

Abstract
Murine mAbs reactive with the surface of Mycobacterium tuberculosis were assayed for their ability to affect the course of infection in mice challenged with virulent organisms. An IgG3 mAb (9d8) specific for arabinomannan and reactive with purified antigen from a clinical isolate of M. tuberculosis conferred partial protection on mice after respiratory challenge (30–60% survival >75 days; P ≤ 0.05). Control mice pretreated with an irrelevant mAb of the same isotype succumbed to tuberculosis within 30 days. Mice with gene disruptions in interferon γ and major histocompatibility complex Class II also were partially protected from challenge. The protective mAb was neither bactericidal nor inhibitory of infection or bacterial replication. Nevertheless, it profoundly altered the nature of the granulomas in the infected lungs. Mice treated with mAb 9d8 and challenged with M. tuberculosis localized the pathogen within granuloma centers, suggesting that the mAb conferred protection by enhancing a cellular immune response.
Mycobacterium tuberculosis causes more deaths annually than any other single human pathogen (1). Individuals infected with HIV are particularly vulnerable to tuberculosis (2). Cell-mediated immunity, including production of interferon γ (IFN-γ) and interleukin-12 and major histocompatibility complex (MHC) Class I and Ia restricted T cells, has been shown to be essential for protection from tuberculosis infection (3–9). A plethora of reports exist in the older tuberculosis literature (10) stating that immune sera contributed to protection against, or progression of, clinical tuberculosis. Unfortunately, much of the data are contradictory, and, as a consequence, a possible role for antibodies in protection against tuberculosis infection has largely been ignored.
Encouragement for the view that antibodies may be able to contribute to protection against an intracellular pathogen come from recent experiments on Cryptococcus neoformans, in which treatment of mice with certain mAbs to the capsular polysaccharide markedly enhanced survival of mice. Protection afforded by the antibodies was isotype-related and dependent upon the presence of intact cell-mediated responses (12). Furthermore, mice genetically deficient in specific components of the cell-mediated immune response were protected by some, but not all, isotypes of the protective mAb (13). These studies reveal that antibodies can participate in the developing immune response to intracellular pathogens. Since human tuberculosis is thought to result from infection with as few as 10–200 tubercle bacilli (14), we sought to explore the possibility that antibodies specific to the surface of M. tuberculosis might prevent infection or modify the course of experimental tuberculosis.
MATERIALS AND METHODS
Mouse Strains.
Six- to eight-week-old female BALB/c and C57BL/6 mice were purchased from Charles River Breeding Laboratories, as were mice genetically disrupted for the IFN-γ gene. Age- and gender-matched Abβ mice genetically disrupted for the Aβ(H2b) gene of the mouse Class II MHC were purchased from Taconic Farms, as were C57BL/6 controls. Mice were housed in the BSL3 facility and fed a standard diet.
Bacterial Strains.
Frozen stocks of known titer of the Erdman strain of M. tuberculosis were prepared as described previously (4).
Antibodies.
The mAbs 9d8, 4f11, and 5c11, which are reactive with M. tuberculosis surface antigens, have been characterized previously (11). The mAb 3E5 to C. neoformans capsular polysaccharide of C. neoformans was used as an isotype-matched irrelevant control (15). Additionally, ascites from the nonproducer NSO cell line was used as a negative control in the pilot experiment. The mAb recognizing the inducible nitric oxide synthase (NOS) protein was a kind gift from Charles Lowenstein, Johns Hopkins University. RB6–8C5, the mAb that recognizes neutrophils, was used as described previously (16).
Antibody Purification.
9d8 ascites fluid was generated in BALB/c mice. Ascites fluid was dialyzed against 0.05 M Tris⋅HCl buffer (pH 7.8). The dialysate was subjected to DE52 anion-exchange column chromatography (Whatman) and subsequently to protein G column chromatography (Pierce). NSO ascites were obtained by paracenthesis of BALB/c mice injected i.p. with NSO myeloma cells.
Characterization of the Specificity of the 9d8 mAb.
A modified ELISA assay was utilized to test several carbohydrate fractions of M. tuberculosis as described previously (11). The 9d8 mAb was tested against purified and fast atom bombardment-mass spectroscopy-confirmed arabinomannan and glucan from M. tuberculosis, as were hybridoma supernatants containing mAb 4f11, an IgM isotype mAb recognizing the mycolyl-arabinogalactan-peptidoglycan complex (mAGP) (11), used as a control.
Polymorphonuclear Leukocyte (PMN) Depletion.
C57BL/6 mice were depleted of the neutrophils by administration of the RB6–8C5 mAb given i.p., on days –2, −1, 0, +1, and +2, and PMN depletion was verified as described (16).
Mouse Infections.
Initial experiments employing the 9d8 antibody were conducted with ascites fluid, while subsequent experiments used purified antibody preparations. Approximately 2 × 105 bacilli were incubated with either 2 ml of 9d8 ascites fluid (at a concentration of 92 μg/ml protein) or an equivalent volume of NSO ascites. Subsequent experiments used 1 × 106 M. tuberculosis incubated with 1.1 mg purified 9d8 or control 3E5 mAb for at least 3 hr, at room temperature. To ensure the study was blinded, color-coded antibody preparations were dispensed to the investigator performing these experiments and collecting all subsequent data. Bacterial suspensions were mixed periodically during the incubation period. Bacilli then were washed twice in PBS and instilled intratracheally (i.t.) to mice. In the pilot experiment, additional groups of mice were given i.p. injections of 1 ml ascites fluid of either 9d8 or NSO ascites fluid 2 days, 1 day, 4 hr before, and 1 day post i.t. infection. For i.t. infections, mice were anesthetized with 70 mg/kg of sodium pentobarbital (Nembutal, Abbott), and a small incision was made at the base of the throat. A 50-μl suspension containing an estimated 5 × 103 virulent M. tuberculosis was instilled via tuberculin syringe. The incision was closed with surgical glue (Nexaband, VPL). Additional groups of mice received 50 μl of the antibody-coated inoculum, estimated to have the same dose of bacilli. At least two mice were harvested from each group 24 hr after infection, and lung homogenates were plated to confirm the initial infecting dose.
Organ Burdens and Histology.
For each experimental group examined, two mice were killed at 24 hr and three mice were killed at 3 weeks postinfection by cervical dislocation. Organs were removed for enumeration of bacilli and pathological analysis. Half of the spleen, liver, and lung tissues were homogenized, and dilutions were plated on media as described previously (4). Remaining mice were left for survival studies, the sample size of which is indicated in the figure legends. Histology was conducted as described previously (4, 17, 18). Sections were viewed on a Zeiss Axiophot compound microscope, using a didymium filter, and photographed with Kodak Lumiere 100 film, to enhance bacterial visualization.
Statistical Analysis.
Two tests were utilized to evaluate differences in survival: the log-rank test was used to assess the course of survival, and χ2 analysis was used to determine the significance of endpoint differences. Student’s t test was used to analyze differences in the number of colony-forming units (cfu) between groups.
RESULTS
A mAb Recognizing the Surface of M. tuberculosis Enhances Survival.
Two mAbs, 5c11 and 9d8, reactive with surface antigens of M. tuberculosis were tested for their ability to affect the survival of BALB/c mice challenged in preliminary experiments with virulent tubercle bacilli by the respiratory route. To maximize the likelihood of antibody associating with M. tuberculosis within the lung, we incubated the bacilli with the mAbs for 4 hr in vitro before challenging the mice with the coated inocula i.t. Mouse survival at a 220-day endpoint was measured (Table 1 A and B). None of the mice receiving bacilli coated with the IgM 5c11 antibody survived. However, 33% of BALB/c mice receiving tubercle bacilli coated with IgG3 mAb 9d8 survived for 220 days, although cfu recovered from lungs and spleens at the time of death of these and control mice were comparable (data not shown). That result led us to infer that (i) not all antibodies reactive with the surface of M. tuberculosis affected the course of infection, and (ii) that the 9d8 mAb appeared to confer some protection against mortality, but apparently did not prevent infection.
Table 1.
Effect of purified mAbs on survival
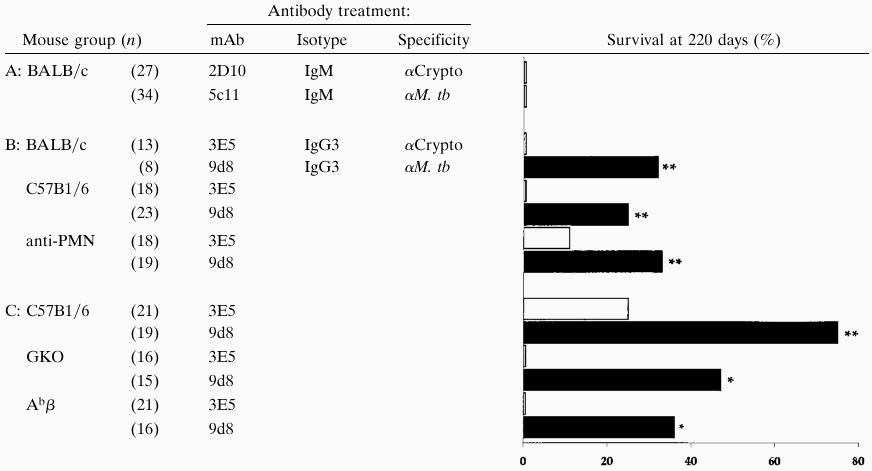 |
Immunocompromised mice were C57B1/6 background; sample size is indicated in parentheses. All mice were challenged i.t. with 5,000 M. tb preincubated with the indicated antibody.
, P ≤ 0.05, by both log-rank and χ2 tests; ∗, P ≤ 0.05 by χ2 test alone.
As a result of these preliminary findings, we proceeded with additional experiments to characterize the protection engendered by the IgG3 9d8 mAb in more detail. For controls we used both ascites derived from the NSO nonproducer hybridoma cell line and the 9d8 IgG3 antibody injected i.p. An additional group of mice received bacilli coated with 9d8 ascites (M. tb-9d8) (Fig. 1A). Only mice infected with M. tb-9d8 survived longer than mice given M. tuberculosis cultured in media alone (P < 0.01, by both log-rank and χ2 tests). I.p. injection of 9d8 did not appear to enhance survival in this study. To verify that the survival advantage afforded was due to specific antibody, purified Ig fractions were used for all subsequent experiments. M. tuberculosis specific mAb 9d8 and the isotype-matched control mAb specific for the surface polysaccharide of Cryptococcus neoformans (3E5) were cultured with the challenge bacilli before mouse infection (M. tb-9d8 and M. tb-3E5, respectively) (Fig. 1B). Again, C57BL/6 mice infected with M. tb-9d8 exhibited a survival advantage relative to the control, M. tb-3E5. Protection afforded by the 9d8 mAb apparently was not dependent upon the host genetic background, since BALB/c mice challenged with M. tb-9d8 (Fig. 1C) were similarly protected. While in four independent experiments, 35–100% of mice given M. tb preincubated with the 9d8 mAb survived, the same antibody given i.p. did not increase survival relative to mice given nonproducer NSO ascites i.p., concomitant with respiratory challenge with virulent M. tuberculosis. Thus, survival consistently was improved by coating the challenge bacilli with the 9d8 mAb.
Figure 1.

M. tb-9d8 enhances survival without affecting bacterial load in mice, irrespective of their genetic background. (A) C57BL/6 mice were given 9d8 (□) or NSO ascites (○) i.p. 48, 24, and 4 hr before i.t. infection with 5 × 103 M. tuberculosis (n = 5). An additional group of mice received the inoculum previously incubated with 9d8 mAb for 4 hr (■) (n = 4). M. tb-9d8-infected mice survived longer than mice given 9d8 or NSO ascites i.p. (P < 0.01, log-rank and χ2 tests). (B) Survival of C57BL/6 mice infected with M. tuberculosis previously cultured with purified mAb 9d8 (■) (n = 23) is prolonged as compared with M. tb-3E5 (□) (n = 18) (P < 0.01, log-rank and χ2 tests). (C) BALB/c mice infected with M. tb-9d8 (■) (n = 9) are protected similarly compared with control M. tb-3E5 (□) (n = 13) infected mice (P < 0.01, log-rank and χ2 tests). (D) Despite enhanced survival, lung bacterial burdens of M. tb-9d8-infected C57BL/6 (▴) and BALB/c (•) did not differ compared with their controls (○ and ▵, respectively) (n = 3 mice per group). (E) Similarly, antibody coating of bacilli in M. tb-9d8 infection did not hamper bacterial dissemination to or replication within extrapulmonary organs. Spleen cfu were comparable in C57BL/6 (▴) and BALB/c (•) M. tb-9d8-infected mice and their controls (▵ and ▴, respectively) (n = 3 mice per group).
Despite the clear prolongation of survival in the mice receiving M. tb-9d8, no significant differences in the lung burden of bacilli were observed. The number of cfu recovered from the lungs of C57BL/6 and BALB/c mice challenged with M. tb-9d8 approximated that obtained from control mice from the outset of infection to the time of death of control mice (Fig. 1D). Incubation with ascites preparations yielded similar findings (data not shown). There similarly were no observable differences in the number of bacilli recovered from spleen homogenates (Fig. 1E) or liver homogenates of the antibody-treated animals (data not shown), indicating that the mAb had little effect on extrapulmonary dissemination. Thus, the increased survival engendered by the 9d8 mAb cannot be explained by inhibition of lung infection or of bacterial replication in the lung.
Organized Granuloma Formation Correlates with Survival in M. tb-9d8-Infected Mice.
To evaluate the effects of mAb coating of M. tuberculosis on localization and host pathology, lung tissue was examined for gross morphologic changes and for the presence of acid fast bacilli (Fig. 2). A representative series of sections is shown in Fig. 2. Pathologic examination of lung sections from mice infected with M. tuberculosis coated with 9d8 ascites (Fig. 2D) revealed no overt differences in their tissue morphology compared with the controls (Fig. 2A). Nor were differences seen in the extent of tissue necrosis or edema within the lungs. A striking difference was observed in the localization of tubercle bacilli within lung tissue. Control mice displayed a random distribution of acid-fast bacilli (AFB) throughout the tissue (Fig. 2B). In contrast, mice receiving M. tb-9d8 ascites (Fig. 2E) or purified M. tb-9d8 (data not shown) revealed bacilli concentrated within well organized granulomas.
Figure 2.

Enhanced survival correlates with granuloma formation. (A) Lung sections of mice infected with M. tb-3E5 stained with hematoxylin/eosin reveal typical pathology associated with experimental tuberculosis infection in mice. Multiple granulomas are seen, as well as infiltrating inflammatory cells. (B) Mice infected with M. tb-3E5 reveal a random distribution of AFB (pink bacilli) in lung tissue sections. (C) Immunohistochemistry conducted on subsequent sections reveals moderate production of iNOS in M. tb-3E5-infected mice, paralleling the presence of AFB. (D) Lung sections of mice infected with M. tb-9d8 stained with hematoxylin/eosin reveal similar gross pathology, compared with controls. (E) M. tb-9d8-infected mice reveal a concentration of bacilli within well formed granulomas. (F) Serial sections reveal that iNOS is produced similarly in cells paralleling AFB deposition. In M. tb-9d8-infected mice, however, iNOS production may function to halt bacterial dissemination throughout the tissue, by serving as a barrier to maintain containment within granuloma centers.
NO, a product of activated macrophages, has been shown to be mycobactericidal in vitro and in vivo (19, 20). Lung macrophages of both control and 9d8-coated M. tuberculosis-infected mice produced iNOS (Fig. 2 C and F) in the foci containing AFB. In control sections, the pattern of iNOS staining was distributed randomly across lung tissue, paralleling the presence of AFB. Lung tissue from mice infected with M. tb-9d8 revealed, however, that iNOS staining was more prominent within cells surrounding clusters of AFB, suggesting that the cells localizing the infection and synthesizing iNOS contribute to bacterial containment within the granulomas.
Antibody Treatment Enhances Survival in Immunocompromised Mice.
Because the protective effect of mAbs in C. neoformans infection was found to be dependent upon the presence of cell-mediated immune mechanisms, we sought to ascertain whether elements of the cellular immune response similarly were necessary for the antibody-mediated protection seen in experimental tuberculosis. Mice genetically disrupted for the IFN-γ and MHC class II genes were infected with M. tuberculosis preincubated with either the 9d8 or 3E5 isotype-matched control mAbs. To learn whether the protective effect of antibody was dependent on PMN, an additional group of mice received the RB6–8C5 mAb that has been shown to deplete PMNs and exacerbate listeria infection (16). Mice depleted of PMNs (Table 1B) were no more susceptible to M. tuberculosis infection than untreated C57BL/6 controls. IFN-γ-deficient mice (GKOs) (Table 1C) were, as previously reported (4, 7), markedly more susceptible to M. tuberculosis infection relative to C57BL/6 controls in terms of their survival. Surprisingly, a statistically significant proportion of the GKO mice (47%) was protected by the 9d8 mAb, although not as effectively as the M. tb-9d8-infected C57BL/6 mice in the same experiments. Mice genetically disrupted in the MHC class II gene (Abβs) challenged with M. tb-9d8 similarly showed enhanced survival (Table 1C).
Enhanced survival could not be attributed to a reduction in mycobacterial numbers, as the number of cfu recovered from the lungs of GKO mice infected with M. tb-9d8 or M. tb-3E5 was not significantly different (Fig. 3A). As was the case for their immunocompetent counterparts, M. tb-9d8-infected GKO mice exhibited no differences in cfu recovered from their spleens, compared with M. tb-3E5 GKO mice (Fig. 3B). M. tb-9d8-infected GKO mice, in fact, had elevated spleen cfu relative to their immunocompetent counterparts, consistent with previous reports (4, 7). Similar results were obtained from the Abβs (data not shown). In general, immunocompromised M. tb-9d8-infected mice exhibited similar pathology as their immunocompetent counterparts. AFB localized within discrete, well formed granulomas comparable in character to those of the intact control C57BL/6 mice infected with M. tb-9d8 (data not shown).
Figure 3.

M. tb-9d8 infection partially protects immunocompromised mice without concomitant decrease in bacterial burden. (A) Survival of M. tb-9d8-infected GKO was not mediated through a reduction in lung cfu. Comparable cfu were recovered from GKO mice infected with M. tb-9d8 (•) or M. tb-3E5 (○), as well as M. tb-3E5-infected C57BL/6 mice (□) (n = 3 mice per group). (E) The 9d8 mAb did not affect extrapulmonary seeding of immunocompromised mice. GKO mice infected with M. tb-9d8 (•) yielded similar numbers of cfu as M. tb-3E5-infected GKO mice (○). GKO mice did, however, reveal increased numbers of cfu as compared with M. tb-3E5-infected C57BL/6 mice (□) (P < 0.05, Student’s t test) (n = 3 mice per group).
The 9d8 Antibody Recognizes Carbohydrate Exposed on the Outer Surface of M. tuberculosis.
Previous studies indicated that the 9d8 mAb reacted with a protease-resistant antigen of M. tuberculosis (11). The 5c11 IgM mAb was found previously to bind the M. tuberculosis major lipid antigen, lipoarabinomannan, and the 4f11 IgM mAb recognized mycolyl-arabinogalactan. In the current set of experiments, 4f11 failed to react with either purified arabinomannan (Fig. 4) or glucan (data not shown). The 9d8 mAb bound to purified arabinomannan and failed to bind purified glucan, establishing its specificity for arabinomannan. Lipoarabinomannan is a surface antigen with three domains: a lipid area containing phosphatidylinositol that binds to the surface of the bacterium, an intermediate domain composed of a branched polymer of mannose, and, at the nonreducing end, a polymer capped with mannose oligosaccharides (22). Thus, the protective 9d8 mAb was specific for an apparently unique surface epitope of M. tuberculosis. While ELISA data indicated that both 5c11 (IgM) (data not shown) and 9d8 (IgG) bound to purified arabinomannan, only 9d8 conferred enhanced protection, suggesting that antibody isotype as well as specificity are critical for engendering protection.
Figure 4.

The 9d8 mAb recognizes arabinomannan on the M. tuberculosis surface. Purified arabinomannan was probed with either mAb 9d8 (○) or 4f11 (□) in a modified ELISA assay. Alkaline phosphatase (ALKP)-conjugated goat anti-mouse (GAM) IgG3 or IgM were added to either 9d8- or 4f11-treated wells, respectively. The mAb 9d8 bound to arabinomannan in a concentration-dependent manner, while 4f11 did not bind.
DISCUSSION
Persistence of M. tuberculosis necessitates the maintenance of an intracellular niche within infected cells. For this reason, it has been argued that humoral immunity can have little effect on the control of natural tuberculous infection (23). Despite an extensive older literature suggestive of a possible therapeutic role for immune sera in the control of disease (10), little attention has been paid to a possible role for antibody in protection from M. tuberculosis infection. Arguments against a role for antibodies in affecting tuberculosis have included that: (i) M. tuberculosis reside intracellularly, inaccessible to antibodies; (ii) titers of antibody increase with progression of disease; and (iii) heat-killed bacilli generate antibodies to M. tuberculosis antigens but fail to protect as well as live bacilli, which induce cell-mediated immunity (26). Although M. tuberculosis clearly reside within lung macrophages at early stages of infection, necrotizing granulomas are the hallmark of progressive disease, a stage in which bacilli can be found extracellularly within tissue and, therefore, in potential contact with antibodies. It was our initial hypothesis that antibodies may also have access to tubercle bacilli before bacterial seeding of the lung and, thus, serve to limit initial infection.
Polyclonal immune sera have produced conflicting results in terms of protection against M. tuberculosis (10) and the fungal infections caused by C. neoformans and C. albicans (24). The inconsistent findings with immune sera most likely reflect the variable specificities and isotypes comprising polyclonal preparations. In the present experiments, mice were infected with M. tuberculosis coated with specific mAbs, and mAb 9d8 was found to result in prolonged survival of a percentage of mice challenged with a rapidly lethal dose of M. tuberculosis. These findings indicate that some mAbs to M. tuberculosis are able to modify the course of experimental tuberculosis infection.
In four independent experiments, coating of bacilli with only the 9d8 mAb consistently enhanced survival of between 35 and 100% of animals tested. Despite variation in the percentage of animals protected between experiments, in each experiment a highly significant survival advantage was afforded by mycobacterial coating with the specific 9d8 mAb. Systemic administration of mAb i.p. followed by respiratory challenge did not enhance survival, which may be attributable to the low quantities of antibody produced in ascites preparations of the 9d8 hybridoma (92 μg/ml); normally, ascites fluids yield roughly 1 mg/ml of mAb. It is equally possible that the IgG3 isotype does not accumulate in adequate concentrations in the lung, such that in either case, insufficient concentrations required for protection were achieved in the lung. These results, while not definitive, suggest that both isotype and specificity may be critical for protection.
Since the lungs received comparable numbers of organisms early after infection and retained comparable numbers of bacilli until the time of death, the hypotheses that mAb coating of tubercle bacilli prevented infection or blocked bacterial replication could not be sustained. Antibody was introduced in the experimental system solely on the day of infection, and since necrosis does not occur until later stages of infection, it is highly implausible the mAb could have been redistributed to coat all the increasing numbers of bacilli. It is also unlikely the 9d8 functioned merely to cluster or aggregate bacilli within the lung, since the cfu recovered from M. tb-9d8 and control infected mice were comparable at all times studied. Additionally, evaluation of tissue soon after infection by light microscopy failed to reveal clumps of bacilli within the airway (data not shown). Finally, the fact that M. tuberculosis coated with 9d8 mAb protected mice whose neutrophils had been depleted with a mAb to murine PMNs confirmed that the protective effect of the 9d8 was not PMN-dependent and did not require lysis or killing the infecting organisms. Rather, we are led to infer that 9d8 affected the cell-mediated immune response in the lung, either by affecting tissue localization of the bacilli or antigen presentation by infected macrophages or dendritic cells.
Granuloma formation was markedly enhanced, particularly in lungs, of mice receiving M. tuberculosis coated with the 9d8 antibody. Immune complexes have been demonstrated to stimulate granuloma formation (25), and perhaps the infection protocol facilitated an earlier or more effective granuloma development. Similar observations have been made in a respiratory model of C. neoformans infection (26, 27), where mAb treatment has been shown to prolong survival and enhance granuloma formation without an apparent reduction in cfu. Although the mechanism by which specific mAb promotes granuloma formation against either pathogen is not understood, Fc receptor activation is known to promote cytokine and chemokine release (28–32), superoxide production (33), and NO production (34) and up-regulate B7 expression (35). The corresponding localization of iNOS surrounding centers of bacterial replication in our system may function as a barrier to infection of the surrounding macrophages and to limit bacterial spread across the tissue. Alternatively, mAb binding to the mycobacterial surface itself may neutralize the toxic or immunosuppressive effects of mycobacterial polysaccharides and/or glycolipids.
In previous experiments it was reported that pretreatment of pathogenic mycobacteria with immune rabbit serum enhanced phagocytosis and facilitated lysosomal fusion, but did not induce enhanced killing of the pathogen (36). IFN-γ preactivation of macrophages was reported to function similarly (37). Thus, at some level, coating of M. tuberculosis by mAb may affect bacterial internalization and subsequent intracellular trafficking within infected macrophages similar to the effects of IFN-γ on macrophages. GKO mice were somewhat protected by 9d8 coating of M. tuberculosis, supportive of a parallel role for antibody opsonization and IFN-γ effects on bacterial intracellular localization.
Immunocompromised adults have a markedly increased risk of contracting tuberculosis (38). HIV infection accelerates the course of tuberculosis disease (2). Effective chemotherapy is not available in many developing countries; moreover, the rising emergence of multidrug-resistant strains necessitates alternative strategies for prevention and treatment of tuberculosis infection in immunocompromised populations. In this context, pretreatment of M. tuberculosis with 9d8 mAb enhanced survival even of some GKO mice, whereas the GKO mice receiving bacilli treated with control antibodies all rapidly succumbed to disease. Although the protection afforded by specific mAb was clearly greater in hosts with intact cell-mediated responses, it is nevertheless encouraging that a specific mAb could significantly enhance the survival of immunocompromised mice.
These findings raise a number of questions: by what molecular mechanisms the specific anti-M. tuberculosis mAb induced protection; why only one of two arabinomannan-recognizing mAbs was effective; why mAbs to another surface carbohydrate, glucan, were not protective; why the specific mAb was only effective in a proportion of mice; and why that proportion varied between experiments. Nevertheless, the results offer encouragement that it may be possible to harness specific humoral responses that are not lacking in individuals with HIV/AIDS for developing improved preventive vaccines or therapeutics that will contribute to protection against tuberculosis.
Acknowledgments
We are indebted to Mariane Roeber, Ph.D., of the Hybridoma Reagent Laboratory for purification of the 9d8 mAb. This work has been supported by National Institutes of Health Grants AI07118, 23545 (B.R.B.), AI33142, 33774, and HL59842, the Burroughs–Wellcome fund Scholar Award in Experimental Therapeutics (A.C.), the Aaron Diamond Foundation (A.G.F.), and the Howard Hughes Medical Institute (B.R.B.). The glucan and AM were supplied by J. B. Robbins, Z. D. Dai, R. Schneerson, V. Pozsgay (National Institute of Child Health and Human Development, National Institutes of Health), S. Morris (Center for Biologics Evaluation and Research, Food and Drug Administration), and D. Schulz (Pasteur Merieux, Lyon, France).
ABBREVIATIONS
- IFN-γ
interferon γ
- MHC
major histocompatibility complex
- M. tb
M. tuberculosis
- GKO
IFN-γ-deficient mice
- AFB
acid-fast bacilli
- PMN
polymorphonuclear leukocyte
- i.t.
intratracheally
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Murray C J L, Lopez A D. In: Global Comparative Assessments in the Health Sector: Disease Burden, Expenditures and Intervention Packages. Murray C J L, Lopez A D, editors. Geneva: World Health Organization; 1994. p. 53. [Google Scholar]
- 2.Lucas S, Nelson A M. In: Tuberculosis: Pathogenesis, Protection and Control. Bloom B R, editor. Washington, DC: Am. Soc. Microbiol.; 1994. pp. 503–513. [Google Scholar]
- 3.Flynn J L, Goldstein M M, Triebold K J, Koller B, Bloom B R. Proc Natl Acad Sci USA. 1992;89:12013–12017. doi: 10.1073/pnas.89.24.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flynn J L, Chan J, Triebold K J, Dalton D K, Stewart T A, Bloom B R. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flynn J L, Goldstein M M, Chan J, Triebold K J, Pfeffer K, Lowenstein C J, Schreiber R, Mak T W, Bloom B R. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 6.Flynn J L, Goldstein M M, Triebold K J, Sypek J, Wolf S, Bloom B R. J Immunol. 1995;155:2515–2524. [PubMed] [Google Scholar]
- 7.Cooper A M, Dalton D K, Stewart T A, Griffin J P, Russell D G, Orme I M. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper A M, Roberts A D, Rhoades E R, Callahan J E, Getzy D M, Orme I M. Immunology. 1995;84:423–432. [PMC free article] [PubMed] [Google Scholar]
- 9.Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile J F, Newpost M, Levin M, Blanche S, Fischer A, Casanova J L. N Eng J Med. 1996;335:1956–1961. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- 10.Glatman-Freedman A, Casadevall A. Clin Microbiol Rev. 1998;11:514–532. doi: 10.1128/cmr.11.3.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glatman-Freedman A, Martin J M, Riska P F, Bloom B R, Casadevall A. J Clin Microbiol. 1996;34:2795–2802. doi: 10.1128/jcm.34.11.2795-2802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casadevall A. Trends Microbiol. 1998;6:102–107. doi: 10.1016/s0966-842x(98)01208-6. [DOI] [PubMed] [Google Scholar]
- 13.Yuan R, Casadevall A, Oh J, Scharff M D. Proc Natl Acad Sci USA. 1997;94:2483–2488. doi: 10.1073/pnas.94.6.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dannenberg A M, Jr, Rook G A W. In: Tuberculosis: Pathogenesis, Protection and Control. Bloom B R, editor. Washington, DC: Am. Soc. Microbiol.; 1994. pp. 463–465. [Google Scholar]
- 15.Mukherjee J, Casadevall A, Scharff M D. J Exp Med. 1993;177:1105–1116. doi: 10.1084/jem.177.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers H W, Unanue E R. Infect Immun. 1993;61:5090–5096. doi: 10.1128/iai.61.12.5090-5096.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanson P E. Lab Med. 1994;25:520–525. [Google Scholar]
- 18.Catoretti G M, Becker H G, Key G. J Pathol. 1992;168:357–362. doi: 10.1002/path.1711680404. [DOI] [PubMed] [Google Scholar]
- 19.Chan J, Xing Y, Magliozzo R, Bloom B R. J Exp Med. 1992;175:1111–1122. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacMicking J D, North R J, LaCourse R, Mudgett J S, Shah S K, Nathan C R. Proc Natl Acad Sci USA. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackaness G B, Blanden R V, Collins F M. Am Rev Respir Dis. 1968;97:337–344. doi: 10.1164/arrd.1968.97.3.337. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee D, Khoo K H, McNeil M R, Dell A, Morris H R, Brennan P J. Glycobiology. 1993;5:497–506. doi: 10.1093/glycob/3.5.497. [DOI] [PubMed] [Google Scholar]
- 23.Jenkin C R, Rowley D. Bacteriol Rev. 1963;27:391–402. doi: 10.1128/br.27.4.391-404.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casadevall A. Infect Immun. 1995;63:4211–4218. doi: 10.1128/iai.63.11.4211-4218.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spector W G, Heesom N. J Pathol. 1969;98:31–39. doi: 10.1002/path.1710980105. [DOI] [PubMed] [Google Scholar]
- 26.Feldmesser M, Casadevall A. J Immunol. 1997;158:790–799. [PubMed] [Google Scholar]
- 27.Feldmesser M, Kress Y, Casadevall A. J Infect Dis. 1998;177:1639–1646. doi: 10.1086/515314. [DOI] [PubMed] [Google Scholar]
- 28.Antegon I, Cuturi M A, Trinchieri G, Perussia B. J Exp Med. 1998;167:452–472. doi: 10.1084/jem.167.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marsch C B, Lowe M P, Rovin B H, Parker J M, Liao Z, Knoell D L, Wewers M D. J Immunol. 1998;160:3942–3948. [PubMed] [Google Scholar]
- 30.Vossen A C T M, Tibbe G J M, Kroos M J, van de Winkel J G J, Benner R, Savelkoul H F J. Eur J Immunol. 1995;25:1492–1496. doi: 10.1002/eji.1830250603. [DOI] [PubMed] [Google Scholar]
- 31.Vecchiarelli A, Monari C, Retini C, Pietrella D, Palazzetti B, Pitzurra L, Casadevall A. Infect Immun. 1998;66:1244–1247. doi: 10.1128/iai.66.3.1244-1247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bayon Y, Alonso A, Sanchez Crespo M. J Immunol. 1997;159:887–894. [PubMed] [Google Scholar]
- 33.Fanger N A, Voigtlaender D, Liu C, Swink S, Wardwell K, Fisher J, Graziano R F, Pfefferkorn L C, Guyre P M. J Immunol. 1997;158:3090–3098. [PubMed] [Google Scholar]
- 34.Mozaffarian N, Berman J W, Casadevall A. J Leukocyte Biol. 1995;57:657–662. doi: 10.1002/jlb.57.4.657. [DOI] [PubMed] [Google Scholar]
- 35.Vecchiarelli A, Monari C, Retini C, Pietrella D, Palazetti B, Pitzurra L, Casadevall A. Eur J Immunol. 1998b;28:114–121. doi: 10.1002/(SICI)1521-4141(199801)28:01<114::AID-IMMU114>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 36.Armstrong J A, Hart P D. J Exp Med. 1975;142:1–16. doi: 10.1084/jem.142.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaible U E, Sturgill-Koszycki S, Schlesinger P H, Russell D G. J Immunol. 1998;160:1290–1296. [PubMed] [Google Scholar]
- 38.Selwyn P A. N Engl J Med. 1989;320:545–550. doi: 10.1056/NEJM198903023200901. [DOI] [PubMed] [Google Scholar]