

Abstract
Sodium-calcium antiporter is the primary efflux pathway for Ca2+ in respiring mitochondria, and hence plays an important role in mitochondrial Ca2+ homeostasis. Although experimental data on the kinetics of Na+-Ca2+ antiporter are available, the structure and composition of its functional unit and kinetic mechanisms associated with the Na+-Ca2+ exchange (including the stoichiometry) remains unclear. To gain a quantitative understanding of mitochondrial Ca2+ homeostasis, a biophysical model of Na+-Ca2+ antiporter is introduced that is thermodynamically balanced and satisfactorily describes a number of independent data sets under a variety of experimental conditions. The model is based on a multistate catalytic binding mechanism for carrier-mediated facilitated transport and Eyring's free energy barrier theory for interconversion and electrodiffusion. The model predicts the activating effect of membrane potential on the antiporter function for a 3Na+:1Ca2+ electrogenic exchange as well as the inhibitory effects of both high and low pH seen experimentally. The model is useful for further development of mechanistic integrated models of mitochondrial Ca2+ handling and bioenergetics to understand the mechanisms by which Ca2+ plays a role in mitochondrial signaling pathways and energy metabolism.
Introduction
Calcium is a key regulatory ion, and alteration of mitochondrial Ca2+ homeostasis can lead to mitochondrial dysfunction and cellular injury (1–5). Despite the recognized role of Ca2+ in mitochondrial bioenergetics and cell physiology and pathophysiology, there are still significant gaps in our understanding of the structure, composition, and kinetic properties of the mitochondrial Ca2+ transport systems (e.g., Na+-Ca2+ antiporter).
The Na+-Ca2+ antiporter mediates Ca2+ efflux from mitochondria in exchange of Na+ from cytosol, and hence plays a key role in mitochondrial Ca2+ homeostasis (1,4). This Na+ influx (Ca2+ efflux) is inhibited by both high and low pH with peak activity near pH = 7.0–7.3 (6,7). Although this Na+-dependent Ca2+ exchange has been characterized in a number of experimental studies (6–15), the kinetic mechanism associated with the exchange has not been well characterized as a mechanistic model that can account for thermodynamics of the exchange process and other physiochemical mechanisms such as allosteric, cooperative binding of Na+ to the antiporter and competitive binding and inhibition by other cations (e.g., H+). Another important point is that the stoichiometry of the exchange is not well known. A number of reports indicate that the exchange is electroneutral (7,9,15): 2Na+ for 1Ca2+. Other reports indicate an electrogenic (perhaps 3Na+ for 1Ca2+) exchange (8,13,16). Therefore, the need for a mechanistic model of the antiporter is apparent for quantitative understanding of mitochondrial Ca2+ handling and bioenergetics and determining the stoichiometry of the exchange.
Magnus and Keizer (17) developed a kinetic model of the Na+-Ca2+ antiporter from the experimental data of Wingrove and Gunter (15) as a module of an integrated model of mitochondrial energy metabolism and Ca2+ handling in pancreatic β-cells. The model was based on a single-step binding mechanism of nNa+ and 1Ca2+ to the antiporter and a phenomenological, irreversible Goldman-Hodgkin-Katz constant-field approximation for electrodiffusion. The model was adopted by Cortassa et al. (18) in their computational models of mitochondrial energy metabolism and Ca2+ dynamics in cardiomyocytes. However, the Magnus-Keizer model is not thermodynamically balanced, as it collapses for membrane potential ΔΨ ≤ ΔΨ∗ = 91 mV.
In part to address the limitations of the Magnus-Keizer model, Nguyen et al. (19) developed another model of the antiporter as a module of a computational model of mitochondrial bioenergetics and Ca2+ dynamics in cardiomyocytes. The model was based on a random-ordered, bi-bi binding mechanism of 3Na+ for 1Ca2+ to the antiporter and a reversible Goldman-Hodgkin-Katz equation for electrodiffusion, which makes the model thermodynamically balanced. Recently, this model was adopted by Dash and Beard (16) in their integrated model of mitochondrial bioenergetics and Ca2+ handling, in which they generalized the model to both 2Na+:1Ca2+ and 3Na+:1Ca2+ stoichiometries to evaluate the actual stoichiometry of the antiporter, based on the dynamic data of Cox and Matlib (12) on matrix free [Ca2+] with extramatrix [Na+] perturbations in isolated respiring cardiac mitochondria. The two antiporter models were parameterized based on the kinetic data of Paucek and Jaburek (7) on Na+-Ca2+ fluxes, obtained using proteoliposomes reconstituted with purified cardiac mitochondrial Na+-Ca2+ antiporters. However, as these data were obtained with ΔΨ = 0, the ΔΨ-dependency of the antiporter function was not mechanistically incorporated into the model for a 3Na+:1Ca2+ exchange. In a recent study, Kim and Matsuoka (14) developed another model of the antiporter, driven by their data showing the ΔΨ-dependency of the exchange in isolated permeabilized cardiomyocytes. The model was based on a simplified mechanism of binding and translocation of Na+ and Ca2+via the antiporter, proposed by Crompton et al. (11). However, none of these models account for the proton inhibition of the antiporter function, observed experimentally (6,7).
In this article, we develop a mechanistic model of mitochondrial Na+-Ca2+antiporter which improves upon and generalizes our previous model (16) and provides a biophysical basis for the ΔΨ-dependency of the antiporter function for a 3Na+:1Ca2+ exchange. The model is thermodynamically balanced and adequately describes the independent data of Paucek and Jaburek (7), Cox and Matlib (12), and Kim and Matsuoka (14) on Na+-Ca2+ fluxes via the antiporter. In addition, the model predicts the inhibitory effects of both high and low pH on the antiporter function, seen experimentally (6,7), making the model applicable to pathological conditions, in which the extra- and intramatrix pH vary considerably. The model is based on Michaelis-Menten kinetics for carrier-mediated facilitated transport (20) and Eyring's free energy barrier theory for absolute reaction rates associated with the conformational change and electrodiffusion of the nNa+- and 1Ca2+-bound carrier complex (20–22).
Mathematical Formulation
Experimental data for model development and validation
Experimental data on the kinetics of mitochondrial Na+-dependent Ca2+ exchange are available from a number of studies (6–15). These data include the initial or pseudo-steady rate data on Na+-Ca2+ fluxes via the antiporter as well as the time-course data on matrix free [Ca2+] with extramatrix [Na+] perturbations in isolated respiring mitochondria and permeabilized myocytes. However, the time-course data on matrix free [Ca2+], which represent an integrated response of cation transport system, are not ideally suited to developing kinetic models of individual cation transporters (e.g., Na+-Ca2+ antiporter). For development of a kinetic model of a cation transporter, the initial or pseudo-steady rate data on cation fluxes via the transporter are more useful. Therefore, the initial rate data of Paucek and Jaburek (7), Cox and Matlib (12), and Kim and Matsuoka (14) on Na+-Ca2+ fluxes via mitochondrial Na+-Ca2+ antiporter are chosen for the development of kinetic model of the antiporter.
Paucek and Jaburek report the initial rate of Na+ efflux (Ca2+ influx) in response to variations in external [Ca2+] and initial rate of Ca2+ efflux (Na+ influx) in response to variations in external [Na+] using proteoliposomes reconstituted with purified Na+-Ca2+ antiporters from beef heart mitochondria. These data also reveal inhibition of Na+-Ca2+ fluxes by both high and low pH with optimal activity near pH = 7.0–7.3. However, these data were obtained without an electrostatic potential across the proteoliposomes. In contrast, the data of Cox and Matlib describe the initial rate of decrease of matrix free [Ca2+] in response to addition of Na+ to the suspensions of respiring mitochondria from rabbit heart with Ca2+ uniporter blocked. Furthermore, the data of Kim and Matsuoka show the Na+ dependency of Ca2+ extrusion from the matrix, in which the Na+-Ca2+ fluxes and matrix free [Ca2+] dynamics were measured with varying extramatrix [Na+] in permeabilized cardiomyocytes during state-2 respiration. These data suggest a ΔΨ-dependent electrogenic exchange of 3Na+:1Ca2+ via the antiporter. Our proposed kinetic model is based on basic physical-chemical principles and parameterized to accurately reproduce the data of Paucek and Jaburek, Cox and Matlib, and Kim and Matsuoka.
Mechanism of Na+ -dependent Ca2+ exchange in mitochondria
Using the available data on Na+-dependent Ca2+ efflux from the matrix, a mathematical model for the kinetics of Na+-Ca2+antiporter is developed using a multistate catalytic binding and interconversion mechanism and Eyring's free energy barrier theory for absolute reaction rates (20–22). Fig. 1 schematizes the proposed kinetic mechanism for a 3Na+:1Ca2+ exchange via the antiporter. The 2Na+:1Ca2+ exchange can be similarly developed (see Supporting Material).
Figure 1.
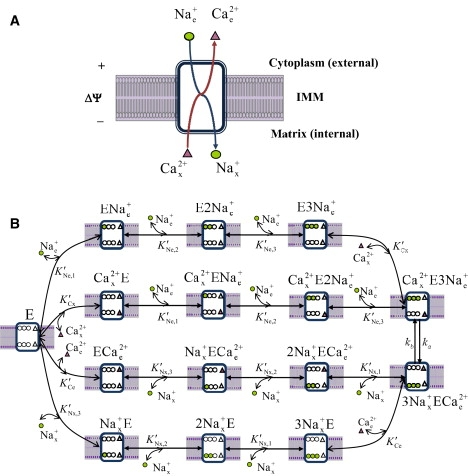
(A, B) Proposed kinetic mechanism of Na+-dependent Ca2+ efflux from mitochondria via Na+-Ca2+ antiporter with a presumed 3Na+:1Ca2+ stoichiometry. The antiporter E has three binding sites for Na+ and one binding site for Ca2+ facing either side of the IMM. In one process, three Na+ ions from cytoplasmic side first cooperatively bind to the unbound antiporter E in three consecutive steps to form the complex E3Nae+. Then one Ca2+ ion from the matrix side binds to the complex E3Nae+ to form the complex Cax2+E3Nae+. In another process, one Ca2+ ion from the matrix side first binds to the unbound antiporter E to form the complex Cax2+E. Then three Na+ ions from the cytoplasmic side cooperatively bind to the complex Cax2+E in three consecutive steps to form the complex Cax2+E3Nae+. The complex Cax2+E3Nae+ then undergoes conformational changes to form the complex 3Nax+ECae2+. The complex 3Nax+ECae2+ then undergoes the reverse processes, where it dissociates in two distinct processes to form three Na+ ions in the matrix side and one Ca2+ ion in the cytoplasmic side, in addition to the unbound antiporter E. K′Ne, p, K′Nx, p, K′Ce, and K′Cx are the apparent dissociation constants associated with the binding of external and internal Na+ and Ca2+ to the antiporter. The 3Na+:1Ca2+ exchange via the interconversion mechanism Cax2+E3Nae+ ↔ 3Nax+ECae2+ is limited by the forward and reverse rate constants ka and kb, which depend on ΔΨ.
The functional unit of the antiporter (E) is assumed to have three binding sites for Na+ and one binding site for Ca2+ on either side of the inner mitochondrial membrane (IMM). The binding of Na+ and Ca2+ to the antiporter is assumed to be via a random-ordered mechanism (e.g., a random-ordered, bi-bi mechanism when three Na+ ions are considered to bind in a single step). If an ionized Na+ from the external side first binds to the unbound antiporter (E) to form the complex ENae+ it then favors binding of another ionized Na+ forming the complex E2Nae+, which further favors the binding of a third ionized Na+ to form the complex E3Nae+. Then an ionized Ca2+ from the internal side binds to the complex E3Nae+ to form the complex Cax2+E3Nae+. On the other hand, if an ionized Ca2+ from the internal side first binds to the unbound antiporter (E) to form the complex Cax2+, it then favors cooperative binding of three ionized Na+ from the external side in three consecutive steps to form the same complex Cax2+E3Nae+. The complex Cax2+E3Nae+ then undergoes a conformational change to form the complex 3Nax+ECae2+. The complex 3Nax+ECae2+ then dissociates three ionized Na+ in the internal side and one ionized Ca2+ in the external side, along with the unbound antiporter (E), by going through the reverse process. The complexes Cax2+E2Nae+, 2Nax+ECae2+, Cax2+ENae+, and Nax+ECae2+ are assumed not to undergo any conformational flipping. The exchange of 3Na+ for 1Ca2+ is limited by the interconversion rate constants ka and kb, which are functions of ΔΨ.
Derivation of mitochondrial Na+-Ca2+ antiporter flux expression
Based on the proposed kinetic mechanism (Fig. 1) and the assumption of quasisteady flux and rapid equilibrium binding of external and internal Na+ and Ca2+ to the antiporter, the binding and translocation reactions for a general nNa+-1Ca2+ antiporter can be summarized as
| (1) |
where (K′Ne, p, K′Nx, p, K′Ce, and K′Cx) are the apparent dissociation constants associated with the binding of external and internal Na+ and Ca2+to the antiporter; n is the stoichiometry (e.g., n = 3 represents a 3Na+:1Ca2+ antiporter); and the dissociation constants are (K′Ne,1, K′Nx,1, K′Ne,2, K′Nx,2, K′Ne,3, K′Nx,3, K′Ce, and K′Cx). The values (ka, kb) are the forward and reverse rate constants in the interconversion of the antiporter complexes Cax2+EnNae+ and nNax+ECae2+. Due to translocation of positive charges across the IMM, the rate constants (ka, kb) are functions of ΔΨ. Depending on the physical locations of Na+ and Ca2+ binding sites on the antiporter, the dissociation constants (K′Ne, p, K′Nx, p, K′Ce, and K′Cx) may also depend on ΔΨ.
Applying rapid equilibrium binding approximations, we have the following relationships between various states of the antiporter,
| (2) |
where the concentrations of various antiporter states are expressed with respect to matrix volume, and [Na+]e, [Na+]x, [Ca2+]e, and [Ca2+]x denote the extramatrix and matrix concentrations of Na+ and Ca2+. As the total antiporter concentration is constant, we have
| (3) |
By substituting Eq. 2 into Eq. 3 and rearranging, the concentration of unbound antiporter ([E]) can be expressed in terms of the concentration of total antiporter ([E]Tot) as
| (4) |
where
| (5) |
Based on the proposed mechanism of Na+-dependent Ca2+ exchange via the antiporter (Fig. 1), the rate of nNa+:1Ca2+ exchange can be expressed as
| (6) |
The generalized nNa+-1Ca2+ antiporter flux expression (6) contains n pairs of dissociation constants for Na+ binding and a pair of dissociation constants for Ca2+ binding (K′Ne, p, K′Nx, p, K′Ce, and K′Cx; p = 1,…,n) as well as two rate constants (ka, kb), resulting in a total of 2n+4 unknown parameters. When n = 3, a total of 10 unknown parameters appears in the flux expression. Further reduction in the number of unknown parameters is obtained under the following assumptions.
Model 1
In one version of the model, the dissociation constant associated with the pth binding step (K′Ne, p, K′Nx, p; p < n) is assumed to be arbitrarily large with the constraints and , which are finite (maximal cooperativity). This is valid when K′Ne, p ≫ 1 μM,…K′Ne, n ≪ 1 μM and K′Nx, p ≫ 1 μM,…K′Nx, n ≪ 1 μM.
Model 2
In another version of the model, the dissociation constants associated with the binding of external and internal Na+ are assumed to be equal to each other: K′Ne, 1 = K′Ne, 2…= K′Ne, n = K′Ne and K′Nx, 1 = K′Nx, 2…= K′Nx, n = K′Nx (partial cooperativity).
Model 3
In the final version of the model, the dissociation constants associated with the binding of external and internal Na+ are assumed to satisfy the constraints: K′Ne, p = (n − p + 1) K′Ne/p and K′Nx, p = (n − p + 1) K′Nx/p; p = 1,2,…n (no cooperativity).
With these three assumptions, the flux expression (6) is reduced to
| (7) |
where
| (8a) |
| (8b) |
| (8c) |
Note that . In each of the three variant models, the reduced flux expression contains only four binding constants (K′Ne, K′Nx, K′Ce, and K′Cx) and two rate constants (ka, kb), for a total of six unknown parameters. Therefore, the three different assumptions on the kinetic mechanism considerably simplify the model. The denominators D1, D2, and D3 in the three reduced flux expressions contain an equal number of unknown parameters and unambiguously account for various kinetic states of the antiporter. D2 and D3 look more complex than D1 due to the nature of the assumption on the kinetic mechanism. Therefore, the three variant models are the three possible minimal models of the antiporter. Even if all three models are able to fit to the same data, the estimates of parameters from these models are expected to differ. Moreover, Model 1 reproduces our previous model (16) with ka = exp(+0.5(n − 2)FΔΨ/RT), kb = exp(−0.5(n − 2)FΔΨ/RT), K′Ce = K′Cx = K′C, and K′Ne = K′Nx = K′N (both independent of ΔΨ).
Kinetic constraint
Under equilibrium conditions, the net nNa+:1Ca2+ exchange flux is zero (JNCE = 0). Thus, the kinetic parameters (K′Ne, K′Nx, K′Ce, ka, and kb) are further constrained by the relationship
| (9) |
where K′eq is the apparent equilibrium constant for the exchange of nNa+ for 1Ca2+ via the antiporter, which is a function of pH and ΔΨ. Given this constraint, the number of unknown parameters is further reduced by one (from six to five).
Effect of pH on mitochondrial Na+-Ca2+ antiporter function
Experimental studies suggest that Na+-Ca2+ antiporter is regulated by protons (H+) on either side of the IMM (6,7). Specifically, these studies reveal that the flux of Na+ or Ca2+ via the antiporter is inhibited by both low and high pH with the minimal effect at an approximate pH = 7.0–7.3. Furthermore, the studies of Paucek and Jaburek (7) suggest that the H+ ions inhibit the antiporter function by exclusively influencing the affinity of Ca2+ binding K′C), without influencing the affinity of Na+ binding (K′N) or the rate constants (ka, kb).
We propose a kinetic mechanism to describe the pH-dependent regulation of Na+ and Ca2+ fluxes via the antiporter (Fig. 2). This is similar to the theory of dibasic acid in enzyme kinetics (23), in which the enzyme is known to become active when a proton is bound at an allosteric site of the enzyme. Otherwise, the enzyme is inactive. In the proposed scheme, H+ ions are assumed to influence the Ca2+ binding steps associated with the unbound antiporter E and Na+-bound antiporter complexes (EnNa+e and nNa+xE) on either side of the IMM. Specifically, each antiporter state C (C: E, EnNa+e, and nNa+xE) is assumed to have multiple (2m, m is unknown) proton binding groups and that only one proton binding group (CmH+) can bind to a Ca2+ ion (active). Other proton binding groups are assumed to have zero affinity for Ca2+ (inactive).
Figure 2.
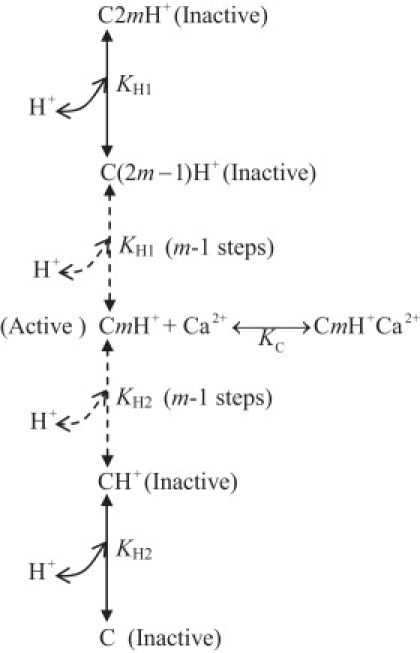
Proposed kinetic mechanism of proton inhibition of nNa+-1Ca2+ antiporter function. The protons are assumed to inhibit the antiporter function by their influences on Ca2+ binding steps, associated with the unbound antiporter E and Na+-bound antiporter complexes (EnNa+e, nNa+xE) in Fig. 1, on either side of the IMM. Each antiporter state (C: E, EnNa+e, nNa+xE) is assumed to have multiple (2m) ionizable groups, but contains only one active ionizable group CmH+ that can bind to a Ca2+ ion. The other ionizable groups are assumed to have zero affinity for Ca2+ (inactive). KH1 and KH2 are the two dissociation constants associated with the binding of protons to the antiporter state C; KC represents the true dissociation constant for Ca2+ binding to the active ionized antiporter state CmH+.
Based on this kinetic mechanism, the rate of nNa+:1Ca2+ exchange can be written as
| (10) |
where
| (11a) |
| (11b) |
| (11c) |
| (11d) |
| (11e) |
Here KH1 and KH2 are the two dissociation constants associated with the binding of protons to the antiporter state C (Fig. 2); KC represents the true dissociation constant for Ca2+ binding to the active ionized antiporter state CmH+; K′C denotes the apparent dissociation constant that lumps the effect of pH, which is experimentally observable (7). We assume that KH1 and KH2 values are the same for both inside and outside of the IMM. Furthermore, as the binding of Na+ to any antiporter state is not influenced by pH, K′Ne = KNe and K′Nx = KNx. Incorporation of pH effects on the antiporter function introduces two more parameters, KH1 and KH2, which are estimated separately. With this formulation, Eq. 9 is reduced to
| (12) |
where Keq is the true equilibrium constant, which is a function of ΔΨ only. Note that when the external and internal pH become equal, Eqs. 10–12 reduce to Eqs. 7–9.
Membrane potential dependence of the kinetic parameters
The electrostatic field of the charged membrane influences the binding of Na+ and Ca2+ to the antiporter and their translocation via the antiporter. To account for this dependency, we assume that the kinetics parameters Keq, KNe, KNx, KCe, KCx, ka, and kb depend on the electrostatic potential difference ΔΨ across the membrane. Our approach is similar to that of Metelkin et al. (24) on the kinetic modeling of mitochondrial adenine nucleotide translocase and Dash et al. (25) on the kinetic modeling of mitochondrial Ca2+ uniporter.
A detailed description of the approach is provided in Appendix A in the Supporting Material. We only provide here the final equations that are used to express Keq, KNe, KNx, KCe, KCx, ka, and kb in terms of ΔΨ,
| (13) |
| (14) |
| (15) |
where αNe or αCe (αNx or αCx) is the ratio of potential difference between Na+ or Ca2+ bound to the site of antiporter facing the external (internal) side of the IMM and Na+ or Ca2+ in the bulk phase to the total membrane potential; βNe or βCe (βNx or βCx) is the displacement of external (internal) Na+ or Ca2+ from the coordinate of maximum potential barrier. For reducing the number of unknown biophysical parameters, we assume αNe = αCe = αe, αNx = αCx = αx, βNe = βCe = βe, and βNx = βCx = βx. Therefore, the four dissociation constants (KNe, KNx, KCe, KCx) and two rate constants (ka, kb) are fully characterized by 10 unknown parameters (K0Ne, K0Nx, K0Ce, K0Cx, K0a, K0b, αe, αx, βe, and βx; note that αe, αx, βe, and βx are estimated separately).
Kinetic and thermodynamic constraints
By substituting Eq. 13 for Keq, Eq. 14 for KNe, KNx, KCe, and KCx, and Eq. 15 for ka and kb into Eq. 12, we obtain the following relationships, which lead to further parameter reduction by two:
| (16) |
Statistical methods and parameter estimation
The 12 antiporter model parameters ϕ = (KNe0, KNx0, KCe0, KCx0, Ka0, Kb0, KH1, KH2, αe, αx, βe, and βx) were estimated in a systematic manner in multiple steps by least-square fitting of the model simulated outputs to the available data,
| (17) |
where Nexp is the number of experiments; Ndata is the number of data points in a particular experiment; are the data on antiporter fluxes; are the corresponding model outputs; and is the maximum value of . A MATLAB-based (The MathWorks, Natick, MA) function optimizer FMINCON is used to minimize the mean residual error E(ϕ) for optimal estimation of the model parameters ϕ, given the constraints of Eq. 16.
Special cases
As in the experiments of Paucek and Jaburek (7), Cox and Matlib (12), and Kim and Matsuoka (14), the internal [Ca2+] or [Na+] was negligible compared to the external [Ca2+] or [Na+], we may not be able to estimate all the 12 model parameters uniquely and accurately, due to nonsensitivity of certain model parameters to the data. Therefore, we explore the parameter estimation process under two special cases: KNe0 = KNx0 and KCe0 = KCx0 so that ka0 = kb0 (Case 1) and KNe0 ≠ KNx0 and KCe0 ≠ KCx0, so that ka0 ≠ kb0 (Case 2). With these simplifications and constraints of Eq. 16, the number of parameters for estimation becomes six for Case 1 and eight for Case 2.
Results
The parameterization and independent validation of the three different kinetic models of 3Na+-1Ca2+ antiporter is illustrated in this section. Specifically, the three variant kinetic models (Model 1, fully cooperativity; Model 2, partial cooperativity; and Model 3, no cooperativity) under two different model assumptions (Case 1, KNe0 = KNx0, KCe0 = KCx0, and ka0 = kb0; and Case 2, KNe0 ≠ KNx0, KCe0 ≠ KCx0, and ka0 ≠ kb0) are used to simulate and fit the independent data sets of Paucek and Jaburek (7), Cox and Matlib (12), and Kim and Matsuoka (14) on the kinetics of Na+-Ca2+ antiporter, which are shown in Figs. 3–5. The estimated model parameter values corresponding to these different model assumptions and data sets are summarized in Table 1. The corresponding analyses and parameter values for a 2Na+-1Ca2+ antiporter are given in the Supporting Material.
Figure 3.
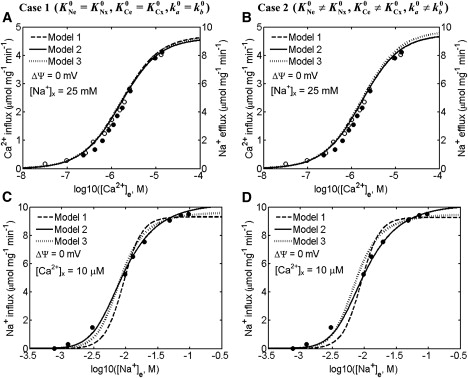
Comparison of the 3Na+-1Ca2+ antiporter models (lines) to the experimental data (points) of Paucek and Jaburek (7) on the kinetics of Na+-Ca2+ fluxes via the antiporter with fixed external pH. Shown are the best fits of three different kinetic models (Model 1, Model 2, and Model 3) under two different model assumptions (Case 1, (A, C); Case 2, (B, D)) to the kinetic data of Paucek and Jaburek, in which the initial rates of Ca2+ influx (Na+ efflux) with variations in external [Ca2+] ([Na+]x = 25 mM, [Na+]e = 0 mM, [Ca2+]x = 0 μM, pHx = 7.3, and pHe = 7.3) (A, B), and the initial rates of Na+ influx (Ca2+ efflux) with variations in external [Na+] ([Na+]x = 0 mM, [Ca2+]x = 10 μM, [Ca2+]e = 0 μM, pHx = 7.3, and pHe = 7.3) (C, D) were measured in proteoliposomes reconstituted with purified Na+-Ca2+ antiporters of beef heart mitochondria. The models were fitted to the data with ΔΨ = 0 mV, in consistence with the experimental protocol. The scaling factor used to scale the model simulated Na+ fluxes to match the data on Na+ fluxes is 2/n = 2/3 (n = 3 is the stoichiometry in nNa+-1Ca2+ exchange).
Figure 4.
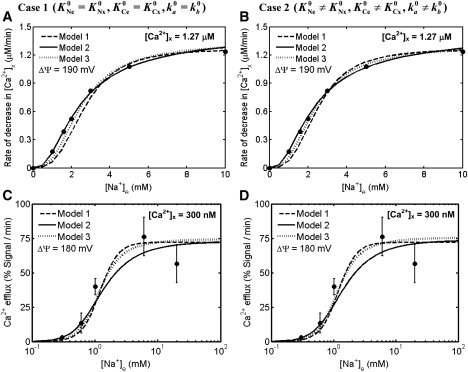
Comparison of the 3Na+-1Ca2+ antiporter models (lines) to the experimental data (points) of Cox and Matlib (12) and Kim and Matsuoka (14) on the kinetics of Na+-Ca2+ fluxes via the antiporter with fixed external pH. Shown are the best fits of three different kinetic models (Model 1, Model 2, and Model 3) under two different model assumptions (Case 1, (A, C); Case 2, (B, D)) to the kinetic data of Cox and Matlib in which the initial rates of decrease of matrix free [Ca2+] with variations in external [Na+] ([Na+]e = 0 mM, free [Ca2+]x = 1.27 μM, free [Ca2+]e = 0.15 μM, pHe = 7.2, and pHx = 7.3) were measured in purified mitochondria from rabbit hearts during state-2 respiration (ΔΨ = 190 mV) using fluorescence probes Fura-2 (A, B), and to the kinetic data of Kim and Matsuoka in which the initial rates of Ca2+ efflux (Na+ influx) with variations in external [Na+] ([Na+]x = [Na+]e/8.6 mM, [Ca2+]x = 300 nM, [Ca2+]e = 0, pHe = 7.2, and pHx = 7.3) were measured in permeabilized cardiomyocytes during state-2 respiration (ΔΨ = 180 mV) using fluorescence probes Rhod-2 (C, D). The models were fitted to the data using the appropriate values of ΔΨ, consistent with the experimental protocols.
Figure 5.
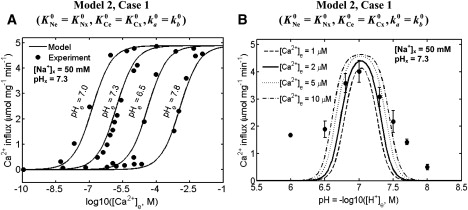
Comparison of the 3Na+-1Ca2+ antiporter models (lines) to the experimental data (points) of Paucek and Jaburek (7) on the kinetics of Na+-Ca2+ fluxes via the antiporter with varying external pH. Shown are the best fits of the best kinetic model (Model 2, Case 1) to the kinetic data of Paucek and Jaburek in which the initial rates of Ca2+ influx (Na+ efflux) with (A) variations in external [Ca2+] at four different levels of external pH ([Na+]x = 50 mM, [Na+]e = 0 mM, [Ca2+]x = 0 μM, pHx = 7.3, and pHe = 7.0, 7.3, 6.5, and 7.8), and (B) variations in external pH and fixed external [Ca2+] ([Na+]x = 50 mM, [Na+]e = 0 mM, [Ca2+]x = 0 μM, [Ca2+]e = 2 μM, and pHx = 7.3) were measured in proteoliposomes reconstituted with purified Na+-Ca2+ antiporters of beef heart mitochondria. Also shown in plot B are the model simulations of the initial rates of Ca2+ influx (Na+ efflux) with variations in external pH at four different levels of external [Ca2+] (1, 2, 5, and 10 μM) with other experimental conditions remaining the same. The model was fitted to the data by setting ΔΨ = 0 mV, in consistent with the experimental protocol.
Table 1.
Estimated parameter values in the models of mitochondrial 3Na+-1Ca2+ antiporter
| Parameter | Values for Model 1 |
Values for Model 2 |
Values for Model 3 |
Reference | |||
|---|---|---|---|---|---|---|---|
| Case 1(K0Ne = K0Nx, K0Ce = K0Cx) | Case 2(K0Ne ≠ K0Nx, K0Ce ≠ K0Cx) | Case 1(K0Ne = K0Nx, K0Ce = K0Cx) | Case 2(K0Ne ≠ K0Nx, K0Ce ≠ K0Cx) | Case 1(K0Ne = K0Nx, K0Ce = K0Cx) | Case 2(K0Ne ≠ K0Nx, K0Ce ≠ K0Cx) | ||
| ka0 | 4.9, 5.56.80, 13.7 | 4.78, 5.46.20, 12.10 | 5.8, 5.87.8, 14.2 | 5.55, 5.737.2, 12.5 | 5.2, 5.657.2, 13.8 | 4.91, 5.66.9, 13.2 | r1, r2r3, r4 |
| kb0 | 4.9, 5.56.80, 13.7 | 4.76, 5.375.70, 14.23 | 5.8, 5.87.8, 14.2 | 5.66, 5.858.5, 13.2 | 5.2, 5.657.2, 13.8 | 5.07, 5.88.55, 13.04 | r1, r2r3, r4 |
| KNe0 | 9.14 × 10−32.2 × 10−31.03 × 10−3 | 8.83 × 10−32.10 × 10−31.02 × 10−3 | 4.8 × 10−31.4 × 10−30.66 × 10−3 | 5.18 × 10−31.36 × 10−30.82 × 10−3 | 6.51 × 10−31.90 × 10−30.90 × 10−3 | 5.87 × 10−31.80 × 10−30.89 × 10−3 | r1, r2r3r4 |
| KNx0 | 9.14 × 10−32.2 × 10−31.03 × 10−3 | 8.82 × 10−31.90 × 10−31.01 × 10−3 | 4.8 × 10−31.4 × 10−30.66 × 10−3 | 4.89 × 10−31.35 × 10−30.77 × 10−3 | 6.51 × 10−31.90 × 10−30.90 × 10−3 | 5.76 × 10−31.90 × 10−30.86 × 10−3 | r1, r2r3r4 |
| KCe0 | 2.28 × 10−9 | 2.29 × 10−9 | 2.26 × 10−9 | 2.29 × 10−9 | 2.23 × 10−9 | 2.30 × 10−9 | all |
| KCx0 | 2.28 × 10−9 | 1.89 × 10−9 | 2.26 × 10−9 | 1.89 × 10−9 | 2.23 × 10−9 | 2.10 × 10−9 | all |
| (pH = 7.0) | 1.36 × 10−7 | 1.37 × 10−7 | 1.38 × 10−7 | 1.37 × 10−7 | 1.35 × 10−7 | 1.43 × 10−7 | r1, r2 |
| (pH = 7.3) | 1.63 × 10−6 | 1.70 × 10−6 | 1.61 × 10−6 | 1.70 × 10−6 | 1.59 × 10−6 | 1.71 × 10−6 | r1, r2 |
| (pH = 6.5) | 3.98 × 10−5 | 3.92 × 10−5 | 4.16 × 10−5 | 3.92 × 10−5 | 4.03 × 10−5 | 4.27 × 10−5 | r1, r2 |
| (pH = 7.8) | 1.17 × 10−3 | 1.22 × 10−3 | 1.16 × 10−3 | 1.22 × 10−3 | 1.14 × 10−3 | 1.23×10−3 | r1, r2 |
| (pH = 7.2) | 4.88 × 10−7 | 5.08 × 10−7 | 4.84 × 10−7 | 5.08 × 10−7 | 4.78 × 10−7 | 5.11 × 10−7 | r3, r4 |
| (pH = 7.3) | 1.63 × 10−6 | 1.40 × 10−6 | 1.61 × 10−6 | 1.40 × 10−6 | 1.59 × 10−6 | 1.56 × 10−6 | all |
| KH1 | 6.45 × 10−8 | 6.47 × 10−8 | 6.39 × 10−8 | 6.47 × 10−8 | 6.41 × 10−8 | 6.38 × 10−8 | all |
| KH2 | 1.39 × 10−7 | 1.40 × 10−7 | 1.39 × 10−7 | 1.40 × 10−7 | 1.39 × 10−7 | 1.40 × 10−7 | all |
| αe = αx = α | 0 | 0 | 0 | 0 | 0 | 0 | all |
| βe = βx = β | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | all |
| Standard physiochemical/thermodynamic parameters used in the model | |||||||
| RT | Gas constant times temperature (298 K) in kJ mol−1 | 2.5775 | |||||
| F | Faraday's constant in kJ mol−1 mV−1 | 0.096484 | |||||
| ZCa, ZNa | Valence of Ca2+ and Na+ (unitless) | 2, 1 | |||||
The rate constants ka0 and kb0 are defined as ka0 = [E]Totka0 and kb0 = [E]Totkb0. The model parameters satisfy the constraints (ka0/kb0)(KCe0/KCx0)(KNx0/KNe0)3 = 1 and 2(α + β) = 1. The rate constants are in the units of μmol/mg/min for Paucek and Jaburek data (7), μM/min in Cox and Matlib data (12), and % Rhod-2 signal/min for Kim and Matsuoka data (14), whereas all dissociation constants are in the units of molar (M). The references r1, r2, r3, and r4 correspond to Figs. 3 and 4, parts A–D, respectively, for each figure.
Figs. 3 and 4 show the model simulations and experimental data of Paucek and Jaburek, Cox and Matlib, and Kim and Matsuoka with fixed external (cytosolic) and internal (matrix) pH. The dashed lines are the simulations from Model 1; the solid lines are the simulations from Model 2; and the dotted lines are the simulations from Model 3. The left panels of Figs. 3 and 4, A and C, show fits for Case 1, whereas the right panels of Figs. 3 and 4, B and D, show fits for Case 2.
In one of the experiments of Paucek and Jaburek (7), the initial rates of Ca2+ influx (Na+ efflux) after variations in external [Ca2+] were measured in proteoliposomes reconstituted with purified Na+-Ca2+ antiporters of bovine heart mitochondria (Fig. 3, A and B). In another experiment, the initial rates of Na+ influx (Ca2+ efflux) after variations in external [Na+] were measured in the same system (Fig. 3, C and D). The measurements were made in the absence of an electrostatic potential across the proteoliposomes. Therefore, for fitting the models to these data, the membrane potential was fixed at ΔΨ = 0 mV. In this case, the biophysical parameters αe = αx = α and βe = βx = β can be arbitrarily chosen to satisfy the thermodynamic constraint of Eq. 16, as these parameters cannot be obtained from these ΔΨ-independent data. Nevertheless, these data provide unique and accurate estimates of the kinetic parameters KNe0, KNx0, KCe0, and KCx0, given KH1 and KH2. However, the rate constants ka and kb estimated from these data differ slightly (Table 1), probably due to discrepancy in these data (Fig. 3, plots A and B, versus plots C and D). As both the external and internal mediums were maintained at a constant pH of 7.3, these data cannot provide accurate estimates of the proton binding constants KH1 and KH2. However, these data provide unique and accurate estimates of the apparent binding constants K′Ce0 and K′Cx0 (Table 1).
In the experiment of Cox and Matlib (12), the initial rates of decrease of matrix free [Ca2+] after variations in external [Na+] were measured in purified mitochondria of rabbit hearts during state-2 respiration (ΔΨ ≈ 190 mV) using the fluorescence probes Fura-2 (Fig. 4, A and B). In the experiment of Kim and Matsuoka (14), the initial rates of Ca2+ efflux (Na+ influx) via the antiporter after variations in external [Na+] were measured in permeabilized cardiomyocytes during state-2 respiration (ΔΨ ≈ 180 mV) using the fluorescence probes Rhod-2 (Fig. 4, C and D). In contrast to the data of Paucek and Jaburek (7), these data depend on nonzero ΔΨ. Therefore, the biophysical parameters αe = αx = α and βe = βx = β are accurately identified from these data, subject to the thermodynamic constraint of Eq. 16. In fact, we obtained the estimates α ≈ 0 and β ≈ 0.5 (Table 1), consistent with the previous estimates of Dash and Beard (16). The values of Ca2+ binding constants KCe0 and KCx0 or (except for pH correction) remained at values estimated from the data of Paucek and Jaburek (7). The values of Na+ binding constants KNe0 and KNx0 or differed from those obtained from the data of Paucek and Jaburek. These differences may be reconciled to the fact that the measured Na+-Ca2+ fluxes are from different experimental systems; proteoliposomes reconstituted with purified cardiac mitochondrial Na+-Ca2+ antiporter of Paucek and Jaburek versus isolated cardiac mitochondria of Cox and Matlib versus isolated permeabilized cardiomyocytes of Kim and Matsuoka. The estimated values of KNe0 and KNx0 are found ∼1 mM, consistent with the reported values in Kim and Matsuoka (14) in isolated permeabilized cardiomyocytes and the estimated values of Dash and Beard (16) from the data of Cox and Matlib (12) in isolated cardiac mitochondria. As the external medium was maintained at a constant pH of 7.2 in both studies, these data do not provide the estimates of the proton binding constants KH1 and KH2. Note that as the Ca2+ efflux (Na+ influx) was expressed in different units (μM/min in Cox and Matlib versus % Rhod-2 signal/min in Kim and Matsuoka), the rate constants were appropriately scaled from the previous estimates to fit to these data (Table 1).
From the fits of the models to the data (Figs. 3 and 4), it is apparent that all three kinetic models (Model 1, Model 2, and Model 3) of the antiporter under either of the conditions (Case 1 or Case 2) can adequately match the data. This indicates that all the variant models were well identified from the available data, with the parameters between the models having equal sensitivities to the data, and that the available data are not suitable to distinguish between the variant models. As the variant models were all effectively derived from the same kinetic mechanism under different assumptions, the estimated parameters expectedly varied between the models, as shown in Table 1. Nevertheless, Model 2 (where the dissociation constants associated with the binding of external and internal Na+ to the antiporter states are assumed to be equal for each of the binding steps: KNe,1 = KNe,2 = KNe,3 = KNe and KNx,1 = KNx,2 = KNx,3 = KNx) fits the data with least error. In addition, Case 1 and Case 2 are indistinguishable, as we obtain KNe0 ≈ KNx0, KCe0 ≈ KCx0, and ka0 ≈ kb0 (Case 1) even with the assumptions KNe0 ≠ KNx0, KCe0 ≠ KCx0, and ka0 ≠ kb0 (Case 2). We therefore consider Model 2 to be the most accurate model, i.e., the one that best describes the data compared to the other two models.
Fig. 5, A and B, shows the fits of Model 2 (Case 1) to the data of Paucek and Jaburek (7) on the kinetics of Ca2+ influx (Na+ efflux) via the antiporter obtained under varying external pH and [Ca2+]. In one of these experiments, the initial rates of Ca2+ influx (Na+ efflux) after variations in external [Ca2+] were measured in proteoliposomes reconstituted with purified Na+-Ca2+ antiporters of beef heart mitochondria with different external pH (pHe = 7.0, 7.3, 6.5, 7.8) (Fig. 5 A). In another experiment, the initial rates of Ca2+ influx (Na+ efflux) after variations in external pH were measured in the same system with fixed external [Ca2+] of 2 μM (Fig. 5 B). The apparent K for Ca2+ binding (K′C) was shifted from 0.55 ± 0.16 μM at pH 7.0, to 2.01 ± 0.16 μM at pH 7.3, to 42 ± 14 μM at pH 6.5, and to 1.0 ± 0.4 mM at pH 7.8.
To simulate the data in Fig. 5 A, the kinetic parameters K′Ne0, K′Nx0, K′Ce0, and K′Cx0 were fixed at values as estimated from the data of Paucek and Jaburek (7), and the biophysical parameters αe = αx = α and βe = βx = β were fixed as estimated from the data of Cox and Matlib (12) and Kim and Matsuoka (14). The maximum number of protons (m) that can bind to the antiporter states (C: E, EnNae+, nNax+E) was determined by trial-and-error until the model successfully reproduces the data. The models fitting to these data also provided accurate estimates of the proton binding constants KH1 and KH2, which are used along with the estimates of the apparent binding constants K′Ce0 and K′Cx0 to obtain the estimates of the true binding constants KCe0 and KCx0 from Eq. 11e. The effect of varying pH on Na+-Ca2+ antiporter function was found to be mediated by a shift in apparent K of Ca2+ (K′C) from 0.138 μM at pH 7.0, to 1.61 μM at pH 7.3, to 41.6 μM at pH 6.5, and to 1.16 mM at pH 7.8, in consistency with the data. The values of Na+-binding constants (KNe0 and KNx0) and the Na+-Ca2+ exchange rate (k) remained the same.
We further validated the antiporter model by comparing the model to the data not used for parameterization. In Fig. 5 B, the model (Model 2, Case 1) with estimated parameter values is used to simulate the Ca2+ influx (Na+ efflux) data of Paucek and Jaburek (7) with fixed external [Ca2+] and varying external pH. For model simulations, four different levels of external [Ca2+] (1 μM, 2 μM, 5 μM, and 10 μM) were used to test the sensitivity of the Ca2+ influx (Na+ efflux) to variations in external [Ca2+]. These simulations show that the model with an external [Ca2+] = 2 μM does not match, accurately, the data for high and low pH (i.e., for pH < 6.5 and pH > 7.5). This is due to inconsistencies in the data sets in Fig. 5, A and B, in these nonphysiological ranges of pH. However, the model adequately predicts the bimodal behavior of Ca2+ influx (Na+ efflux) via the antiporter with varying external pH, observed experimentally.
Discussion
This article provides a detailed, systematic analysis of the kinetics of Na+-Ca2+ antiporter, which is the primary pathway for Na+-mediated Ca2+ extrusion in respiring mitochondria, and hence plays an important role in regulating mitochondrial Ca2+. Specifically, it presents a biophysically based, mathematical model of the antiporter, which is developed based on a multistate catalytic-binding mechanism for carrier-mediated facilitated transport (20) and Eyring's free energy barrier theory for interconversion and electrodiffusion (20–22), and validated based on comparison of model outputs to several independent data sets on Na+-Ca2+ fluxes via the antiporter from the literature (7,12,14). The model adequately describes the ΔΨ-independent data on Na+-Ca2+ fluxes of Paucek and Jaburek (7) in proteoliposomes reconstituted with purified Na+-Ca2+ antiporters from beef heart mitochondria as well as the ΔΨ-dependent data on Na+-dependent Ca2+ effluxes of Cox and Matlib (12) in purified cardiac mitochondria and Kim and Matsuoka (14) in permeabilized cardiomyocytes during state-2 respiration. The model differs from the previous attempts (16–19) in that it is thermodynamically balanced and incorporates the observed pH-dependency of the antiporter function (6,7). The generalization of the model to both 2Na+:1Ca2+ and 3Na+:1Ca2+ stoichiometry is another important feature, making the model accessible to study both electroneutral and electrogenic Na+:1Ca2+ exchange via the antiporter.
An important aspect of this study is modular model development. Once the kinetic model of the antiporter is derived and evaluated (with fixed matrix and extra-matrix pH), the proton inhibition mechanism of the antiporter function is incorporated into the model, requiring estimation of only two additional kinetic parameters based on additional kinetic data (ΔΨ-independent, pH-dependent) from the same system (7). The pH-dependence is introduced here for the first time, making the model applicable to study antiporter function under pathological conditions (e.g., myocardial ischemia) in which the extra- and intramatrix pH vary considerably. Once the pH-dependence is established, the ΔΨ-dependence of the antiporter function is incorporated into the model by introducing the ΔΨ-dependence of the kinetic parameters based on biophysical theory of ion binding and translocation, making the model thermodynamically balanced. The additional parameters introduced by this theory are evaluated based on additional ΔΨ-dependent kinetic data from the same system (12,14). As the resulting model is developed and parameterized in a modular fashion, representing many independent kinetic data sets on the antiporter function, the developed model is expected to best describe the antiporter function under both physiological and pathophysiological conditions.
There is no direct evidence regarding the structure and composition of Na+-Ca2+ antiporter functional unit as well as the mechanism of Na+-dependent Ca2+ efflux from mitochondria. Our model assumes the antiporter to have a general n number of Na+ binding sites (n = 2 or 3) and explores various Na+ binding mechanisms (fully cooperativity: Model 1, partial cooperativity: Model 2, and no cooperativity: Model 3) to determine the most possible Na+ binding mechanism that explains the observed data on the kinetics of Na+-Ca2+ exchange via the antiporter under a variety of experimental conditions. Specifically, with our model analyses of the available kinetic data (7,12,14), we are able to distinguish between three versions of the model, and show that Model 2 predicts the data more accurately compared to Model 1 and Model 3, with both 2Na+:1Ca2+ electroneutral and 3Na+:1Ca2+ electrogenic exchange.
The analyses of the available kinetic data (7,12,14) with three different kinetic models of the antiporter shows that for these models to fit the data, the dissociation constants associated with the binding of external and internal Na+ and Ca2+ to the antiporter in the absence of electric field (ΔΨ = 0) have to be of equal magnitudes (i.e., KNe0 and KCe0 have to be equal to KNx0 and KCx0). In fact, we obtain the estimates KNe0 ≈ KNx0 and KCe0 ≈ KCx0 even with the assumptions KNe0 ≠ KNx0 and KCe0 ≠ KCx0 (Table 1), making the two cases (Case 1 and Case 2) indistinguishable. The estimates of the biophysical parameters αe = αx = α ≈ 0 and βe = βx = β ≈ 0.5, which provide the best fit of the models to the data (7,12,14), suggest that:
1. The charge distribution on the antiporter during Na+(Ca2+) binding to the antiporter and Na+(Ca2+) translocation via the antiporter is linearly decreasing (increasing) along the direction of Na+(Ca2+) translocation from the outside (inside) to inside (outside) of the antiporter, and
2. The free energy barrier that impedes the Na+(Ca2+) translocation via the antiporter seems to be symmetric (because βe = βx = β ≈ 0.5).
By considering two Na+ binding sites on the antiporter (n = 2), we are also able to fit the resulting model to the data of Paucek and Jaburek (7) (Appendix B in the Supporting Material), which suggests an electroneutral exchange of 2Na+ for Ca2+ via the antiporter. This indicates that these data are not suitable to identify the stoichiometry of the nNa+:1Ca2+ exchange. The data (7,12,14) that are used for model parameterization are mostly the initial Na+-Ca2+ fluxes via the antiporter. In these experiments, matrix Na+ or Ca2+ was typically negligible compared to extramatrix Na+ or Ca2+ for various initial Na+ or Ca2+ flux measurements. As a result, these data were not adequate to fully characterize the stoichiometry of the nNa+:1Ca2+ exchange. However, with the help of an integrated model analysis of the dynamic data on matrix free [Ca2+] with varying extramatrix [Na+] in isolated respiring cardiac mitochondria (12), we are able show an electrogenic exchange of 3Na+ for Ca2+ via the antiporter, consistent with earlier experimental observations (8,13) and our recent model conclusion (16) (Appendix C in the Supporting Material).
The Na+-Ca2+ antiporter model derived here is able to explain the data of Cox and Matlib (12) and Kim and Matsuoka (14) on mitochondrial Na+-mediated Ca2+ effluxes, which are dependent on membrane potential ΔΨ (Fig. 4), without considering the nonphysical assumptions of previous models (17,18). In particular, these models have considered an offset potential ΔΨ∗ (≈ 91 mV) and flux expressions appropriate for potential measured relative to this offset potential, and hence are not thermodynamically balanced. These models were justified based on the explanation that the electrical potential across the antiporter may not fall to zero concomitantly with the bulk membrane potential, possibly because of fixed charges producing electric field gradients localized to the antiporter. However, such models cannot be reconciled with measurements of bulk Na+ and Ca2+ movement between the matrix and extramatrix space. Our biophysical model of the antiporter is able to account for the observed kinetic data based on a mechanistic formulation that is thermodynamically feasible. In doing this the singularity that occurs at ΔΨ = ΔΨ∗ ≈ 91 mV in previous models (17,18) does not exist in our model. Furthermore, Model 1 and Case 1 of our model are analogous to the antiporter model of Dash and Beard (16) and Nguyen et al. (19) in their integrated model of mitochondrial bioenergetics and Ca2+ handling.
The proposed mechanism of proton inhibition on the antiporter function is one of the most striking features of this work. Our nNa+ or 1Ca2+ antiporter model (n = 2 or 3), with proton inhibition mechanism incorporated, is able to explain accurately the data of Paucek and Jaburek (7) on proton inhibition of Na+-mediated Ca2+ exchange via the antiporter (Fig. 5). The binding of m protons (m = 6) to the antiporter state C (C: E, EnNae+, nNax+E) (Fig. 2) is shown to influence the affinity of Ca2+ binding to the antiporter, without affecting the affinity of Na+ binding to the antiporter and maximal nNa+:1Ca2+ exchange rate (Fig. 5 A). The bimodal response of the antiporter function (Ca2+ influx) to external pH for a given external [Ca2+] observed from the model simulation is consistent with the response seen experimentally (6,7) (Fig. 5 B); the inhibition of the antiporter activity is shown to be at both low and high pH with the minimal effect (optimal activity) at ∼pH = 7.0.
A bell-shaped pH-dependence of enzyme activity is usually modeled based on the theory of dibasic acid (23), in which the enzyme is known to become active when a proton is bound at an allosteric site of the enzyme. Without any proton bound or with two protons bound, the enzyme is usually inactive. In the case of a Na+-Ca2+ antiporter, this bell-shaped pH-dependence phenomenon seems to be somewhat complex. We tried to incorporate the proton inhibition mechanism of the antiporter function into the model by an induction process to fit the model to the transport flux data in Fig. 5, A and B. In this process, 2m (m = 1, 2, 3, …) proton-bound states of the fully unloaded or fully nNa+ loaded antiporter (C: E, EnNae+, nNa+xE) are considered, with exactly m proton-bound states, which are considered to be active for Ca2+ binding. Only when m = 6, we were able to fit the model to the data. A more appropriate mechanism of proton inhibition of the antiporter function might be that protons must bind to the antiporter at allosteric sites to enable Na+-Ca2+ exchange (active antiporter state) but also that protons competitively bind to the Ca2+ binding sites of the antiporter. In this case, high proton concentration will inhibit Ca2+ binding and low proton concentration will render the antiporter inactive. We also mention here that, as per the proposed mechanism of pH-dependence of the antiporter function, m protons are cotransported in a cycle of Na+-Ca2+ exchange.
This model of nNa+-1Ca2+ antiporter is based on a general, random-ordered, bi-bi binding and transformation mechanism (Fig. 1). In this kinetic scheme, the probability of both the Ca2+ binding after, and that preceding, the binding of all nNa+ is considered, due to lack of sufficient information about their order of binding. Various assumptions on the binding mechanism resulted in several (six) alternative minimal models, all having an equal number of unknown parameters. Alternative kinetic schemes can be formulated in which either the inner loop (Ca2+ binding precedes the nNa+ binding) or the outer loop (Ca2+ binding follows the nNa+ binding) in Fig. 1 can be considered. Accordingly, the denominator D in the flux expression will be modified. However, we note here that the number of unknown parameters in these two possible models would be the same as that in the proposed model of the antiporter. These models may also be able to fit the available kinetic data with appropriate changes in the parameter values, compared to the parameter values in our model. Therefore, selecting any specific mechanism or model will certainly require additional kinetic data on the antiporter function and information regarding the structure of the antiporter.
In addition to the general kinetic scheme shown in Fig. 1 and its two variant schemes described above, another possible general kinetic scheme and its two possible variant schemes for the antiporter function would be to consider the unbound antiporter E to have two conformational states Ee and Ex, depending on the orientation and the position of Ca2+ and nNa+ binding sites on the external or the internal side of the IMM. Each of these models would involve two additional rate constants for this conformational change, which are distinct from the rate constants ka and kb for the conformational change of the fully loaded antiporter states Cax2+EenNae+ and nNax+ExCae2+. Specifically, they will not depend on the transmembrane potential ΔΨ, as no net charge is translocated via this conformational change. Therefore, applying these models would involve estimating a larger number of adjustable parameters than would be required by our model.
Present understanding of cardiomyocytes Ca2+ handling suggests the existence of intracellular Ca2+ subdomains (junctional cleft or submembrane space, where higher Na+ or Ca2+ levels, compared to the average Na+ or Ca2+ concentrations in the cytosol, may develop during cell excitation) (26,27). These Ca2+ subdomains are believed to be essential in some critical aspects of cell signaling and cell cycling. Consequently, the mitochondrial population situated near to these Ca2+ subdomains is expected to have different behavior (e.g., higher Ca2+ uptake; also may be increased redox states and increased respiration) compared to those mitochondrial populations that are far away from these Ca2+ subdomains. Therefore, our biophysical model of mitochondrial Na+-Ca2+ antiporter will form the basis for constructing biophysically based, integrated models of mitochondrial bioenergetics and Ca2+ handling (by integrating the Na+-Ca2+ antiporter model to our existing models of the tricarboxylic acid cycle, oxidative phosphorylation, cation handling, and electrophysiology (28)), which may be helpful in understanding the mechanisms by which Ca2+ plays a role in mediating signaling pathways and modulating energy metabolism, both locally as well as over the whole cell.
Acknowledgments
The authors are thankful to the two anonymous reviewers for very helpful comments. The authors are also thankful to Feng Qi, Kalyan Vinnakota, Fan Wu, and Brian Carlson for helpful discussions.
This research was supported by grants No. SDG-0735093N (to R.K.D.) from the American Heart Association and No. R01-HL072011 (to D.A.B.) from the National Institutes of Health, Bethesda, MD.
Supporting Material
References
- 1.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 2.Brookes P.S., Yoon Y., Sheu S.S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 3.Duchen M.R. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- 4.Gunter T.E., Gunter K.K., Gavin C.E. Mitochondrial calcium transport: physiological and pathological relevance. Am. J. Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- 5.O'Rourke B., Cortassa S., Aon M.A. Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 2005;20:303–315. doi: 10.1152/physiol.00020.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baysal K., Brierley G.P., Jung D.W. Regulation of the mitochondrial Na+/Ca2+ antiport by matrix pH. Arch. Biochem. Biophys. 1991;291:383–389. doi: 10.1016/0003-9861(91)90150-h. [DOI] [PubMed] [Google Scholar]
- 7.Paucek P., Jaburek M. Kinetics and ion specificity of Na+/Ca2+ exchange mediated by the reconstituted beef heart mitochondrial Na+/Ca2+ antiporter. Biochim. Biophys. Acta. 2004;1659:83–91. doi: 10.1016/j.bbabio.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 8.Baysal K., Jung D.W., Brierley G.P. Na+-dependent Ca2+ efflux mechanism of heart mitochondria is not a passive Ca2+/2Na+ exchanger. Am. J. Physiol. 1994;266:C800–C808. doi: 10.1152/ajpcell.1994.266.3.C800. [DOI] [PubMed] [Google Scholar]
- 9.Brand M.D. The stoichiometry of the exchange catalyzed by the mitochondrial calcium/sodium antiporter. Biochem. J. 1985;229:161–166. doi: 10.1042/bj2290161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crompton M., Capano M., Carafoli E. The sodium-induced efflux of calcium from heart mitochondria. A possible mechanism for the regulation of mitochondrial calcium. Eur. J. Biochem. 1976;69:453–462. [Google Scholar]
- 11.Crompton M., Kunzi M., Carafoli E. The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur. J. Biochem. 1977;79:549–558. doi: 10.1111/j.1432-1033.1977.tb11839.x. [DOI] [PubMed] [Google Scholar]
- 12.Cox D.A., Matlib M.A. A role for the mitochondrial Na+-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J. Biol. Chem. 1993;268:938–947. [PubMed] [Google Scholar]
- 13.Jung D.W., Baysal K., Brierley G.P. The sodium-calcium antiport of heart mitochondria is not electroneutral. J. Biol. Chem. 1995;270:672–678. doi: 10.1074/jbc.270.2.672. [DOI] [PubMed] [Google Scholar]
- 14.Kim B., Matsuoka S. Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange. J. Physiol. 2008;586:1683–1697. doi: 10.1113/jphysiol.2007.148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wingrove D.E., Gunter T.E. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J. Biol. Chem. 1986;261:15166–15171. [PubMed] [Google Scholar]
- 16.Dash R.K., Beard D.A. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 2008;586:3267–3285. doi: 10.1113/jphysiol.2008.151977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magnus G., Keizer J. Minimal model of β-cell mitochondrial Ca2+ handling. Am. J. Physiol. 1997;273:C717–C733. doi: 10.1152/ajpcell.1997.273.2.C717. [DOI] [PubMed] [Google Scholar]
- 18.Cortassa S., Aon M.A., O'Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 2003;84:2734–2755. doi: 10.1016/S0006-3495(03)75079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen M.H., Dudycha S.J., Jafri M.S. The effects of Ca2+ on cardiac mitochondrial energy production is modulated by Na+ and H+ dynamics. Am. J. Physiol. Cell Physiol. 2007;292:2004–2020. doi: 10.1152/ajpcell.00271.2006. [DOI] [PubMed] [Google Scholar]
- 20.Keener J.P., Sneyd J. Springer; New York: 1998. Mathematical Physiology. [Google Scholar]
- 21.Lauger P. Ion transport through pores: a rate-theory analysis. Biochim. Biophys. Acta. 1973;311:423–441. doi: 10.1016/0005-2736(73)90323-4. [DOI] [PubMed] [Google Scholar]
- 22.Woodbury J.W. Eyring rate theory model of the current-voltage relationship of ion channels in excitable membranes. In: Hirschfelder J., editor. Chemical Dynamics: Papers in Honor of Henry Eyring. John Wiley and Sons; New York: 1971. [Google Scholar]
- 23.Cornish-Bowden A. Portland Press; London: 2004. Fundamentals of Enzyme Kinetics. [Google Scholar]
- 24.Metelkin E., Goryanin I., Demin O. Mathematical modeling of mitochondrial adenine nucleotide translocase. Biophys. J. 2006;90:423–432. doi: 10.1529/biophysj.105.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dash R.K., Qi F., Beard D.A. A biophysically based mathematical model for the kinetics of mitochondrial calcium uniporter. Biophys. J. 2009;96:1318–1332. doi: 10.1016/j.bpj.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michailova A., McCulloch A. Model study of ATP and ADP buffering, transport of Ca2+ and Mg2+, and regulation of ion pumps in ventricular myocyte. Biophys. J. 2001;81:614–629. doi: 10.1016/S0006-3495(01)75727-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bers D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 28.Wu F., Yang F., Beard D.A. Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J. Biol. Chem. 2007;282:24525–24537. doi: 10.1074/jbc.M701024200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.