

Abstract
Background and purpose:
α7-Nicotinic acetylcholine receptors (α7 nAChRs) play an important role in cognitive function. Positive allosteric modulators (PAMs) amplify effects of α7 nAChR agonist and could provide an approach for treatment of cognitive deficits in neuropsychiatric diseases. PAMs can either predominantly affect the apparent peak current response (type I) or increase both the apparent peak current response and duration of channel opening, due to prolonged desensitization (type II). The delay of receptor desensitization by type II PAMs raises the possibility of Ca2+-induced toxicity through prolonged activation of α7 nAChRs. The present study addresses whether type I and II PAMs exhibit different cytotoxicity profiles.
Experimental approach:
The present studies evaluated cytotoxic effects of type I PAM [N-(4-chlorophenyl)]-α-[(4-chloro-phenyl)-aminomethylene]-3-methyl-5-isoxazoleacet-amide (CCMI) and type II PAM 1-[5-chloro-2,4-dimethoxy-phenyl]-3-[5-methyl-isoxazol-3-yl]-urea (PNU-120596), or 4-[5-(4chloro-phenyl)-2-methyl-3-propionyl-pyrrol-1-yl]-benzenesulphonamide (A-867744). The studies used cultures of PC12 cells and primary cultures of rat cortical neuronal cells.
Key results:
Our results showed that neither type I nor type II PAMs had any detrimental effect on cell integrity or cell viability. In particular, type II PAMs did not affect neuron number and neurite outgrowth under conditions when α7 nAChR activity was measured by Ca2+ influx and extracellular signal-regulated kinases 1 and 2 phosphorylation, following exposure to α7 nAChR agonists.
Conclusions and implications:
This study demonstrated that both type I and type II α7 nAChR selective PAMs, although exhibiting differential electrophysiological profiles, did not exert cytotoxic effects in cells endogenously expressing α7 nAChRs.
Keywords: α7 nAChR, positive allosteric modulator, ERK1/2, cytotoxicity
Introduction
α7-Nicotinic acetylcholine receptors (nAChRs; nomenclature follows Alexander et al., 2008) are acetylcholine-gated cation channels that exhibit biophysical and pharmacological profiles distinct from other nAChR subtypes and are located in brain regions critical to learning and memory (see Dajas-Bailador and Wonnacott, 2004; Hogg and Bertrand, 2004). Preclinical validation of α7 nAChRs as a drug target for potential treatment of cognitive deficits such as in Alzheimer's disease and schizophrenia largely stems from pharmacological studies in animal models. For example, α7 nAChR selective agonists have been shown to improve cognitive performance across a variety of preclinical models relevant to Alzheimer's disease and schizophrenia (Van Kampen et al., 2004; Bodnar et al., 2005; Wishka et al. 2006; Bitner et al. 2007; Boess et al. 2007; Pichat et al. 2007) whereas antagonists or antisense have been shown to impair cognitive function (Felix and Levin, 1997; Bettany and Levin, 2001; Curzon et al., 2006). More recently, α7 nAChR selective positive allosteric modulators (PAMs) have also been described and shown to be effective in animal models, further validating the role of α7 nAChRs.
PAMs are chemically heterogeneous and capable of differentially modifying the biophysical properties of α7 nAChRs without interacting at the orthosteric binding site (for a review, see Bertrand and Gopalakrishnan, 2007). PAMs, in principle, can enhance α7 nAChR functions without altering the temporal integrity of neurotransmission. Based on biophysical properties, at least two different profiles of α7 PAMs can be distinguished: type I – exemplified by 5-hydroxyindole, genistein, NS-1738 and CCMI (Compound 6 in Ng et al., 2007) – predominantly increases peak current amplitude response, and type II – represented by PNU-120596 (Hurst et al., 2005), TQS (Grønlien et al., 2007) and A-867744 (Malysz et al., 2009) – affecting both the peak current response and the current decay profile. Both type I and II PAMs have shown efficacy in cognition and sensory gating deficit models in vivo (Hurst et al., 2005; Timmermann et al., 2007; Ng et al. 2007; Faghih et al. 2009).
Although types I and II PAMs with different electrophysiological profiles have demonstrated in vivo behavioural efficacy, the physiological and pharmacological effects following the allosteric modulation needs to be further defined. Especially, changes in desensitization profiles of α7 nAChR and the associated changes in Ca2+ dynamics can be triggered by type II PAMs through delaying desensitization of the receptor, but not by type I PAMs (see Bertrand and Gopalakrishnan, 2007). It has been suggested that the altered kinetics and increased sustained levels of Ca2+ may lead to activation of various cytotoxic events. For example, in the characterization of a type I PAM CCMI, it was shown that PNU-120596 was cytotoxic, whereas CCMI did not significantly alter nAChR kinetics and had no effect on cell viability (Ng et al., 2007). With the emergence of many type I and II PAMs, the question remains whether Ca2+ entry resulted from delayed α7 nAChR desensitization by type II PAMs leads to Ca2+-induced cytotoxicity. This study aimed to address whether type I and type II PAMs can reveal differential cytotoxicity profiles. Our data suggest that both type I and type II PAMs do not cause toxic effect when enhancing the activation of α7 nAChR in PC12 and rat cortical neuronal cells.
Methods
Cell culture and treatment
All animal care and experimental procedures complied with Institutional guidelines.
Rat cortical neuronal cells were isolated and cultured as previously described (Hu et al., 2007). Briefly, postnatal day 0 (P0) Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA, USA) were anesthetized with inhaled CO2 and killed by decapitation. The meninges were removed and the cortices were excised and subsequently incubated at 37°C for 30 min in Hanks' Balanced Salt Solutions (HBSS) containing 35 U·mL−1 papain before the addition of 0.2 mg·mL−1 DNase I buffer in HBSS. Tissues were then mechanically triturated by repeated passages through a 5-mL pipette. Cell debris was removed by passing cells through a 70 µm nylon filter. The cell suspension was centrifuged at 200 ×g for 5 min at room temperature and the resulting pellet was resuspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum. Cortical neuronal cell cultures were plated at a density of 5 × 105 cells·mL−1 in poly-D-lysine coated 96-well plates and maintained at 37°C with 5% CO2.
PC12 cells (American Type Culture Collection, Manassas, VA) were cultured as previously described (Hu et al., 2008). Briefly, PC12 cells were plated on poly-D-lysine coated 96-well plates, cultured in Ham's F12K medium supplemented with 15% horse serum, 2.5% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin at 37°C with 5% CO2.
Calcium mobilization assay (FLIPR)
α7 nAChR-mediated elevation of intracellular Ca2+ levels was measured using the FLIPR calcium 4 assay kit. PC12 cells and rat P0 cortical neuronal cells were grown in the poly-D-lysine coated 96-well plates for 3 and 6 days respectively. Prior to the assay, the culture media were removed and cells were loaded with 100 µL of calcium-4 no-wash dye prepared in N-methyl D-glucosamine (NMDG) buffer (10 mM HEPES, pH 7.4, 140 mM NMDG, 5 mM KCl, 1 mM MgCl2, 10 mM CaCl2) for 1 h at room temperature. α7 nAChR PAMs and PNU-282987 were also prepared in NMDG buffer. The effects of α7 nAChR PAMs were determined by first adding PAMs for 5 min followed by addition of 10 µM PNU-282 987 for another 5 min. Changes in fluorescence intensity were recorded over time at 25°C in FLIPR (λEX= 488 nm, λEM= 540 nm). Concentration–response data were generated.
Extracellular signal-regulated kinases 1 and 2 (ERK1/2) in-cell Western blot assays
Undifferentiated PC12 cells were cultured for 3 days. Prior to experiments, the culture media were replaced with serum-free media, to starve cells overnight. On the day of the experiment, cell media were removed and cells were treated with α7 nAChR PAMs prepared in Dulbecco's phosphate buffer saline (D-PBS) for 10 min at 37°C followed by addition of 10 µM α7 nAChR agonist PNU-282987 for 5 min. Cells were then fixed with 4% paraformaldehyde for 60 min, followed by four washes using phosphate-buffered saline (PBS) (pH 7.4), permeabilized with 0.1% Triton X-100/PBS and blocked with the Odyssey blocking buffer for 3 h at room temperature. The cells were then incubated overnight at 4°C with mouse anti-phospho-ERK (pERK) monoclonal antibody (1:500) and rabbit anti-total ERK (tERK) antibody (1:1000). On the following day, cells were washed three times with 0.1% Tween-20/PBS and incubated with the Alexa Fluor 680 anti-rabbit antibody (1:1000) for detection of total ERK (tERK) or with IRDye 800CW anti-mouse IgG antibody (1:1000) for 1 h at room temperature for detection of pERK. Cells were washed three times, and target signals were simultaneously visualized using Odyssey Infrared Imaging Scanner (Li-Cor Biosciences, Lincoln, NE, USA) with the 680-nm fluorophore emitting an image of red color (tERK) and the 800-nm fluorophore emitting an image of green color (pERK). The integrated fluorescence intensities were calculated and analysed using the Odyssey Infrared Imaging System Application Software version 1.2.15 (Li-Cor Biosciences). Data analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA). tERK and pERK signals were quantitated and pERK/tERK ratio were normalized to the control response of 10 µM PNU-120596 in presence of 10 µM PNU-282987.
Cytotoxicity assays
Cytotoxicity was measured by cell integrity and cell viability. PC12 cells and P0 cortical neuronal cells were cultured in complete growth medium for 3 and 6 days, respectively, prior to treatment with varying concentrations of PAMs for 24 h. Cell integrity was assessed using Toxilight assay (Cambrex, Rockland, ME, USA) that measures activity of the enzyme adenylate kinase (AK) released into the medium from damaged cells. Toxilight bioassay was conducted as previously described (Miret et al., 2006). Briefly, 20 µL of cell culture media were transferred to a white walled 96-well plate, and 100 µL of AK working solution was added to each well. The plates were incubated for 5 min at room temperature. Luminescence was measured in the Wallac Victor 21 420 multilabel counter (PerkinElmer, Waltham, MA, USA). Cell viability was assessed using the Cell Titer 96 Aqueous One Solution kit (Promega, Madison, WI, USA) that measures activity of mitochondrial dehydrogenase in metabolically active cells. Cell Titer 96 Aqueous One Solution assay was conducted as previously described (Woods et al., 2008). The colorimetric methyl tetrazolium salt reagent was added to each well of the plate, and incubated for 1–4 h. The absorbance values were read at 490 nm.
High content screen (HCS) microscopy analysis
Rat P0 primary cortical neuronal cells were cultured for 6 days and treated with varying concentrations of either PNU-120 596 or A-867744 alone and in presence of 10 µM PNU-282987 for 3 days. Cells were then fixed and stained as previously described (Hu et al., 2007). Briefly, neurons were stained by mouse anti-tubulin monoclonal antibody (1:100) and then recognized by FITC-labelled anti-mouse antibody (1:1000). Nuclei (360/40 nm excitation and 465/30 nm emission filters) and neuron (475/35 nm excitation and 535/40 nm emission filters) images were collected using the ImageXpress Micro automatic fluorescent microscopy system. Typically, four wells were used for each treatment condition on a 96-well plate, and images were collected from four fields within each well. The neurite outgrowth module in the Imaging software was used to automatically count neuron numbers and the extent of neurite outgrowth. Experiments were conducted independently three times, in quadruplicate for each treatment group.
Data analysis
Data are expressed as mean± SEM. Student's t-tests (paired and two-tailed) were performed to assess significant differences. Wherever applicable, P values less then 0.05 were considered statistically significant.
Materials
DMEM, Ham's F12K medium, PBS, D-PBS, FBS, horse serum, L-glutamine and penicillin/streptomycin were purchased from Gibco Life Technologies Inc. (Grand Island, NY, USA). Papain (Worthington, Lakewood, NJ, USA), Deoxyribonuclease I (DNase I), monoclonal mouse anti-phospho-ERK 1/2 and polyclonal rabbit anti-ERK1/2 antibodies were purchased from Sigma-Aldrich (St. Louis, MO, USA). IRDye 800CW conjugated affinity-purified anti-mouse IgG antibody (Rockland Immunochemicals, Gilbertsville, PA, USA), Alexa Fluor 680 goat anti-rabbit IgG antibody (H+L) (Molecular Probes, Eugene, OR, USA), Hoechst 33 342 (Molecular Probes), Anti-tubulin monoclonal antibody (Upstate, Lake Placid, NY, USA), Rabbit anti-glial fibrillary acidic protein (GFAP) antibody (Chemicon International Inc, Temecula, CA, USA), Fluorescein (FITC)-conjugated affinity-purified donkey anti-mouse lgG antibody and rhodamine (TRITC)-conjugated affinity-purified donkey anti-rabbit lgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were purchased from indicated sources. Poly-D-lysine coated 96-well plates were purchased from Becton Dickinson Labware (Bedford, MA, USA). Nylon filters (70 mm) were purchased from BD Biosciences Discovery Labware (Bedford, MA, USA). Test compounds including A-867744, PNU-120596, PNU-282987 and CCMI were synthesized in house. Odyssey Infrared Imaging Scanner and blocking buffer were from Li-Cor Biosciences. The ImageXpress Micro automated fluorescent microscopy system, flurometric imaging plate reader (FLIPR) and FLIPR calcium4 assay kit were from Molecular Devices Corporation (Sunnyvale, CA, USA).
Results
Effect of PAMs on intracellular Ca2+ changes in PC12 and cortical neurons
PC12 and rat cortical neuronal cells have been shown to express endogenous functional α7 nAChRs either by high affinity binding of α7 nAChR ligands or by functional responsiveness to α7 nAChR ligands (Dickinson et al., 2007; Anderson et al., 2008; El Kouhen et al., 2009), or by α7 nAChR-mediated calcium signalling (Ren et al., 2005; Dickinson et al. 2007). Initially, we determined the differential modulation of α7 nAChR agonist activity by type I and II PAMs by both FLIPR-based Ca2+ and ERK1/2 phosphorylation measurements. Both PNU-120596 and A-867 744 evoked robust increases in intracellular Ca2+ in the presence of α7 nAChR agonist PNU-282987 in both PC12 [Figure 1A, EC50= 0.2 µM (0.13–0.27 µM, 95% CI, n= 3) and EC50= 0.3 µM (0.20–0.39 µM, 95% CI, n= 3) respectively] and rat cortical neuronal cells [Figure 1B, EC50= 0.3 µM (0.14–0.60 µM, 95% CI, n= 3) and EC50= 0.1 µM (0.07–0.16 µM, 95% CI, n= 3) respectively]. No robust intracellular Ca2+ changes were observed with PAM or agonist alone as expected (data not shown). When applied with agonist, CCMI did not induce substantial intracellular Ca2+ changes in either cell line (Figure 1A,B). These observations indicated that types I and II PAMs differentially modulated α7 nAChR agonist activity in the PC12 and rat cortical neuronal cells, which was consistent with the differential effects of type I and type II PAMs on agonist-evoked Ca2+ dynamics.
Figure 1.
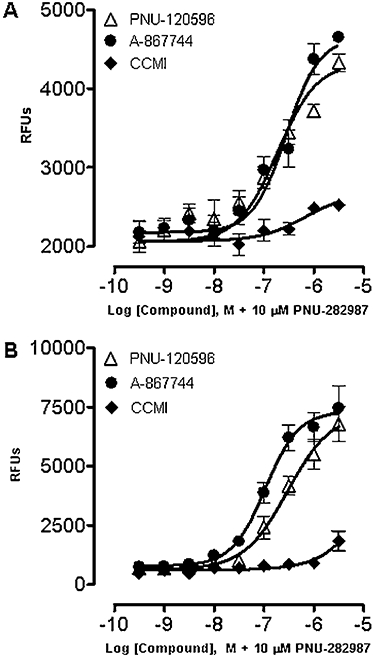
Effect of α7 nAChR type I and type II PAMs on intracellular Ca2+ levels in PC12 cells and rat cortical neuronal cells in culture. PC12 cells (A) or rat cortical neuronal cells (B) were treated with a range of concentrations of PNU-120596, A-867744 or CCMI for 10 min before the addition of 10 µM PNU-282987 for 5 min. Ca2+ influx was measured by FLIPR calcium 4 assay and expressed as relative fluorescence units (RFUs). Each point represents the mean ± SEM of three independent experiments.
Effect of PAMs on phosphorylation of ERK1/2 in PC12 cells
Next, we assessed the effects of PAMs on ERK1/2 phosphorylation in PC12 cells. A robust increase in ERK1/2 phosphorylation was induced by α7 nAChR agonists in the presence of type II PAMs (El Kouhen et al., 2009). As illustrated in Figure 2, ERK phosphorylation was enhanced in a concentration-dependent manner by both PNU-120596 and A-867744 in the presence of the agonist PNU-282987 [Figure 2A,B, EC50= 1.7 µM (1.1–2.5 µM, 95% CI, n= 3) and EC50= 0.4 µM (0.35–0.52 µM, 95% CI, n= 3 respectively]. However, CCMI did not increase pERK levels in the present of PNU-282987. The agonist or PAM alone did not increase pERK, as described previously (El Kouhen et al., 2009). These observations, consistent with the differential effects on agonist-evoked Ca2+ dynamics, further indicate distinctions between type I and type II PAMs in modulating α7 nAChR functions.
Figure 2.
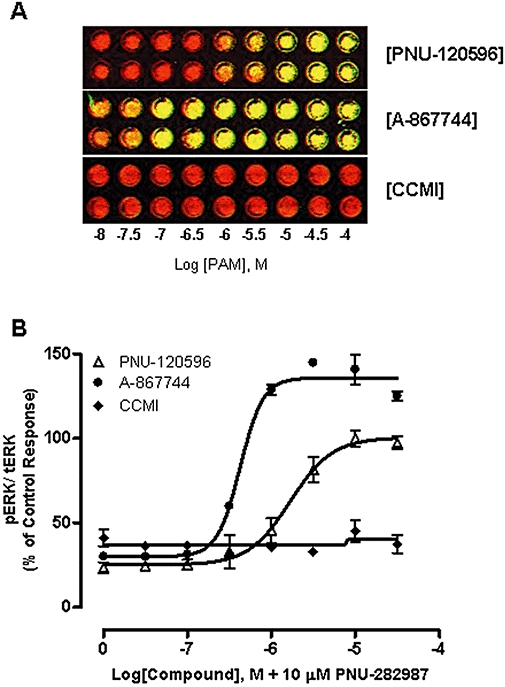
Effect of α7 nAChR type I and type II PAMs on ERK1/2 phosphorylation in PC12 cells. (A) Representative images of ERK1/2 phosphorylation measured by InCell Western in a 96-well format. PC12 cells were treated with varying concentrations of PNU-120596, A-867744 or CCMI for 10 min at 37°C prior to the addition of 10 µM PNU-282987 for 5 min. Cells were then imaged for ERK1/2 phosphorylation signals. The superimposed image indicates co-staining of pERK (green) and tERK (red). (B) ERK1/2 phosphorylation. tERK and pERK signals were quantified and pERK/tERK ratio were normalized to the control response to 10 µM PNU-120596 + 10 µM PNU-282987. Each point represents the mean ± SEM of three independent experiments.
Effects of PAMs on cell integrity and viability in PC12 cells and cortical neurons
Potential cytotoxic effects of PAMs were assessed in PC12 and rat cortical neurons by two measurements: cell integrity and viability (see Methods). Thapsigargin was used as a positive toxicity control for both assays.
PC12 cells treated with varying concentrations of PAMs in the absence or presence of the agonist PNU-282987 for 24 h did not show significant loss of cell integrity (Figure 3A) or significant decreases in cell viability (Figure 3B). Under these conditions, concentration-dependent cytotoxicity was observed with thapsigargin in both assays (Figure 3A,B).
Figure 3.
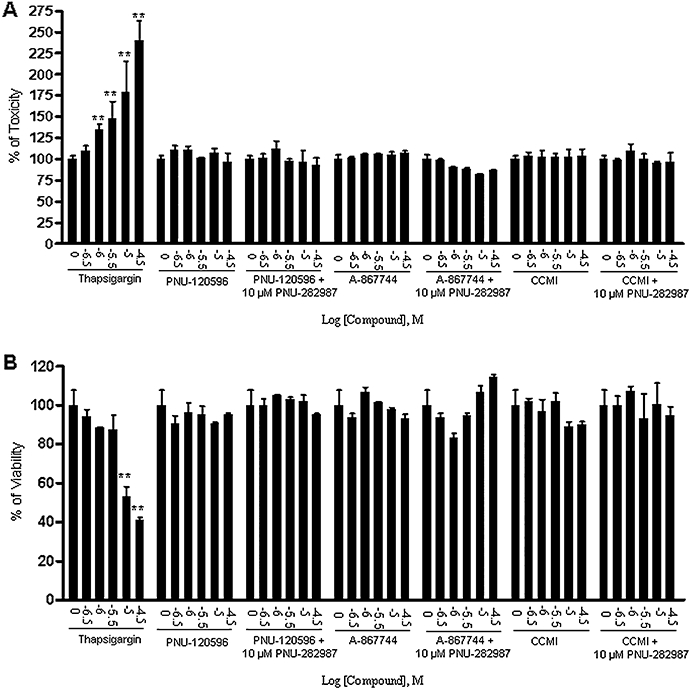
α7 nAChR type I and type II PAMs did not show cytotoxic effect on cell integrity or cell viability of PC12 cells. PC12 cells were cultured for 3 days and then treated with a range of concentrations of PNU-120596, A-867744 or CCMI alone or in the presence of 10 µM PNU-282987 for 24 h followed by measurements of cell integrity determined by the release of adenylate kinas and cell viability determined by mitochondrial dehydrogenase enzyme activity. (A) Cell integrity: PAMs alone or in combination with agonist did not increase the release of adenylate kinase, whereas thapsigargin reduced cell integrity in a concentration-dependant manner. (B) Cell viability: PAMs alone or in combination with the agonist did not affect mitochondrial dehydrogenase enzyme activity, whereas thapsigargin significantly reduced cell viability in a concentration-dependent manner. The values are shown as the percentage of untreated cells. Each point represents the mean ± SEM of 5 independent experiments. **P < 0.01 versus the untreated group.
Cell integrity and cell viability were also measured in rat primary cortical neuronal cells following treatment with varying concentrations of PAMs (PNU-120596 or CCMI) in the absence or presence of the agonist PNU-282987. Again, PAMs alone or in combination with PNU-282987 revealed no cytotoxicity in both assays, while thapsigargin showed concentration-dependent cytotoxic effects under these conditions (Figure 4A,B).
Figure 4.
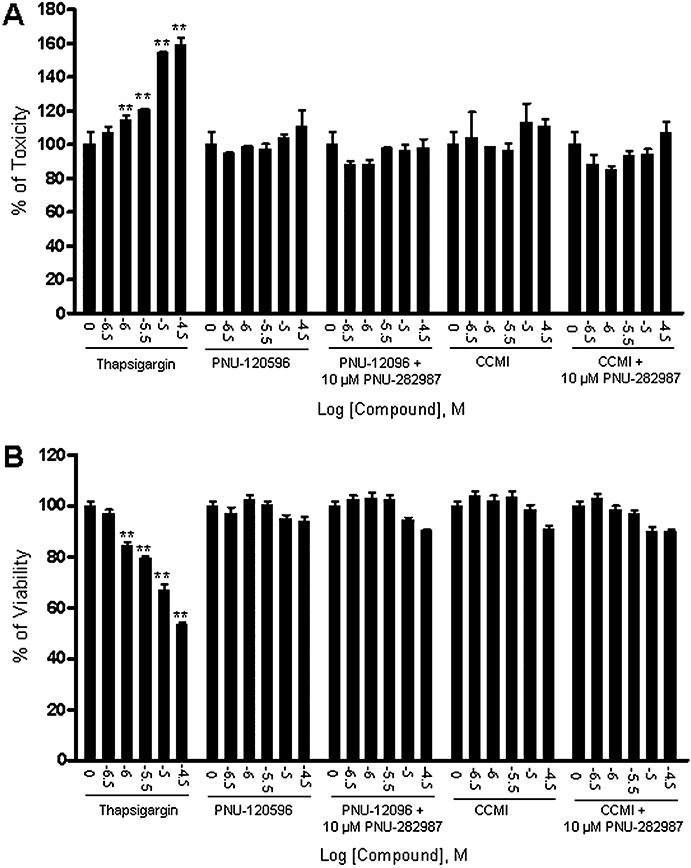
α7 nAChR type I and type II PAMs did not show cytotoxic effect on integrity or viability of rat cortical neuronal cells. Rat primary cortical neuronal cells were cultured for 6 days and then treated with a range of concentrations of PNU-120596 or CCMI alone or in the presence of 10 µM PNU-282987 for 24 h. Cell integrity was determined by the release of adenylate kinase and cell viability was determined by mitochondrial dehydrogenase enzyme activity. (A) Cell integrity: PAMs alone or in combination with the agonist did not increase the release of adenylate kinase, whereas thapsigargin reduced cell integrity in a concentration-dependent manner. (B) Cell viability: PAMs alone or in combination with the agonist did not affect mitochondrial dehydrogenase enzyme activity, whereas thapsigargin significantly reduced cell viability in a concentration-dependent manner. The values are shown as the percentage of the untreated group. Each point represents the mean ± SEM of four independent experiments. **P < 0.01 versus the untreated group.
To further assess for potentially subtle effects of the PAMs, we utilized high-content microscopy techniques to determine changes in neuron number and neurite outgrowth in rat primary cortical neuronal cell cultures. Cells were treated with varying concentrations of PNU-120596 or A-867744 in the presence or absence of the α7 nAChR agonist PNU-282987 for 3 days. Figure 5 demonstrates that PNU-120596 or A-867744 (up to 30 µM) alone or in combination with PNU-282987 did not alter morphology of the cells (Figure 5A), neuron number (Figure 5B) or neurite outgrowth (Figure 5C).
Figure 5.
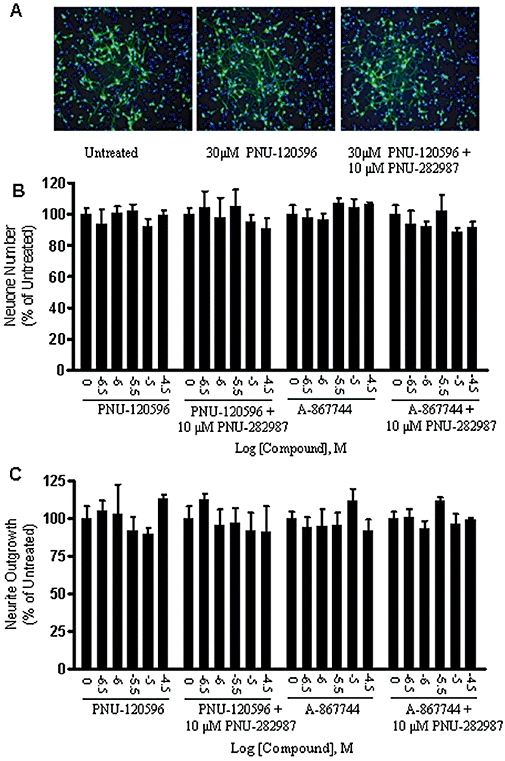
α7 nAChR type II PAMs did not show cytotoxic effect on morphology, cell number or neurite outgrowth of rat primary cortical neurons. Rat primary cortical neuronal cells were cultured for 6 days and then treated with a range of concentrations of PNU-120596 or A-867744 in the presence or absence of the α7 nAChR agonist PNU-282987 for 3 days. Neurons were stained with anti-tubulin antibody (green) and nuclei were stained with Hoechst 33342 (blue). (A) Representative superimposed images of neurons and nuclei treated with buffer control, 30 µM PNU-120596 or 30 µM PNU-120596 in combination with 10 µM PNU-282987 respectively. The PAMs alone or in combination with the agonist did not affect neuron morphology (A), neuron number (B), or neurite outgrowth (C). The values are shown as the percentage of the untreated group. Each point represents the mean ± SEM of three independent experiments.
Discussion
In the present study, we investigated the effect of type I and type II PAMs in PC12 and rat cortical neuronal cells expressing endogenous α7 nAChR. Our results showed that type II PAMs PNU-120596 and A-867744 did not exhibit detrimental effects on cell integrity, cell viability, neuron number or neurite outgrowth under conditions when receptor activity was measured by downstream cellular responses such as α7 nAChR-sensitive Ca2+ influx and ERK1/2 phosphorylation.
In the recent years, a chemically heterogenous class of molecules has been identified as selective PAMs of α7 nAChRs (Bertrand and Gopalakrishnan, 2007). Initial reports have suggested that important considerations be given regarding development of PAMs, particularly those belonging to type II, as therapeutic agents, as the effects of type II PAMs on desensitization could lead to cytotoxic effects. For example, Ng et al. (2007) showed that PNU-120 596 was cytotoxic in SHSY-5Y cells overexpressing α7 nAChRs, whereas CCMI was not. In addition, in L250T mutant mice, the mutation in the α7 nAChR that reduced desensitization promoted apoptosis (Orr-Urtreger et al., 2000). With the emergence of many more type I and II PAMs, the question remains whether Ca2+ entry due to preventing or reversing α7 nAChR desensitization by type II PAMs leads to Ca2+-induced cytotoxicity.
PNU-120596 and A-867744 are examples of prototypical type II PAMs that increases both agonist-evoked peak currents and prolongs the evoked response in the continued presence of the agonist (Hurst et al., 2005; Grønlien et al., 2007) whereas compounds like CCMI and NS-1738 are examples of type I PAMs with no effect on desensitization kinetics. In the present study, we showed that both of the type II PAMs dramatically enhanced Ca2+ responses and ERK phosphorylation in a concentration-dependent manner whereas CCMI, a type I PAM, did not. ERK1/2 phosphorylation in response to α7 nAChR agonists has been recently demonstrated both in vitro (Steiner et al., 2007; Dickinson et al., 2008; El Kouhen et al., 2009) and in vivo (Bitner et al., 2007). For example, in PC12 cells, robust increase in ERK1/2 phosphorylation can be revealed by α7 nAChR agonists in the presence of type II PAMs (El Kouhen et al., 2009). The observation that type I PAMs do not increase Ca2+ response or ERK phosphorylation changes in vitro is attributable to the lack of effect of such compounds on receptor desensitization, in addition to amplification of peak current responses, detected using electrophysiological approaches (Ng et al., 2007; Timmermann et al., 2007).
No cytotoxic effects were detected when type I or type II PAMs were applied alone or combined with a selective α7 agonist, PNU-282987, as assessed by changes in adenylate kinase release or mitochondrial dehydrogenase activity in both PC12 cells and in cortical neurons, under conditions where thapsigargin evoked concentration-dependent cytotoxic effects. We also employed HCS, a sensitive method for detecting neuronal degeneration and enabling quantitative analysis of a larger amount of morphology data than conventional microscopy (Hu et al., 2008). Treatment of cortical neuronal cells with type II PAMs in the absence or presence of α7 nAChR agonist PNU-282987 for 3 days did not alter cell morphology, neuron number or neurite outgrowth (Figure 5). These observations indicating lack of cytotoxicity with PAMs of either profile is in contrast to the report showing that PNU-120596 was cytotoxic whereas CCMI had no effect on cell viability in SH-SY5Y cells transfected with α7 nAChRs (Ng et al., 2007). The basis of this discrepancy is unclear at present, but might be attributed to the potential differences between rodent and human cell lines, overexpression of α7 nAChRs per se or limitations in the assays. For example, the cytotoxic effect of thapsigargin on SH-SY5Y cells [10 µM yielding 20–30% viability; Ng et al. (2007)] appears to be greater than that on PC12 cells (10 µM yielding 50–70% viability) examined in this study. In addition to cell viability, our present study thoroughly analysed cell integrity and morphology in both PC12 and primary rat cortical neuronal cells. Thus, these approaches may be more relevant to physiological conditions although it should be noted that in vivo studies following chronic treatment with PAMs will be necessary to define their longer-term effects on safety and tolerability. In conclusion, our study demonstrated that α7 nAChR PAMs do not exhibit cytotoxic properties in cells expressing α7 nAChRs. PAMs derived from both profiles could represent viable opportunities as therapeutic approaches for treatment of cognitive dysfunction and other indications.
Glossary
Abbreviations:
- a7 nAChRs
a7 nicotinic acetylcholine receptors
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- HCS
high content screen
- PAM
positive allosteric modulators
References
- Anderson DJ, Bunnelle WH, Surber BW, Du J, Surowy CS, Tribollet E. 3H]A-585539[(1S,4S)-2,2-dimethyl-5-(6-phenylpyridazin-3-yl)-5-aza-2-azoniabicyclo [2.2.1]heptane], a novel high-affinity alpha7 neuronal nicotinic receptor agonist: radioligand binding characterization to rat and human brain. J Pharmacol Exp Ther. 2008;324:179–187. doi: 10.1124/jpet.107.130062. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd) 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;74:1155–1163. doi: 10.1016/j.bcp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Bettany JH, Levin ED. Ventral hippocampal alpha 7 nicotinic receptor blockade and chronic nicotine effects on memory performance in the radial-arm maze. Pharmacol Biochem Behav. 2001;70:467–474. doi: 10.1016/s0091-3057(01)00643-8. [DOI] [PubMed] [Google Scholar]
- Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusc OJ, Curzon P, et al. Broad-spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J Neurosci. 2007;27:10578–10587. doi: 10.1523/JNEUROSCI.2444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M, et al. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem. 2005;48:905–908. doi: 10.1021/jm049363q. [DOI] [PubMed] [Google Scholar]
- Boess FG, De Vry J, Erb C, Flessner T, Hendrix M, Luithle J, et al. The novel alpha7 nicotinic acetylcholine receptor agonist N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2-carboxamide improves working and recognition memory in rodents. J Pharmacol Exp Ther. 2007;321:716–725. doi: 10.1124/jpet.106.118976. [DOI] [PubMed] [Google Scholar]
- Curzon P, Anderson DJ, Nikkel AL, Fox GB, Gopalakrishnan M, Decker MW, et al. Antisense knockdown of the rat alpha7 nicotinic acetylcholine. Neurosci Lett. 2006;410:15–19. doi: 10.1016/j.neulet.2006.09.061. receptor produces spatial memory impairment. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Hanrott KE, Mok MH, Kew JN, Wonnacott S. Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC12 cells. J Neurochem. 2007;100:1089–1096. doi: 10.1111/j.1471-4159.2006.04273.x. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- El Kouhen R, Hu M, Anderson DJ, Li J, Gopalakrishnan M. Pharmacology of α7 Nicotinic acetylcholine receptor mediated extracellular signal-regulated kinase signaling in pc12 cells. Br J Pharmacol. 2009;156:638–648. doi: 10.1111/j.1476-5381.2008.00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix R, Levin ED. Nicotinic antagonist administration into the ventral hippocampus and spatial working memory in rats. Neuroscience. 1997;81:1009–1017. doi: 10.1016/s0306-4522(97)00224-8. [DOI] [PubMed] [Google Scholar]
- Faghih R, Gopalakrishnan SM, Gronlien JH, Malysz J, Briggs CA, Wetterstrand C, et al. Discovery of A-867744 as a novel positive allosteric modulator of the a7 nicotinic acetylcholine Receptor. J Med Chem. 2009;52(10):3377–3384. doi: 10.1021/jm9003818. [DOI] [PubMed] [Google Scholar]
- Grønlien JH, Håkerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, et al. Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. 2007;72(3):715–724. doi: 10.1124/mol.107.035410. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Bertrand D. Nicotinic acetylcholine receptors as drug targets. Curr Drug Targets CNS Neurol Disord. 2004;3:123–130. doi: 10.2174/1568007043482507. Review. [DOI] [PubMed] [Google Scholar]
- Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J. High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007;1151:227–235. doi: 10.1016/j.brainres.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hu M, Waring JF, Gopalakrishnan M, Li J. Role of GSK-3beta activation and alpha7 nAChRs in Abeta(1-42)-induced tau phosphorylation in PC12 cells. J Neurochem. 2008;106:1371–1377. doi: 10.1111/j.1471-4159.2008.05483.x. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Hajós M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J. Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malysz J, Gronlien JH, Anderson DJ, Haakerud M, Thorin-Hagene K, Ween H, et al. In vitro pharmacological characterization of a novel allosteric modulator of alpha 7 nAChR, A-867744. J Pharmacol Exp Ther. 2009;330(1):257–267. doi: 10.1124/jpet.109.151886. exhibiting unique pharmacological profile. [DOI] [PubMed] [Google Scholar]
- Miret S, De Groene EM, Klaffke W. Comparison of in vitro assays of cellular toxicity in the human hepatic cell line HepG2. J Biomol Screen. 2006;11:184–193. doi: 10.1177/1087057105283787. [DOI] [PubMed] [Google Scholar]
- Ng HJ, Whittemore ER, Tran MB, Hogenkamp DJ, Broide RS, Johnstone TB, et al. Proc Natl Acad Sci USA. 2007;104:8059–8064. doi: 10.1073/pnas.0701321104. Nootropic alpha7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Broide RS, Kasten MR, Dang H, Dani JA, Beaudet AL, et al. Mice homozygous for the L250T mutation in the alpha7 nicotinic acetylcholine receptor show increased neuronal apoptosis and die within 1 day of birth. J Neurochem. 2000;74:2154–2166. doi: 10.1046/j.1471-4159.2000.0742154.x. [DOI] [PubMed] [Google Scholar]
- Pichat P, Bergis OE, Terranova JP, Urani A, Duarte C, Santucci V, et al. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (II) efficacy in experimental models predictive of activity against cognitive symptoms of schizophrenia. Neuropsychopharmacology. 2007;32:17–34. doi: 10.1038/sj.npp.1301188. [DOI] [PubMed] [Google Scholar]
- Ren K, Puig V, Papke RL, Itoh Y, Hughes JA, Meyer EM. Multiple calcium channels and kinases mediate alpha7 nicotinic receptor neuroprotection in PC12 cells. J Neurochem. 2005;94:926–933. doi: 10.1111/j.1471-4159.2005.03223.x. [DOI] [PubMed] [Google Scholar]
- Steiner RC, Heath CJ, Picciotto MR. Nicotine-induced phosphorylation of ERK in mouse primary cortical neurons: evidence for involvement of glutamatergic signaling and CaMKII. J Neurochem. 2007;103:666–678. doi: 10.1111/j.1471-4159.2007.04799.x. [DOI] [PubMed] [Google Scholar]
- Timmermann DB, Grønlien JH, Kohlhaas KL, Nielsen EØ, Dam E, Jørgensen TD, et al. An allosteric modulator of the α7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in. vivo. J Pharmaco Exp Ther. 2007;323:294–307. doi: 10.1124/jpet.107.120436. [DOI] [PubMed] [Google Scholar]
- Van Kampen M, Selbach K, Schneider R, Schiegel E, Boess F, Schreiber R. AR-R 17779 improves social recognition in rats by activation of nicotinic alpha7 receptors. Psychopharmacology (Berl) 2004;172:375–383. doi: 10.1007/s00213-003-1668-7. [DOI] [PubMed] [Google Scholar]
- Wishka DG, Walker DP, Yates KM, Reitz SC, Jia S, Myers JK, et al. Discovery of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide, an agonist of the alpha7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: synthesis and structure – activity relationship. J Med Chem. 2006;49:4425–4436. doi: 10.1021/jm0602413. [DOI] [PubMed] [Google Scholar]
- Woods DC, Liu HK, Nishi Y, Yanase T, Johnson AL. Inhibition of proteosome activity sensitizes human granulosa tumor cells to TRAIL-induced cell death. Cancer Lett. 2008;260:20–27. doi: 10.1016/j.canlet.2007.10.016. [DOI] [PubMed] [Google Scholar]