

Abstract
AMP-activated protein kinase (AMPK) is a serine/threonine kinase activated by conditions that increase the AMP : ATP ratio. In carotid body glomus cells, AMPK is thought to link changes in arterial O2 with activation of glomus cells by inhibition of unidentified background K+ channels. Modulation by AMPK of individual background K+ channels has not been described. Here, we characterize effects of activated AMPK on recombinant TASK-1, TASK-3, TREK-1 and TREK-2 background K+ channels expressed in HEK293 cells. We found that TREK-1 and TREK-2 channels but not TASK-1 or TASK-3 channels are inhibited by AMPK. AMPK-mediated inhibition of TREK involves key serine residues in the C-terminus that are also known to be important for PKA and PKC channel modulation; inhibition of TREK-1 requires Ser-300 and Ser-333 and inhibition of TREK-2 requires Ser-326 and Ser-359. Metabolic inhibition by sodium azide can also inhibit both TREK and TASK channels. The effects of azide on TREK occlude subsequent channel inhibition by AMPK and are attenuated by expression of a dominant negative catalytic subunit of AMPK (dnAMPK), suggesting that metabolic stress modulates TREK channels by an AMPK mechanism. By contrast, inhibition of TASK channels by azide was unaffected by expression of dnAMPK, suggesting an AMPK-independent mechanism. In addition, prolonged exposure (6–7 min) to hypoxia (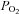 = 11 ± 1 mmHg) inhibits TREK channels and this response was blocked by expression of dnAMPK. Our results identify a novel modulation of TREK channels by AMPK and indicate that select residues in the C-terminus of TREK are points of convergence for multiple signalling cascades including AMPK, PKA and PKC. To the extent that carotid body O2 sensitivity is dependent on AMPK, our finding that TREK-1 and TREK-2 channels are inhibited by AMPK suggests that TREK channels may represent the AMPK-inhibited background K+ channels that mediate activation of glomus cells by hypoxia.
= 11 ± 1 mmHg) inhibits TREK channels and this response was blocked by expression of dnAMPK. Our results identify a novel modulation of TREK channels by AMPK and indicate that select residues in the C-terminus of TREK are points of convergence for multiple signalling cascades including AMPK, PKA and PKC. To the extent that carotid body O2 sensitivity is dependent on AMPK, our finding that TREK-1 and TREK-2 channels are inhibited by AMPK suggests that TREK channels may represent the AMPK-inhibited background K+ channels that mediate activation of glomus cells by hypoxia.
Introduction
AMP-activated protein kinase (AMPK) is a ubiquitously expressed serine/threonine kinase activated by conditions that increase the AMP : ATP ratio. As such, AMPK is considered an important sensor of cellular energy state and metabolic stress (Carling, 2004; Hardie, 2007). Activated AMPK is known to modulate a variety of downstream effectors to increase ATP production and limit cellular energy expenditure (Carling, 2004; Hardie, 2007). In carotid body glomus cells – peripheral chemoreceptors that sense changes in arterial O2 to regulate breathing – AMPK has been shown to link hypoxia-induced decrease in metabolic activity to increased cell excitability (Wyatt et al. 2007). The mechanism by which AMPK modulates excitability of glomus cells and contributes to O2-sensing appears to involve inhibition of a Ba2+-sensitive resting K+ conductance most similar to that produced by the KCNK gene family of background K+ channels (Wyatt et al. 2007).
Several members of the KNCK family of background K+ channels are expressed in the carotid body and one or more of these channels may contribute to O2 sensing. For example, two closely associated members of the TASK (TWIK-related acid sensitive K+ channel) subgroup of channels – TASK-1 (K2P3.1; KCNK3) and TASK-3 (K2P9.1; KNCK9) – and two related members of the TREK (TWIK-related K+ channel) subfamily – TREK-1 (K2P2.1; KCNK2) and TREK-2 (K2P10.1; KCNK10) – are expressed in the carotid body (Buckler et al. 2000; Yamamoto et al. 2002; Yamamoto & Taniguchi, 2006). In addition, the O2 sensitive background K+ current measured in glomus cells has voltage-dependent (i.e. mild outward rectification) and pharmacological properties (i.e. activation by inhaled anaesthetics and weak inhibition by Ba2+) that are consistent with involvement of either TASK or TREK channels (Buckler et al. 2000; Goldstein et al. 2005; Buckler, 2007; Duprat et al. 2007). The relatively low unitary conductance (∼14 pS) and acid sensitivity of the intrinsically O2-sensitive channels in glomus cells (Buckler et al. 2000) favour TASK-1 among these candidates. Indeed, carotid bodies isolated from TASK-1 and TASK-1/TASK-3 double knockout mice showed reduced hypoxia sensitivity compared to wild-type animals and, furthermore, the hypoxic ventilatory response was correspondingly attenuated in TASK-1 knockout mice in vivo (Trapp et al. 2008). Although these results indicate that TASK-1 likely contributes to O2-sensing by carotid body, it is noteworthy that approximately half of the carotid body O2 response was retained in the double TASK knockout animals (Trapp et al. 2008), indicating that there is a TASK-independent component of the carotid body O2-sensing mechanism. The mechanism responsible for this residual chemoafferent response observed in TASK knockout mice remains to be determined, and we hypothesize that this may reflect a separate AMPK-dependent O2-sensing process in glomus cells.
The specific background K+ channel sensitive to AMPK in carotid body cells has not been determined, in large part because the pharmacological tools available to identify these channels in native contexts are extremely limited. In addition, there is no information regarding AMPK sensitivity of the TASK or TREK channels expressed in carotid body. Thus, the purpose of this study was to determine which of these candidate channels can be modulated by AMPK. Our results indicate that AMPK suppresses activity of both TREK-1 and TREK-2, but not TASK-1 or TASK-3, and it mediates TREK channel modulation by the metabolic inhibitor sodium azide and hypoxia. AMPK-dependent TREK channel inhibition involves C-terminal phosphorylation sites that are also crucial for PKA- and PKC-dependent channel modulation. Together, these results suggest that TREK-1 and/or TREK-2 channels represent viable candidates to account for the background K+ channels that underlie AMPK-dependent O2 sensing in carotid body glomus cells.
Methods
Cell culture and transfection
Mouse TREK-1 (GenBank accession no. U73488.2) and rat TREK-2 (NM207261) channel mutants were prepared in mammalian expression vectors (pRK5, TREK-1; pcDNA3.1, TREK-2) as described (Murbartian et al. 2005; Kang et al. 2006). We used a polymorphic variant of the long version of mTREK-1 with three amino acid differences (A63T, K84A, E101K) (Murbartian et al. 2005). Rat TASK-1 (GenBank accession number AF031384), TASK-3 (GenBank accession number AF391084) and tandem-linked TASK-1/TASK-3 heterodimeric channels in pcDNA3 have been described (Talley & Bayliss, 2002). The cDNA for a dominant negative catalytic subunit (α1) of AMPK (dnAMPK) was kindly provided by the laboratory of David Carling (Woods et al. 2000).
Human embryonic kidney (HEK) 293 cells stably expressing the thyrotropin-releasing hormone receptor (E2 cells; obtained from Graeme Milligan; Kim et al. 1994) were maintained in Dulbecco's modified Eagle's medium (DMEM)–F-12 containing 10% fetal bovine serum (FBS), penicillin (100 U ml−1), and streptomycin (100 μg ml−1) and supplemented with G418 (400 μg ml−1). In some experiments, HEK 293 cells not expressing thyrotropin-releasing hormone receptor were also used. All constructs were transfected using LipofectAMINE 2000 according to the manufacturer's instructions. Cells were co-transfected with channel clones and dnAMPK, together with EGFP, plated onto poly-l-lysine (100 μg ml−1)-coated glass coverslips ∼15–20 h after transfection and allowed to adhere for 1 h at 37°C before recording.
Electrophysiology
Coverslips containing transfected cells were placed in a recording chamber positioned on a microscope (Zeiss Axioskop FS) and visualized using DIC optics and fluorescence. Cells were continuously perfused at 2 ml min−1 with a bath solution (equilibrated with room air, 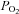 = 160 mmHg) composed of (in mm): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 Hepes, 10 glucose; pH was adjusted to 7.3 by addition of NaOH. Whole cell patch clamp recordings were established using Sylgard-coated borosilicate pipettes which had a DC resistance of 3–5 MΩ when filled with internal solution composed of (mm): 120 KCH4SO3, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 10 Hepes, 10 EGTA, 3 Mg-ATP, 0.3 GTP-Tris, pH 7.2. All recordings were obtained at room temperature (∼24°C), except where noted.
= 160 mmHg) composed of (in mm): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 Hepes, 10 glucose; pH was adjusted to 7.3 by addition of NaOH. Whole cell patch clamp recordings were established using Sylgard-coated borosilicate pipettes which had a DC resistance of 3–5 MΩ when filled with internal solution composed of (mm): 120 KCH4SO3, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 10 Hepes, 10 EGTA, 3 Mg-ATP, 0.3 GTP-Tris, pH 7.2. All recordings were obtained at room temperature (∼24°C), except where noted.
Drugs
All drugs were purchased from Sigma. The membrane permeant adenosine analogue 5-aminoimidazole-4-carboxamide-1-b-d-ribofuranoside (AICAR; 2 mm) was used to activate AMPK (Corton et al. 1995; Hardie, 2007). As previously described (Murbartian et al. 2005), we inhibited PKC with bisindoylmaleimide (1 μm) added to the bath and inhibited PKA with (Rp)-cAMP-S (1 mm) added to the pipette solution. It is well known that metabolic poisons (e.g. cyanide or azide) mimic effects of hypoxia on the carotid body (Mulligan & Lahiri, 1981; Wyatt & Buckler, 2004). Therefore, to mimic mild hypoxia we used low concentrations of sodium azide (10 μm) at a bath temperature of 30°C. Hypoxic medium was made by equilibrating bath solution with 100% N2. We measured oxygen partial pressure (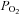 ) at the base of the tissue chamber (∼2 mm from the room air-liquid interface) using a polarographic O2 electrode (ISO-OXY-2) and the Apollo 1000 amplifier with DataTrax2 software (WPI). The O2 electrode was calibrated before each experiment with solutions equilibrated with 95% O2 (
) at the base of the tissue chamber (∼2 mm from the room air-liquid interface) using a polarographic O2 electrode (ISO-OXY-2) and the Apollo 1000 amplifier with DataTrax2 software (WPI). The O2 electrode was calibrated before each experiment with solutions equilibrated with 95% O2 (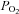 = 722 mmHg), 21% O2 (
= 722 mmHg), 21% O2 (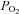 = 160 mmHg) and 100% N2 plus 1 mm dithionite (
= 160 mmHg) and 100% N2 plus 1 mm dithionite (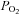 = 0 mmHg); O2 sensitivity of this electrode was > 100 pA mmHg−1.
= 0 mmHg); O2 sensitivity of this electrode was > 100 pA mmHg−1.
Data acquisition and analysis
Recordings were obtained in whole cell voltage clamp using an Axopatch 200B amplifier and a Digidata 1322A analog digital converter with pCLAMP v.10 (Molecular Devices). Series resistance was compensated by ∼70% and a liquid junction potential of 10 mV was corrected offline. Depolarizing ramps from −130 to +20 mV (0.2 V s−1) were applied every 5 s to generate current–voltage (I–V) curves. For analysis, holding current was measured at −60 mV and slope conductance was calculated from linear fits to ramp currents obtained between −30 and +5 mV. Statistical significance was determined at P < 0.05 by ANOVA and Newman–Keuls multiple comparison test or Student's t test. Data are presented as means ±s.e.m.
Results
TASK and TREK currents were recorded from transiently transfected HEK 293 cells in the whole cell voltage clamp configuration. All recordings were maintained for several minutes to establish stable baseline conditions before exposure to experimental challenges.
TASK channels are not modulated by AMP-activated protein kinase
Recent evidence indicates that AMPK is required for O2 sensing by the carotid body (Wyatt et al. 2007) and there is considerable evidence that TASK channels contribute to the O2-sensitive current in glomus cells (Buckler et al. 2000; Buckler, 2007; Duprat et al. 2007; Williams & Buckler, 2004). We therefore tested the possibility that TASK channels are modulated by AMPK. In order to determine if AMPK can modulate channel activity we activated endogenous AMPK with AICAR, a membrane permeant adenosine analogue. Surprisingly, we found that neither TASK-1 nor TASK-3 was inhibited by AICAR. Figure 1 shows the characteristic response of TASK-1 and TASK-3 to changes in bath pH; as expected, acidification to pH 5.9 caused a near complete inhibition of TASK current whereas alkalization to pH 8.4 activated TASK channels. At pH 7.3, bath application of AICAR (2 mm) inhibited TASK-1 conductance by only 7.8% (n= 5) (Fig. 1A) and TASK-3 conductance by only 9.7% (n= 5) (Fig. 1B). We also tested the effects of AICAR under experimental conditions designed to increase TASK conductance; at pH 8.4, exposure to AICAR also had no effect on TASK-1 or TASK-3 (data not shown). In addition, it was recently shown that TASK-1/TASK-3 heterodimers are the predominant conformation of TASK-like O2-sensitive background K+ channel in glomus cells (Kim et al. 2009). Therefore, we tested effects of AICAR on heteromeric TASK channels. As shown in Fig. 1C, bath application of AICAR at pH 7.3 had no discernable effect on heteromeric TASK channels (inhibition of only 8.7 ± 3%, n= 6). Our finding that recombinant TASK channels are insensitive to AICAR indicates that these channels are not downstream targets of AMPK and therefore are unlikely to mediate the AMPK-dependent component of O2 sensing in glomus cells (Wyatt et al. 2007).
Figure 1. TASK-1 and TASK-3 channels are not modulated by activated AMPK.

Whole cell currents were recorded from HEK 293 cells following transient transfection with TASK-1 or TASK-3. Traces of conductance (measured as a linear slope between −30 and +5 mV) as a function of time show characteristic responses of TASK-1 (A), TASK-3 (B) and heteromeric TASK channels (C) to changes in bath pH; acidification from pH 7.3 to pH 5.9 inhibits TASK channels whereas alkalization to pH 8.4 activates TASK channels. At pH 7.3, 3–5 min applications of AICAR (2 mm) had no discernable effect on TASK channel conductance. Insets, current–voltage (I–V) relationships characteristic of TASK-1 and TASK-3 were obtained from depolarizing voltage ramp commands (−130 to +20 mV at 0.2 V s−1) during control conditions (a) and exposure to AICAR (b). Bar graphs show averaged conductance (n= 5) of the relevant TASK channels at pH 7.3 in the absence and presence of AICAR. These results indicate that AICAR-mediated activation of AMPK does not affect activity of recombinant TASK channels.
TREK channels are inhibited by AMPK
TREK channels are another subgroup of background K+ channel expressed by carotid body glomus cells (Yamamoto & Taniguchi, 2006). Although not inherently O2 sensitive (Buckler & Honore, 2005; Caley et al. 2005), they present a number of characteristics consistent with the O2-sensitive current in type I cells (e.g. they are active at resting membrane potential, show only modest voltage dependence and are activated by halothane (Patel & Honore, 2001; Kim, 2005; Honore, 2007). Therefore, we tested the possibility that TREK-1 or TREK-2 channels are modulated by AMPK.
We found that AICAR-mediated activation of AMPK inhibited both TREK-1 and TREK-2 channel activity. Bath application of AICAR (2 mm) caused a strong and reversible inhibition of TREK-1 conductance (Fig. 2A); averaged data show that AICAR decreased conductance from 14.5 ± 1.2 to 6.6 ± 0.6 nS (n= 13). Likewise, AICAR also inhibited TREK-2 conductance (Fig. 2B); averaged data show that AICAR decreased conductance from 14.3 ± 1.7 to 9.2 ± 0.9 nS (n= 7). AICAR-mediated inhibition reached a maximum of 51.6 ± 4.8% (TREK-1) or 33.7 ± 4.6% (TREK-2) within ∼2 min and was typically fully reversed within 3–5 min during wash.
Figure 2. TREK channels are inhibited by AMPK.

A, trace of whole cell conductance shows a typical time course for inhibition of TREK-1 by bath application of AICAR (2 mm). Inset, I–V plots of whole cell TREK-1 current evoked by ramp commands (−130 to +20 mV at 0.2 V s−1) during control (a) and in the presence of AICAR (b). Bar graph of averaged data (n= 13) shows that 2 min exposure to AICAR decreases TREK-1 conductance. B, representative conductance trace shows the time course of TREK-2 inhibition by AICAR (2 mm). Inset, I–V plots of whole cell TREK-2 ramp currents during control (a) and in the presence of AICAR (b). Bar graph of averaged data (n= 7) shows that 2 min exposure to AICAR decreases TREK-2 conductance. The effect of AICAR on TREK-1 and TREK-2 was reversible in the majority of cells tested.
TREK channels are inhibited by PKA and PKC (Patel et al. 1998; Murbartian et al. 2005; Kang et al. 2006, 2007). Therefore, to exclude the possibility that effects of AMPK involve PKA or PKC we tested effects of AICAR under conditions previously shown to inhibit PKA and PKC (Murbartian et al. 2005); cells were pre-incubated and recorded in bath solution containing the PKC inhibitor bisindoylmaleimide (1 μm) and with the PKA blocker (Rp)-cAMP-S (1 mm) added to the pipette solution. We found that AMPK-mediated inhibition of TREK-1 was retained in the presence of these PKA and PKC blockers; under these conditions AICAR decreased TREK-1 conductance by 31.3 ± 4.4% (n= 5) (data not shown). These results indicate that PKA and PKC do not contribute to AMPK-mediated inhibition of TREK channels and demonstrate that TREK-1 and TREK-2 channels are inhibited by AMPK.
C-terminal phosphorylation sites are required for TREK channel inhibition by AMPK
The C-terminal domains of TREK channels are important for channel regulation by physical and chemical signals including protons, temperature and stretch (Patel & Honore, 2001; Kim, 2005; Honore, 2007). In addition, the C-termini of TREK channels contain phosphorylation sites required for channel modulation by receptor-associated metabotropic signalling mechanisms. For example, activation of Gαs-coupled receptors inhibit TREK-1 currents by PKA mediated phosphorylation of Ser-333 located in the C-terminus (Patel et al. 1998; Murbartian et al. 2005). Further, activation of Gαq-coupled receptors and subsequent activation of PKC inhibits TREK-1 channels by sequential phosphorylation of Ser-300 and Ser-333 (Murbartian et al. 2005) and PKC mediated inhibition of TREK-2 requires Ser-326 and Ser-359 (Kang et al. 2006, 2007). To determine if these or other putative PKA and/or PKC phosphorylation sites are required for AMPK-mediated inhibition of TREK, we tested effects of AICAR on TREK-1 and TREK-2 channels with alanine substitutions in putative PKA and PKC phosphorylation sites.
We found that alanine substitution at Ser-333 (S333A) blocked AMPK-mediated inhibition of TREK-1 (Fig. 3A). As expected of a mutation that mimics the dephosphorylated state, cells transfected with S333A mutant channels produced basal currents of 19.3 ± 1.9 nS (n= 6) that are significantly larger than cells expressing wild-type TREK-1 channels. Exposure to the same concentration of AICAR that inhibited wild-type TREK-1 current within 2 min had no effect on these S333A mutant channels; averaged data (n= 6) show that AICAR inhibited conductance by only 4.3 ± 2.4% (Fig. 3A). In addition, alanine substitution at Ser-300 (S300A) also blocked AMPK-mediated inhibition of TREK-1 (Fig. 3B). Under control conditions, S300A mutant channels produced the largest levels of basal conductance (45.0 ± 11.5 nS, n= 9) and 3 min exposure to AICAR had no effect on these channels; averaged data (n= 9) show that AICAR decreased conductance by only 10.5 ± 3.6% (Fig. 3B). We also examined other putative phosphorylation sites in the C-terminal domain of TREK-1, including Thr-303, Thr-328 and Ser-345; AMPK-mediated inhibition of TREK-1 was fully retained in channels with alanine substitutions at these sites (all > 42 ± 4% inhibition, n= 9; data not shown).
Figure 3. Select serine residues in the C-terminus of TREK-1 and TREK-2 are required for AMPK-modulation.

A, representative conductance trace shows that Ala substitution at Ser-333 (S333A) blocks AICAR-mediated inhibition of TREK-1. Inset, I–V plot of the S333A mutant TREK-1 current during control (a) and exposure to AICAR (b). Averaged data (n= 6) shows that 2 min exposure to AICAR had no effect on S333A mutant TREK-1 conductance. B, conductance trace shows that Ala substitution at Ser-300 (S300A) also blocked AICAR-mediated inhibition of TREK-1. Inset, I–V plot of the S300A mutant TREK-1 current during control (a) and exposure to AICAR (b). Averaged data (n= 9) show that 2 min exposure to AICAR had no effect on S300A mutant TREK-1 conductance. C, representative conductance trace show that Ala substitutions at Ser-326 and Ser-359 (S326A/S359A) of TREK-2 channels blocks AICAR-mediated inhibition of TREK-2. Inset, I–V plot of the S326A/S359A mutant TREK-2 current during control (a) and exposure to AICAR (b). Averaged data (n= 5) show that 3 min exposure to AICAR had no effect on S326A/S359A mutant TREK-1 conductance.
Mutations of PKC phosphorylation sites in TREK-2 also disrupted AMPK modulation of TREK-2. Alanine substitutions at Ser-326 and Ser-359 (S326A/S359A) decreased AMPK-mediated inhibition of TREK-2 (Fig. 3C). Under control conditions, S326A/S359A mutant channels produced a basal conductance of 19.5 ± 3.7 nS (n= 5) and 3 min exposure to AICAR had no effect on these channels; averaged data (n= 5) show that AICAR decreased conductance by only 7.3 ± 1.3% (Fig. 3C). These results are consistent with the previously defined roles of Ser-333 and Ser-300 in TREK-1 and Ser-326 and Ser-359 in TREK-2 as important phosphorylation sites for kinase-dependent modulation of TREK channels and indicate that these residues are also important for AMPK-mediated TREK channels.
AMPK mediates TREK channel inhibition by metabolic stress
AMPK is thought to confer O2 sensitivity to type I cells in the carotid body by acting as an intermediary between metabolic state and cellular activity (Wyatt et al. 2007). Background K+ channels (e.g. TASK) are thought to be important determinants of O2-induced depolarization (Buckler et al. 2000; Buckler, 2007; Duprat et al. 2007), but our data indicate that TASK channels are not modulated by AMPK (i.e. acute exposure to AICAR). On the other hand, previous studies show that TREK channels are not directly sensitive to hypoxia (Buckler & Honore, 2005; Caley et al. 2005), i.e. short exposure to 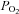 as low as < 4 mmHg did not affect TREK conductance. However, because our results indicate that activation of AMPK inhibits TREK-1 and TREK-2 channel activity, we hypothesized that AMPK might confer metabolic sensitivity to cells expressing TREK channels. To test this hypothesis, we determined the effects on TREK channel activity of metabolic stress in the form of sodium azide or prolonged hypoxia (6–7 min); we performed these experiments at 30°C in order to increase metabolic rate and the activity of temperature-sensitive TREK channels (Maingret et al. 2000; Alloui et al. 2006).
as low as < 4 mmHg did not affect TREK conductance. However, because our results indicate that activation of AMPK inhibits TREK-1 and TREK-2 channel activity, we hypothesized that AMPK might confer metabolic sensitivity to cells expressing TREK channels. To test this hypothesis, we determined the effects on TREK channel activity of metabolic stress in the form of sodium azide or prolonged hypoxia (6–7 min); we performed these experiments at 30°C in order to increase metabolic rate and the activity of temperature-sensitive TREK channels (Maingret et al. 2000; Alloui et al. 2006).
As shown in Fig. 4Aa, exposure to sodium azide (10 μm) at 30°C inhibited wild-type TREK-1 conductance by 39.9 ± 7.0% (n= 6). We observed similar results in response to cyanide (10 μm) (33 ± 3% inhibition, n= 4, P < 0.03; data not shown). We also found that the same alanine substitutions that rendered TREK-1 unresponsive to AICAR (i.e. S333A and S300A) also left cells expressing these mutant channels unresponsive to sodium azide (10 μm) (Fig. 4Ab and c and C). As shown in Fig. 4Ba, sodium azide also inhibited wild-type TREK-2 conductance by 45.4 ± 2.8% (n= 3) but had no effect on the S326A/S359A mutant channel (Fig. 4Bb and C). These results indicate that Ser-333 and/or Ser-300 of TREK-1 and Ser-326 and/or Ser-359 of TREK-2 are important residues for sodium azide-mediated inhibition of TREK channels and suggest that AMPK contributes to the mechanism by which sodium azide inhibits TREK channels.
Figure 4. Inhibition of TREK channels by sodium azide require AMPK phosphorylation sites.

Whole cell currents were recorded from HEK cells transfected with wild-type or AICAR-resistant mutant TREK-1 (S333A, S300A) or TREK-2 (S326A/S359A) channels during exposure to metabolic stress in the form of sodium azide. Aa, trace of wild-type TREK-1 conductance and bath temperature show that heating from 24°C to 30°C increases TREK-1 activity. At 30°C bath application of sodium azide (10 μm) caused a rapid and reversible inhibition of TREK-1 conductance. Inset, I–V plots of TREK-1 current obtained at 30°C during control conditions (a) and in the presence of azide (b). Ab and Ac, bath temperature (top) and whole cell conductance from S300A (Ab) and S333A (Ac) mutant TREK-1 channels show that heating from 24°C to 30°C increases whole cell conductance of cells expressing either mutant channel. At 30°C bath application of sodium azide (10 μm) had no effect on S300A (Ab) or S333A (Ac) conductance. Inset, I–V relationships of S300A and S333A mutant TREK-1 channels obtained at 30°C during control conditions (a) and in the presence of azide (b). Ba, trace of wild-type TREK-2 conductance and bath temperature show that heating from 24°C to 30°C increases TREK-2 activity. At 30°C exposure to sodium azide (10 μm) caused a rapid and reversible inhibition of TREK-2 conductance. Inset, I–V plots of TREK-2 current obtained at 30°C during control conditions (a) and in the presence of azide (b). Bb, bath temperature (top) and conductance trace from a cell expressing the TREK-2 S326A/S359A mutant channel (bottom) show that the mutant channel is activated by warming but exposure to sodium azide (10 μm) at 30°C had no effect on conductance. Inset, I–V relationships of TREK-2 S326A/S359A mutant obtained at 30°C during control conditions (a) and in the presence of azide (b). C, average data summarize the effects of sodium azide on wild-type (n= 6) and mutant TREK-1 channels (S333A, n= 6; S300A, n= 3) as well as wild-type (n= 3) and S326A/S359A mutant (n= 4) TREK-2 channels.
Two additional experimental approaches were used to confirm that the effect of metabolic stress on activity of TREK channels is mediated by an AMPK-dependent mechanism. First, we tested effects of sodium azide on wild-type TREK channels in cells co-transfected with dnAMPK. If azide-mediated inhibition of TREK channels involves AMPK, then we expect expression of dnAMPK will suppress endogenous AMPK activity and decrease effects of azide on TREK channels. We found that expression of dnAMPK attenuated azide-mediated inhibition of TREK-1 (Fig. 5A) and TREK-2 (Fig. 5B); averaged data show that, in the presence of dnAMPK, sodium azide inhibited TREK-1 and TREK-2 conductance by only 3.8 ± 2% (n= 4) and 4.6 ± 3% (n= 3), respectively. We also attempted to use compound C to pharmacologically block AMPK, but that drug had non-specific inhibitory effects on TREK channels expressed in HEK cells; e.g. compound C (20 μm) alone inhibited TREK-1 conductance from 4.9 ± 1.1 to 3.2 ± 0.8 nS (37 ± 2%, n= 5, P < 0.01). Since blocking AMPK action would be expected to disinhibit TREK channels (and increase channel activity), we consider the decrease in channel activity observed with compound C to be non-specific.
Figure 5. Expression of dnAMPK blocks the effects of sodium azide on TREK channels.

A, trace of wild-type TREK-1 conductance in the presence of dnAMPK and bath temperature show that heating from 24°C to 30°C increases TREK-1 activity. At 30°C bath application of sodium azide (10 μm) had no discernable effect on channel conductance. Inset, I–V plots of TREK-1 current in the presence of dnAMP obtained at 30°C during control conditions (a) and in the presence of azide (b). Bar graph of average data (n= 4) shows the percentage inhibition of TREK-1 by azide alone and in the presence of dnAMPK. B, trace of wild-type TREK-2 conductance in the presence of dnAMPK and bath temperature show that heating from 24°C to 30°C increases TREK-1 activity. At 30°C bath application of sodium azide (10 μm) had no discernable effect on channel conductance. Inset, I–V plots of TREK-2 current in the presence of dnAMP obtained at 30°C during control conditions (a) and in the presence of azide (b). Bar graph of average data (n= 3) shows the percentage inhibition of TREK-1 by azide alone and in the presence of dnAMPK.
We used an occlusion protocol in a second series of experiments. For this, we exposed cells expressing wild-type TREK channels to AICAR during sustained sodium azide application. We reasoned that if AMPK is a common signalling intermediary shared by both AICAR and sodium azide, then TREK channel inhibition by AICAR would be occluded by sodium azide. Sodium azide (10 μm) inhibited wild-type TREK-1 channel activity (Fig. 6A). In the continued presence of azide, effects of AICAR were indeed occluded; under these conditions, exposure to AICAR had no effect on TREK-1 channel activity. Similar results were observed with TREK-2; sodium azide inhibited wild-type channel activity and in the continued presence of azide, exposure to AICAR had no additional effect on TREK-2 channel activity (Fig. 6B). These results further support the conclusion that AMPK mediates effects of metabolic stress on TREK-1 and TREK-2 channels.
Figure 6. Sodium azide occludes AICAR-mediated inhibition of TREK channels.

A, traces of wild-type TREK-1 conductance and bath temperature show that heating from 24°C to 30°C increases TREK-1 conductance. At 30°C exposure to sodium azide (10 μm) inhibited TREK-1 conductance. In the continued presence of sodium azide, bath application of AICAR had no effect on TREK-1 conductance. Inset, I–V relationship of TREK-1 current obtained under control conditions at 30°C (a) and during exposure to azide alone (b) and azide in the presence of AICAR (c). Bar graph summarizes the percentage inhibition by azide alone and AICAR in the presence of azide. B, traces of wild-type TREK-2 conductance and bath temperature show that heating from 24°C to 30°C increases TREK-1 conductance. At 30°C exposure to sodium azide (10 μm) inhibited TREK-2 conductance. In the continued presence of sodium azide, bath application of AICAR had no effect on TREK-2 conductance. Inset, I–V relationship of TREK-2 current obtained under control conditions at 30°C (a) and during exposure to azide alone (b) and azide in the presence of AICAR (c). Bar graph summarizes the percentage inhibition by azide alone and AICAR in the presence of azide.
We also found that sodium azide inhibited heteromeric TASK channels by 32 ± 8% under control conditions (Fig. 7A); however, unlike the case for TREK channels, inhibitory effects of azide on these TASK channels was retained in cells expressing dnAMPK (31 ± 4% inhibition; Fig. 7B). These results are consistent with the known effects of metabolic stress on TASK-like currents in glomus cells (Williams & Buckler, 2004), and support the possibility that metabolic stress inhibits TASK channels by an AMPK-independent mechanism.
Figure 7. Sodium azide inhibits heteromeric TASK channels by an AMPK-independent mechanism.

A, trace of TASK1–TASK-3 conductance and bath temperature show that heating from 24°C to 30°C increases activity of TASK channels. At 30°C 3 min exposure to sodium azide (10 μm) inhibits channel conductance. Inset, I–V plots of heteromeric TASK current obtained at 30°C during control conditions (a) and in the presence of sodium azide (b). B, trace of TASK1–TASK-3 conductance in the presence of dnAMPK and bath temperature show that heating from 24°C to 30°C increases activity of TASK channels. At 30°C exposure to sodium azide (10 μm) inhibits channel conductance. Inset, I–V plots of heteromeric TASK current in the presence of dnAMP obtained at 30°C during control conditions (a) and in the presence of sodium azide (b). C, bar graph of average data (n= 4) shows the percentage inhibition of heteromeric TASK channels by sodium azide alone and in the presence of dnAMPK.
To determine if AMPK can modulate TREK channels in response to a physiologically relevant signal, we tested effects of hypoxia on TREK channels. Previous studies have established that TREK channels are resistant to relatively brief bouts of hypoxia (Buckler & Honore, 2005; Caley et al. 2005), indicating that TREK channels are not directly O2 sensitive. However, short exposure to hypoxia may not be sufficient to inhibit metabolic activity of HEK cells. Therefore, for these experiments we examined effects of more prolonged hypoxia (6–7 min). We measured 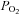 in the tissue bath (∼2 mm from the gas–liquid interface) and determined that our hypoxic medium has a
in the tissue bath (∼2 mm from the gas–liquid interface) and determined that our hypoxic medium has a 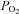 of 11 ± 1 mmHg. Initially, exposure to this level of hypoxia had no effect on activity of TREK channels (Fig. 8A). However, with continued exposure to hypoxia, TREK conductance decreased from 6.8 ± 0.8 nS at 4 min hypoxia to 4.0 ± 0.4 nS at 6 min hypoxia (39 ± 7% inhibition, n= 4, P < 0.04). Channel conductance returned to 6.8 ± 0.8 nS within ∼10 min when
of 11 ± 1 mmHg. Initially, exposure to this level of hypoxia had no effect on activity of TREK channels (Fig. 8A). However, with continued exposure to hypoxia, TREK conductance decreased from 6.8 ± 0.8 nS at 4 min hypoxia to 4.0 ± 0.4 nS at 6 min hypoxia (39 ± 7% inhibition, n= 4, P < 0.04). Channel conductance returned to 6.8 ± 0.8 nS within ∼10 min when 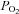 was returned to control levels. In some recordings the cells did not recover from hypoxia, presumably due to current rundown; those cells were omitted from the study. As was observed with sodium azide, the effects of hypoxia on TREK channels were attenuated by expression of dnAMPK; under these conditions hypoxia decreased TREK conductance by only 1 ± 1% (n= 4) (Fig. 8B and C).
was returned to control levels. In some recordings the cells did not recover from hypoxia, presumably due to current rundown; those cells were omitted from the study. As was observed with sodium azide, the effects of hypoxia on TREK channels were attenuated by expression of dnAMPK; under these conditions hypoxia decreased TREK conductance by only 1 ± 1% (n= 4) (Fig. 8B and C).
Figure 8. Hypoxia inhibits TREK channels by an AMPK-dependent mechanism.

A, trace of wild-type TREK-1 conductance and bath temperature show that heating from 24°C to 30°C increases TREK-1 activity. At 30°C exposure to hypoxia (measured bath 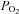 = 11 ± 1 mmHg) for 6 min inhibited channel conductance. Inset, I–V plots of TREK-1 current obtained at 30°C under control conditions (a) and during exposure to hypoxia (b). B, trace of wild-type TREK-1 conductance in the presence of dnAMPK and bath temperature show that heating from 24°C to 30°C increases TREK-1 activity. At 30°C exposure to hypoxia (
= 11 ± 1 mmHg) for 6 min inhibited channel conductance. Inset, I–V plots of TREK-1 current obtained at 30°C under control conditions (a) and during exposure to hypoxia (b). B, trace of wild-type TREK-1 conductance in the presence of dnAMPK and bath temperature show that heating from 24°C to 30°C increases TREK-1 activity. At 30°C exposure to hypoxia (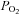 = 11 ± 1 mmHg) for 6 min had no effect on channel conductance. Inset, I–V plots of TREK-1 current in the presence of dnAMP obtained at 30°C under control conditions (a) and during exposure to hypoxia (b). C, bar graph of average data (n= 4) shows the percentage inhibition of TREK-1 by hypoxia alone and in the presence of dnAMPK.
= 11 ± 1 mmHg) for 6 min had no effect on channel conductance. Inset, I–V plots of TREK-1 current in the presence of dnAMP obtained at 30°C under control conditions (a) and during exposure to hypoxia (b). C, bar graph of average data (n= 4) shows the percentage inhibition of TREK-1 by hypoxia alone and in the presence of dnAMPK.
Discussion
The ability to sense decreases in arterial 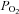 levels to stimulate breathing is a critically important function of peripheral chemoreceptors. AMPK links changes in O2 with activation of glomus cells in the carotid body by inhibition of an unidentified background K+ channel (Wyatt et al. 2007). In an effort to identify potential downstream targets of hypoxia-activated AMPK, we characterized AMPK sensitivity of candidate background K+ channels in a heterologous expression system. We found that TREK-1 and TREK-2, but not TASK-1 and TASK-3, are inhibited by AMPK. TREK channel inhibition requires select serine residues in the C-terminus that are also known to be important for PKA- and PKC-mediated channel modulation. We also found that inhibiting metabolism with sodium azide or hypoxia decreased activity of TREK-1 and TREK-2 channels in an AMPK-dependent manner. Sodium azide-mediated channel inhibition was not observed in AMPK-insensitive mutant TREK channels, it was abrogated by dnAMPK, and it blocked AICAR-mediated TREK inhibition. Likewise, TREK channel inhibition by prolonged hypoxia was blocked in cells expressing dnAMPK. These results identify AMPK as a regulator of TREK channel activity and suggest a novel mechanism by which metabolic activity can control cell excitability.
levels to stimulate breathing is a critically important function of peripheral chemoreceptors. AMPK links changes in O2 with activation of glomus cells in the carotid body by inhibition of an unidentified background K+ channel (Wyatt et al. 2007). In an effort to identify potential downstream targets of hypoxia-activated AMPK, we characterized AMPK sensitivity of candidate background K+ channels in a heterologous expression system. We found that TREK-1 and TREK-2, but not TASK-1 and TASK-3, are inhibited by AMPK. TREK channel inhibition requires select serine residues in the C-terminus that are also known to be important for PKA- and PKC-mediated channel modulation. We also found that inhibiting metabolism with sodium azide or hypoxia decreased activity of TREK-1 and TREK-2 channels in an AMPK-dependent manner. Sodium azide-mediated channel inhibition was not observed in AMPK-insensitive mutant TREK channels, it was abrogated by dnAMPK, and it blocked AICAR-mediated TREK inhibition. Likewise, TREK channel inhibition by prolonged hypoxia was blocked in cells expressing dnAMPK. These results identify AMPK as a regulator of TREK channel activity and suggest a novel mechanism by which metabolic activity can control cell excitability.
In the carotid body, hypoxia-mediated activation of AMPK and inhibition of background K+ channels are thought to determine glomus cell O2 sensitivity. In situ hybridization and immunohistochemistry show that glomus cells express several closely associated members of the TASK and TREK subfamilies of KNCK background K+ channels (Buckler et al. 2000; Yamamoto et al. 2002; Yamamoto & Taniguchi, 2006). Of these, there is considerable evidence that TASK channels contribute to glomus cell O2 sensitivity, as described above. However, since the hypoxic ventilatory response is only partially reduced in TASK-1 and double TASK knockout mice while remaining unaffected in TASK-3 knockout mice (Trapp et al. 2008), it seems clear that background K+ channels in addition to TASK contribute to O2 sensing by carotid body glomus cells. Here we present evidence that recombinant TREK-1 and TREK-2 channels are modulated by AMPK, suggesting that if TREK channels are expressed in glomus cells they may contribute to O2 signalling by serving as downstream targets for hypoxia-activated AMPK.
There has been some controversy regarding O2 sensitivity of TREK channels. Miller et al. (2004) reported that TREK-1 channels were rapidly inhibited by hypoxia with a  threshold of 60 mmHg, whereas other studies showed that TREK channels were unaffected by ∼3 min exposure to
threshold of 60 mmHg, whereas other studies showed that TREK channels were unaffected by ∼3 min exposure to  levels as low as < 4 mmHg (Buckler & Honore, 2005; Caley et al. 2005). We also found that TREK channels are resistant to brief (< 3 min) bouts of hypoxia (
levels as low as < 4 mmHg (Buckler & Honore, 2005; Caley et al. 2005). We also found that TREK channels are resistant to brief (< 3 min) bouts of hypoxia (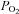 = 9 ± 1 mmHg) but that prolonged exposure can inhibit TREK channel activity. This protracted time course is consistent with the hypothesis that hypoxic stress inhibits TREK channels by an indirect metabolic mechanism that involves activation of AMPK. Although we found that channel inhibition by both hypoxia and by azide were AMPK dependent, it is interesting to note that recovery of channel activity following hypoxia was relatively slow by comparison to that after sodium azide treatment. The reason for this is not clear but it may reflect additional effects of hypoxia on some aspect of the recovery mechanism that are not shared by azide-induced metabolic inhibition.
= 9 ± 1 mmHg) but that prolonged exposure can inhibit TREK channel activity. This protracted time course is consistent with the hypothesis that hypoxic stress inhibits TREK channels by an indirect metabolic mechanism that involves activation of AMPK. Although we found that channel inhibition by both hypoxia and by azide were AMPK dependent, it is interesting to note that recovery of channel activity following hypoxia was relatively slow by comparison to that after sodium azide treatment. The reason for this is not clear but it may reflect additional effects of hypoxia on some aspect of the recovery mechanism that are not shared by azide-induced metabolic inhibition.
TREK-1 channels are regulated by a variety of factors, including kinase-dependent phosphorylation. Previous studies demonstrate that neurotransmitter activation of G-protein coupled receptors leads to inhibition of TREK-1 by PKA- or PKC-mediated phosphorylation of select serine residues located in the C-terminus of TREK. For example, stimulation of Gαs-coupled receptors inhibits TREK-1 by PKA-mediated phosphorylation of Ser-333 (Patel et al. 1998) and stimulation of Gq-coupled receptors inhibits TREK-1 by sequential phosphorylation of Ser-333 followed by Ser-300 (Murbartian et al. 2005). Activation of Gq-coupled receptors also inhibits TREK-2 by PKC mediated phosphorylation of Ser-326 or Ser-359 (Kang et al. 2006, 2007). The data presented here identify a novel AMPK kinase-dependent inhibition of TREK-1 and TREK-2 channels involving the same two previously identified PKA and/or PKC phosphorylation sites. Alanine substitution of these sites on TREK-1 and TREK-2 increased basal conductance, as expected for a mutation that mimics the dephosphorylated state of the channel. Importantly, these mutations also disrupted AMPK-mediated channel inhibition and prevented the effects of metabolic stress on TREK-1 channel activity.
It is possible that we have underestimated the inhibitory effects of AMPK on TREK channels because both AICAR and sodium azide can cause an intracellular acidification that may activate TREK channels and partially offset the inhibitory effects of AMPK. For example, it is well known that metabolic stress can result in the accumulation of lactic acid, and it has also been shown that AICAR can inhibit activity of the Na+/H+ exchanger (NHE) (Moopanar et al. 2006). Both of these scenarios could result in a drop in intracellular pH, and since TREK channels are activated by intracellular acidification, this could antagonize the inhibitory effects of AMPK on TREK channels.
As mentioned, TASK channels have been implicated in responses to hypoxia by various cell types, including the carotid body. This is thought to be due, in part, to an intrinsic O2 sensitivity of the channels. We also found that sodium azide decreased activity of TASK channels, suggesting that those channels also respond to metabolic stress. However, TASK channels were unaffected by AICAR and were inhibited by azide even in the presence of dnAMPK, indicating that TASK channel inhibition by metabolic stress occurs via a mechanism that is independent of AMPK. Previous studies have shown in excised patches from carotid body glomus cells that a TASK-like current can be activated by physiologically relevant levels of ATP, whereas rotenone (which inhibits complex 1 of the mitochondrial electron transport chain) decreased intercellular ATP levels and inhibited the TASK-like current (Varas et al. 2007; Williams & Buckler, 2004). Together these observations indicate that metabolic stress inhibits TASK channels by an AMPK-independent mechanism that may involve disruption of a direct ATP interaction with TASK channels.
In summary, our results indicate that activated AMPK can inhibit recombinant TREK-1 and TREK-2 channels but not TASK-1 or TASK-3 channels. We identified serine residues in the C-terminus of TREK-1 (Ser-300 and Ser-333) and TREK-2 (Ser-326 and Ser-359) that are required for AMPK modulation of these channels. To the extent that AMPK modulation of background K+ channels is required for glomus cell O2 sensing (Wyatt et al. 2007), our results suggest that TREK-1 and/or TREK-2 channels are potential downstream targets for hypoxia-activated AMPK and peripheral O2 sensing.
Acknowledgments
We thank Dr Donghee Kim for providing the TREK-2 S326A/S359A mutant. We thank Dr David Carling for providing us with the dominant negative AMPK. This work was supported in part by grants F32 HL80890 (DKM), and NS33583 (DAB) from the National Institutes of Health.
Glossary
Abbreviations
- AMPK
AMP-activated protein kinase
- dnAMPK
dominant negative AMPK
- HEK cells
human embryonic kidney cells
- PKA
protein kinase A
- PKC
protein kinase C
- TASK
TWIK-related acid sensitive K+ channel
- TREK
TWIK-related K+ channel
- TWIK
two-pore, weakly inwardly rectifying K+ channel
Author contributions
O.K.: conception and design of the experimental protocol; collection, analysis and interpretation of data; revising the manuscript; final approval of the manuscript. J.P.B.: collection, analysis and interpretation of data; revising the manuscript; final approval of the manuscript. D.A.B.: conception and design of the experimental protocol; revising the manuscript; final approval of the manuscript. D.K.M.: conception and design of the experimental protocol; collection, analysis and interpretation of data; drafting the manuscript; final approval of the manuscript.
References
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, Borsotto M, Romey G, Heurteaux C, Reeh P, Eschalier A, Lazdunski M. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006;25:2368–2376. doi: 10.1038/sj.emboj.7601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. TASK-like potassium channels and oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2007;157:55–64. doi: 10.1016/j.resp.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Honore E. The lipid-activated two-pore domain K+ channel TREK-1 is resistant to hypoxia: implication for ischaemic neuroprotection. J Physiol. 2005;562:213–222. doi: 10.1113/jphysiol.2004.077503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caley AJ, Gruss M, Franks NP. The effects of hypoxia on the modulation of human TREK-1 potassium channels. J Physiol. 2005;562:205–212. doi: 10.1113/jphysiol.2004.076240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade–a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lauritzen I, Patel A, Honore E. The TASK background K2P channels: chemo- and nutrient sensors. Trends Neurosci. 2007;30:573–580. doi: 10.1016/j.tins.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev. 2005;57:527–540. doi: 10.1124/pr.57.4.12. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Honore E. The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci. 2007;8:251–261. doi: 10.1038/nrn2117. [DOI] [PubMed] [Google Scholar]
- Kang D, Choe C, Cavanaugh E, Kim D. Properties of single two-pore domain TREK-2 channels expressed in mammalian cells. J Physiol. 2007;583:57–69. doi: 10.1113/jphysiol.2007.136150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Han J, Kim D. Mechanism of inhibition of TREK-2 (K2P10.1) by the Gq-coupled M3 muscarinic receptor. Am J Physiol Cell Physiol. 2006;291:C649–C656. doi: 10.1152/ajpcell.00047.2006. [DOI] [PubMed] [Google Scholar]
- Kim D. Physiology and pharmacology of two-pore domain potassium channels. Curr Pharm Des. 2005;11:2717–2736. doi: 10.2174/1381612054546824. [DOI] [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I, Carroll JL. Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. J Physiol. 2009;587:2963–2975. doi: 10.1113/jphysiol.2009.171181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GD, Carr IC, Anderson LA, Zabavnik J, Eidne KA, Milligan G. The long isoform of the rat thyrotropin-releasing hormone receptor down-regulates Gq proteins. J Biol Chem. 1994;269:19933–19940. [PubMed] [Google Scholar]
- Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honore E. TREK-1 is a heat-activated background K+ channel. EMBO J. 2000;19:2483–2491. doi: 10.1093/emboj/19.11.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P, Peers C, Kemp PJ. Polymodal regulation of hTREK1 by pH, arachidonic acid, and hypoxia: physiological impact in acidosis and alkalosis. Am J Physiol Cell Physiol. 2004;286:C272–C282. doi: 10.1152/ajpcell.00334.2003. [DOI] [PubMed] [Google Scholar]
- Moopanar TR, Xiao XH, Jiang L, Chen ZP, Kemp BE, Allen DG. AICAR inhibits the Na+/H+ exchanger in rat hearts: possible contribution to cardioprotection. Pflugers Arch. 2006;453:147–156. doi: 10.1007/s00424-006-0124-z. [DOI] [PubMed] [Google Scholar]
- Mulligan E, Lahiri S. Dependence of carotid chemoreceptor stimulation by metabolic agents on PaO2 and PaCO2. J Appl Physiol. 1981;50:884–891. doi: 10.1152/jappl.1981.50.4.884. [DOI] [PubMed] [Google Scholar]
- Murbartian J, Lei Q, Sando JJ, Bayliss DA. Sequential phosphorylation mediates receptor- and kinase-induced inhibition of TREK-1 background potassium channels. J Biol Chem. 2005;280:30175–30184. doi: 10.1074/jbc.M503862200. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998;17:4283–4290. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Bayliss DA. Modulation of TASK-1 (Kcnk3) and TASK-3 (Kcnk9) potassium channels: volatile anaesthetics and neurotransmitters share a molecular site of action. J Biol Chem. 2002;277:17733–17742. doi: 10.1074/jbc.M200502200. [DOI] [PubMed] [Google Scholar]
- Trapp S, Isabel AM, Wisden W, Gourine AV. A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J Neurosci. 2008;28:8844–8850. doi: 10.1523/JNEUROSCI.1810-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varas R, Wyatt CN, Buckler KJ. Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. J Physiol. 2007;583:521–536. doi: 10.1113/jphysiol.2007.135657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BA, Buckler KJ. Biophysical properties and metabolic regulation of a TASK-like potassium channel in rat carotid body type 1 cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L221–L230. doi: 10.1152/ajplung.00010.2003. [DOI] [PubMed] [Google Scholar]
- Woods A, Azzout-Marniche D, Foretz M, Stein SC, Lemarchand P, Ferre P, Foufelle F, Carling D. Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol Cell Biol. 2000;20:6704–6711. doi: 10.1128/mcb.20.18.6704-6711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt CN, Buckler KJ. The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. J Physiol. 2004;556:175–191. doi: 10.1113/jphysiol.2003.058131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt CN, Mustard KJ, Pearson SA, Dallas ML, Atkinson L, Kumar P, Peers C, Hardie DG, Evans AM. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J Biol Chem. 2007;282:8092–8098. doi: 10.1074/jbc.M608742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Kummer W, Atoji Y, Suzuki Y. TASK-1, TASK-2, TASK-3 and TRAAK immunoreactivities in the rat carotid body. Brain Res. 2002;950:304–307. doi: 10.1016/s0006-8993(02)03181-5. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Taniguchi K. Expression of tandem P domain K+ channel, TREK-1, in the rat carotid body. J Histochem Cytochem. 2006;54:467–472. doi: 10.1369/jhc.5A6755.2005. [DOI] [PubMed] [Google Scholar]