

Abstract
Ca2+ channel β subunits (Cavβs) are essential for regulating the surface expression and gating of high voltage-activated Ca2+ channels through their interaction with Ca2+ channel α1 subunits. In efforts to uncover new interacting partners and new functions for Cavβ, we identified a new splicing isoform of Pax6, a transcription factor crucial for the development of the eye, nose, brain, and pancreas. Pax6 contains two DNA binding domains (paired domain and homeodomain), a glycine-rich linker connecting these two domains and a C-terminal proline-, serine-, and threonine-rich transactivation domain. The protein sequence and function of Pax6 are highly conserved from invertebrate to human. The newly isolated isoform, named Pax6(S), retains the paired domain, linker, and homeodomain of Pax6, but its C terminus is composed of a truncated classic proline, serine, and threonine domain and a unique S tail. Pax6(S) shows a similar level of transcriptional activity in vitro as does Pax6, but only in primates is the protein sequence highly conserved. Its spatial-temporal expression profiles are also different from those of Pax6. These divergences suggest a noncanonical role of Pax6(S) during development. The interaction between Pax6(S) and Cavβ is mainly endowed by the S tail. Co-expression of Pax6(S) with a Ca2+ channel complex containing the β3 subunit in Xenopus oocytes does not affect channel properties. Conversely, however, β3 is able to suppress the transcriptional activity of Pax6(S). Furthermore, in the presence of Pax6(S), β3 is translocated from the cytoplasm to the nucleus. These results suggest that full-length Cavβ may act directly as a transcription regulator independent of its role in regulating Ca2+ channel activity.
Keywords: Channels/Calcium, RNA/Splicing, Transcription, Transcription/General Factors, Transcription/Regulation, Transcription/Repressor, Pax6, transactivation
Introduction
Ca2+ channel β subunit (Cavβ)4 is a cytosolic auxiliary protein of multimeric high voltage-activated (HVA) Ca2+ channel complexes, which include L-, N-, P/Q-, and R-type Ca2+ channels. It plays an essential role in chaperoning the channel complex to the plasma membrane and normalizing its gating properties (1–4). Crystal structures of Cavβ in complex with its high affinity binding site in the principal pore-forming α1 subunit (Cavα1) show that much of the exposed surface of Cavβ is unoccupied and is available to engage in interactions with other regions of Cavα1 or with other proteins (5–7). Indeed, an increasing number of proteins has been shown to directly interact with Cavβ, including the Rem/Rad/Gem/Kir (RGK) family of small monomeric GTPases (8, 9), RIM1 (10), ryanodine receptors (11), Ahnak (12, 13), bestrophin-1 (14), and dynamin (15). Many of these proteins have been reported to regulate the activity of HVA Ca2+ channels. To search for other potential Cavβ-interacting proteins, we carried out yeast two-hybrid screens using the β3 subunit as bait. Among the candidate target proteins we isolated, one was related to Pax6.
Pax6 is a transcription factor that belongs to the paired box (Pax) family (16–25). It is widely expressed in the eye, nose, pancreas, and the central nervous system in both embryonic and adult mammals, and it plays important roles in regulating the development of these tissues and organs (18, 20, 26–31). The protein sequence of Pax6 is highly conserved throughout vertebrates, lower chordates, and invertebrates (24). The function of Pax6 is also highly conserved, as suggested by the induction of ectopic eye structures after the overexpression of Drosophila or mouse Pax6 genes in Drosophila or Xenopus laevis embryos (32–35).
The human Pax6 gene is located on chromosome 11p13 and occupies 14 exons (exons 1–13 plus exon 5α between exons 5 and 6) in a 22-kb genomic region (36). There are at least three Pax6 isoforms produced by alternative splicing (24). The canonical Pax6 is generated from a transcript composed of exons 1–13 (Fig. 1A). It contains a paired domain (PD), a homeodomain (HD), a glycine-rich linker connecting the above two domains, and a C-terminal proline, serine, and threonine (PST)-rich domain (Fig. 1A). The second Pax6 isoform contains 14 extra amino acids encoded by exon 5α in the PD and is named Pax6(5α). Protein translation of the above two isoforms begins in exon 4 and terminates in exon 13 (Fig. 1A). The third isoform is generated by translational initiation in exon 8. It does not contain the PD and, therefore, is named paired-less Pax6 (Pax6(ΔPD)).
FIGURE 1.

Composition of Pax6 and No. 8 cDNAs and subdomains of their protein products. A, upper, canonical Pax6 cDNA is composed of 13 exons. Translation starts in exon 4 and ends in exon 13. lower, Pax6 protein contains the PD, linker region, HD, and PST domain. The boarders between the domains are marked by the numbers below. B, upper, the No. 8 cDNA spans the major part of exon 7, entire exons 8–11, and the unique exon 11α (filled black box) that is the intron between exons 11 and 12 in the Pax6 gene. Note that exon 11α contains a stop codon in fame with the preceding exons. Lower, the predicted protein product of No. 8 is shown. It includes the C-terminal end of the PD, linker region, HD, the N-terminal portion of the canonical PST domain (PSTN), and an S tail (filled black box) encoded by 11α. Gray indicates the regions that are present in full-length Pax6(S) cDNA and protein but are missing in No. 8.
The PD and HD are two domains where Pax6 interacts with its DNA targets. The C-terminal PST domain plays a key role in regulating Pax6 transcriptional activity but does not bind DNA directly. Missense mutations and partial or complete truncation of the PST domain decrease the transcriptional activity of Pax6 (37, 38). Fusion of the Pax6 PST with the transcription factor GAL4 increases the activity of GAL4 (37, 39, 40), suggesting that the transactivity of the PST domain can be independent of the PD and HD. The PST domain encompasses 152 amino acids encoded by exons 10–13 (Fig. 1A). Studies have revealed that these four exons synergistically stimulate transcriptional activation and that the transactivation potential is not localized but spread throughout the PST domain (37). It has been suggested that the transactivity of the PST domain stems from its interaction with other regulatory proteins, which enhances the assembly of the transcriptional preinitiation complex (41). Recent studies demonstrate that the high proportions of serine and threonine residues in the PST domain allow phosphoryl and dephosphoryl modulation (42–44), which may fine-tune the protein-protein interactions.
In this study we identified a novel splicing isoform of Pax6 named Pax6(S). It contains the canonical PD and HD, but it has a different C terminus composed of the N-terminal half of the canonical PST and a unique S tail encoded by the intron between exons 11 and 12. In contrast to Pax6, Pax6(S) is completely conserved only in human and chimpanzee, and it seems to be expressed only at the early stages of development, suggesting a yet-to-be-defined and perhaps noncanonical function during development. Pax6(S) retains transcriptional activity, but its C terminus shows less transactivity compared with the canonical PST domain. In addition, we found that Pax6(S) interacted with a full-length Cavβ through its S tail. This interaction did not alter Ca2+ channel properties, but it decreased Pax6(S) activity in vitro and resulted in the translocation of Cavβ from the cytoplasm to the nucleus. Our results suggest a novel function of full-length Cavβ as a suppressor of Pax6(S).
EXPERIMENTAL PROCEDURES
Plasmid Constructs
For yeast two-hybrid library screens and pairwise interaction assay, β3 (GenBankTM accession number M88751) core Gly-16–Gly-366 or full-length was cloned into the pGBKT7 vector (Clontech). Different fragments of Pax6(S) were cloned into the pGADT7 vector (Clontech). For glutathione S-transferase (GST) pulldown assay, β3 full-length was cloned into a modified pGEX4T-1 vector (GE Healthcare). For electrophysiology, α1 (X57477), β3, α2-δ (M21948), Pax6(S), and No. 8 were individually cloned into an oocyte expression vector, pGEMHE (modified from pGEM-3Z, Promega), or its variants. cDNA encoding the S tail of Pax6(S) (Val-345—Asp-401) was subcloned into a modified pET26b vector (Novagen). For luciferase assays, the Pax6 consensus DNA binding sequence, CD19-2, was cloned into the promoter region of a modified pGL3-OFLuc vector (Promega) to produce the reporter construct. Pax6 (M93650), Pax6(S), or β3 was cloned into the expression vector p3XFLAG-CMV-7.1 (referred to as FLAG vector; Sigma) (referred to as FLAG-Pax6, FLAG-Pax(S), and FLAG-β3, respectively). FLAG-Pax6, FLAG-Pax(S), and β3 cloned in pEGFP-C3 (Clontech) (referred to as pEGFP-C3-β3) were also used for immunofluorescence imaging. Different fragments of PST or PSTNS tails were fused with GAL4 in a modified pCG vector that expressed residues 1–147 of GAL4. All constructs were generated by PCR and confirmed by sequencing.
Yeast Two-hybrid Assay
All vectors, yeast strains, reagents, and methods were adopted from the BD MATCHMAKERTM screening kit (Clontech). The yeast strains Saccharomyces cerevisiae AH109 (MATα, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1UAS-GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2, URA3::MEL1UAS-MEL1TATA-lacZ, MEL1) and Y187 (MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, met-, gal80Δ, URA3::GAL1UAS-GAL1TATA- lacZ, MEL1) were employed as hosts in the two-hybrid assay. AH109 contains two nutritional reporter genes for adenine and histidine. Both AH109 and Y187 harbor the LacZ and MEL1 reporter genes.
A human adult brain cDNA library (Clontech) was screened with the pGBKT7-β3_ core construct. The library was constructed in the pGADT7-rec vector. All procedures were carried out according to the manufacturer's instructions (Clontech). Briefly, pGBKT7-β3_ core was transformed into Y187 and grown in medium lacking tryptophan. The AH109 yeasts pretransformed with the human adult brain cDNA library were then mated to these Y187 cells and grown in a medium lacking adenine, histidine, tryptophan, and leucine. After growing for 10 days, the cells were plated on selective plates lacking adenine, histidine, tryptophan, and leucine with 5-bromo-4-chloro-3-indolyl-α-d-galactopyranoside (X-α-gal) and incubated at 30 °C for 2 weeks. The colonies were then picked, and the plasmids were extracted and transformed into the bacterial strain Escherichia coli DH5α for amplification. The plasmids were extracted from DH5α and checked with sequencing. The specificity of the interactions was tested by transforming competent AH109 yeast cells with one bait construct (in pGBKT7) and one target construct (in pGADT7) and examining the resulting colonies for activation of the ADE2, HIS3, and MEL1 reporters on selective plates as described above.
Protein Synthesis and GST Pulldown Assay
The S tail of Pax6(S) (Val-345—Asp-401) subcloned into a modified pET26b vector was expressed in BL21(DE3) to obtain the S tail protein. cDNA encoding β3 was subcloned into a modified pGEX4T-1 vector and expressed in BL21(DE3) bacteria to obtain the GST_β3 protein. The No. 8 protein was synthesized in vitro with the TNT® Coupled Transcription/Translation Systems (Promega). GST_β3 was immobilized on glutathione-Sepharose 4B beads (Novagen). The No. 8 protein bound to the immobilized GST_β3 was eluted from the beads with glutathione and detected with Coomassie Blue staining on SDS-PAGE.
5′ Rapid Amplification of cDNA Ends
The full-length Pax6(S) was obtained by 5′-rapid amplification of cDNA end (5′ RACE) reaction with the 5′/3′ RACE kit (Roche Applied Science). The procedures were carried out according to the manufacturer's instruction. The following primers were used: SP1 (5′-GGGCATGAATTAATGAGT-3′), SP2 (5′-TCTCCGACTTGACTGGTC-3′), SP3 primer (5′-GGGAAAGUCCACCACCAGCCGCACTTAC-3′), oligo(dT) anchor primer (5′-GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTTV-3′, V = A, C, or G), and PCR anchor primer (5′-GGAGACAUGACCACGCGTATCGATGT-3′). The product of the second round of PCR was inserted into the lacZα gene in the NEB206A vector according to the instruction of USERTM Friendly Cloning kit (New England Biolabs). The ligation mixture was transformed into DH5α cells and plated on a LB plate with isopropyl 1-thio-β-d-galactopyranoside and X-α-gal to perform a white/blue selection. The white colonies, which indicate an insertion in the vector, were selected. The plasmids were extracted and sequenced.
BLAST Search of Nucleotide Databases
The nucleotide sequence of No. 8 and Pax6(S) was scanned against the human genomic plus transcript database, human Pax6 mRNA (GenBankTM accession number M93650), human DNA sequence from clone XX-A1280 on chromosome 11(Z83307), and databases of high throughout genomic sequences and whole-genome shotgun reads.
Cell Culture and Transfection
Human embryonic kidney (HEK) 293T cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen). CHO cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2 mm glutamine. Both cell cultures were incubated in an incubator at 37 °C under 5% CO2 and were subcultivated every 2–3 days with a ratio of 1:5.
HEK 293T or CHO cells were transfected at a confluence of over 90%. Transfections were performed with plasmid DNA coated with LipofectamineTM 2000 (Invitrogen) according to the manufacturer's instructions. For HEK 293T cells, which were plated on poly-d-lysine-coated coverslips immersed in 35-mm dishes (BD Biosciences), 10 μl of LipofectamineTM 2000 and 4.0 μg of total DNAs were transfected per dish. For CHO cells, which were plated onto 24-well plates (Corning), 2 μl of LipofectamineTM 2000 and 0.8 μg of total DNAs were transfected per well.
Cell and Tissue Slide Imaging
24–40 h after transfection, cells transfected with FLAG-Pax6 or FLAG-Pax6(S) (with or without pEGFP-C3-β3 co-transfected) were fixed with 5% polyformaldehyde and permeabilized with 0.5% Triton X-100. They were then subjected to immunostaining. 1 μg/ml anti-FLAG monoclonal antibody (Sigma) or anti-Pax6(S) polyclonal antibody with a dilution of 1:100 was used. After staining, cells were mounted on glass slides with ProLong® Gold Antifade reagent (with DAPI) (Invitrogen). Cells transfected with pEGFP-C3-β3 were only fixed with 5% polyformaldehyde and mounted with the same reagent. Paraffin-embedded human retina and brain tissue slides (BioChain) were deparaffinized with xylene and then rehydrated through a series of graded ethanol (100, 95, 70, and 50%). The slides were subsequently immersed in an antigen retrieval solution containing 10 mm Tris base, 1 mm EDTA, and 0.05% Tween 20 (pH 9.0) at 60 °C overnight. In the next 2 days the slides were permeabilized and stained with the same procedures described above. Anti-Pax6 polyclonal antibody (Covance) with a dilution of 1:500 was used to stain the endogenous Pax6. All the above cells or tissue slides were imaged with an Olympus FluoView 500 confocal microscope (Olympus) or Nikon 80i upright epifluorescence microscope (Nikon).
Western Blot Assay
A preblotted polyvinylidene difluoride membrane containing ∼50 μg of protein lysates per lane from eight different human tissues (brain, kidney, lung, small intestine, heart, liver, skeletal muscle, and placenta) was obtained from BioChain and then analyzed by hybridization with antibodies according to the instruction of Odyssey® Infrared Imaging System (LI-COR Biosciences). Anti-Pax6(S) polyclonal antibody with a dilution of 1:100 was used as the primary antibody. 0.2 μg/ml IR Dye® 800 (LI-COR Biosciences) was used as the secondary antibody. The membrane was scanned with Odyssey® Infrared Imager under 800-nm channels at 169-μm resolution (LI-COR Biosciences).
Luciferase Assay
To test the transcriptional activity of Pax6 and Pax6(S), pGL3-OFLuc-CD19-2, FLAG-Pax6 or FLAG-Pax6(S), and pRL-SV40 were transfected into CHO cells with a ratio of 2:14:0.1. To examine the effects of different fragments of the PSTNS tail on GAL4 activity, p5XGAL4-E1b-Luc, pCG-GAL4 containing PSTNS fragment, or pCG-GAL4 itself and pRL-SV40 were transfected into CHO cells with a ratio of 20:1:0.4. To examine the effects of β3 on Pax6(S) activity, pGL3-OFLuc-CD19-2, FLAG-Pax6(S) (or FLAG-Pax6(ΔS) or empty FLAG vector), FLAG-β3 (or empty FLAG vector), and pRL-SV40 were co-transfected into CHO cells with a ratio of 10:40:15:0.8. pRL-SV40, which constitutively expresses Renilla firefly driven by a SV40 promoter, was used as an internal control. 24–40 h after transfection the medium was removed, and CHO cells were briefly washed with phosphate-buffered saline solution twice. Luciferase assay was performed at room temperature using the Dual-Luciferase Reporter Assay System (Promega).
Electrophysiology and Data Analysis
Xenopus oocytes were prepared and maintained as described before (45). cRNAs of α1, α2-δ, β3, Pax6(S), and No. 8 were synthesized in vitro. Each oocyte was injected with ∼6 ng of α1, 6 ng of α2-δ, and 4 ng of β3 with or without 6 ng of Pax6(S) or No. 8 co-injected. Recordings were performed 4–6 days after injection. In the case of inside-out macropatch recordings, 20 μm S tail protein was applied.
The solutions and protocols for two-electrode voltage clamp and patch clamp are as described before (45). Currents were sampled at 10 kHz and filtered at 2.5 kHz. The holding potential for all the following protocols was −80 mV. Macroscopic currents were evoked by 20-ms depolarizations ranging from −40 mV to +100 mV in 10-mV increments at a 6-s interval. Tail currents were always recorded by repolarization to −30 mV regardless of the preceding test pulse. To obtain the current-voltage relationship, peak currents evoked by depolarizations were plotted against the test potentials. To obtain the activation curves, tail currents were normalized by that after the depolarization to +100 mV and plotted against the test potentials. To examine deactivation, a 10-ms depolarization to +100 mV was applied to fully open the channels followed by repolarizations ranging from −80 mV to +80 mV in 10-mV increments at a 5- s interval to obtain tail currents. Steady-state inactivation was determined by a three-pulse protocol in which a 20-ms normalizing pulse to +30 mV (pulse A) was followed sequentially by a 25-s conditioning pulse (ranging from −80 mV to +50 mV) and a 20-ms test pulse to +30 mV (pulse B). The interval between each protocol was 2 min. Peak current evoked by pulse B, normalized by that evoked by pulse A, was plotted against the conditioning potentials to obtain the voltage dependence of inactivation.
The voltage dependence of activation and inactivation was fitted with the Boltzmann function 1/(1 + exp(−(V − V½)/k), where V½ and k are the midpoint of activation or inactivation and the slope factor, respectively. To obtain the time constant of deactivation (τdeact), tail currents were fitted with a single exponential function. τdeact was then plotted against the test potentials to produce the voltage-dependent deactivation curve. Data were represented as the mean ± S.D. (number of observations). Significance was determined using two-tailed Student's t tests.
RESULTS
Cloning of Full-length Pax6(S)
In an yeast two-hybrid screen of a human adult brain cDNA library using the core domain Gly-16–Gly-366 of Ca2+ channel β3 subunit as bait, 12 targets were identified (data not shown). A BLAST search of the human genome revealed that the cDNA sequence of No. 8 matched part of Pax6. More careful comparison between No. 8 and Pax6 cDNA sequences showed that the No. 8 cDNA spans the major part of exon 7, entire exons 8–11, and the 5′ half of the intron between exons 11 and 12 (renamed exon 11α, Fig. 1B) of the Pax6 gene. Exon 11α contains a stop codon that is in-frame with the preceding exons. Hence, the protein encoded by No. 8 contains the last 12 amino acids of the PD, the linker region, the HD, and the N-terminal half of the PST domain (PSTN) present in the canonical Pax6 plus a novel C-terminal tail encoded by exon 11α (Fig. 1B). Compared with the canonical PST, which contains 15.1% proline, 17.8% serine, and 12.5% threonine in mammalian species, this new tail also has a high proportion of serine (19.3%) but a lower proportion of proline (7%) and threonine (1.8%). It was, thus, named the S tail, and S tail plus the PSTN was named PSTNS (Fig. 1B).
The amino acid sequence of No. 8 suggests the existence of a new splicing isoform of Pax6 that was designated as Pax6(S). The DNA sequence preceding exon 7 was missing in No. 8 (Fig. 1B). We next used 5′ rapid amplification of cDNA ends to identify the nucleotide sequence of full-length Pax6(S) (for details, see “Experimental Procedures”). As shown in Figs. 1B and 2, the Pax6(S) transcript includes exons 4–11α, producing a 401-amino acid protein with a calculated molecular mass of ∼45 kDa. Thus, the only difference between Pax6(S) and Pax6 proteins lies in the C terminus (compare the lower panels of Fig. 1, A and B). The 3′-untranslated region (UTR) of Pax6(S) is different from that of Pax6; however, it does contain a classic polyadenylation signal (Fig. 2). The 5′-UTR of the Pax6(S) transcript is composed of only 48 nucleotides (Fig. 2), whereas Pax6 has a 417-nucleotide 5′-UTR. Because UTRs carry information for regulation of translation and mRNA stability (46–53), the divergence in the UTR sequence implies that translation and/or mRNA stability of Pax6(S) and Pax6 are differentially modulated.
FIGURE 2.

The nucleotide and protein sequences of full-length Pax6(S). Nucleotides are in lowercase letters, and amino acids are capitalized. Boundaries between exons are labeled by vertical lines with numbers on both sides indicating the preceding and following exons, separately. Exons are numbered the same as in Pax6 gene (except for exon 11α). Exon 11α and the S tail are in red. The cDNA sequences to which the SP1, SP2, and SP3 primers annealed (see “Experimental Procedures”) are framed. The epitope that the Pax6(S) antibody recognizes is in shadow. 5′- and 3′-UTRs are in blue. The polyadenylation signal in the 3′-UTR is underlined.
The protein sequence of the canonical PST domain is 100% conserved from rodents to human. Do different species, if they produce the S tail homologues, also share a high identity in this region? We performed a BLAST search with the DNA sequence of exon 11α and indeed pulled out homologues from various species. However, the protein sequences of these homologues were 100% conserved only between human and chimpanzee (supplemental Fig. S1). The conservation remained high among the primates we examined, decreasing to 92% for sumatran orangutan and 91% for olive baboon and rhesus monkey, but it decreased dramatically in rabbit (25%) and mouse (31%) (supplemental Fig. S1). In fact, the predicted rabbit and mouse S tails are 14 and 22 amino acids shorter than the human S tail, respectively (supplemental Fig. S1). Using embryonic and adult mouse brain cDNA libraries, we performed reverse transcription-PCR analysis with primers that specifically annealed to the DNA encoding the predicted mouse S tail. We did not detect any reverse transcription-PCR products (data not shown), suggesting low, if any, expression of the S tail in mouse.
Expression of Pax6(S) Protein in Situ
To study Pax6(S) protein expression, a rabbit polyclonal antibody against an epitope (HNPGPREVRSGSGP) located within the unique S tail (Fig. 2) was synthesized. To test the specificity of this Pax6(S) antibody, Pax6(S) or Pax6 (both tagged with FLAG on the N terminus) were individually expressed in HEK 293T cells. The Pax6(S) antibody stained the cells expressing FLAG-Pax6(S) (Fig. 3A, upper leftmost) but not those expressing FLAG-Pax6 (Fig. 3A, lower leftmost). The lack of staining in the latter was not due to a lack of protein expression, as strong FLAG signals were detected with an anti-FLAG antibody (Fig. 3A, the second left panel in the lower row).
FIGURE 3.

Nuclear localization and in situ expression of Pax6(S). A, HEK 293T cells expressing FLAG-Pax6(S) (upper row) or FLAG-Pax6 (lower row) were stained with (from left to right) an anti-Pax6(S) antibody, an anti-FLAG antibody, and a nuclear dye DAPI. Immunofluorescence from these three stainings is overlaid in the rightmost panels. B, Pax6 (left) but not Pax6(S) (right) was detected in the inner nuclear layer (arrow) of the human retina. C, staining of Pax6(S) in 4-month (left) and 5-month (right) embryonic human brain slides is shown. Arrows point out three positive signals. D, staining of Pax6(S) in various human embryonic tissue lysates with Western blot is shown. Bar, 10 μm.
Two nuclear localization signals are present in Pax6, one in the PD and the other in the linker region (54). The PST domain has been suggested to contain glycosylation sites (55) that may also be involved in the nuclear transport of Pax6 (56, 57). Overexpressed Pax6(S) was localized in the cell nucleus as was Pax6, as indicated by colocalization with a nuclear dye, DAPI (Fig. 3A), indicating that the novel S tail of Pax6(S) does not change the subcellular localization of Pax6(S).
With a specific anti-Pax6(S) antibody in hand, we went on to examine Pax6(S) expression in situ. The critical role of Pax6 in eye development has been well established (32–35). We indeed detected Pax6 in the inner nuclear layer of human retina (Fig. 3B, left), in agreement with previous reports (58). However, we did not detect Pax6(S) in retina (Fig. 3B, right). Because the Pax6(S) transcript was originally isolated from a human adult brain cDNA library, we then screened Pax6(S) expression in various regions of human adult brain, including the frontal, temporal, parietal, and occipital lobes, pons, thalamus, and corpus callosum. None of these regions showed any detectable Pax6(S) signal (data not shown), suggesting that Pax6(S) expression is low in adult brain. We next examined Pax6(S) expression in human embryonic brain. Interestingly, nucleus-like signals were detected in 4-month-old (Fig. 3C, left) but not in 5-month-old (Fig. 3C, right) embryonic brain. Western blots detected a protein band of ∼45 kDa in the lysate of human embryonic brain (Fig. 3D). In addition, protein bands of ∼50 kDa were detected in lysates of various human embryonic tissues including the lung, small intestine, heart, liver, skeletal muscle, and placenta (Fig. 3D). This higher-than-calculated molecular weight could be due to phosphoryl or glycosyl modification of Pax6(S) in these tissues. In fact, it has been reported by several groups that phosphorylation or glycosylation of Pax6 upper-shifted its protein band in Western blot (42–44, 55). Extra bands of ∼80 or ∼37 kDa were present in the small intestine, heart, skeletal muscle, and placenta, suggesting that apart from Pax6(S), Pax6 might have other isoforms sharing part of the S tail that can be recognized by the Pax6(S) antibody. Alternatively, these bands may simply be generated by a nonspecific binding of the antibody. However, because they were not universally stained in every tissue (Fig. 3D) and were completely absent in human cell line lysates we tested (data not shown), they are less likely to be artifacts. To verify this, further experiments such as peptide competition assays need to be performed.
Pax6(S) Retains Transcriptional Activity in Vitro
As a transcription factor, Pax6 governs the expression of diverse genes through binding to their cis-elements via its PD or HD. The C-terminal PST domain is not directly involved in DNA binding but plays a role in regulating the transcriptional activity (37–40). We investigated whether Pax6(S) retains transcriptional activity and, if it does, whether its unique S tail confers a different level of transcriptional activity. The activity of Pax6(S) and Pax6 was examined in vitro with a luciferase assay. CD19-2, one of the consensus DNA binding sequences for the PD (24), was used as the promoter of the luciferase reporter gene. As shown in Fig. 4A, Pax6(S) enhanced the luciferase-induced fluorescence by ∼4-fold, reflecting its ability in driving luciferase transcription. This ability is as potent as that of Pax6 (Fig. 4A), indicating that the S tail does not confer a different transcriptional activity to Pax6(S) in this context.
FIGURE 4.

In vitro transcriptional activity of Pax6(S) and transactivity of PSTNS. A, the reporter construct, pGL3-OFLuc-CD19-2, was present in all groups. The basal transcriptional activity detected in the control group (C) could be caused by endogenous Pax6 or Pax6(S). B, exons encoding the PST or PSTNS were dissected and fused to GAL4 gene. The reporter construct, p5XGAL4-E1b-Luc, was present in all groups. PSTC refers to the C-terminal half of the PST domain, which is missing in Pax6(S) and is the S tail counterpart in Pax6. *, p < 0.001. n = 6 for all. Error bars indicate the mean ± S.D.
The PSTNS Domain Shows Transactivity
The PST domain of Pax6 has been shown to have a transactivity independent of the PD and HD, as it increases the activity of GAL4 when fused to GAL4 (37, 39, 40). Using a luciferase assay, we investigated the transactivity of the PSTNS domain of Pax6(S) by fusing its coding sequence, exons 10–11α (Fig. 1B), to the GAL4 coding sequence. As a control, exons 10–13, which encode the canonical PST domain (Fig. 1A), were also fused to the GAL4 coding sequence in a separate expression construct. In the reporter construct p5XGAL4-E1b-Luc, a GAL4 binding sequence was introduced as the promoter of the luciferase gene. GAL4 itself was able to slightly drive the expression of luciferase (Fig. 4B, compare bars a and b). Its activity was greatly increased when GAL4 was fused with PST (encoded by exons 10–13) (Fig. 4B, compare bars c and b). PSTNS (encoded by exons 10–11α) also enhanced the activity of GAL4 (Fig. 4B, compare bars d and b), albeit to a much less degree than PST did (Fig. 4B, compare bars d and c). Consistent with its inability to bind DNA by itself, neither PST nor PSTNS was able to induce luciferase expression in the absence of GAL4 (Fig. 4B, bars h and i).
A study has shown that exons 10–13 synergistically stimulate transcriptional activation and that the transactivation potential is not localized but spread throughout PST (37). We confirmed this finding by individually fusing exons 10–11 or exons 12–13 to the GAL4 coding sequence. Both were able to stimulate the GAL4 activity, with exons 12–13 being more potent than exons 10–11 (Fig. 4B, compare bars e and f). However, neither shows a transactivity comparable with that produced by exons 10–13 (Fig. 4B, compare bars c, e, and f). Exon 11α, which is uniquely present in Pax6(S), by itself enhanced the GAL4 activity but to a lesser extent than exons 10–11α did (Fig. 4B, compare bars g and d), suggesting that exon 11α also works synergistically with exons 10–11 to stimulate transcriptional activation. In the absence of GAL4, exons 10–11, 12–13, and 11α had no effect (Fig. 4B, bars j, k, and l).
Why did the PSTNS domain show a lower transactivity than the PST when fused with GAL4 (Fig. 4B, bars c and d), whereas Pax6(S) exhibited transcriptional activity as potent as that of Pax6 (Fig. 4A)? One explanation is that PST and PSTNS produce different allosteric effects on GAL4 but not in Pax6 and Pax6(S) and, thus, lead to different regulations of GAL4 activity.
Pax6(S) Interacts with Cavβ Mainly through the S Tail
The interaction between Pax6(S) and β3 was first identified from a yeast two-hybrid assay (Fig. 5A) and was subsequently confirmed by a pulldown assay. The No. 8 protein synthesized in vitro was pulled down by GST-tagged β3 (Fig. 5B). This effect was not an artifact caused by the GST tag, as the No. 8 protein could not be pulled down by GST itself (Fig. 5B).
FIGURE 5.
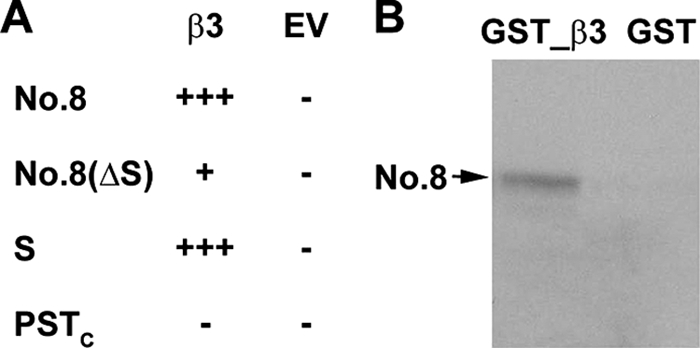
Interaction between No. 8 and Ca2+ channel β subunit. A, a summary of the results from yeast two-hybrid assay is shown. Positive and negative results are shown by plus and minus signs, respectively. Compared with one plus sign, three plus signs indicate more yeast cell growing on the selective plates, suggesting a stronger interaction. EV, empty vector. B, re-test the interaction between No. 8 and β3 with the GST pulldown assay.
By performing a pairwise yeast two-hybrid assay, we next examined whether the S tail of Pax6(S) was involved in this interaction. No. 8 was dissected into two fragments. Fragment 1 (Fig. 5A, No. 8(ΔS)) contains the regions that are common in both No. 8 and Pax6. Fragment 2 (Fig. 5A, S) is entirely the S tail. Fragment 1 showed a weak interaction with β3, whereas fragment 2 (i.e. the S tail) conferred a much stronger interaction. On the contrary, the C-terminal half of the PST domain (PSTC), which is missing in Pax6(S) and is the S tail counterpart in Pax6, did not seem to interact with β3 (Fig. 5A).
Pax6(S) Does Not Affect the Biophysical Properties of Ca2+ Channels
It is surprising that Pax6(S), a transcription factor, can interact with a channel protein. Cavβs profoundly regulate gating and the surface expression of HVA Ca2+ channels (1–4). To test whether the interaction between Pax6(S) and Cavβ affects HVA Ca2+ channel properties, we expressed the channel complex containing Cav2.1, α2-δ, and β3 in Xenopus oocytes with or without Pax6(S) being present and examined a host of channel biophysical properties with cell-attached patch clamp. Pax6(S) did not affect the current-voltage relationship, voltage-dependent activation, deactivation speed (indicated by deactivation constant, τdeact), voltage-dependent inactivation, and inactivation speed (Fig. 6, A–E). Similarly, No. 8 did not affect these biophysical properties (supplemental Fig. S2). However, Pax6(S) moderately reduced the current amplitude measured by two-electrode voltage clamp (Fig. 6F), suggesting that Pax6(S) may interfere with channel trafficking to the plasma membrane. In contrast, co-expression of No. 8 did not significantly change the current amplitude (Fig. 6F), suggesting that No. 8 does not interfere with channel trafficking. The current reduction caused by Pax6(S) could also be due to toxicity of Pax6(S) RNA or protein, as it occurred only when large amounts of Pax6(S) cRNA were injected.
FIGURE 6.

Pax6(S) does not affect Ca2+ channel biophysical properties. For A–F, cRNA of Pax6(S) or No. 8 was co-injected into Xenopus oocytes with the channel complex cRNAs, and cell-attached patch clamps (A–E) or two-electrode voltage clamp (F) were performed. For G and H, channel complex cRNAs were injected into Xenopus oocytes, and purified S tail protein was applied to the intracellular side of the channels in the inside-out macropatch configuration. A, current-voltage relationship is shown. B, voltage-dependent activation is shown. C, deactivation constant τdeact is shown. D, voltage-dependent inactivation is shown. E, representative inactivation traces is shown. F, whole cell current is shown. G, time course of normalized current under S tail treatment is shown. The current was evoked by a 20-mV depolarization. H, voltage-dependent activation under the indicated conditions is shown. Data points represent normalized tail currents recorded at −30 mV after depolarization to the indicated test potential. *, p < 0.05. n = 5–8 for all. Error bars indicate the mean ± S.D.
One could argue that the lack of effects of Pax6(S) on the channel properties in the above experiments could be because Pax6(S) cRNA failed to produce protein products in Xenopus oocytes or Pax6(S) proteins were completely sequestered in the nucleus. To exclude these possibilities, purified S tail protein, which by itself is capable of interacting with Cavβ (Fig. 5A), was directly applied to the intracellular side of the channels in the inside-out macropatch configuration. This treatment did not affect the current amplitude and voltage-dependent activation (Fig. 6, G and H). Altogether, these results suggest that the interaction between Pax6(S) and Cavβ does not affect the activity of HVA Ca2+ channels.
Cavβ Decreases the Transcriptional Activity of Pax6(S) in Vitro
We next investigated whether Cavβ affects the transcriptional activity of Pax6(S), as determined by the luciferase assay described in Fig. 4A. Pax6(S) robustly stimulated luciferase expression (Fig. 7, compare bars a and b), and co-expression of β3 decreased this activity by ∼50% (Fig. 7, compare bars b and c). Suppression of Pax6(S) activity by β3 was a specific effect, because the activity of Pax6(S) was unchanged in the presence of bacterial maltose-binding protein (MBP) (Fig. 7, compare bars b and d).
FIGURE 7.

Cavβ decreases Pax6(S) transcriptional activity. Pax6(S) or Pax6(ΔS) was expressed individually, with β3, or with maltose-binding protein (MBP). Reporter construct, pGL3-OFLuc-CD19-2, and internal control, pRLSV40, were present in all groups. *, p < 0.01. n = 6 for all. Error bars indicate the mean ± S.D.
We further investigated whether the suppression of Pax6(S) activity by β3 is through the S tail. A Pax6(ΔS) construct, in which the S tail was truncated from Pax6(S), was capable of stimulating luciferase activity to a larger level than did Pax6(S) (Fig. 7, compare bars b and e). However, β3 did not dampen the transcriptional activity of Pax6(ΔS) (Fig. 7, compare bars f and e). These results indicate that β3 can suppress Pax6(S) activity, and it does so through interacting with the unique S tail of Pax6(S).
Overexpressed Pax6(S) and Cavβ Co-localize in the Nuclei of HEK 293T Cells
Does Cavβ regulate Pax6(S) activity in cells? As an initial step to address this question, we investigated the subcellular localization of Cavβ in the absence and presence of Pax6(S) or No. 8, the Cavβ-interacting component of Pax6(S). Pax6(S) is localized in the nuclei, as shown in Fig. 3A. As a subunit of HVA Ca2+ channels, Cavβ is predominantly distributed in the cytoplasm (1–4). Does the interaction between Pax6(S) and Cavβ change their respective subcellular localization? Pax6(S) and β3 were tagged with FLAG and EGFP, respectively. When they were expressed in HEK 293T cells separately, Pax6(S) was always restricted in the nucleus (Fig. 8A, left) and β3 in the cytoplasm (Fig. 8A, right) as expected. However, in the presence of Pax6(S), β3 was translocated into the nucleus and co-localized with Pax6(S) (Fig. 8B). Similar observations were obtained when β3 was with coexpressed with No. 8 (supplemental Fig. S3). In the presence of β3, a larger amount of No. 8 became aggregated along the nuclear membrane and exhibited a punctuate expression pattern (supplemental Fig. S3B). This, however, was not the case for Pax6(S).
FIGURE 8.

Co-localization of overexpressed Pax6(S) and β3 in the nuclei of HEK 293T cells. A, HEK 293T cells expressing Pax6(S) (fused with FLAG, left column) or β3 (fused with EGFP, right column) separately were stained with an anti-FLAG antibody (top left) and a nuclear dye DAPI (middle). The bottom panels show the overlaid immunofluorescence. B, three individual examples of HEK 293T cells transfected with both Pax6(S) fused with FLAG and β3 (fused with EGFP) are displayed in three columns, showing the co-localization of Pax6(S) and β3 in the nuclei. Bar, 10 μm.
DISCUSSION
Pax6(S) Is a Novel Splicing Isoform of Pax6
Pax6 is critical for the development of various tissues and organs, particularly the eye and the nervous system (18, 20, 27–31). Here we reported a new splicing isoform of Pax6, Pax6(S), whose C terminus (PSTNS) is composed of a truncated canonical PST domain and a unique S tail. Compared with Pax6, Pax6(S) differed in tissue distribution, temporal expression profile, and the transactivity of the PSTNS domain. Our results suggest a yet-to-be-defined noncanonical role of Pax6(S) during development.
The protein sequence of classic Pax6 is highly conserved from invertebrates to human, which also leads to functional conservation. For example, overexpression of the mouse or Drosophila Pax6 gene induces an ectopic eye structure in both vertebrates and invertebrates (32–35). In contrast, the protein sequence of Pax6(S) is highly conserved only among primates (supplemental Fig. S1). The critical role of Pax6 in eye development was consistent with its expression in the inner nuclear layer of the human retina (Fig. 3B, left (58)). However, we did not detect any Pax6(S) expression in the human retina (Fig. 3B, right). The divergence in sequence conservation between Pax6 and Pax6(S) and the lack of Pax6(S) expression in the retina imply that whereas Pax6 is involved in developmental events common in both lower and higher animals, Pax6(S) probably plays roles in regulating more specific functions existing solely in primates.
The temporal expression profile of Pax6(S) during development also seems to be different. Classic Pax6 is expressed in both embryos and adults (24). However, Pax6(S) seems to be preferably expressed in embryonic tissues. Even during embryonic development, Pax6(S) expression appears to be strictly regulated. It was detected in 4-month- but not in 5-month-old embryonic human brain (Fig. 3C). This different expression profile of Pax6(S) could be at least partially related to the unique 5′- and 3′-UTRs of its mRNA (Fig. 2).
Cavβ Acts as a Transcription Regulator
As a channel accessory subunit, Cavβ is predominantly localized in the cytoplasm (1–4) (also see Fig. 8A). However, co-expression of Pax6(S) redistributed Cavβ into the nucleus (Fig. 8B). This could be a direct effect of Pax6(S) (i.e. Pax6(S) enters the cytoplasm and transport Cavβ to the nucleus) or involves an unknown chaperone protein(s). The first scenario requires colocalization of Pax6(S) and Cavβ in the cytoplasm, which we never observed under our experimental conditions; however, it could be that the cytoplasmic expression of Pax6(S) was too transient to be detected.
We also showed that the transcriptional activity of Pax6(S) was reduced through its interaction with Cavβ. One could propose at least two scenarios to explain how Cavβ reduces Pax6(S) activity. First, Cavβ, Pax6(S), and Pax6(S)-regulated DNA form a complex. The binding of Cavβ allosterically suppresses Pax6(S) activity. Second, instead of associating with the Pax6(S)-DNA complex, Cavβ forms a complex with Pax6(S) and removes Pax6(S) from its DNA targets. These scenarios need to be examined in further studies.
The potential role of Cavβ in directly regulating gene expression was first suggested by the study of Hibino et al. (59). In that study a short splice variant of β4, named β4c, in the chicken cochlea and brain was found to interact directly with the chromo shadow domain of chromobox protein 2/heterochromatin protein 1γ (CHCB2/HP1γ), a nuclear protein involved in gene silencing and transcriptional regulation. Co-expression of this protein specifically recruited β4c to the nuclei of mammalian cells. Furthermore, β4c dramatically attenuated the gene-silencing activity of CHCB2/HP1γ. This effect was β4c-specific, as a longer isoform, β4a, did not affect CHCB2/HP1γ activity (59). These findings establish β4c as a likely transcription regulator. However, β4c is severely truncated and lacks all the amino acids that are involved in the high affinity interaction between a full-length Cavβ and the I-II loop of the Ca2+ channel α1 subunit (5–7), an interaction that is critical for Cavβ regulation of Ca2+ channel surface expression and biophysical properties. Indeed, β4c has little effect on Ca2+ channel activity (59).
Our results provide evidence that a full-length Cavβ can directly interact with a transcription factor and regulate its activity. Transcriptional regulation may be a general function of Cavβ as, apart from β3, all other Cavβ species (β1b, β2a, and β4) we tested were also able to interact with Pax6(S) (data not shown). Consistent with this notion, β4, and to a lesser extent, β1b and β3, are translocated into the nucleus when exogenously expressed in cardiac cells (60), and β3 localizes in the nucleus when it is co-expressed in PC12 cells with Rad and Rem, two members of the RGK family of Ras-related monomeric small GTP-binding proteins (61).
CASK, a membrane-associated guanylate kinase involved in cell junction, has been shown to have a transcription regulation function that lies in its guanylate kinase domain (62, 63). This further suggests a general role of Cavβ in transcriptional modulation, as all Cavβs contain a homologous guanylate kinase domain (5–7). CASK can either act as a co-activator of Tbr-1, a T-box transcription factor, or as an independent transcription factor through binding to a specific DNA sequence (the T-element) (62). Can Cavβ also act as an independent transcription factor? In our luciferase assay system, where pGL3-OFLuc-CD19-2 was used as the reporter construct, Cavβ was unable to induce luciferase expression in the absence of Pax6(S) (Fig. 7, bar h). However, it cannot be excluded that Cavβ could govern gene expression through binding to an unknown, specific DNA region, the sequence of which could be identified using chromatin immunoprecipitation.
Function and Regulation of Pax6(S)
Pax6(S) is fully conserved only in human and chimpanzee. Therefore, to examine the function of Pax6(S) under physiological conditions, cell lines must be utilized because animal models are impractical. We have screened more than 10 cell lines developed from diverse human tissues including the eye, lung, brain, and colon. Unfortunately, none of them showed detectable expression of Pax6(S) (data not shown). If a suitable cell line is found, the endogenous Pax6(S) can be knocked down with the RNA interference technique, and its physiological functions can be explored by examining changes in gene expression, cell differentiation, and cell morphology.
Phosphorylation and dephosphorylation of Pax6 are important modulations that fine-tune its functions (42–44). Several phosphorylation sites have been identified within the PST domain, including Thr-281, Thr-304, and Thr-373 (42). On the other hand, protein serine/threonine phosphatase-1 dephosphorylates Pax6 and attenuates its activity in human lens epithelial cells (44). Because the S tail of Pax6(S) has a similar proportion of serine but a much lower proportion of proline and threonine compared with the PST tail in Pax6, it is likely that Pax6(S) is regulated differently by protein kinases or phosphatases. In addition, protein phosphorylation has been shown to affect nuclear import or export of various proteins, including the SV40 large T antigen (64), cyclin B1 (65), nucleolin (66), mitogen-activated protein kinase-activated protein kinase 2 (67), tumor suppressor protein 53 (68), and human double minute 2 oncogene (69). It will be interesting to examine whether phosphorylation of Pax6(S) affects its subcellular localization and, if it does, whether this plays a role in the cytoplasm-to-nucleus redistribution of Cavβ (Fig. 8). These studies will provide further insights into the mechanism and physiological regulation of the interaction between Pax6(S) and Cavβ.
Supplementary Material
Acknowledgments
We are grateful to Dr. Ron M. Prywes for luciferase assay constructs. We thank Kathryn Abele and Zafir Buraei for reading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants NS045819 and NS053494 (to J. Y.). This work was also supported by an Established Investigator award from the American Heart Association (to J. Y.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) GQ141695.
- Cavβ
- Ca2+ channel β subunit
- CHCB2/HP1γ
- chromobox protein 2/heterochromatin protein 1γ
- GST
- glutathione S-transferase
- HD
- homeodomain
- HVA
- high voltage-activated
- Pax6
- paired box protein 6
- PD
- paired domain
- PST
- proline, serine, and threonine
- 5′-UTR
- untranslated region
- X-α-gal
- 5-bromo-4-chloro-3-indolyl-α-d-galactopyranoside
- DAPI
- 4′,6-diamidino-2-phenylindole
- CHO
- Chinese hamster ovary
- HEK
- human embryonic kidney
- RGK
- Rem/Rad/Gem/Kir.
REFERENCES
- 1.Arikkath J., Campbell K. P. (2003) Curr. Opin. Neurobiol. 13, 298–307 [DOI] [PubMed] [Google Scholar]
- 2.Birnbaumer L., Qin N., Olcese R., Tareilus E., Platano D., Costantin J., Stefani E. (1998) J. Bioenerg. Biomembr. 30, 357–375 [DOI] [PubMed] [Google Scholar]
- 3.Dolphin A. C. (2003) J. Bioenerg. Biomembr. 35, 599–620 [DOI] [PubMed] [Google Scholar]
- 4.Hidalgo P., Neely A. (2007) Cell Calcium 42, 389–396 [DOI] [PubMed] [Google Scholar]
- 5.Chen Y. H., Li M. H., Zhang Y., He L. L., Yamada Y., Fitzmaurice A., Shen Y., Zhang H., Tong L., Yang J. (2004) Nature 429, 675–680 [DOI] [PubMed] [Google Scholar]
- 6.Opatowsky Y., Chen C. C., Campbell K. P., Hirsch J. A. (2004) Neuron 42, 387–399 [DOI] [PubMed] [Google Scholar]
- 7.Van Petegem F., Clark K. A., Chatelain F. C., Minor D. L., Jr. (2004) Nature 429, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Béguin P., Nagashima K., Gonoi T., Shibasaki T., Takahashi K., Kashima Y., Ozaki N., Geering K., Iwanaga T., Seino S. (2001) Nature 411, 701–706 [DOI] [PubMed] [Google Scholar]
- 9.Finlin B. S., Crump S. M., Satin J., Andres D. A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 14469–14474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiyonaka S., Wakamori M., Miki T., Uriu Y., Nonaka M., Bito H., Beedle A. M., Mori E., Hara Y., De Waard M., Kanagawa M., Itakura M., Takahashi M., Campbell K. P., Mori Y. (2007) Nat. Neurosci. 10, 691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng W., Altafaj X., Ronjat M., Coronado R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 19225–19230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haase H., Podzuweit T., Lutsch G., Hohaus A., Kostka S., Lindschau C., Kott M., Kraft R., Morano I. (1999) FASEB J. 13, 2161–2172 [DOI] [PubMed] [Google Scholar]
- 13.Haase H., Alvarez J., Petzhold D., Doller A., Behlke J., Erdmann J., Hetzer R., Regitz-Zagrosek V., Vassort G., Morano I. (2005) FASEB J. 19, 1969–1977 [DOI] [PubMed] [Google Scholar]
- 14.Yu K., Xiao Q., Cui G., Lee A., Hartzell H. C. (2008) J. Neurosci. 28, 5660–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Gutierrez G., Miranda-Laferte E., Neely A., Hidalgo P. (2007) J. Biol. Chem. 282, 2156–2162 [DOI] [PubMed] [Google Scholar]
- 16.Walther C., Guenet J. L., Simon D., Deutsch U., Jostes B., Goulding M. D., Plachov D., Balling R., Gruss P. (1991) Genomics 11, 424–434 [DOI] [PubMed] [Google Scholar]
- 17.Dahl E., Koseki H., Balling R. (1997) BioEssays 19, 755–765 [DOI] [PubMed] [Google Scholar]
- 18.Dohrmann C., Gruss P., Lemaire L. (2000) Mech. Dev. 92, 47–54 [DOI] [PubMed] [Google Scholar]
- 19.Kozmik Z. (2005) Curr. Opin. Genet. Dev. 15, 430–438 [DOI] [PubMed] [Google Scholar]
- 20.Kozmik Z. (2008) Brain Res. Bull. 75, 335–339 [DOI] [PubMed] [Google Scholar]
- 21.Pichaud F., Desplan C. (2002) Curr. Opin. Genet. Dev. 12, 430–434 [DOI] [PubMed] [Google Scholar]
- 22.Strachan T., Read A. P. (1994) Curr. Opin. Genet. Dev. 4, 427–438 [DOI] [PubMed] [Google Scholar]
- 23.Ziman M. R., Rodger J., Chen P., Papadimitriou J. M., Dunlop S. A., Beazley L. D. (2001) Histol. Histopathol. 16, 239–249 [DOI] [PubMed] [Google Scholar]
- 24.Callaerts P., Halder G., Gehring W. J. (1997) Annu. Rev. Neurosci. 20, 483–532 [DOI] [PubMed] [Google Scholar]
- 25.Ton C. C., Hirvonen H., Miwa H., Weil M. M., Monaghan P., Jordan T., van Heyningen V., Hastie N. D., Meijers-Heijboer H., Drechsler M. (1991) Cell 67, 1059–1074 [DOI] [PubMed] [Google Scholar]
- 26.Sakurai K., Osumi N. (2008) J. Neurosci. 28, 4604–4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osumi N., Shinohara H., Numayama-Tsuruta K., Maekawa M. (2008) Stem Cells 26, 1663–1672 [DOI] [PubMed] [Google Scholar]
- 28.Nomura T., Haba H., Osumi N. (2007) Dev. Growth Differ. 49, 683–690 [DOI] [PubMed] [Google Scholar]
- 29.Manuel M., Price D. J. (2005) Brain Res. Bull. 66, 387–393 [DOI] [PubMed] [Google Scholar]
- 30.Brink C. (2003) Cell. Mol. Life Sci. 60, 1033–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Habener J. F., Stoffers D. A. (1998) Proc. Assoc. Am. Physicians 110, 12–21 [PubMed] [Google Scholar]
- 32.Altmann C. R., Chow R. L., Lang R. A., Hemmati-Brivanlou A. (1997) Dev. Biol. 185, 119–123 [DOI] [PubMed] [Google Scholar]
- 33.Chow R. L., Altmann C. R., Lang R. A., Hemmati-Brivanlou A. (1999) Development 126, 4213–4222 [DOI] [PubMed] [Google Scholar]
- 34.Halder G., Callaerts P., Gehring W. J. (1995) Science 267, 1788–1792 [DOI] [PubMed] [Google Scholar]
- 35.Onuma Y., Takahashi S., Asashima M., Kurata S., Gehring W. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 2020–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walther C., Gruss P. (1991) Development 113, 1435–1449 [DOI] [PubMed] [Google Scholar]
- 37.Tang H. K., Singh S., Saunders G. F. (1998) J. Biol. Chem. 273, 7210–7221 [DOI] [PubMed] [Google Scholar]
- 38.Singh S., Chao L. Y., Mishra R., Davies J., Saunders G. F. (2001) Hum. Mol. Genet. 10, 911–918 [DOI] [PubMed] [Google Scholar]
- 39.Czerny T., Busslinger M. (1995) Mol. Cell. Biol. 15, 2858–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glaser T., Jepeal L., Edwards J. G., Young S. R., Favor J., Maas R. L. (1994) Nat. Genet. 7, 463–471 [DOI] [PubMed] [Google Scholar]
- 41.Tjian R., Maniatis T. (1994) Cell 77, 5–8 [DOI] [PubMed] [Google Scholar]
- 42.Kim E. A., Noh Y. T., Ryu M. J., Kim H. T., Lee S. E., Kim C. H., Lee C., Kim Y. H., Choi C. Y. (2006) J. Biol. Chem. 281, 7489–7497 [DOI] [PubMed] [Google Scholar]
- 43.Mikkola I., Bruun J. A., Bjorkoy G., Holm T., Johansen T. (1999) J. Biol. Chem. 274, 15115–15126 [DOI] [PubMed] [Google Scholar]
- 44.Yan Q., Liu W. B., Qin J., Liu J., Chen H. G., Huang X., Chen L., Sun S., Deng M., Gong L., Li Y., Zhang L., Liu Y., Feng H., Xiao Y., Liu Y., Li D. W. (2007) J. Biol. Chem. 282, 13954–13965 [DOI] [PubMed] [Google Scholar]
- 45.He L. L., Zhang Y., Chen Y. H., Yamada Y., Yang J. (2007) Biophys. J. 93, 834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolognani F., Perrone-Bizzozero N. I. (2008) J. Neurosci. Res. 86, 481–489 [DOI] [PubMed] [Google Scholar]
- 47.Cannell I. G., Kong Y. W., Bushell M. (2008) Biochem. Soc. Trans. 36, 1224–1231 [DOI] [PubMed] [Google Scholar]
- 48.Evans T. C., Hunter C. P. (2005) WormBook, (C. elegans Research Community, ed) WormBook, doi/10.1895/wormbook.1.34.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.López de Silanes I., Quesada M. P., Esteller M. (2007) Cell. Oncol. 29, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shyu A. B., Wilkinson M. F., van Hoof A. (2008) EMBO J. 27, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steinman R. A. (2007) Leukemia 21, 1158–1171 [DOI] [PubMed] [Google Scholar]
- 52.Pickering B. M., Willis A. E. (2005) Semin. Cell Dev. Biol. 16, 39–47 [DOI] [PubMed] [Google Scholar]
- 53.van der Velden A. W., Thomas A. A. (1999) Int. J. Biochem. Cell Biol. 31, 87–106 [DOI] [PubMed] [Google Scholar]
- 54.Carrière C., Plaza S., Caboche J., Dozier C., Bailly M., Martin P., Saule S. (1995) Cell Growth Differ. 6, 1531–1540 [PubMed] [Google Scholar]
- 55.Lefebvre T., Planque N., Leleu D., Bailly M., Caillet-Boudin M. L., Saule S., Michalski J. C. (2002) J. Cell. Biochem. 85, 208–218 [PubMed] [Google Scholar]
- 56.Guinez C., Morelle W., Michalski J. C., Lefebvre T. (2005) Int. J. Biochem. Cell Biol. 37, 765–774 [DOI] [PubMed] [Google Scholar]
- 57.Monsigny M., Rondanino C., Duverger E., Fajac I., Roche A. C. (2004) Biochim. Biophys. Acta 1673, 94–103 [DOI] [PubMed] [Google Scholar]
- 58.Stanescu D., Iseli H. P., Schwerdtfeger K., Ittner L. M., Remé C. E., Hafezi F. (2007) Eye 21, 90–93 [DOI] [PubMed] [Google Scholar]
- 59.Hibino H., Pironkova R., Onwumere O., Rousset M., Charnet P., Hudspeth A. J., Lesage F. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 307–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colecraft H. M., Alseikhan B., Takahashi S. X., Chaudhuri D., Mittman S., Yegnasubramanian V., Alvania R. S., Johns D. C., Marbán E., Yue D. T. (2002) J. Physiol. 541, 435–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Béguin P., Mahalakshmi R. N., Nagashima K., Cher D. H., Ikeda H., Yamada Y., Seino Y., Hunziker W. (2006) J. Mol. Biol. 355, 34–46 [DOI] [PubMed] [Google Scholar]
- 62.Hsueh Y. P., Wang T. F., Yang F. C., Sheng M. (2000) Nature 404, 298–302 [DOI] [PubMed] [Google Scholar]
- 63.Hsueh Y. P. (2006) Curr. Med. Chem. 13, 1915–1927 [DOI] [PubMed] [Google Scholar]
- 64.Jans D. A., Ackermann M. J., Bischoff J. R., Beach D. H., Peters R. (1991) J. Cell Biol. 115, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang J., Bardes E. S., Moore J. D., Brennan J., Powers M. A., Kornbluth S. (1998) Genes Dev. 12, 2131–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwab M. S., Dreyer C. (1997) Eur. J. Cell Biol. 73, 287–297 [PubMed] [Google Scholar]
- 67.Engel K., Kotlyarov A., Gaestel M. (1998) EMBO J. 17, 3363–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takahashi K., Suzuki K. (1993) Int. J. Cancer 55, 453–458 [DOI] [PubMed] [Google Scholar]
- 69.Jackson M. W., Patt L. E., LaRusch G. A., Donner D. B., Stark G. R., Mayo L. D. (2006) J. Biol. Chem. 281, 16814–16820 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.