds. The first additional set of simulations identified 11 basins to within 19 degrees on average from the basins identified by replica exchange: the 9 basins identified after iteration 40 in Table II, plus the minima (αR, C7ax) and (C7ax, C7ax); in addition, it identified 4 artificial minima. The second additional set of simulations identified 11 basins to within 23 degrees on average from the basins found by replica exchange: the 9 basins identified after iteration 40 in Table II, plus the minima (αR, C7ax) and (h, C7eq); in addition, it identified 1 artificial minimum. Of all possible pairings of the 7 artificial minima found in the 3 sets of 40 GAMUS simulations, in only one instance is the distance between the two elements of a pair less than 60 degrees.
We have observed that the subset of common minima that GAMUS obtains in multiple independent runs with different random seeds tend to agree with the lowest free energy minima that result from a costly established, method, such as replica exchange or adaptive umbrella sampling. Although it is inconsistent with the purpose of an efficient exploration of a free energy surface, one might consider an ensemble of sequences of GAMUS simulations of certain duration, each starting from different random seeds or from different initial conditions. Following a classification of all the minima obtained at the end of each GAMUS sequence, one can cluster the common minima and approximate a probability of finding each minimum in this ensembles as the ratio of the number of sequences that found this minimum over the number of sequences in the ensemble. If this probability is very close to 1, the minimum is safely considered a real minimum of the underlying free energy landscape. If this probability is considerably less than one, the minimum is questionable.
CONCLUSION
We introduced GAMUS, an adaptive umbrella sampling method that uses Gaussian-mixture models of the density to escape free energy basins in high dimensional systems. We implemented the method for dihedral angles and analyzed its performance for the alanine dipeptide and alanine tripeptide. For the alanine dipeptide, the location and shape of all five basins were quickly identified; the accurate assessment of the relative free energies took slightly longer. For the tripeptide, the location of several low basins were quickly established, but only approximate relative free energies were obtained. In each case, a few (1-2) artificial basins were identified as well. Since the location of these minima changes from run to run, the location of the true basins can be established by repeating the calculations multiple times using different random number seeds.
The method requires little computer memory and avoids areas of high free energy, so that we expect it to be useful for the location of free energy basins in complex biomolecular systems. Although in the present example we only tested the sampling of basins in two and four dimensions, it is known [39] that Gaussian mixture models can extend to a few more dimensions. In analogy to related developments to the metadynamics methodology [61], it should be possible to use multiple GAMUS simulations in a Hamiltonian replica exchange simulation.
Although GAMUS helps expand the number of correlated coordinates that can be sampled simultaneously, it does not address the question of which reaction coordinates to use when those coordinates are not obvious. In problems where conformational change involves hinge-bending type motions, the choice of coordinates is relatively well defined and sampling may be easier with GAMUS than with previously available methods, because the total number of relevant coordinates is limited so that being able to include more of them should help. GAMUS has essentially the same limitations as other sampling methods: if the time required to sample the space is such that convergence can be achieved, good results can be obtained in multidimensional problems; if not, not.
Acknowledgments
PM thanks Kevin Bowers for discussions. Partial support for the work done at Harvard University was provided by the National Institute of Health, and computer time for the work done at Harvard University was provided by the Bauer Center for Genomics Research. Computer time for the work done at Arizona State University was provided by the Fulton High Performance Computing Initiative.
Appendix A
The metadynamics [9, 17] and GAMUS methods have common characteristics and a few clear differences. Both methods have as one major objective the escape from free energy minima, and do so by the use of a Gaussian mixture model. The difference between the methods is three fold. First, in metadynamics the Gaussian mixture models the free energy surface, while in GAMUS the Gaussians mixture models the probability density. Second, GAMUS employs a fitting procedure for the Gaussians at predefined stages where all Gaussians are evaluated based on all available data; in metadynamics, a single new Gaussian is added to the mixture model at predefined time intervals. Third, and most importantly, GAMUS optimizes the covariance and the weight of the Gaussians, while metadynamics uses identical Gaussians with identical weights. By optimizing the covariance and weight of the Gaussians, GAMUS is less dependent on initial conditions and can more efficiently fill the basins. To illustrate this difference, we will use the simple analogy of filling a house with balls. While the metadynamics method would use the same size ball to fill up all the rooms (including bookshelves and open cabinets), GAMUS would use spherical and ellipsoidal balls of different sizes and with different orientation to maximize the coverage of available volume with as few balls as possible. We expect that GAMUS’ flexibility to fill up basins is even more important for high dimensional systems, especially when those basins have non-uniform shapes.
The conformational flooding method [8] that preceded metadynamics used a single Gaussian biasing potential to escape from the current free energy minimum. In conformational flooding, the potential acts on the lowest frequency principal components only. These collective eigenmodes are obtained from a principal component analysis of short MD runs; the latter analysis corresponds to fitting a single multi-dimensional Gaussian to the probability density in configuration space. In order to avoid extrapolation errors, the strength and width of the Gaussian biasing potential are determined by a user-defined parameter. Although conformational flooding adapts the covariance of the (single) Gaussian biasing to the local probability density, it is clearly different from GAMUS: the latter biases the probabilities with a mixture model of multiple Gaussians that are iteratively fit to all available data of a sequence of simulations.
Appendix B
In the multi-state acceptance ratio method [45], the work Wij associated with switching a system at configuration q, Hamiltonian Hi and temperature Ti to the state with configuration q, Hamiltonian Hj and temperature Tj is measured as:
| (16) |
Using Crooks fluctuation theorem [62], the likelihood of measuring the work Wij given that the switch is in the i to j direction equals:
| (17) |
where Fij is the free energy difference between simulation i and j. Ultimately, we will obtain the ratio of partition functions (needed in Eq. 4) from the optimal estimate of this free energy difference:
| (18) |
Using Bayes’ theorem [63], the probability of measuring a switch in the i to j direction, given that the work equals Wij, can be expressed as a Fermi-function [64-66]:
| (19) |
where Mij = kT ln(ni→j/nj→i), and ni→j and nj→i are the number of forward and reverse switches, respectively. Rather than using only two simulations for our estimate of the free energy difference (or, equivalently, the ratio in partition functions, Eq. 18), we use the work data for all possible switches between all states. Assuming that the data is independent, the likelihood of observing forward switches from all states i to every other state j is given by [45]:
| (20) |
The optimal estimate for the free energy differences are obtained from the maximization of the likelihood p(all i → all j). Since all derivatives of the likelihood are available in closed form, this optimization can be done efficiently by the Newton-Raphson algorithm [45].
Appendix C
The memory-efficient implementation of the expectation maximization algorithm to fit Gaussian mixture models on sampled data qn (n = 1, …, N) with weights wn has been described by Bowers et al [39] (see also Baggenstoss [46]) and is summarized below. This algorithm determines the weights πm, centers μm, and variance-covariance matrices Σm of the M Gaussians through iterations of the equations below. In the expectation (E) step we estimate the probability that the m-th component of the mixture model at iteration k generates the n-th sample:
| (21) |
With the definition of:
| (22) |
in the maximization (M) step, we formally obtain the weights, centers, and variance-covariance matrices from:
| (23) |
| (24) |
| (25) |
where N is the total number of sampled points from the various simulations that are included in the fit. The weights wn of configuration qn correspond to Eq. 4:
| (26) |
where j is the index of the simulation in which configuration qn was sampled.
To increase the accuracy of the estimation, all arithmetic is performed in the log-domain and only the right Cholesky factor of the variance-covariance matrix is evaluated in the implementation of the mixture model [46]. To minimize round-off errors, this Cholesky factor is updated with a row-wise economy QR factorization [39] based on a Givens rotation [67]. We further imposed constraints on the variance-covariance matrix [46], such that the Gaussians would not shrink to less than one degree in any direction. These constraints avoid overfitting that could otherwise result from the collapse of a multi-dimensional Gaussian to a single data point.
Contributor Information
Paul Maragakis, Email: paul.maragakis@DEShawResearch.com.
Arjan van der Vaart, Email: vandervaart@asu.edu.
Martin Karplus, Email: marci@tammy.harvard.edu.
References
- 1.Elber R, Karplus M. Science. 1987;235:318–321. doi: 10.1126/science.3798113. [DOI] [PubMed] [Google Scholar]
- 2.Frauenfelder H, Parak F, Young R. Ann Rev Biophys Biophys Chem. 1988;17:451–479. doi: 10.1146/annurev.bb.17.060188.002315. [DOI] [PubMed] [Google Scholar]
- 3.Krivov S, Karplus M. Proc Natl Acad Sci USA. 2004;101:14766–14770. doi: 10.1073/pnas.0406234101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singhal N, Snow C, Pande V. J Chem Phys. 2004;121:415–425. doi: 10.1063/1.1738647. [DOI] [PubMed] [Google Scholar]
- 5.Oliveberg M, Wolynes P. Quart Rev Biophys. 2005;38:245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 6.Noe F, Horenko I, Schuette C, Smith J. J Chem Phys. 2007;126:155102. doi: 10.1063/1.2714539. [DOI] [PubMed] [Google Scholar]
- 7.Buchete N-V, Hummer G. J Phys Chem B. 2008;112:6057–6069. doi: 10.1021/jp0761665. [DOI] [PubMed] [Google Scholar]
- 8.Grubmüller H. Phys Rev E. 1995;52:2893–2906. doi: 10.1103/physreve.52.2893. [DOI] [PubMed] [Google Scholar]
- 9.Laio A, Parrinello M. Proc Natl Acad Sci USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hooft R, van Eijck B, Kroon J. J Chem Phys. 1992;97:6690–6694. [Google Scholar]
- 11.Bartels C, Karplus M. J Comp Chem. 1997;18:1450–1462. [Google Scholar]
- 12.Hamelberg D, Mongan J, McCammon J. J Chem Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 13.Krivov S, Chekmarev S, Karplus M. Phys Rev Lett. 2002;88:038101. doi: 10.1103/PhysRevLett.88.038101. [DOI] [PubMed] [Google Scholar]
- 14.van der Vaart A. Theor Chem Acc. 2006;116:183–193. [Google Scholar]
- 15.Torrie G, Valleau J. J Comput Phys. 1977;23:187–199. [Google Scholar]
- 16.Voter AF. Phys Rev Lett. 1997;78:3908–3911. [Google Scholar]
- 17.Bussi G, Laio A, Parrinello M. Phys Rev Lett. 2006;96:090601. doi: 10.1103/PhysRevLett.96.090601. [DOI] [PubMed] [Google Scholar]
- 18.Kirkwood J. J Chem Phys. 1935;3:300–313. [Google Scholar]
- 19.Darve E, Pohorille A. J of Chem Phys. 2001;115:9169–9183. [Google Scholar]
- 20.Rosso L, Minary P, Zhu ZW, Tuckerman ME. J Chem Phys. 2002;116:4389–4402. [Google Scholar]
- 21.Carter EA, Ciccotti G, Hynes JT, Kapral R. Chem Phys Lett. 1989;156:472–477. [Google Scholar]
- 22.Ciccotti G, Kapral R, Vanden-Eijnden E. Chem Phys Chem. 2005;6:1809–1814. doi: 10.1002/cphc.200400669. [DOI] [PubMed] [Google Scholar]
- 23.Ren W, Vanden-Eijnden E, Maragakis P, W E. J Chem Phys. 2005;123:134109. doi: 10.1063/1.2013256. [DOI] [PubMed] [Google Scholar]
- 24.Maragliano L, Fischer A, Vanden-Eijnden E, Ciccotti G. J Chem Phys. 2006;125:024106. doi: 10.1063/1.2212942. [DOI] [PubMed] [Google Scholar]
- 25.Maragliano L, Vanden-Eijnden E. Chem Phys Lett. 2007;446:182–190. [Google Scholar]
- 26.Pan AC, Sezer D, Roux B. J Phys Chem B. 2008;112:3432–3440. doi: 10.1021/jp0777059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrenberg A, Swendsen R. Phys Rev Lett. 1989;63:1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 28.Frenkel D, Smit B. Understanding Molecular Simulation. 2. Academic Press; Elsevier USA: 2002. [Google Scholar]
- 29.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman P. J Comp Chem. 1992;13:1011–1021. [Google Scholar]
- 30.Roux B. Comp Phys Commun. 1995;91:275–282. [Google Scholar]
- 31.Souaille M, Roux B. Comp Phys Commun. 2001;135:40–57. [Google Scholar]
- 32.Berezhkovskii A, Szabo A. J Chem Phys. 2005;122:014503. doi: 10.1063/1.1818091. [DOI] [PubMed] [Google Scholar]
- 33.Krivov S, Karplus M. J Phys Chem B. 2006;110:12689–12698. doi: 10.1021/jp060039b. [DOI] [PubMed] [Google Scholar]
- 34.W E, Vanden-Eijnden E. J Stat Phys. 2006;123:503–523. [Google Scholar]
- 35.Krivov S, Karplus M. Proc Natl Acad Sci USA. 2008;105:13841–13846. doi: 10.1073/pnas.0800228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson A, Hermans J. Proteins. 1988;3:262–265. doi: 10.1002/prot.340030408. [DOI] [PubMed] [Google Scholar]
- 37.van der Vaart A, Karplus M. J Chem Phys. 2007;126:164106. doi: 10.1063/1.2719697. [DOI] [PubMed] [Google Scholar]
- 38.Dempster AP, Laird NM, R D. J Roy Stat Soc B. 1977;39:1–38. [Google Scholar]
- 39.Bowers K, Devolder B, Yin L, Kwan T. Comp Phys Comm. 2004;164:311–317. [Google Scholar]
- 40.Maragakis P, Spichty M, Karplus M. J Phys Chem B. 2008;112:6168–6174. doi: 10.1021/jp077037r. [DOI] [PubMed] [Google Scholar]
- 41.Minh DDL, Adib AB. Phys Rev Lett. 2008;100:180602. doi: 10.1103/PhysRevLett.100.180602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaynes ET. Probability Theory: The Logic of Science. Cambridge University Press; Cambridge: 2003. [Google Scholar]
- 43.Gallicchio E, Andrec M, Felts A, Levy R. J Phys Chem B. 2005;109:6722–6731. doi: 10.1021/jp045294f. [DOI] [PubMed] [Google Scholar]
- 44.Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. 2008 doi: 10.1063/1.2978177. arXiv.org:0801.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maragakis P, Spichty M, Karplus M. Phys Rev Lett. 2006;96:100602. doi: 10.1103/PhysRevLett.96.100602. [DOI] [PubMed] [Google Scholar]
- 46.Baggenstoss PM. Statistical Modeling Using Gaussian Mixtures and HMMs with Matlab. 2002 http://www.npt.nuwc.navy.mil/Csf/htmldoc/pdf/pdf.html.
- 47.Abramowitz M, Stegun IA. Handbook of Mathematical Functions with Formulas, Graphs, and Mathematical Tables. Dover; New York: 1964. [Google Scholar]
- 48.Wu CFJ. The Annals of Statistics. 1983;11:95–103. [Google Scholar]
- 49.Gelman A, Carlin JB, Stern HS, Rubin DB. Bayesian Data Analysis. 2. Chapman & Hall/CRC; New York: 2004. [Google Scholar]
- 50.Kass RE, Wasserman L. J Am Stat Assoc. 1996;91:1343–1370. [Google Scholar]
- 51.Brooks B, Bruccoleri R, Olafson B, States D, Swaninathan S, Karplus M. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 52.Neria E, Fischer S, Karplus M. J Chem Phys. 1996;105:1902–1921. [Google Scholar]
- 53.Schaefer M, Karplus M. J Phys Chem. 1996;100:1578–1599. [Google Scholar]
- 54.Apostolakis J, Ferrara P, Caflisch A. J Chem Phys. 1999;110:2099–2108. [Google Scholar]
- 55.Brooks C, Karplus M. J Chem Phys. 1984;79:6312–6325. [Google Scholar]
- 56.Ryckaert J, Ciccotti G, Berendsen H. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 57.Marinari E, Parisi G. Europhys Lett. 1992;19:451–458. [Google Scholar]
- 58.Lyubartsev A, Martsinovski A, Shevkunov S, Vorontsov-Velyaminov P. J Chem Phys. 1992;96:1776–1783. [Google Scholar]
- 59.Sugita Y, Okamoto Y. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 60.Michielin O, Zoete V, Gierasch T, Eckstein J, Napper A, Verdine G, Karplus M. J Am Chem Soc. 2002;124:11131–11141. doi: 10.1021/ja012695p. [DOI] [PubMed] [Google Scholar]
- 61.Piana S, Laio A. J Phys Chem B. 2007;111:4553–4559. doi: 10.1021/jp067873l. [DOI] [PubMed] [Google Scholar]
- 62.Crooks G. Phys Rev E. 1999;60:2721–2726. doi: 10.1103/physreve.60.2721. [DOI] [PubMed] [Google Scholar]
- 63.Bayes T. Phil Trans. 1763;53:370–418. [Google Scholar]
- 64.Shirts M, Bair E, Hooker G, Pande V. Phys Rev Lett. 2003;91:140601. doi: 10.1103/PhysRevLett.91.140601. [DOI] [PubMed] [Google Scholar]
- 65.Bennett C. J Comp Phys. 1976;22:245–268. [Google Scholar]
- 66.Maragakis P, Ritort F, Bustamante C, Karplus M, Crooks GE. J Chem Phys. 2008;129:024102. doi: 10.1063/1.2937892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Golub GH, Van Loan Charles F. Matrix Computations. 3. The Johns Hopkins University Press; Baltimore, MD: 1996. [Google Scholar]