

Abstract
The crystal structure of human carbonic anhydrase II (HCA II) obtained at 0.9 Å resolution reveals that a water molecule, termed deep water, Dw, and bound in a hydrophobic pocket of the active site forms a short, strong hydrogen bond with the zinc-bound solvent molecule, a conclusion based on the observed oxygen-oxygen distance of 2.45 Å. This water structure has similarities with hydrated hydroxide found in crystals of certain inorganic complexes. The energy required to displace Dw contributes in significant part to the weak binding of CO2 in the enzyme-substrate complex, a weak binding that enhances kcat for the conversion of CO2 into bicarbonate. In addition, this short, strong hydrogen bond is expected to contribute to the low pKa of the zinc-bound water and to promote proton transfer in catalysis.
The hydration of CO2 to produce bicarbonate and a proton is catalyzed by the carbonic anhydrases (CAs) and plays a significant role in a number of physiological processes including respiration, fluid secretion, and pH control. There are 14 human gene products classified as CAs including HCA II which is wide spread in tissues and heavily concentrated in red cells. The most efficient of these enzymes, including HCA II, proceed near diffusion control with kcat/Km for hydration at 108 M-1s-1(1, 2).
Our understanding of the steps in this catalysis is based in significant part on the structure of the active site revealed by x-ray crystallography studies. The first HCA II structures were determined to 2.0 Å resolution and identified the key features of the enzyme mechanism (3), whereas subsequent structures obtained between 2.3 and 1.1 Å resolution have focused on detailed understanding of the geometry about the zinc, orientations of the proton shuttle residue His64, and solvation of residues the active site (4-6). Recent structural analysis of HCA II at 0.9 Å resolution reported here allows enhanced interpretation with application to understanding the catalytic mechanism, in particular additional understanding of the role of solvent.
The active-site cavity of HCA II is conical and 15 Å deep having one side lined with predominantly hydrophobic residues and the other lined by hydrophilic residues. At the bottom of the cavity is a zinc ion coordinated in a tetrahedral geometry to three histidine residues (His94, 96, and 119) and a solvent ligand (Figure 1). A wide body of spectroscopic and kinetic data are consistent with a pKa near 7 describing the protolysis of the aqueous ligand of the metal forming zinc-bound hydroxide (1, 2). The mechanism of catalysis comprises nucleophilic attack of zinc-bound hydroxide on CO2, followed by transfer of a proton from zinc-bound water to solution to regenerate the active form (Figure 2). A network of apparently hydrogen bonded water molecules is observed in crystal structures extending from the zinc-bound solvent to the inwardly oriented proton shuttle residue His64 located about 8 Å from the metal (4, 5). This structure of ordered water molecules is likely closely related to viable pathways of proton transfer during catalysis (7, 8).
Figure 1.
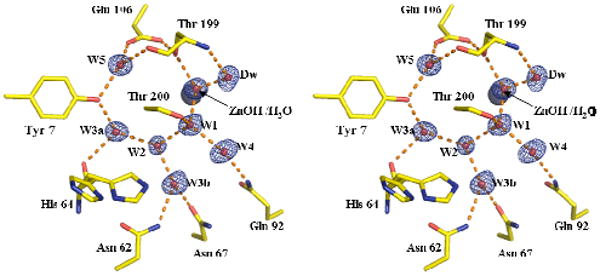
Stereoview of the active site of HCA II. The zinc is represented by a gray sphere and the oxygen atoms of water molecules as smaller red spheres. Dotted lines are presumed hydrogen bonds. Stick figures are selected amino acids of the active site with both the inward and outward orientations of His64 shown. The electron density 2Fo-Fc Fourier map is contoured at 2.0σ. This figure was created using PyMOL(28).
Figure 2.
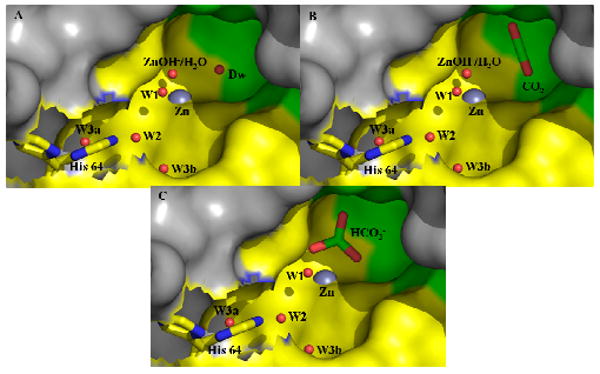
Catalytic mechanism, (A) the active site without bound CO2 showing the deep water; (B) the active site showing bound CO2 occupying the site of the deep water; (C) the binding of bicarbonate at the active site of T200H HCA II (23). Surface and stick representation; water molecules depicted as red spheres, the green shaded region are hydrophobic residues, and the yellow region hydrophilic residues. This figure was created using PyMOL(28).
The final refined 0.9 Å resolution model, 258 residues and 486 water molecules, was refined to an Rcryst of 12.5% and Rfree of 13.1%. A full description of the structure determination and data collection and refinement statistics is given in Table S1 (Supporting Information).
Of particular interest for this report is the structure of the apparently hydrogen bonded solvent water network that includes the zinc-bound solvent. This network emanates from the deep water (Dw) in the hydrophobic pocket formed in part by the side chains of Val121, Val143, Trp209, and Leu198 to the water molecules labeled W1, W2, W3a, W3b and W4 shown in Figures 1, 2, and 3. In crystal structures, this chain extends to but is not in hydrogen bond contact with the proton shuttle residue His64. The zinc-bound solvent appears to form a hydrogen bond with the side chain of Thr199, and the deep water molecule Dw appears to participate in hydrogen bonds with the backbone amide of Thr199 and with the zinc-bound water molecule. The mechanism of the proton transfer utilizing pathways such as this has been the subject of considerable investigations (2, 7-12).
Figure 3.
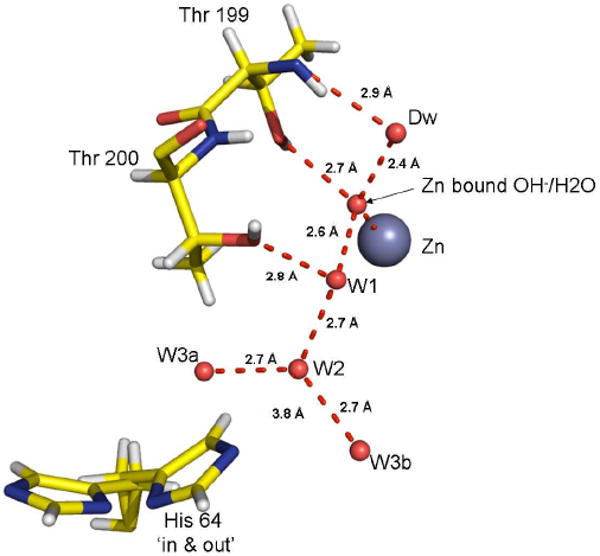
The ordered water network in the active site of HCA II. The zinc is represented by a gray sphere and the oxygen atoms of water molecules as smaller red spheres. Dotted lines are presumed hydrogen bonds with heavy atom distances given. Stick figures are selected amino acids of the active site with both the inward and outward orientations of His64 shown. This figure was created using PyMOL (28).
The current high resolution structure provides a clearer view of the solvation at the active site (Figure 1). The hydrogen bonds in this water network have distances typical of solvent water, with O-O distances near 2.7 to 2.9 Å. A more detailed picture of distances and bond angles involving active site solvent is provided in Figures S1 and S2 (Supporting Information). However, there is a short hydrogen bond with O-O distance estimated at 2.45 (±0.03) Å between Dw and the zinc-bound solvent (Figure 3). The crystallographic occupancy is near 100% for Dw, and both this water molecule and the zinc-bound solvent have B factors that are low (near 10 Å2) and close in value to the B factors of the surrounding amino acids. Analysis of the anisotropic thermal motions of Dw indicates that the bulk of its movement is perpendicular to the hydrogen bond with the zinc-bound solvent (Table of Contents figure).
Under specific and well described conditions, short hydrogen bonds involving water with O-O distances close to 2.4 Å have been observed (13). These are designated low barrier hydrogen bonds (LBHB) reflecting the low-barrier for hydrogen movement between the heteroatoms. Such LBHBs are usually observed in nonprotic solvents and involve closely matched values of pKa for the heteroatoms of the hydrogen bond (13).
The Dw is bound in a hydrophobic pocket of the active site. Moreover, with a solution pKa near 7.0, the zinc-bound solvent in the crystal structure is probably in large part zinc-bound hydroxide under the conditions of crystallization (pH 7.0)(14). This structure has similarities with the identification by crystallography of the LBHB of the hydrated hydroxide anion HOHOH− formed in the hydrophobic region between sheets of phenyl rings in trimethyl ammonium salts of tris (thiobenzohydroximato)-chromate(III)(15). In this case the O-O atom distance is 2.3 Å in a structure the authors describe as a central proton surrounded by two OH− groups. This is probably a good model for the observed LBHB in HCA II, in which the deep water is in a hydrophobic environment and likely involves the zinc-bound hydroxide.
Weak hydrogen bonds typical of water molecules in solution have a favorable enthalpy of formation near 5 kcal/mol; however, LBHBs can have such enthalpies near 15 - 25 kcal/mol (13). This has significance in the catalysis by HCA II since the binding of CO2 to its catalytically productive binding site displaces the deep water molecule Dw (Figure 2)(16, 17) and thus requires the cleavage of the LBHB contributing to the very weak binding of CO2 at this site. A dissociation constant for CO2 at its catalytic site in HCA II has been estimated at 100 mM measured by infrared spectroscopy (18, 19).
A tight binding of substrate at the reactive site is a disadvantage for catalysis by HCA II; it adversely affects its physiological function which requires it to enhance catalysis for maximum velocity of kcat = 106 s-1 and near diffusion-controlled second-order rate constants for hydration. In arguments elucidated by Fersht (20), the tight binding of substrate (without affecting the transition state) lowers the energy level of the substrate-enzyme complex thereby increasing the activation energy of kcat. By providing a thermodynamic well or pit that accumulates tightly-bound substrate, the rate of catalysis is decreased. For an enzyme that requires rapid catalysis like carbonic anhydrase, it is advantageous for substrate binding to be weak and the active site to remain largely unbound at physiological levels of substrate CO2. The concentration of CO2 in plasma for example is near 1 mM, the value of Km for hydration is 10 mM, and the estimated dissociation constant of CO2 is 100 mM. It appears that HCA II evolved weak substrate binding by having it displace the Dw which participates in a LBHB.
There is likely another role for the LBHB as it may contribute to the low pKa near 7 for the zinc-bound water molecule, the protolysis of which is enhanced using the energy of formation of the LBHB. In this aspect, the role of the LBHB has an analogy with the catalytic mechanism of liver alcohol dehydrogenase in which proton removal from the Zn-coordinated alcohol is promoted by forming a LBHB with Ser48 in the reactant state (21). The alkoxide then undergoes hydride transfer to generate product. In each case, forming the LBHB provides the energy to pump the proton to His64 in HCA II and to His51 in horse liver alcohol dehydrogenase (21).
It is unclear whether these arguments will apply in the dehydration direction as well for which the maximal catalytic rates are slower than in hydration (maximal steady-state constants are kcat/Km = 2 × 107 M-1s-1 and kcat = 0.6 μs-1 (22)). Crystal structures of bicarbonate bound at the active-site metal, the presumed catalytic site, have been obtained for the mutant of HCA II with Thr200 replaced by His (23), with Thr199 replaced with Ala (24), and for HCA II which Zn(II) is replaced by Co(II) (25). Although the orientation of the bound bicarbonate is somewhat different in each of these examples, in all three cases the binding of bicarbonate displaces the deep water. The dissociation constant of bicarbonate at the active site of HCA II is estimated near 100 mM by 13C NMR measurements (26) with a similar Ki value estimated by inhibition by bicarbonate of the esterase capacity of HCA II (27). The value of Km for dehydration is 32 mM (22), and concentration of bicarbonate in plasma is near 24 mM. However, the form of the enzyme that is catalytic in the dehydration direction contains zinc-bound water. This configuration is not comparable to a hydrated hydroxide, and a low barrier hydrogen bond will likely not be found in this case. Hence, at present we cannot make the argument that weak binding of bicarbonate is caused in part by the displacement of the deep water which participates in a LBHB.
This effect of the LBHB in catalysis to weaken substrate binding in HCA II is different than the effect shown in examples for which the formation of a LBHB not in the enzyme-substrate complex but in the transition state lowers the overall free energy of activation (13).
Supplementary Material
Acknowledgments
We thank Professor W. Wallace Cleland for helpful comments. This work was funded by grants from the National Institutes of Health (GM25154 D.N.S and R.M.) and the Thomas Maren Foundation (R.M.), and the MacCHESS grant (US NIH grant RR001646), by US DOE grant DE-FG02-97ER62443, and CHESS, which is supported by the US NSF and NIH-NIGMS through NSF grant DMR- 0225180.
Footnotes
Supporting Information Available: Text on the material and methods. Table of refinement statistics for the structure of human carbonic anhydrase II, a figure of bond lengths in the active site, and a figure of bond angles in the active site. This material is available free of charge via the Internet at http://pubs.acs.org. Coordinates are deposited in the Protein Data Bank with accession code 3KS3.
References
- 1.Lindskog S. Pharmacol Ther. 1997;74:1–20. doi: 10.1016/s0163-7258(96)00198-2. [DOI] [PubMed] [Google Scholar]
- 2.Silverman DN, McKenna R. Acc Chem Res. 2007;40:669–675. doi: 10.1021/ar7000588. [DOI] [PubMed] [Google Scholar]
- 3.Liljas A, Lovgren S, Bergsten PC, Carlbom U, Petef M, Waara I, Strandbe B, Fridborg K, Jarup L, Kannan KK. Nature-New Biology. 1972;235:131–137. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
- 4.Nair SK, Christianson DW. Journal of the American Chemical Society. 1991;113:9455–9458. [Google Scholar]
- 5.Fisher SZ, Maupin CM, Budayova-Spano M, Govindasamy L, Tu C, Agbandje-McKenna M, Silverman DN, Voth GA, McKenna R. Biochemistry. 2007;46:2930–2937. doi: 10.1021/bi062066y. [DOI] [PubMed] [Google Scholar]
- 6.Christianson DW, Fierke CA. Accounts of Chemical Research. 1996;29:331–339. [Google Scholar]
- 7.Maupin CM, McKenna R, Silverman DN, Voth GA. Journal of the American Chemical Society. 2009;131:7598–7608. doi: 10.1021/ja8091938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy A, Taraphder S. Journal of Physical Chemistry B. 2007;111:10563–10576. doi: 10.1021/jp073499t. [DOI] [PubMed] [Google Scholar]
- 9.Riccardi D, Konig P, Guo H, Cui Q. Biochemistry. 2008;47:2369–2378. doi: 10.1021/bi701950j. [DOI] [PubMed] [Google Scholar]
- 10.Cui Q, Karplus M. Journal of Physical Chemistry B. 2003;107:1071–1078. [Google Scholar]
- 11.Smedarchina Z, Siebrand W, Fernandez-Ramos A, Cui Q. Journal of the American Chemical Society. 2003;125:243–251. doi: 10.1021/ja0210594. [DOI] [PubMed] [Google Scholar]
- 12.Fisher SZ, Tu CK, Bhatt D, Govindasamy L, Agbandje-McKenna M, McKenna R, Silverman DN. Biochemistry. 2007;42:3803–3813. doi: 10.1021/bi602620k. [DOI] [PubMed] [Google Scholar]
- 13.Cleland WW. Biochemistry. 2000;39:1580–1580. [Google Scholar]
- 14.Avvaru BS, Arenas DJ, Tu CK, Tanner DB, McKenna R, Silverman DN. 2009 doi: 10.1016/j.abb.2010.07.010. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abudari K, Raymond KN, Freyberg DP. Journal of the American Chemical Society. 1979;101:3688–3689. [Google Scholar]
- 16.Domsic JF, Avvaru BS, Kim CU, Gruner SM, Agbandje-McKenna M, Silverman DN, Mckenna R. Journal of Biological Chemistry. 2008;283:30766–30771. doi: 10.1074/jbc.M805353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sjoblom B, Polentarutti M, Djinovic-Carugo K. Proc Natl Acad Sci USA. 2009;106:10609–10613. doi: 10.1073/pnas.0904184106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krebs JF, Rana F, Dluhy RA, Fierke CA. Biochemistry. 1993;32:4496–4505. doi: 10.1021/bi00068a004. [DOI] [PubMed] [Google Scholar]
- 19.Riepe ME, Wang JH. Journal of Biological Chemistry. 1968;243:2779–&. [PubMed] [Google Scholar]
- 20.Fersht A. Structure and Mechanism in Protein Science. W. H. Freeman & Co.; New York: 1999. [Google Scholar]
- 21.Ramaswamy S, Park DH, Plapp BV. Biochemistry. 1999;38:13951–13959. doi: 10.1021/bi991731i. [DOI] [PubMed] [Google Scholar]
- 22.Steiner H, Jonsson BH, Lindskog S. European Journal of Biochemistry. 1975;59:253–259. doi: 10.1111/j.1432-1033.1975.tb02449.x. [DOI] [PubMed] [Google Scholar]
- 23.Xue YF, Vidgren J, Svensson LA, Liljas A, Jonsson BH, Lindskog S. Proteins-Structure Function and Genetics. 1993;15:80–87. doi: 10.1002/prot.340150110. [DOI] [PubMed] [Google Scholar]
- 24.Xue YF, Liljas A, Jonsson BH, Lindskog S. Proteins-Structure Function and Genetics. 1993;17:93–106. doi: 10.1002/prot.340170112. [DOI] [PubMed] [Google Scholar]
- 25.Hakansson K, Wehnert A. J Mol Biol. 1992;228:1212–1218. doi: 10.1016/0022-2836(92)90327-g. [DOI] [PubMed] [Google Scholar]
- 26.Simonsson I, Jonsson BH, Lindskog S. European Journal of Biochemistry. 1979;93:409–417. doi: 10.1111/j.1432-1033.1979.tb12837.x. [DOI] [PubMed] [Google Scholar]
- 27.Steiner H, Jonsson BH, Lindskog S. Febs Letters. 1976;62:16–20. doi: 10.1016/0014-5793(76)80006-3. [DOI] [PubMed] [Google Scholar]
- 28.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; Palo Alto CA USA.: 2008. http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.