

Abstract
Pin1 plays a key role in various biological cellular processes via the recognition of phosphorylated Ser/Thr-Proline motifs. Moreover, high expression levels of Pin1 are correlated to tumorgenesis in some cancer types. Here, we identify a novel series of small molecular weight compounds with a core structure mimicking the phoshorylated serine. The binding affinity and binding mode of the compounds for Pin1 are analyzed via NMR spectroscopy and computational studies. The reported chemical probes and relative binding data to Pin1 represent valuable stepping stones for the validation of Pin1 as target for drug discovery and for eventually the development of possible lead compounds.
Keywords: comparative molecular field analysis, NMR, Pin1, WW-domain
Peptidyl-prolyl cis/trans isomerases (PPIase) are enzymes catalyzing proline cis/trans isomerization, a crucial step for protein folding (1–3). To date, three families of highly conserved PPIase have been identified, namely, cyclophilins (Cyp) (4), FK506-binding proteins (FKBP) (5) and parvulins (6). Unlike Cyp and FKBP, the parvulin family of proteins has a unique feature to selectively recognize a phosphorylated Ser/Thr-Pro motif (p-Ser/Thr-Pro) (7,8).
Human Pin1 is the most widely studied protein in the parvulin family. Isomerization by Pin1 modulates a multitude of biological processes, including protein folding and biological activity (9), protein stability (8,10) as well as subcellular localization (11). As phosphorylation is a major regulation mechanism in cell, Pin1 is also involved in cycle regulation, oncogenesis, signal transduction and neurodegeration in Alzheimer’s disease via targeting several key proteins, such as cyclin D1 (12–15), c-jun (16), c-Myc (17), β-catenin (11), p53 (9,18,19) and tau (9). Pin1 is constituted by two domains, an N-terminal WW domain (Pin1ww) and a C-terminal catalytic domain (Pin1cat), both of which have a specificity for the p-Ser/Thr-Pro motif. Pin1ww is a member of the WW family proteins (20) and has a much higher binding affinity for the motif than Pin1cat (21), hence is thought as a docking site mediating the substrate recognition of Pin1. Pin1cat displays a weaker binding affinity against p-Ser/Thr-Pro motif but possesses a full isomerase activity even without the WW domain (10). Two different crystal structures of Pin1-peptide complexes have been reported (8,21). The first structure of Pin1 containing a Ala-Pro peptide in the catalytic domain (PDB code: 1PIN) exhibits a ‘close’ active site conformation, in which a phosphate binding loop β1/α1 (residues 70–83) is folded backwards the binding pocket via specific interactions with a phosphate-mimic sulfate ion. The second structure of Pin1 contains a phosphopeptide bound to the WW domain (PDB code: 1F8A) in which the loop β1/α1 is positioned away from the catalytic domain thus leading to an ‘open’ conformation in the binding pocket. Moreover, the WW domain twists towards the catalytic domain to form a deep clamp that constitutes the phosphopeptide docking site. In solution, two domains of Pin1 are loosely connected (22) but their mobility can be differently modulated by different phosphopeptides (23).
The fact that Pin1 is prevalently overexpressed in several human tumors including breast (16), colorectal (24), prostate (25) and thyroid cancers (26) makes Pin1 an attractive therapeutic target for cancer treatment. To date, relevant Pin1 inhibitors reported are phosphopeptide mimics targeting on Pin1cat at the nanomolar level (27,28). However, poor cell membrane permeability of the peptide mimics limits their application for drug development and target validation (29). Considering its substrate recognition capability, Pin1ww is potentially also a valid target for inhibitor design. So far, the only known Pin1ww inhibitors are pThr-Pro analogs (30).
In this study, we report a novel series of compounds that bind to Pinww. The compound series BI-81 was found via a virtual screening approach of a commercial library followed by experimental binding data using NMR spectroscopy techniques. To explain the differences in compound binding affinities, a three-dimensional quantitative structure activity relationship (3D QSAR) model was established and comparative molecular field analysis (CoMFA) (31) studies were subsequently performed. The generated 3D QSAR model gives insights into the contribution of various substituents on the binding affinity of the compounds and provides information in designing and predicting the binding ability of novel derivatives combined with docking studies. These studies resulted in the identification of compound 29 which, possessing a comparable binding affinity but a higher stability and membrane permeability to the known phosphopeptide, represents a valid stepping stone for further validation studies and eventually hit to lead optimizations.
Materials and Methods
Protein expression and production
The gene coding for the human Pin1 was amplified via PCR and subcloned into pET28α vector using the Nde I and EcoR I clone sites. The resulting proteins contain an extra His-taque (GSSHHHHHHSSGLVPRGSH) on the N-terminus. The protein was expressed in the Escherichia coli strain BL21(DE3) and purified using Ni2+ affinity chromatography. The uniformly N15-labeled Pin1 was produced by growing the bacteria in M9 minimal media containing 15NH4Cl as the sole nitrogen source. The NMR samples were dissolved in 50 mM potassium phosphate buffer (pH 6.5) containing 5 mM Deuterium–dithioerythritol, 0.03% NaN3 and 90%/10% (H2O/D2O).
NMR spectroscopy
NMR spectra were acquired on a 600 MHz Bruker Avance spectrometer equipped with either a TXI probe and z-shielded gradient coils or a TCI cryoprobe. All NMR data were processed and analyzed using TOPSPIN2.0 (Bruker Biospin Corp, Billerica, MA, USA) and SPARKY (32). 2D [15N, 1H]-HSQC experiments were acquired using 32 scans with 2048 and 128 complex data points in the 1H and 15N dimensions at 300 K. 3D 15N-NOESY experiments were acquired using 16 scans with 2048 × 96 × 128 data points. Compound binding was detected at 27 °C by comparing the 2D [15N, 1H]-HSQC spectra of 100 μM Pin1 in the absence and presence of compounds at mole ratios of 5:1 and 20:1. The chemical shift changes were calculated using the following equation (33):
Dissociation equilibrium constants (Kd) of compounds against Pin1 were determined by monitoring the protein chemical shift perturbations as function of compound concentration. For instance, equivalent amounts of BI-81 compounds were added to a 100 μM sample of Pin1 to yield 1:1 and 1:2 stoichiometries of protein/ligand concentration. Titration analysis was done by fitting chemical shift data into the equation:
where X is the mole ratio of ligand/protein, K represents Kd, Y is the observed chemical shift perturbation value at each titration point and P is the maximum chemical shift perturbation value of the fully complexed protein (30).
3D QSAR model and CoMFA analysis
A library of BI-81 containing 39 1,2,4-triazole analogs was built for the following QSAR studies. The BI-81 compounds were divided into a training set (32 compounds) for generating a 3D QSAR model and a test set (seven compounds) for validating the quality of the model. Selection of the training set and test set compounds was done based on the structural diversity and wide range of activity such that the test set compounds represent a range of biological activity similar to that of the training set. The activity of compounds was represented by the induced chemical shift perturbation value of Trp34NE in Pin1, which was then converted to log value and used as dependent variables in the 3D QSAR calculations.
As the spatial alignment of compounds in 3D QSAR study is one of the most sensitive and determining factors in obtaining a reliable model, we manually aligned the structures of BI-81 compounds. First, we adopted the docked conformation of compound 29 as a template after energy minimization using the standard Tripos force field. Second, we sketched the structures of the remaining compounds based on the template using SYBYL 7.0 (Tripos, St Louis, MO, USA) and fitted the atoms of 1,2,4-triazole core structure into that of the template. The structural energy minimization was performed using standard Tripos force field and Gasteiger–Huckel partial charges calculation with an energy gradient convergence criterion of 0.05 kcal/mol.
For the CoMFA studies, the regression analysis was carried out using the full cross-validated partial least-squares (PLS) method (leave-one-out) using standard options after the compound structure alignment. The final model was developed via non-cross-validated conventional analysis with the optimum number of components equal to 5 that yields the highest q2.
Steric and electrostatic interactions were calculated using an sp3 carbon atom as steric probe and a +1 charge as electrostatic probe with the Tripos force field. The CoMFA grid spacing was 2.0 Å in the x, y and z directions. The default value of 30 kcal/mol was set as the maximum steric and electrostatic energy cutoff.
Docking studies
The virtual screening model of Pin1 was prepared using the crystal structure of Pin1 in complex with phosphopeptide (PDB code 1F8A). A library of 420 000 compounds (Chembridge Corp, San Diego, CA, USA) was docked into this model using the program GOLD version 2.1 (CCDC Software Ltd, Cambridge, UK) and ranked using Gold-score. The compound binding site was defined within a 10 Å radius around the binding sites of phosphopeptide. Standard default parameter settings were used. In virtual screening study, 10 genetic algorithm (GA) steps were used for each compound. The docking of the best compound 29 was also carried out using GOLD program under the same parameters used in virtual screening except that GA value was set 500 and the best 50 structures were selected.
Plasma stability assay
Test compound solution was incubated (20 μM final concentration) with fresh rat plasma at 37 °C. The reactions were terminated at 0, 30 and 60 min by the addition of two volumes of methanol containing internal standard. Following protein precipitation and centrifugation, the samples were analyzed by LC-MS. The percentage of parent compound remaining at each time-point relative to the 0 min sample is calculated from peak area ratios in relation to the internal standard. Compounds were run in duplicate with a positive control known to be degraded in plasma.
Parallel artificial membrane permeation assay (PAMPA) (34)
A 96-well microtiter plate (Millipore, #MSSACCEPTOR; Billerica, MA, USA) was completely filled with aqueous buffer solution (pH 7.2) and covered with a microtiter filterplate (Millipore, #MAP-BMN310) to create a sort of sandwich construction. The hydrophobic filter material was impregnated with a lipid solution (Avanti Polar Lipids) in chloroform and the organic solvent was allowed to completely evaporate. Permeation studies were started by the transfer of 200 μL of a 100 μM test compound solution on top of the filterplate. In general phosphate pH 7.2 buffer was used. The maximum DMSO content of the stock solutions was <5%. In parallel, an equilibrium solution lacking a membrane was prepared using the exact concentrations and specifications but lacking the membrane. The concentrations of the acceptor and equilibrium solutions were determined using the Shimadzu LCMS-2010EV and AUC methods. The permeation of a compound through the membrane layer is described by the percentage permeation (% flux). The flux values were calculated considering the concentration of the acceptor compartment after 8 h and that of a reference well with the same concentration containing no membrane barrier.
Results and Discussion
Identification of small molecular weight compounds binding to Pin1
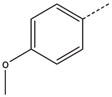
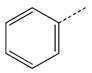
To identify low molecular weight compounds binding to Pin1, a library of ~420 000 compounds (Chembridge Corp) was screened in silico against Pin1 using GOLD (35,36). From the 200 top ranking hits according to goldscore, we found a series that has a core structure of 1,2,4-triazole with an extended acidic arm presenting a putative phosphoserine mimic (Table 1). Although it is well known that Pin1 recognizes p-Ser/Thr-Pro motif, early studies also indicated that using a glutamate rather than p-Ser/Thr group before the critical proline is well tolerated in Pin1 enzyme activity assays (37,38). Furthermore, latter studies on the interactions between Pin1 and amyloid peptides (38,39) indicated that the side chain of a glutamate residue is also recognized by Pin1ww. These results suggest the possibility of using an acidic group in the design of p-Ser/Thr-Pro mimics. Hence, nearly 110 commercially available analogs of the initially selected virtual hits were found with similarity above 70%, 39 of which were then selected (library BI-81) for experimental verification.
Table 1.
Structures of BI-81 compounds
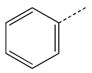
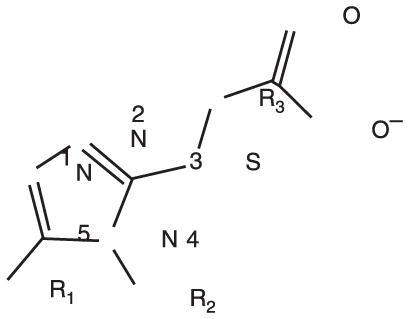 | |||||||
|---|---|---|---|---|---|---|---|
| ID | R1 | R2 | R3 | ID | R1 | R2 | R3 |
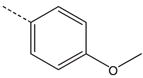
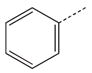
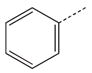
| 1a | 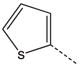 |
 |
CH2 | 21 | 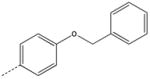 |
 |
CH(CH3) |
| 2 |  |
 |
CH2 | 22 |  |
CH2(CH=CH2) | CH2 |
| 3 | 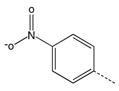 |
CH2(CH=CH2) | CH2 | 23 | 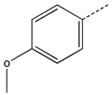 |
CH2(CH=CH2) | CH(CH3) |
| 4 | 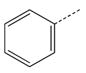 |
CH2(CCH3=CH2) | CH2 | 24 | 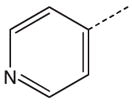 |
 |
CH2CH2 |
| 5 a | 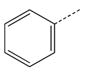 |
CH2(CH=CH2) | CH2 | 25 | 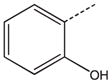 |
 |
CH2 |
| 6 |  |
 |
CH2 | 26 |  |
CH3 | CH2CH2 |
| 7 |  |
CH2(CCH3=CH2) | CH2 | 27 | 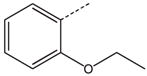 |
 |
CH2 |
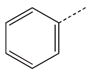
| 8 | 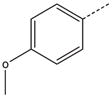 |
CH2(CCH3=CH2) | CH2 | 28 | 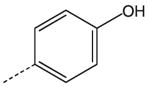 |
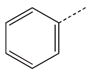 |
CH2 |
| 9 | 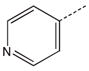 |
CH2(CH=CH2) | CH2 | 29 | 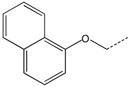 |
CH2(CH=CH2) | CH2 |
| 10 | 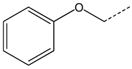 |
CH2(CH=CH2) | CH2 | 30 a | 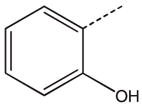 |
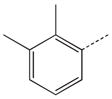 |
CH2 |
| 11 a | 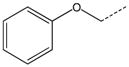 |
CH2(CCH3=CH2) | CH2 | 31 | 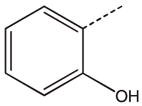 |
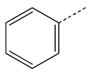 |
CH(CH3) |
| 12 | 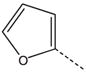 |
CH2(CH=CH2) | CH2 | 32 | 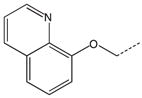 |
CH2CH3 | CH2 |
| 13 | 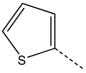 |
 |
CH2 | 33 a | 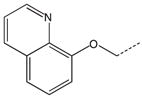 |
CH2(CH=CH2) | CH2 |
| 14 | 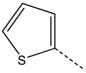 |
CH2(CH=CH2) | CH2 | 34 | 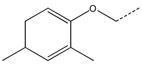 |
CH2(CH=CH2) | CH2 |
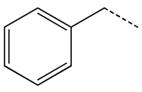
| 15 | 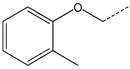 |
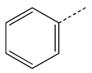 |
CH2CH2 | 35 | 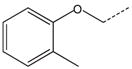 |
 |
CH2 |
| 16 | 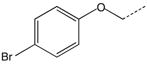 |
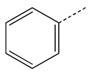 |
CH(CH3) | 36 | 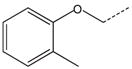 |
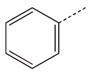 |
CH2 |
| 17 | 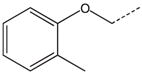 |
 |
CH(CH3) | 37 | 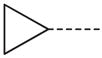 |
 |
CH2 |
| 18 a | 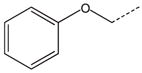 |
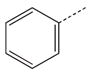 |
CH2 | 38 | 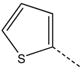 |
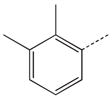 |
CH2 |
| 19 | 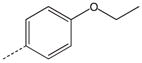 |
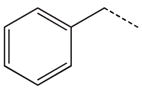 |
CH2 | 39 | 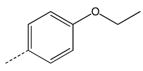 |
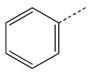 |
CH2 |
| 20 | 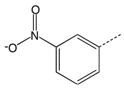 |
CH2(CH=CH2) | CH2 | ||||
Compounds in the training set.
The ability of BI-81 compounds to interact with Pin1 was determined via NMR chemical shift perturbation experiments. The chemical shift perturbation experiment is a widely used approach to detect protein–ligand interactions that is detected by observing cross-peak shifts in 2D [15N, 1H]-HSQC spectra of the target recorded after the addition of test ligands (33,40–42). Therefore, the 2D [ 15N, 1H]-HSQC spectra of 100 μM Pin1 mixed with each of BI-81 compounds at various concentrations (500 μM to 4 mM) were recorded. When the HSQC spectra of Pin1 in the presence of BI-81 compounds were superimposed on the spectrum of Pin1 alone (Figure 1A), significant shifts of selected cross-peak were observed indicative of specific binding. To further delineate the site of binding for the hits, the induced chemical shift perturbation values of four effective BI-81 compounds were mapped on the primary sequence of Pin1 (Figure 1B). Consistent to our virtual screening strategy, most of the significant cross-peak movements caused by the hits (Δppm > 0.1) correspond to residues located the WW domain. The two most significantly affected cross peaks are from the side chain amide of Trp34 (Trp34HE) and the backbone amide of Arg17. Considering their highly structural similarity, it is not surprising that BI-81 compounds exhibit similar binding behaviors and induce qualitatively similar perturbation in the HSQC spectrum of Pin1 binding. Hence, differences in the observed perturbation values are indicative of the relative binding affinities of individual compounds against Pin1. These measurements can therefore be used to monitor improvement in binding affinity of subsequent compounds.
Figure 1.

(A) The 2D [ 15N, 1H]-HSQC spectra of Pin1 recorded in the absence of compounds (red), and in presence of compounds 7 (purple), 10 (red), 11 (cyan) and 29 (blue) are overlaid. Two most affected cross-peaks from the side chain of Trp34 (Trp34HE) and the backbone amide of Arg17 respectively were boxed and labeled. (B) The chemical shift perturbation values of the overall Pin1 residues in the presence of compounds 7 (filled triangle), 10 (open triangle), 11 (open circle) and 29 (filled circle) are plotted against the primary sequence of Pin1.
CoMFA analysis of BI-81 compounds
To elucidate and rationalize the structural features of BI-81 compounds required for binding to Pin1, we built a 3D QSAR model and performed CoMFA analyses. The initial compound activity data were obtained by testing three compounds, 7, 11 and 29, which induced a relatively large chemical shift perturbation on Trp34HE (Figure 2A). Compound dissociation constants (Kd) for the three compounds were determined via a 2D HSQC titration method (Figure 2B,C). The Kd values of the three compounds are 2.5 mM, 1 mM and 300 μM respectively. Therefore, the three compounds can be ranked as 29 > 11 > 7 according to the binding affinities from high to low. However, using the 2D HSQC titration to determine Kd value is a time-consuming and protein-consuming task to be performed for all BI-81 compounds. Comparing the chemical shift perturbation values of 7, 11 and 29 with the measured Kd values indicates that the chemical shift values at a given protein/ligand ratio can be used to estimate relative Kd values. Hence, the binding affinity data can be estimated by measurements of the compound induced chemical shift perturbation of Trp34HE at a given ligand to protein ratio.
Figure 2.

(A) The chemical shift perturbation values of Trp34HE in the presence of BI-81 compounds are summarized. The three compounds the Kd of which were tested via 2D NMR titration are labeled via asterisk. (B) The Kd values are calculated by fitting the chemical shift perturbation values for compounds 29 (●), 11 (○) and 7 (×) at different ligand/protein ratios. The fit curves are displayed in black line. The 95% confidence bands are displayed in dash line. (C) The cross-peak movements of Trp34HE are displayed in the presence of compounds 29, 11 and 7. For compounds 29 and 11, five titration points at ligand/protein ratios 2:1 (red), 4:1 (blue), 6:1 (orange), 10:1 (cyan) and 15:1 (pink) are displayed. For compound 7, six titration points at ligand/protein ratios 1:1 (red), 2:1(blue), 3:1 (orange), 5:1(cyan), 7:1 (pink), 10:1 (purple) and 15:1 (coral) are displayed. All the peaks without the addition of the compounds were displayed in green.
By using these values, we performed a CoMFA analysis to dissect the contribution of each substituent to the observed binding affinities. Of the total 39 BI-81 compounds, 32 were randomly selected as a training set to build a CoMFA model and the remaining seven were selected as a test set for model validation. As the spatial alignment of compounds in 3D QSAR study is one of the most sensitive and determining factors in obtaining a reliable model, we manually aligned the energy minimized conformers of BI-81 compounds via fitting the five atoms in 1,2,4-trizole ring (Figure 3A). After the alignment, the leave-one-out PLS analysis was carried out and yielded a cross-validation q2 value of 0.606 with five components. As the cross-validated correlation coefficient (q2) is used as a measure of reliability of prediction, the q2 value above 0.6 suggests that our model should posses a reliable predictive ability. Subsequently, internal non-cross-validated PLS regression was carried out using the previously obtained optimum number of components and yielded a regression coefficient (r 2) of 0.946. The r2 value represents the goodness of 3D QSAR model. The statistical parameters associated with the CoMFA analysis are reported in Table 2.
Figure 3.

(A) Alignment of the BI-81 compounds in the training set by fitting the atoms in 1,2,4-triazole ring. (B) Plots of the calculated versus experimental chemical shift perturbation values of Trp34HE. The values of compounds in the training set, test set and application set are displayed in black circle, red circle and yellow dot, respectively. (C) The Kd values for the compounds, compounds 47 and 48 are calculated by fitting the titration points. The structures of compounds 47 and 48 are displayed as insert. The dashed lines represent 95% confidence fitting.
Table 2.
Summary of 3D QSAR analysis statistics
| CoMFA analyses based on training set | |
|---|---|
| PLS statistics | |
| q2 (CV correlation coefficient) | 0.605 |
| n (number of component) | 5 |
| S (standard error of prediction) | 0.133 |
| r2 (correlation coefficient) | 0.9415 |
| F (F-ratio) | 88.259 |
| Field distribution (%) | |
| Steric | 0.834 |
| Electrostatic | 0.166 |
To evaluate the predictive ability of this model, we subsequently calculated the chemical shift perturbation values of the compounds in the test set. The experimental values versus predicted values of BI-81 compounds in the training set and test set are plotted in Figure 3B. As shown, the predicted values from the constructed 3D QSAR model are in very good agreement with the experimental data, again confirming the good predictive ability of the derived QSAR model.
To further utilize and evaluate this model, the affinities of a total of 64 commercially available analogs (application set) were predicted. Based on the built 3D QSAR model, the chemical shift perturbation values of the application set were predicted and displayed in Figure 3B. In the application set, two compounds (47 and 48) were predicted having the highest chemical shift perturbation values close to the best compound, 29 in the training set, suggesting that they are expected to have a similar or even higher binding affinities for Pin1 compared with compound 29. The actual Kd value of compounds 47 and 48 were measured via 2D HSQC titrations. In agreement with the predictions from the 3D QSAR model, the Kd values of compounds 47 and 48 were 326 and 423 μM respectively (Figure 3C), hence close to the value obtained for compound 29. Therefore, the predictive ability of the derived 3D QSAR model was proved to be successful. As a result, this model should be useful in identifying more potent compounds and in guiding further medical chemistry efforts.
Visualization of CoMFA maps
To obtain a pictorial view of the information included in the 3D QSAR model, CoMFA contour maps were generated. Contour maps display how the variation of steric or electrostatic properties in the structural features of molecules contained in the training set lead to increase or decrease activity. Figure 4A,B show the contour maps of CoMFA steric and electrostatic fields, respectively. For convenience, all the main positions in Figure 4 were labeled and shown with the compound 29. As indicated from Figure 4A, the large green contour around the naphthalene ring indicates that bulky groups are favored for activity in this area. This illustrates why the compounds of large substituents in this area, such as compounds 29, 32 and 33 are more potent than the compounds of small R1 substituents such as compounds 11 and 12. On the other hand, as most of R1 substituents contain linker connecting hydrophilic heads to the C5 on the core structure, the linkers play key roles for increasing activity via delivering the bulky group to the proper position (green contour). This explains why compound 11 has a stronger binding affinity than compound 4 even though they have same R1 substituent. The yellow contours labeled 2′ and 3′ in Figure 4A represent sterically disfavored areas, which mean that the addition of bulky modifications near these regions will decrease activity. The closeness of the green and yellow contours suggests tight hit for the R1 substituent. Consistent with this observation, some compounds with a long chain modification on R1 hydrophobic head exhibit weak binding affinities for Pin1 (data not shown).
Figure 4.

(A) CoMFA steric contour map displayed together with compound 29. Green contour indicates favorable steric regions and yellow contours indicate disfavorable steric regions. (B) CoMFA electrostatic contour maps displayed together with compound 29. Red contours indicate region where more electronegative substituents are favorite, whereas blue contours indicate regions where such substituents are disfavored. (C, D) Comparison of the contour maps from the CoMFA analysis and the binding pocket of Pin1ww in the crystal structure. Compound 29 is in magenta. Residues Arg14, Arg17, Tyr23 and Trp34 are in orange and their respective position are reported on the surface. (E) Comparison of binding modes of compound 29 in the docking complex (magenta) and the CoMFA model. (F) Comparison of binding modes of docked compound 29 and the CTD phosphopeptide in the crystal structure (cyan).
Red CoMFA contour maps represent electronegative favorite regions. As shown in Figure 4B, contours 4′ and 5′ correspond to the acidic group on compound 29. In fact, compounds lacking the acidic head linked to 1,2,4-triazole cannot appreciably bind to Pin1. The red contour 6′ corresponds to the nitro group in compound 3 and 7 which illustrates why these compounds have higher binding affinities than compounds 4 and 5. Blue contours in Figure 4B represent electronegative disfavorable regions. Overlaying BI-81 compounds indicates that the position 7′ corresponds to the R1 substituents without linkers. However, compared with the steric fields, electrostatic fields only contribute 17% to the overall activity. Moreover, the compounds without linkers display very weak binding affinity. In conclusion, the information included in the contour maps provides a visual guide that could be useful for future medicinal chemistry optimizations to increase potency. For instance, compound activity can be improved via increasing the hydrophilic property on R1 substituents or modifying R1 with more electronegative groups.
Binding mode of BI81 compounds to Pin1
To further ascertain the binding mode of compound 29 to Pin1, we performed docking studies using GOLD. The binding orientation of compound 29 in the docking complex is shown in Figure 4C,D as the same orientation as that in the Figure 4A,B. In the docking complex, the acidic extension of compound 29 functions as a hook to interact with the side chain of Arg14 via two electrostatic interactions. The importance of the acidic hook is supported by the fact that the other 1,2,4-triazole analogs without this acidic hook cannot interact with Pin1 (data not shown). The naphthalene substituent of compound 29 is located in the hydrophobic patch of the WW domain and stabilized via hydrophobic interactions with the side chain of Trp34. The 1,2,4-triazole functioning as a linker to connect two functional important components, the acidic group and the naphthalene ring, is located in the narrow tunnel under Arg17 and stabilized via hydrogen bonds with the backbone and side chain of Arg17. The short R2 substituent pointing towards the wall of the hydrophobic pocket seems to be stabilized via hydrophobic interactions with the side chain of Tyr23. These important residues including Arg14, Arg17, Tyr23 and Trp34 are indicated in Figure 4C,D. To compare whether this binding mode is consistent with the CoMFA and docking studies, the QSAR model was loaded into the protein surface by fitting the orientations of compound 29. As indicated in Figure 4C,E, compound 29 displays very similar orientations within the contour maps which accurately describe the overall properties of the binding surface of the WW domain. This parallel provides further confidence in the predictive ability of the 3D QSAR model.
To further verify the docking results, we performed further NMR studies with compound 29. Figure 5 displays the 1D spectra of compound 29 (200 μM) recorded in presence and absence of Pin1 (20 μM). Apart from peaks from position 9 and 10 in the R2 substituent that are invisible because of the overlap with the water resonance (data not shown), the NMR spectrum nicely corresponds with the structure of the compound. After the addition of Pin1, the peaks from naphthalene are those that within the observable resonances of compound 29 experience the most significant shifts (Figure 5). The fact that both, the peaks from the naphthalene of compound 29 and those of Trp34HE of Pin1 experience the most significant chemical shift perturbation upon complexation, would suggest the two moieties possibly directly interact with each other. To verify this hypothesis, we also collected 3D 15N-resolved [H 1, H 1]-NOESY spectra of Pin1 in the absence and presence of compound 29. After the addition of compound 29, significant NOE cross-peaks changes nearby the Trp34 have been observed and additional peaks corresponding to the naphthalene of compound 29 have been detected (Figure S1). These data and evidences support a direct interaction between the naphthalene of compound 29 and the indole of Trp34 as anticipated by the docking studies.
Figure 5.

Comparison of 1D NMR spectra of compound 29 (200 μM) in the absence (black) and presence (red) of Pin1 (20 μM).
A crystal structure of Pin1 in complex with a phoshorylated peptide (CTD phosphopeptide: YpSPTpSPS) binding in the WW domain was recently reported (21). Three important factors function as main forces to maintain the stability of the complex include hydrophobic interactions between CTD phosphopeptide and the side chains of Trp34 and Tyr23, and hydrogen bonds between two phosphates from the peptide and the side chains of Arg14 and Arg17. Comparing the binding orientation of compound 29 in the docking complex and CTD phosphopeptide orientation in the crystal structure as shown in Figure 4F indicates that compound 29 mimics CTD phosphopeptide in its interaction with Pin1ww via the same three main relations. First, the large hydrophobic pocket composed of the side chain of Trp34 in the WW domain corresponds to the green contour which makes it possible to accommodate the bulky naphthalene group in compound 29. Second, the red contours 1′ and 2′ correspond to the side chain of Arg14 forming electrostatic interactions with the acidic hook in compound 29. Third, the core structure of 1,2,4-triazole forms hydrogen bonds with the side chain and backbone amine of Arg17.
Comparing the docking complex structure with the CoMFA contour maps discloses new perspectives for improvement of binding affinities. It has been reported that to bind Pin1, the peptides of four residues or longer (43) have significantly increased binding affinities. The reported crystal structure also indicated that except for three main interactions, other possible interactions can take place between the tail of the peptide and the upper wall of the pocket. NMR studies of a bivalent peptide and Pin1 interactions (44) indicated that, aside from residues 17 and 34, the region from residues 27–31 also experienced apparent chemical shift perturbation in the HSQC spectra upon the addition of the bivalent peptide. Residues 27–31 constitute the wall of the hydrophobic pocket corresponding to the position of the R2 substituent in our CoMFA analysis and docking model. Because of lack of sufficient diversity on R2 substituent in the commercial compounds, contributions of R2 to the activity could not be further exploited in this study. However, these observations should provide valid insights for the design of compounds to be synthesized.
Comparison with the known peptide inhibitor
Interactions between Pin1 and peptides have been reported in the previous studies and the Kd values for the peptides were determined via various approaches. Hence, we selected and synthesized Pintide and its Kd value was measured under the same approach and conditions used in this study. We repeated 2D HSQC titration experiments and calculated a Kd value of 323 μM for this peptide (data not shown), which is similar to the reported value of ~200 to 400 μM (23). Likewise, the locations of chemical shift perturbations induced by Pintide are similar to those induced by compound 29, e.g. strong chemical shift perturbations in the WW domain and weak chemical shift perturbations in the catalytic domain. According to the previous study, the strong chemical shift perturbations derives from the direct interactions between Pintide and the WW domain while the weak chemical shift perturbations in the catalytic domain results from the composite effects including the direct weak interaction an indirect conformational changes of the catalytic domain. Therefore, compound 29 displays a similar binding affinity than Pintide.
Some potent peptide mimic and small molecular weight inhibitors of Pin1 have also been recently reported (27,28,30,44). However, as for peptide mimics, the permeability and stability issues limit their applications as chemical probes for target validation. To evaluate the stability and permeability of our compounds, we performed molecular stability and membrane permeability assays. As shown in Figure 6, Pintide is rapidly degraded in plasma after 30 min while compounds 29 and 47 exhibit fourfolds greater stability after 60 min. Likewise, compounds 29 and 47 exhibit two times higher permeability than Pintide (30% versus 14%, Table 3). The reported chemical probes and relative binding data to Pin1 represent valuable stepping stones for the validation of Pin1 as target for drug discovery and for eventually the development of possible lead compounds.
Figure 6.

Plots of the plasma stabilities of compounds 29, 47 and Pintide.
Table 3.
Results of parallel artificial membrane permeation assay (PAMPA)
| Compound ID | 29 | 47 | Pintide |
|---|---|---|---|
| PAMPA FLUX (%) | 30 | 34 | 14 |
Conclusions
A novel scaffold of Pin1 inhibitor presenting a putative phosphoserine mimic was found based on a virtual screening approach. Subsequently, an expanded library of compounds was assembled by collecting the commercial available analogs. The binding affinities and binding mode of these compounds against Pin1 were investigated via NMR chemical shift perturbation experiments. Compound 29 exhibited an effective dissociation constant of 300 μM, similar to that of the reported phosphorylated peptide. Moreover, compound 29 possesses a higher stability and cell membrane permeation than the peptide. Furthermore, the CoMFA analysis was carried out to further highlight the contributions of each substituent in the tested compounds to their binding affinity. Combined with docking studies and NMR data, the resulting 3D QSAR model provides an invaluable tool to evaluate the binding affinity of additional compounds. With the employment of all of these results, further design, synthesis and activity evaluation will be performed in our laboratory.
Supplementary Material
Figure S1. Comparison of strips from 3D-N 15-NOESY spectra of Pin1 in absence (A) and in presence (B) of compound 29 (1:10 molar ratio Pin1/compound).
Acknowledgments
This work was supported in part by NIH grants and HL082574, AI059572 (to M.P.).
Footnotes
Additional Supporting Information may be found in the online version of this article:
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Gothel SF, Marahiel MA. Peptidyl-prolyl cis–trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol Life Sci. 1999;55:423–436. doi: 10.1007/s000180050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmid FX, Mayr LM, Mucke M, Schonbrunner ER. Prolyl isomerases: role in protein folding. Adv Protein Chem. 1993;44:25–66. doi: 10.1016/s0065-3233(08)60563-x. [DOI] [PubMed] [Google Scholar]
- 3.Shaw PE. Peptidyl-prolyl isomerases: a new twist to transcription. EMBO Rep. 2002;3:521–526. doi: 10.1093/embo-reports/kvf118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 5.Harding MW, Galat A, Uehling DE, Schreiber SL. A receptor for the immunosuppressant FK506 is a cis–trans peptidyl-prolyl isomerase. Nature. 1989;341:758–760. doi: 10.1038/341758a0. [DOI] [PubMed] [Google Scholar]
- 6.Rahfeld JU, Schierhorn A, Mann K, Fischer G. A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 1994;343:65–69. doi: 10.1016/0014-5793(94)80608-x. [DOI] [PubMed] [Google Scholar]
- 7.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 8.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 9.Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZX. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–853. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 10.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 11.Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- 12.Lu KP, Liou YC, Vincent I. Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer’s disease. BioEssays. 2003;25:174–181. doi: 10.1002/bies.10223. [DOI] [PubMed] [Google Scholar]
- 13.Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liou YC, Ryo A, Huang HK, Lu PJ, Bronson R, Fujimori F, Uchida T, Hunter T, Lu KP. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci USA. 2002;99:1335–1340. doi: 10.1073/pnas.032404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dominguez-Sola D, Dalla-Favera R. PINning down the c-Myc oncoprotein. Nat Cell Biol. 2004;6:288–289. doi: 10.1038/ncb0404-288. [DOI] [PubMed] [Google Scholar]
- 18.Ryan KM, Vousden KH. Cancer: pinning a change on p53. Nature. 2002;419:795–797. doi: 10.1038/419795a. [DOI] [PubMed] [Google Scholar]
- 19.Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature. 2002;419:853–857. doi: 10.1038/nature01120. [DOI] [PubMed] [Google Scholar]
- 20.Wintjens R, Wieruszeski JM, Drobecq H, Rousselot-Pailley P, Buee L, Lippens G, Landrieu I. 1H NMR study on the binding of Pin1 Trp-Trp domain with phosphothreonine peptides. J Biol Chem. 2001;276:25150–25156. doi: 10.1074/jbc.M010327200. [DOI] [PubMed] [Google Scholar]
- 21.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 22.Bayer E, Goettsch S, Mueller JW, Griewel B, Guiberman E, Mayr LM, Bayer P. Structural analysis of the mitotic regulator hPin1 in solution: insights into domain architecture and substrate binding. J Biol Chem. 2003;278:26183–26193. doi: 10.1074/jbc.M300721200. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs DM, Saxena K, Vogtherr M, Bernado P, Pons M, Fiebig KM. Peptide binding induces large scale changes in inter-domain mobility in human Pin1. J Biol Chem. 2003;278:26174–26182. doi: 10.1074/jbc.M300796200. [DOI] [PubMed] [Google Scholar]
- 24.Kim CJ, Cho YG, Park YG, Nam SW, Kim SY, Lee SH, Yoo NJ, Lee JY, Park WS. Pin1 overexpression in colorectal cancer and its correlation with aberrant beta-catenin expression. World J Gastroenterol. 2005;11:5006–5009. doi: 10.3748/wjg.v11.i32.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, Wheeler TM, Lu KP, Bao L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 26.Nakashima M, et al. Cyclin D1 overexpression in thyroid tumours from a radio-contaminated area and its correlation with Pin1 and aberrant beta-catenin expression. J Pathol. 2004;202:446–455. doi: 10.1002/path.1534. [DOI] [PubMed] [Google Scholar]
- 27.Wildemann D, Erdmann F, Alvarez BH, Stoller G, Zhou XZ, Fanghanel J, Schutkowski M, Lu KP, Fischer G. Nanomolar inhibitors of the peptidyl prolyl cis/trans isomerase Pin1 from combinatorial peptide libraries. J Med Chem. 2006;49:2147–2150. doi: 10.1021/jm060036n. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Daum S, Wildemann D, Zhou XZ, Verdecia MA, Bowman ME, Lucke C, Hunter T, Lu KP, Fischer G, Noel JP. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem Biol. 2007;2:320–328. doi: 10.1021/cb7000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 30.Smet C, Duckert JF, Wieruszeski JM, Landrieu I, Buee L, Lippens G, Deprez B. Control of protein-protein interactions: structure-based discovery of low molecular weight inhibitors of the interactions between Pin1 WW domain and phosphopeptides. J Med Chem. 2005;48:4815–4823. doi: 10.1021/jm0500119. [DOI] [PubMed] [Google Scholar]
- 31.Cramer RD, III, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steriods to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 32.Goddard TD, Kneller DG. SPARKY 3. San Francisco: University of California; 2008. [Google Scholar]
- 33.Farmer BT, II, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Jr, Robertson JG, Mueller L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat Struct Biol. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 34.Seydel JK, Wiese M. Drug–Membrane Interactions: Analysis, Drug Distribution, Modeling. Weinheim: Wiley-VCH; 2002. [Google Scholar]
- 35.Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 36.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 37.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 38.Ramelot TA, Nicholson LK. Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J Mol Biol. 2001;307:871–884. doi: 10.1006/jmbi.2001.4535. [DOI] [PubMed] [Google Scholar]
- 39.Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 40.Lian LY, Barsukov IL, Derrick JP, Roberts GC. Mapping the interactions between streptococcal protein G and the Fab fragment of IgG in solution. Nat Struct Biol. 1994;1:355–357. doi: 10.1038/nsb0694-355. [DOI] [PubMed] [Google Scholar]
- 41.Williamson RA, Carr MD, Frenkiel TA, Feeney J, Freedman RB. Mapping the binding site for matrix metalloproteinase on the N-terminal domain of the tissue inhibitor of metalloproteinases-2 by NMR chemical shift perturbation. Biochemistry. 1997;36:13882–13889. doi: 10.1021/bi9712091. [DOI] [PubMed] [Google Scholar]
- 42.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Fussel S, Reimer U, Schutkowski M, Fischer G. Substrate-based design of reversible Pin1 inhibitors. Biochemistry. 2002;41:11868–11877. doi: 10.1021/bi0262395. [DOI] [PubMed] [Google Scholar]
- 44.Daum S, Lucke C, Wildemann D, Schiene-Fischer C. On the benefit of bivalency in peptide ligand/pin1 interactions. J Mol Biol. 2007;374(1):147–161. doi: 10.1016/j.jmb.2007.09.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparison of strips from 3D-N 15-NOESY spectra of Pin1 in absence (A) and in presence (B) of compound 29 (1:10 molar ratio Pin1/compound).