

Abstract
Expression of cyclins D1 (cD1) and D2 (cD2) in ventricular zone and subventricular zone (SVZ), respectively, suggests that a switch to cD2 could be a requisite step in the generation of cortical intermediate progenitor cells (IPCs). However, direct evidence is lacking. Here, cD1 or cD2 was seen to colabel subsets of Pax6-expressing radial glial cells (RGCs), whereas only cD2 colabeled with Tbr2. Loss of IPCs in cD2−/− embryonic cortex and analysis of expression patterns in mutant embryos lacking cD2 or Tbr2 indicate that cD2 is used as progenitors transition from RGCs to IPCs and is important for the expansion of the IPC pool. This was further supported by the laminar thinning, microcephaly, and selective reduction in the cortical SVZ population in the cD2−/−cortex. Cell cycle dynamics between embryonic day 14–16 in knock-out lines showed preserved parameters in cD1 mutants that induced cD2 expression, but absence of cD2 was not compensated by cD1. Loss of cD2 was associated with reduced proliferation and enhanced cell cycle exit in embryonic cortical progenitors, indicating a crucial role of cD2 for the support of cortical IPC divisions. In addition, knock-out of cD2, but not cD1, affected both G1-phase and also S-phase duration, implicating the importance of these phases for division cycles that expand the progenitor pool. That cD2 was the predominant D-cyclin expressed in the human SVZ at 19–20 weeks gestation indicated the evolutionary importance of cD2 in larger mammals for whom expansive intermediate progenitor divisions are thought to enable generation of larger, convoluted, cerebral cortices.
Introduction
The cerebral cortex arises through a precisely timed and orchestrated series of cell divisions to generate an astonishing array of neuronal and glial cell types, ordered in a highly stereotyped laminar organization (Takahashi et al., 1995a; Caviness et al., 2003; Tarui et al., 2005). Not only does progenitor proliferation set the size of the cerebral cortex, but it also determines cortical morphogenesis, whether brain contours will be smooth or convoluted, and whether the cerebral ventricles will be large or small (Chenn and Walsh, 2003; Kriegstein et al., 2006). Cortical divisions are subject to external factors and intrinsically programmed transcriptional subroutines that impact proliferation behavior of neural progenitors in a determination-stage-specific manner (Noctor et al., 2004, 2008; Englund et al., 2005; Hevner et al., 2006; Shen et al., 2006).
The cortical germinal epithelium was once considered relatively homogeneous, with neurogenesis occurring in the pseudostratified ventricular zone (VZ) and gliogenesis in the later arising subventricular zone (SVZ) or secondary proliferative population (Levison and Goldman, 1993; Takahashi et al., 1995b). It has since been established that neurons may arise from either the VZ or SVZ, from asymmetric neurogenic divisions (primarily in the VZ) that yield a neuronal daughter and a replenished progenitor, or from symmetric neurogenic divisions (primarily in the SVZ) in which both daughters are neurons (Noctor et al., 2008). Thus, the prevailing outcomes of cytokinesis in the VZ (asymmetric daughter cells) and SVZ (symmetric daughter cells) suggest differences in the molecular machinery implementing these divisions.
We showed that two G1-phase active cyclins, cyclin D1 (cD1) and cyclin D2 (cD2), are differentially expressed in neural progenitor cells of the VZ (cD1) and SVZ (cD2) (Glickstein et al., 2007a,b). Absence of either cyclin is associated with different histogenetic outcomes. Mice lacking cD2 are microcephalic despite normal body mass and parvalbumin-expressing interneurons are disproportionately missing in cerebellum and cerebral cortex (Huard et al., 1999; Glickstein et al., 2007b). In contrast, mice lacking cD1 are runted, but brain size is essentially normal, with no selective deficits in specific neuronal subsets (Chen et al., 2005; Glickstein et al., 2007b; and present study). Although cD2 expression in the SVZ cells suggested that cD2 may be important for the establishment of intermediate progenitors (Glickstein et al., 2007a), there is no direct evidence that cD2 is indeed expressed in transit amplifying progenitors or indication of its impact on cycling in either cortex or ganglionic eminence populations. Here, the relationships between cD1 or cD2 protein expression and radial glial cells (RGCs) or intermediate progenitor cells (IPCs) are explored. The data indicate that cD2 is critically important for establishing a full complement of IPCs, whereas cD1 expression is not found in this expansive population. cD2 deficiency alters cell cycle parameters in the VZ, primarily affecting G1- and S-phases, but cD1 deficiency leaves cell cycle measures unchanged. Thus, cD2 is a major determinant of brain size and is more essential than cD1 because of the ability of cD2 to promote amplifying divisions.
Materials and Methods
Animals
Mice were housed in climate-controlled Thoren units with a 12 h light/dark cycle. The cD2 gene was inactivated by disruption of exons I and II (Sicinski et al., 1996). The cD1 gene was inactivated by the deletion of exons I–III (Sicinski et al., 1995). Heterozygous cD1+/− and cD2+/− mice, maintained on a congenic C57BL/6J background, were bred to produce wild-type (WT) and homozygous null littermates for all studies. Tbr2 was conditionally inactivated in cortex starting at embryonic day 9.5 (E9.5) by Foxg1–Cre-mediated excision of Tbr2 exon 3 that was flanked by loxP (fx) sites (Sessa et al., 2008). Tbr2fx/+::Foxg1–Cre and Tbr2fx/fx mice were mated to produce Tbr2fx/fx::Foxg1–Cre conditional knock-out (CKO) and Tbr2fx/fx control sibling embryos harvested at E13 and E14.5 (kindly provided by C.-A. Mao and W. Klein, University of Texas, M. D. Anderson Cancer Center, Houston, TX). All procedures involving animals were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Genotyping
Genomic DNA was extracted from tail snips using standard protocols and examined by PCR-based genotyping using primers for WT and null alleles as published previously (Sicinski et al., 1995, 1996; Sessa et al., 2008).
Human tissues
Fetal brain tissue was obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders under contracts N01-HD-4-3368 and N01-HD-4-3383. Tissue was collected with proper consent and with the approval of ethics committees. There was no evidence of disease or developmental abnormality. Samples (n = 2) were obtained from Caucasian males at 19 and 20 gestational weeks after postmortem intervals of 1 and 4 h, respectively, and storage for 11–12 years. Tissues were paraffinized, coronally sectioned (10 μm), and immunostained as described below for mouse tissues.
Immunohistochemistry
Adult animals were anesthetized with ketamine/xylazine and perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer, pH 7.4. Embryo heads were drop fixed in 4% PFA overnight at 4°C. Brains tissues were processed (Tissue Tech 2000; Bayer Corp.) for paraffin embedding in the coronal plane. Brains were sectioned at 4–10 μm (embryo to adult), mounted on adhesive-coated capillary gap slides (Thermo Fisher Scientific), and immunostained on a TechMate 500 semiautomated stainer (Ventana Medical Systems) after antigen retrieval by steam in Reveal buffer, pH 5.9 (Biocare Medical) for 30 min and cooling for 10 min. Sections were treated with 3% H2O2 for 10 min, blocked in Sniper (Biocare Medical) for 30 min, and then incubated with primary antibody overnight at +4°C. Sections were then sequentially incubated in species/isotype-appropriate secondary antibody and peroxidase-conjugated tertiary antibody (Signet Rabbit or Murine kit; Covance) or Vectastain Elite kit (Vector Laboratories) for 30 min each, followed by reaction with 3,3-diaminobenzidine (DAB) (Covance). When comparing between experimental groups or across genotypes, slides were processed together, thereby minimizing variability.
Primary antibodies included the following: rabbit anti-cD1 (α-cD1) (1:500; RM-9104; LabVision), mouse α-cD2 (1:100; MS-221; LabVision), rabbit α-Tbr2 (1:1000; Millipore Corporation), rabbit α-cleaved caspase-3 (1:1000; Millipore Corporation), mouse α-Pax-6 (1:400; Developmental Studies Hybridoma Bank), rabbit α-Ki67 (1:1000; SP6; LabVision), mouse anti-bromodeoxyuridine (BrdU) (1:50; GE Healthcare), mouse α-p27 (1:10,000; Sigma-Aldrich), rabbit α-phosphorylated retinoblastoma (pRb) (1:200; Cell Signaling Technology), rabbit α-phosphohistone 3 (PH3) (1:1000; Millipore Corporation), mouse α-reelin (1:500; Millipore Corporation), rabbit α-Tbr1 (1:2000; courtesy of Robert F. Hevner, University of Washington, Seattle, WA), mouse α-Tuj1 (β-ΙΙΙ tubulin; 1:2000; Covance), and mouse α-neuronal-specific nuclear protein (NeuN) (1:5000; Millipore Corporation). A terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) kit (S7101; Millipore Corporation) was used for in situ labeling of nuclei with DNA strand breaks. The specificity of the α-cD2 and α-cD1 antibodies used here has been established previously (Glickstein et al., 2007a).
For dual labeling, standard reaction for cD1 or cD2 immunoperoxidase labeling was followed by Sniper block and incubation with primary antibodies against Tbr2 or Pax6 at +4°C overnight. After washing, sections were incubated in Alexa Fluor-conjugated secondary antibodies (1:500; Invitrogen) for 1 h, washed, and coverslipped with Vectashield (Vector Laboratories). At least 20 tissue sections from multiple embryos were examined to confirm or refute the coexpression of cDs with Pax6 or Tbr2 at E12.5, E14.5, and E16.5. Images of DAB-labeled nuclei were inverted and pseudocolored and subsequently overlaid with immunofluorescent images. Antibodies for BrdU and Tbr2, PH3, or Ki67 were combined for traditional dual-immunofluorescent labeling. Fluorescently labeled sections were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories) and immunoperoxidase-only labeled sections were counterstained with hematoxylin. Sections were photographed digitally at 4×, 10×, and 20× magnification using a SPOT camera (Diagnostic Instruments). Images were not modified in any way, except for adjustments of brightness and contrast.
BrdU pulse labeling
BrdU was administered (50 mg/kg, i.p.) to pregnant mice 1 or 24 h before harvest date to assess the S-phase labeling index and the proliferative fraction, respectively, as enumerated in Results. A cumulative BrdU labeling protocol was followed to determine the growth fraction (GF) (fraction of the cell population that is proliferative), to determine the time to reach the growth fraction (Tc − Ts), and, subsequently, to estimate the length of the cell cycle (Tc) and the duration of S-phase (Ts) and G1-phase (TG1) (Takahashi et al., 1995a). Pregnant mice were injected with BrdU (50 mg/kg, i.p.) starting at E14.5–E15 with survival times of 0.5 and 3.5 h (after single injection at t = 0 h), 6.5 and 9.5 h (after doses at t = 0, 3 h), 12.5 and 15.5 h (after doses at t = 0, 3, 6 h), and 18 h (after doses at t = 0, 3, 6, 9 h). Other mice were killed at 1.5, 2, and 2.5 h after BrdU injection to estimate the duration of G2-M-phase (TG2+M).
Quantification of cell cycle dynamics and statistics
All quantification was performed blind to genotype and using a standard field of view that consisted of 100 μm linear distance of the dorsomedial cortex at the mid-hemispheric level. Three fields of view for each case were averaged, and all groups contained sets of WT and null littermates (tissues from three or more litters were examined for each parameter). Genotype averages were compared using a Student's t test (p < 0.05 level of significance). As detailed below, some quantification paradigms used SVZ-located PH3-labeled nuclei to define the abventricular limit of the VZ equivalently in all genotypes.
Distribution of PH3-labeled nuclei.
PH3-immunopositive nuclei were counted in the VZ (those at the ventricular wall) or SVZ (those labeled cells positioned abventricularly, typically >70 μm from ventricle).
S-phase labeling index.
Tissues were labeled for BrdU and PH3 dual-immunofluorescence. The fraction of DAPI-labeled cells having BrdU labeling (1 h after BrdU application) was calculated for the VZ (ventricular wall to the diameter of one cell ventral to PH3-labeled cells) or the SVZ (tissue above the VZ).
Proliferative fraction.
Determined 24 h after BrdU injection, this was the fraction of BrdU-positive cells that dual immunolabel for Ki67. Thus, the “in-cycle fraction” represents the cells that remain in the cell cycle over a 24 h period after BrdU pulse.
The quiescent fraction.
The reciprocal of the in-cycle fraction, the “cycle-exited fraction” evaluates the cells labeled with BrdU only and not Ki67 and represents the proportion of cells leaving the cycle over a 24 h period after BrdU injection.
Cell cycle duration.
The average fraction of Nissl-labeled cells having BrdU labeling in the VZ (as defined above using PH3 labeling) was calculated for each group/genotype, and the linear least squares algorithm for linear regression analysis (Excel; Microsoft) was performed to estimate Tc − Ts and Ts for each genotype. This analysis was performed as described in detail previously (Takahashi et al., 1993, 1995a; Lu et al., 2007). The fraction of PH3-labeled cells having BrdU labeling in the VZ in dual-labeled sections was calculated, and the least squares algorithm was used to estimate TG2+M. TG1 was then deduced, and estimates of Tc, Ts, TG1, and TG2+M and their SEs [TG1 SE estimated as described previously (Lu et al., 2007)] were statistically compared using a Z-test statistic (significance at p < 0.05 if Z > 1.96 on two-sided test). Cell cycle parameters estimated here were in line with those reported previously for C57BL/6J mice (Hodge et al., 2004; Chakrabarti et al., 2007).
Proportion of Tbr2-immunopositive cells labeled with BrdU.
Tissue from the cumulative BrdU labeling experiment (pulses at t = 0, 3 h and harvest at t = 6.5 h) was labeled for BrdU and Tbr2 dual immunofluorescence. The fraction of all Tbr2-labeled cells having BrdU labeling was calculated.
Quantification of Tbr2- and Pax6-labeled cells.
Single- and dual-labeled nuclei were counted in Tbr2 and Pax6 dual-immunofluorescently labeled tissue sections in a field of view that consisted of 50 μm linear distance of the dorsomedial cortex at the mid-hemispheric level in three sections per subject, which were then averaged for each case.
Quantitative stereology
Two-dimensional and stereological counting methods were used to estimate neuron numbers and density in adult cD2−/−, cD1−/−, and WT littermates using the optical fractionator (West et al., 1991) and Stereoinvestigator software (MicroBrightField). Nissl-labeled neurons in the somatosensory barrel field cortex of 40 μm sections (collected from fixed brains using a sliding microtome) were assessed by morphological criteria that characterize neuronal nuclei (Peters et al., 1991) and were counted in superficial (layers II–III) or deep (layers IV–VI) cortical layers. The barrel fields were analyzed in one hemisphere for each section in the series in which it was present (6–8 sections collected every 200 μm). The barrel fields were easily identifiable by the densely packed cell columns of granule cells in layer IV characteristic of this cortical region and as described previously (Glickstein et al., 2005). The medial and lateral limits included only those regions of somatosensory cortex having clearly visible barrels. Optical dissector frame and counting grid sizes of 30 and 250 μm2, respectively, permitted systematic random sampling of three to five neurons within an 8 μm depth focusing range for each sampling field and >200 total neurons for each subregion per case. Intra-sample coefficients of error (Schmitz and Hof, 2000) were always <0.05 and were equivalent across genotypes. All regions were sampled at 63× magnification in Koehler illumination conditions. The volume of the different laminar domains of interest in each subregion was estimated using the Cavalieri principle. The estimated total neuron numbers were expressed as the density per subregion, thereby controlling for the reduced brain volume in cD2−/− mice. cD2−/− and cD2+/+ littermates (from three litters) were examined in adulthood (8–10 weeks) and cD1−/− and cD1+/+ littermates (from four litters) were examined at postnatal day 21 attributable to premature death of cD1−/− mice thought to be a result of starvation (Sicinski et al., 1995). In WT mice, the average barrel field volumes were 1.58 ± 0.05 mm3 (8–10 weeks) and 1.50 ± 0.14 mm3 (3 weeks), and the average estimated number of neurons was 2.73 × 105 ± 1.8 × 104 (8–10 weeks) and 2.67 × 105 ± 2.6 × 104 (3 weeks). The diameters of 20 randomly selected neurons per case were measured to compare neuronal size between genotypes, and no differences in neuronal diameter between genotypes in any group were detected. Genotype averages were compared using a Student's t test (p < 0.05).
Results
Aberrant development of cyclin D2 (cD2−/−) but not cyclin D1 (cD1−/−) null neocortex
Developmental defects in mice deficient for cD2 result in microcephaly and selective parvalbumin interneuron deficits in forebrain and cerebellum (Huard et al., 1999; Glickstein et al., 2007b). In contrast, mice lacking cD1 are normocephalic and have no interneuronopathies (Chen et al., 2005; Glickstein et al., 2007b). The volume and neuronal packing density within the somatosensory barrel field cortex was stereologically analyzed in cD2−/−, cD1−/−, and WT littermates. The cortex was thinner in cD2−/− mice compared with cD2+/+ (Fig. 1A, B), and volume reductions were greater in the superficial (↓42%) than in the deeper (↓31%) cortical layers (Fig. 1E). In contrast, the volume of cD1−/− barrel field cortex was only modestly reduced with no exceptional shrinkage in superficial cortical layers (Fig. 1C–E). Neuronal packing density was comparable in cD2−/− and cD2+/+; therefore, glutamatergic neuron numbers must be diminished to account for the loss in volume in cD2 nulls, because they make up the great majority of cortical neurons. Interneurons make up only a small fraction of the cortical cell population, and interneuron loss alone would not be sufficient to account for a marked cortical volume reduction (Glickstein et al., 2007b). In contrast, neuronal packing density in cD1−/− was mildly elevated relative to cD1+/+ mice (Fig. 1F). Therefore, although cD1 loss was primarily compensated for developmentally, cD2 deficiency was not. Moreover, the considerable deficit in superficial cortical layers suggested that cD2 was needed to maintain the progenitor pool and/or support later neurogenic events.
Figure 1.
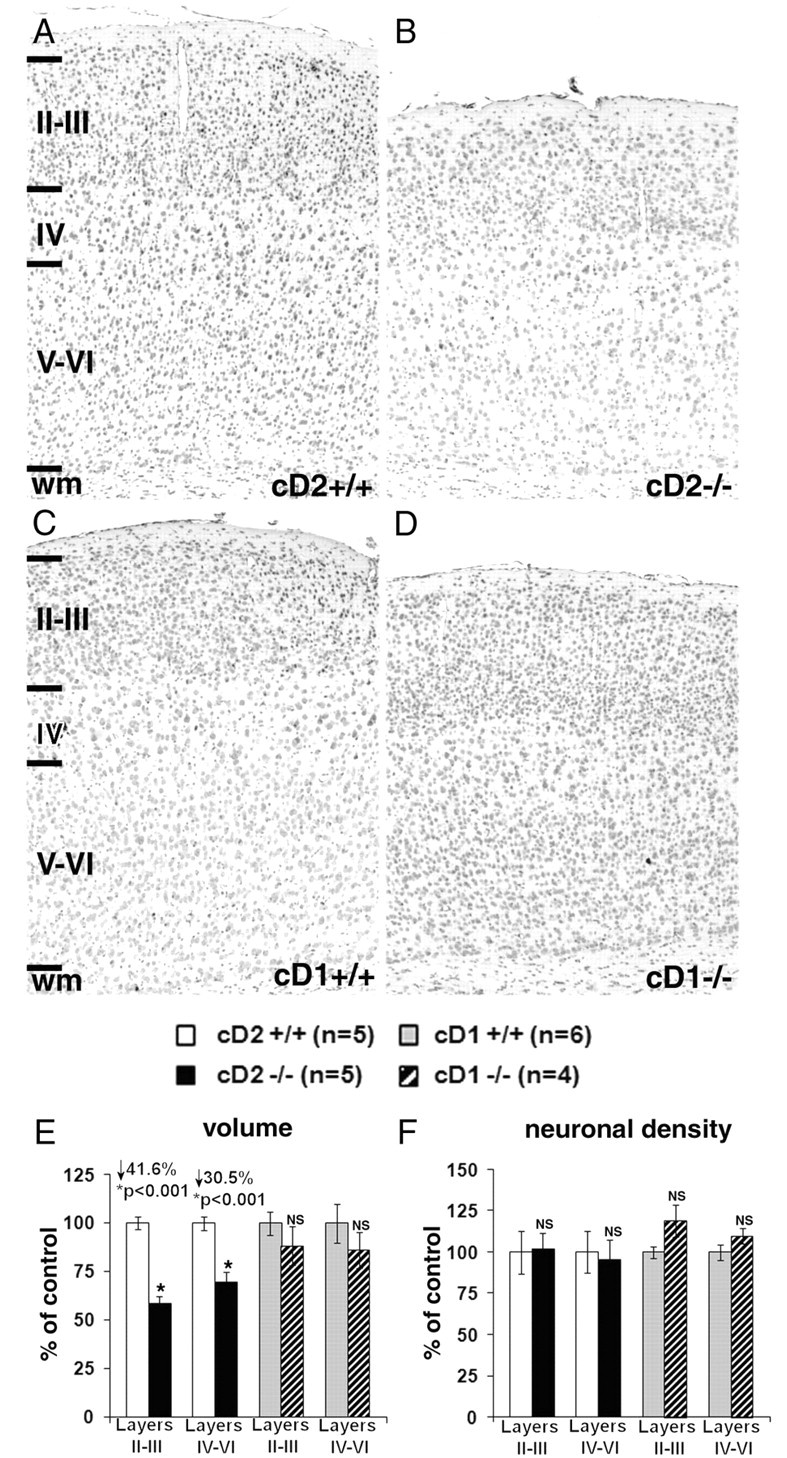
Cortical volume and neuronal density in the somatosensory barrel field cortex of cD2−/−, cD1−/−, and wild-type mice. A–D, Nissl-stained sections (10 μm) show decreased cortical thickness in both superficial (II–III) and deep (IV–VI) layers of 8-week-old cD2−/− (B) compared with littermate cD2+/+ (A). Cortical thickness of 3-week-old cD1−/− (D) is only slightly reduced compared with littermate cD1+/+ (C). E, Somatosensory cortical volume is significantly reduced in cD2−/− compared with cD2+/+ (layers II–III > IV–VI) and unchanged in cD1−/− relative to cD1+/+ mice. F, Neuronal density is unchanged in cD2−/− compared with cD2+/+ cortex despite reduced volume and is mildly and nonsignificantly increased in cD1−/− relative to cD1+/+. Volumes and densities in cD2−/− and cD1−/− are expressed as percentage of control (cD2+/+ and cD1+/+, respectively; mean ± SEM). wm, White matter.
Selectively disturbed cell cycle regulation in cD2−/− embryonic neocortex
Cyclins D1 and D2 have distinct expression patterns in the embryonic forebrain with limited overlap (Glickstein et al., 2007a). Across the neurogenic period, cD1 cells were mainly localized to the VZ, whereas cD2-labeled cells were more prevalent in the SVZ (Fig. 2A–F). In cD2−/− mice, there were fewer cD1-immunoreactive nuclei abventricularly with paler labeling of nuclei at the ventricular wall (Fig. 2B). In cD1−/− cortex, the distribution of cD2-labeled nuclei was more extensive in the VZ relative to that observed in cD1+/+ (Fig. 2E). In WT, these labeling patterns were consistent in nearly all cortical regions except rostromedial (nascent cingulate) cortex, in which the SVZ is less distinct at this age and cD2 immunoreactivity was less apparent. Because of the SVZ-biased expression pattern of cD2, the remaining analyses were focused on the E14.5 dorsomedial cortex (mid-hemispheric level), in which the SVZ is well established, although other times in the neurogenic period were examined as noted.
Figure 2.
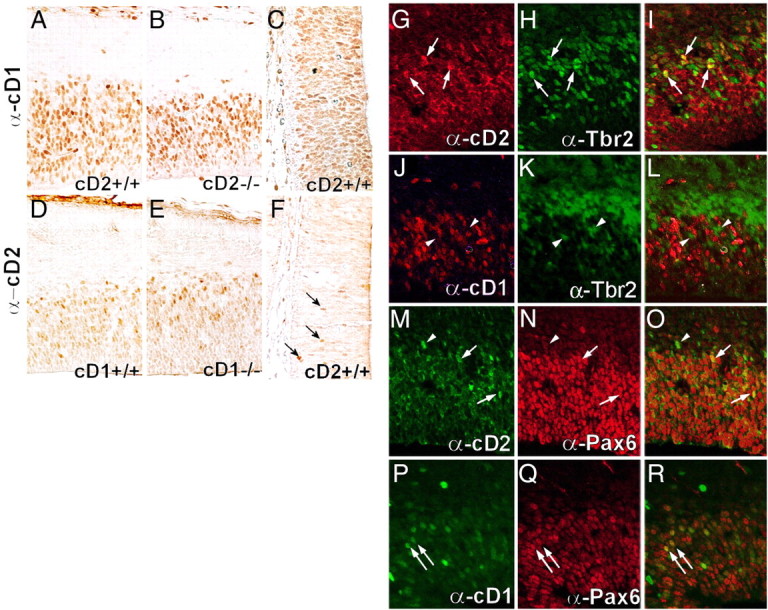
The expression of cD2 is characteristic of intermediate progenitor cells in the cortex. A–C, cD1-immunolabeled cells are predominantly in the VZ in the dorsomedial cortex (at mid-hemisphere) in WT mice at E14.5 (A, C). cD1 labeling in the SVZ and near the ventricular wall is reduced in cD2−/− mice (B) compared with cD2+/+ (A). cD1 immunolabeling is abundant in the rostromedial cortex of cD2+/+ mice (C). D–F, Labeling for cD2 is restricted to abventricular regions of dorsomedial cortical proliferative zone in cD1+/+ (D) and cD1−/− (E) mice and is just detectable in cD2+/+ rostromedial (cingulate) cortex (F), in which the SVZ is less prominent at this age. Arrows identify cD2-immunopositive nuclei in the adjacent septal cortex. G–L, The expression of cD2 (G) and Tbr2 (H) overlap (I) in many nuclei in the cortex. However, cD1 (J) and Tbr2 (K) immunolabeling mark different nuclei (L). M–R, The expression of cD2 (M) or cD1 (P) and Pax6 (N, Q) overlap (O, R). Arrows identify nuclei that coexpress markers, and arrowheads denote non-colabeled cells.
Cortical progenitor specification toward glutamatergic neurons proceeds from Pax6 to Tbr2 expression (Hevner et al., 2006; Quinn et al., 2007). In the developing cortex, Pax6-expressing radial glia can divide asymmetrically to yield an RGC and an early SVZ progenitor, which is thought to undergo terminal symmetric division (Noctor et al., 2004; Götz and Barde, 2005; Noctor et al., 2008). The transition to intermediate progenitor (transit amplifying) cells is characterized by dwindling expression of Pax6 and burgeoning expression of Tbr2 (Englund et al., 2005; Hodge et al., 2008; Pontious et al., 2008). There is not an abrupt switch from Pax6+ to Tbr2+ expression but rather a gradient, with a subset of cells producing both Pax6 and Tbr2 simultaneously. This may account for why conditional inactivation of Tbr2 starting at E9.5 affects both superficial and deep cortical layers, although outer layers are more severely affected (Sessa et al., 2008). Nuclei expressing cD2, but not cD1, also expressed Tbr2 (Fig. 2G–L), indicating that cD2 was selectively used in IPCs. Both cD2 and cD1 immunolabeling overlapped with Pax6 immunofluorescence (Fig. 2M–R), which allowed for both cDs to have a role in radial glia. Alternatively, cD2 and Pax6 dual-labeled nuclei might represent cells that were about to or had just become intermediate progenitors, which themselves comprise a minority of cells in the VZ (Hevner et al., 2006; Noctor et al., 2008). Whether nuclei in transition (i.e., those that coexpressed Pax6 and Tbr2) selectively used cD2 was explored, but conclusions regarding triple labeling could not be drawn because of the transient nature of cyclin expression (Sherr, 1994; Sakaue-Sawano et al., 2008) and confidence in the detection of slight immunoreactivity. Notably, no cD1 and Tbr2 colabeled nuclei were observed in cD2−/− mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), which suggested that cD1 could not compensate for cD2 in intermediate progenitor cell cycling.
Although their population was reduced, cD2 was not an absolute requirement for the emergence of glutamatergic neurons, which were present in the cortices of cD2 mutants. However, in E14.5 cortical neuroepithelium lacking cD2, numbers of Pax6+, Tbr2+, and Pax6/Tbr2 dual-labeled cells were significantly reduced compared with wild type (Fig. 3A, B, E) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In contrast, embryonic cortex lacking cD1 showed no reduction and indeed tended toward increased numbers of Pax6+, Tbr2+, or Pax6/Tbr2-labeled cells compared with normal siblings (Fig. 3C–E). Despite the differences in cell numbers in the cD2−/− embryos, the proportions of Pax6+, Tbr2+, and Pax6/Tbr2 dual-labeled cells were essentially the same in wild-type, cD2−/−, and cD1−/− brains (Fig. 3F) and compared well with the distribution observed previously in wild-type mice (Englund et al., 2005). Therefore, loss of cD2 reduces Pax6+, Pax6+/Tbr2+ dual-labeled, and Tbr2+ progenitors proportionately, whereas cortical progenitor numbers are unchanged by the loss of cD1 expression. If so, the numbers of intermediate progenitors (Tbr2+ cells that pulse label with BrdU) should also be diminished in cD2 nulls. This latter hypothesis was confirmed by significant reductions (↓38%) in the fraction of Tbr2 cells that dual labeled with BrdU 6.5 h after the application of BrdU in cD2−/− mice (Fig. 3G); this pulse duration (two pulses were administered 3.5 and 6.5 h before harvest) allowed for labeling of transitioning cells.
Figure 3.
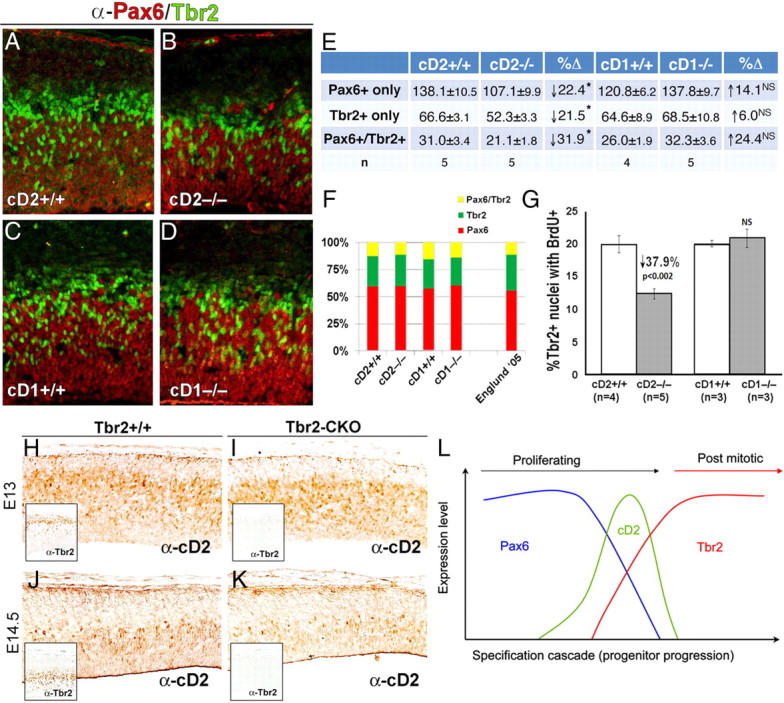
Evidence that cD2 functions in cortical progenitors transitioning from radial glia into IPCs. A–E, Pax-6- and Tbr2-immunofluorescent cell numbers are significantly reduced in cD2−/− (A) compared with cD2+/+ mice (B) and are equivalent in cD1+/+ (C) and cD1−/− (D) littermates. E, Quantification of Pax6+, Tbr2+, and dual-labeled Pax6+/Tbr2+ cell numbers in the E14.5 neuroepithelium. *p < 0.05. NS, Not significant. F, Proportion of Pax6, Tbr2 single, and dual-labeled cells in E14.5 neuroepithelium. n = 4–5 embryos per group. G, The number of intermediate progenitors, identified as Tbr2 cells dual immunolabeled with BrdU immunoreactivity (BrdU-IR; administered 3.5 and 6.5 h before harvest) is significantly reduced in cD2−/− compared with cD2+/+ and cD1+/+ and unchanged in cD1−/− mice. H–K, Appearance of anti-cD2 immunostaining (α-cD2) in control (H, J) and Tbr2-CKO (I, K) embryonic cortex at E13 and E14.5. L, Relative timing of Pax6, cD2, and Tbr2 expression based on results in Figures 2 and 3. cD2 spans the temporal progression of cortical progenitors from Pax6 to Tbr2 expression, as cells transition from radial glial cells to intermediate progenitors. Tbr2 expression continues beyond cD2, because cD2 is detected only in proliferative cells (Glickstein et al., 2007a) and Tbr2 persists briefly in postmitotic progenitors (Hevner et al., 2006).
The proportional loss of Pax6+ only, Tbr2+ only, and Pax6+/Tbr2+ cells in cD2 null embryos raises the question as to where cD2 is induced during the temporal progression of cortical progenitor development. Gene expression arrays validated by real-time PCR studies have shown previously that, in the absence of Pax6, E15 mouse cortex displays a twofold induction of cD2 compared with controls (Holm et al., 2007). Thus, Pax6 is not required to induce cD2 expression in cortical progenitors. We therefore tested whether Tbr2 expression is required for cD2 expression to appear in embryonic cortex (Fig. 3H–K). Conditional inactivation of Tbr2 in Tbr2fx/fx::Foxg1–Cre (Tbr2-CKO) embryonic cortex was compared with littermate Tbr2fx/fx (Tbr2+/+) controls at E13 and E14.5. Immunostaining for cD2 was equivalent to control at E13 but appeared to label fewer cells in the E14.5 cortex. This cD2 pattern in theTbr2-CKO parallels an enhanced cell cycle exit documented in the conditional Tbr2 mutant (Arnold et al., 2008; Sessa et al., 2008) and suggests an earlier peak in the transitioning population. Diminished cD2 expression in the E14.5 Tbr2-CKO could result if Tbr2 promoted cD2 expression or simply because, as shown previously, by this age, there were fewer basal progenitor cells to express cD2 (Arnold et al., 2008; Sessa et al., 2008). Thus, although it could not distinguish whether Tbr2 has a negative or positive modulatory effect on cD2, the data clearly showed that Tbr2 expression was not required for cD2 expression in cortical progenitors. Together, the data in Figures 2 and 3 positioned the timing of cD2 expression in cortical progenitors as they transition from Pax6+ RGCs into IPCs (Fig. 3L).
To understand the impact of cD2 deficiency on the cell cycle, three G1-S regulatory proteins, the cyclin-dependent kinase inhibitors p27-Kip1 and p57-Kip2, and phosphorylated (Ser807/811) retinoblastoma (pRb) were examined in the cortex of cD2−/−, cD1−/−, and WT littermates (Fig. 4). No p57-immunoreactive nuclei were observed in the cortical plate (data not shown), suggesting the importance of p27 in the cortical wall during neurogenesis. Compared with cD2+/+ embryos, p27 immunoreactivity was elevated in cD2−/− cortex across the neurogenic period (Fig. 4A,B,E,F,I,J), whereas p27 was reduced in the SVZ/VZ of cD1−/− embryos compared with cD1+/+ or cD2+/+ tissues (Fig. 4M,N,Q,R). pRb immunolabeling was reduced in cD2−/−, most notably in the SVZ (Fig. 4C,D,G,H,K,L), and unchanged in cD1−/− (Fig. 4O,P,S,T) from E12.5 through E16.5. The increased expression of p27 and decreased expression of pRb in cD2−/− cerebral wall were consistent with slower progression through G1-phase in the absence of cD2. Overexpression or knockdown of Rb and p27 has selective effects on cell cycle duration in G1-S-phase and neuron production (Sherr, 2000; Geng et al., 2001; Mitsuhashi et al., 2001; Ferguson et al., 2002). The immunolabeling results presented here indicated that cD1 and cD2 are poised to differentially regulate cell cycle progression and exert distinct effects on cortical development.
Figure 4.
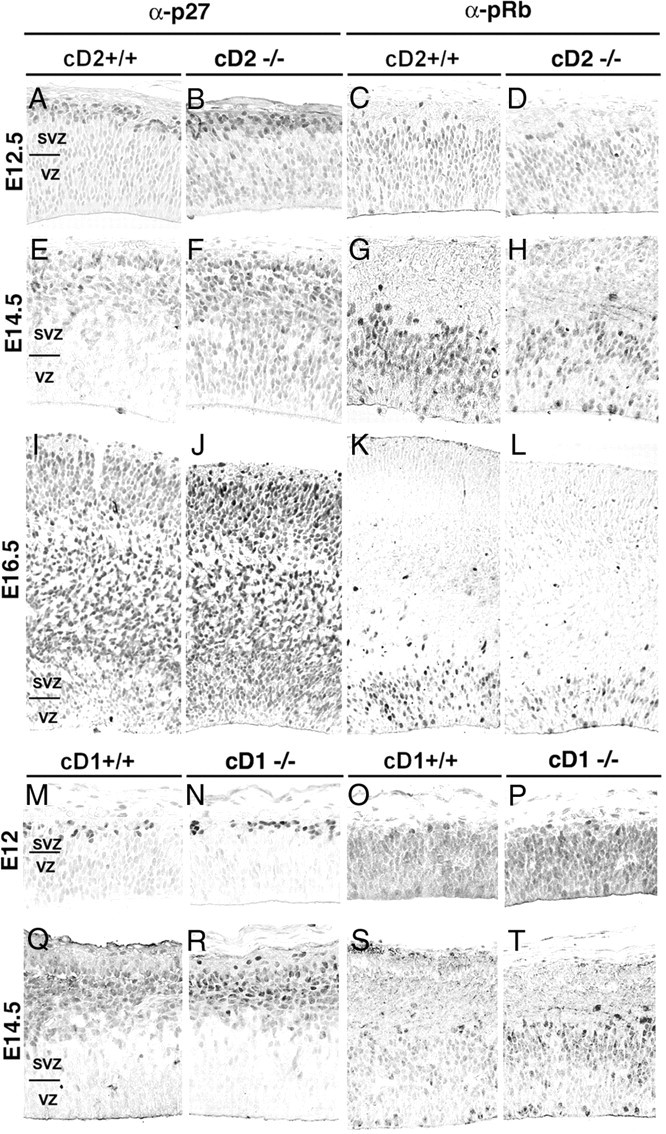
Cdk-inhibitor p27 and cD-Cdk effector retinoblastoma (pRb) in the cortex of cD2−/− and cD1−/− mice and WT littermates. A–L, The labeling intensity of p27 is increased in cD2−/− mice (B, F, J) compared with cD2+/+ (A, E, I) at early and later ages in the neurogenic period. Nuclei immunostained for pRb were diminished in abventricular cortical regions in cD2−/− (D, H, L) relative to cD2+/+ (C, G, K) across the neurogenic period. M–T, Immunolabeling for p27 was paler in the ventricular zone of cD1−/− (N, R) compared with WT (M, Q) at all ages examined. Labeling for pRb was unchanged in cD1−/− (P, T) compared with WT (O, S). Labeling was examined in three to five cases per genotype.
The expression of cell cycle markers was compared in mutant and WT cortex. In cD2−/− mice, numbers of PH3-positive, M-phase nuclei were unchanged at the ventricular surface at E12.5, E14.5, and E16.5 but were substantially reduced (as much as −58%) abventricularly at E12.5 and in the SVZ at E14.5 (Figs. 5A,B,E,F,I,J, 6A). In contrast, PH3-labeled cell numbers were unaffected in cD1−/− (Figs. 5M,N, 6A). Ki67 labeling (S-G2-M-phase cells) was also reduced abventricularly in cD2−/− at E12.5, E14.5, and E16.5 (Fig. 5C,D,G,H,K,L) but was mildly increased in cD1−/− (Fig. 5O, P). Thus, the proliferating progenitor pool was diminished in the cD2−/− cortex, notably in the SVZ. Despite this reduction in the cD2−/− progenitor pool, no qualitative changes with respect to wild-type siblings were observed at these embryonic ages in the distribution of postmitotic cells expressing Tbr1, a transcription factor expressed in glutamatergic neurons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) or reelin (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), expressed in Cajal–Retzius neurons. Thus, the output of postmitotic neurons from the neuroepithelium was likely increased in the early-mid neurogenic period. TUNEL and activated caspase-3 immunolabeling detected no differences in apoptosis among cD2−/−, cD1−/−, and WT cortices during the neurogenic period (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) or postnatally (Glickstein et al., 2007b).
Figure 5.
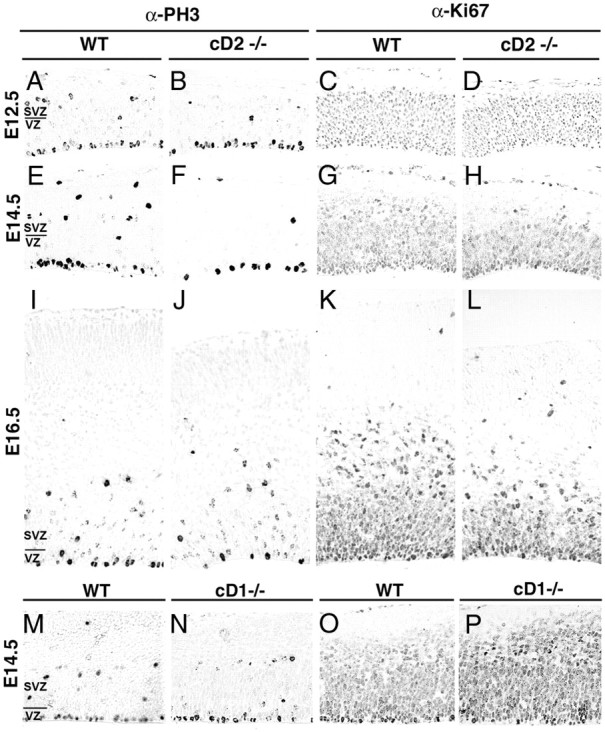
Proliferation markers PH3 and Ki67 are altered in the cortex of cD2−/− relative to cD1−/− and WT littermates. A–L, The numbers of PH3-labeled M-phase nuclei are reduced in the SVZ but unchanged in the VZ of cD2−/− (B, F, J) compared with cD2+/+ (A, E, I) at E12.5 and E14.5 but not at E16.5. Numbers of Ki67-labeled cells (S-G2-M-phases) are also reduced in the SVZ of cD2−/− (D, H, L) compared with cD2+/+ littermates (C, G, K) across the neurogenic period. M–P, The numbers of PH3- and Ki67-immunolabeled nuclei are unchanged in cD1−/− mice (N, P) compared with cD1+/+ (M, O). Labeling was compared in three or more cases per genotype.
Figure 6.
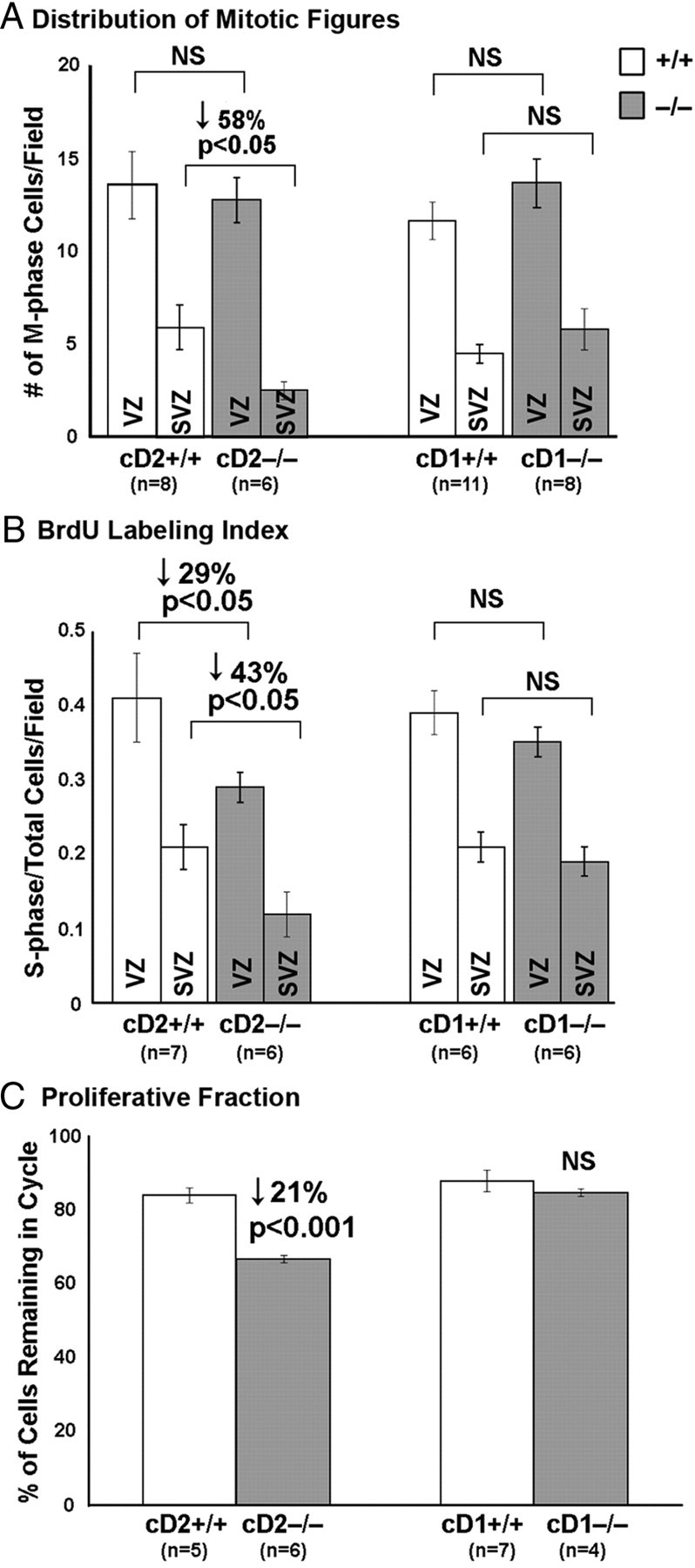
Reduced proliferation and early progenitor cell cycle exit is observed in cD2−/− cortex at E14.5. A, Fewer M-phase (PH3-immunopositive) nuclei in the SVZ but not the VZ of cD2−/− compared with cD2+/+. Equivalent numbers of M-phase nuclei are in the VZ and SVZ of cD1+/+ and cD1−/− mice. B, BrdU (1 h) pulse labels fewer S-phase nuclei in cD2−/− mice. The proportion of total (DAPI) nuclei labeled by anti-BrdU is significantly reduced in the VZ and SVZ of cD2−/− compared with cD2+/+ mice and is unchanged in cD1−/−. C, Dual immunolabeling with BrdU and Ki67 (to label S-G1-M phase cells) identifies those cells still in cycle 24 h after the application of BrdU (proliferative fraction). The fraction of dual-labeled cells remaining in cycle is decreased in cD2−/− compared with cD2+/+ mice and is unchanged in cD1−/−. This suggests that progenitors are prematurely depleted in cD2−/− mice. Field is standard field of view.
Cell cycle dynamics were further explored (Fig. 6, Tables 1, 2) (supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material). At E14.5, the number of PH3-labeled, M-phase cells in the VZ did not significantly differ in cD2−/− and cD1−/− compared with wild-type siblings (Fig. 6A). However, compared with WT and cD1−/− mice, the number of abventricular M-phase cells was sharply decreased in the cD2 nulls, showing almost 60% fewer PH3+ cells in the SVZ but no significant decrease in VZ mitoses (Fig. 6A). In addition, fewer BrdU pulse-labeled, S-phase nuclei (1 h after BrdU administration) were present in both the VZ and the SVZ in cD2 with respect to either wild-type controls or cD1 nulls (Fig. 6B) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Compared with wild type, the S-phase labeling index was reduced more severely in the cD2−/− SVZ than VZ (43 vs 29%) (Fig. 6B). Therefore, although cell cycle dynamics were affected in both VZ and SVZ compartments of cD2 nulls, the SVZ was more severely impacted by the loss of cD2 expression.
Table 1.
Duration of cell cycle phases in the VZ of cD2−/−, cD1−/−, and WT littermates (cD2+/+, cD1+/+)
| cD2+/+ (n = 37) | cD2−/− (n = 38) | cD1+/+ (n = 38) | cD1−/− (n = 36) | |
|---|---|---|---|---|
| GF (%) | 92.5 ± 0.3 | 91.6 ± 2.1 | 91.7 ± 4.0 | 92.6 ± 0.7 |
| Tc (h) | 22.1 ± 0.9 | 23.8 ± 0.8 | 22.8 ± 1.4 | 23.1 ± 1.7 |
| TG2+M (h) | 2.4 ± 0.1 | 2.4 ± 0.1 | 2.4 ± 0.1 | 2.4 ± 0.2 |
| TG1 (h) | 12.2 ± 0.6 | 16.5 ± 0.6* | 12.4 ± 0.9 | 12.4 ± 1.1 |
| Ts (h) | 7.5 ± 0.3 | 4.8 ± 0.2* | 8.0 ± 0.5 | 8.3 ± 0.6 |
| Model fit: SSR/MSE | 0.275/0.001 | 0.234/0.001 | 0.254/0.004 | 0.253/0.006 |
Data represent estimated parameter ± SE. SSR, Sum of squared residuals; MSE, mean squared error. No significant differences between cD1+/+ and cD1−/− or cD2+/+ estimates.
*p < 0.05; z-statistic, 7.98 (Ts), −5.1 (TG1) cD2+/+ versus cD2−/−.
Table 2.
Summary of G1-modulator expression and cell cycle analyses in cD2−/− and cD1−/− cortex compared with WT littermates
| α-cD1 | α-cD2 | α-Tbr2/Pax6 | α-p27 | α-pRb | #M-phase | S-phase labeling | In-cycle fraction | Exited-cycle fraction | Cell cycle duration | |
|---|---|---|---|---|---|---|---|---|---|---|
| cD2−/− | ||||||||||
| VZ | ↑ | ↓ | ↑↑ | ↓ | ↔ | ↓ | ↓↓ | ↑↑ | ↑Tc, ↑TG1, ↓Ts | |
| SVZ | ↔ | ↓ | ↑↑ | ↓ | ↓ | ↓↓ | ↓↓ | ↑↑ | ↑Tc, ↑TG1, ↓Ts | |
| cD1−/− | ||||||||||
| VZ | ↑↑ | ↔ | ↓ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | |
| SVZ | ↑ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ |
To explore whether there was an enhanced rate of terminal divisions to account for depletion of the progenitor pool in the cD2−/− cortex, we measured the fraction of BrdU+ progenitors expressing Ki67 and therefore remaining in cycle 24 h after BrdU administration. The in-cycle fraction was reduced in cD2−/− but was unchanged in cD1−/− compared with WT at all times examined (reductions in the in-cycle fraction: E16.5 ≫ E12.5, E14.5) (Fig. 6C) (supplemental Table 2, available at www.jneurosci.org as supplemental material). Conversely, the cycle-exited fraction was increased, indicating that more progenitor cells exited the cell cycle in cD2−/− than in WT or cD1−/−, consistent with premature terminal differentiation of progenitors in the absence of cD2 but not cD1. The expression of neuroblast and neuron markers Tuj1 and NeuN, which encroach on the VZ in cD2−/− compared with WT and cD1−/−, further supported this conclusion (supplemental Fig. 6, available at www.jneurosci.org as supplemental material).
To further examine neocortical cell cycle dynamics in the absence of D2 or D1 cyclins, cell cycle phase durations were analyzed in cD2 and cD1 null mutants (Table 1). Measuring the duration of individual phases of the cell cycle is not possible in the SVZ (Takahashi et al., 1995b). However, in the VZ, the length of the cell cycle phases was estimated between E14.5 and E15.5 in cD2−/−, cD1−/−, and WT using a cumulative BrdU pulse-labeling paradigm (Takahashi et al., 1995a). There were no significant differences in the GF between genotypes, which supported that delayed migration in any genotype did not unduly influence results. The duration of each phase did not significantly differ in cD1−/− compared with cD1+/+ or cD2+/+. In contrast, the duration of G1 (TG1) was substantially prolonged and the duration of S phase (Ts) was significantly reduced in cD2−/− tissues (Table 1). As a result, the duration of the cell cycle (Tc) was mildly yet nonsignificantly increased in cD2−/− VZ. The prolonged G1-phase duration (Table 1) was consistent with the observation of enhanced cell cycle exit in Figure 6C and supplemental Table 2 (available at www.jneurosci.org as supplemental material), because lengthening of G1 has been associated with progenitor exit from the cell cycle (Calegari and Huttner, 2003; Tarui et al., 2005).
cD2 is the predominant cyclin D in the human SVZ
The SVZ is thought to serve a fundamental role in the massive expansion of the cerebral hemispheres, the development of upper cortical layers, and, potentially, in gyrification in the second trimester of human fetal gestation (Kriegstein et al., 2006). We explored whether cD2 was also the predominant D-type cyclin in the human SVZ at 19 gestational weeks. At this age, the VZ is marginal with respect to the substantially expanded SVZ, which is bisected into an inner and outer SVZ by incoming fibers (Zecevic et al., 2005) (Fig. 7B,C). cD1 showed scant labeling throughout cerebrum, and the few nuclei observed were always in the inner SVZ (Fig. 7D). In contrast, immunolabeling for cD2 was robust in both the VZ and the SVZ, in both medial and lateral SVZ and was observed in both the outer and the inner SVZ (inner SVZ ≫ outer SVZ) (Fig. 7E). The same areas showing robust cD2 immunoreactivity were colabeled for Tbr2 (Fig. 7F). cD2 may thus serve an important evolutionary role in brain development, because it plays a vital role in the cycling of Tbr2-immunopositive cells that can modulate human brain size (Baala et al., 2007).
Figure 7.
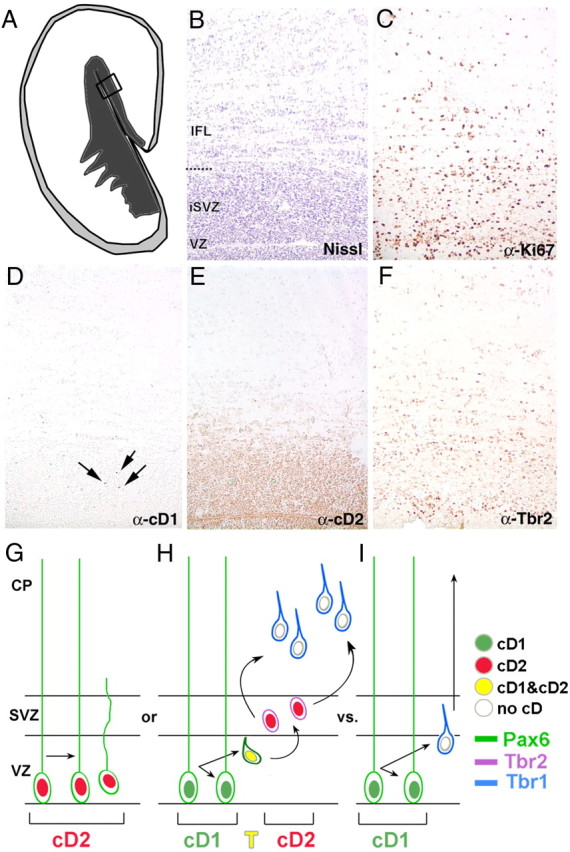
cD2 is the predominant cyclin D in the human SVZ. A–F, The proliferative zones of adjacent sections (10 μm) of human fetal cortex at 19 gestational weeks shown (B–F) are indicated by the box in the schema (A). The general architecture of the region is revealed with Nissl staining (B), and the distribution of proliferative cells is discerned with Ki67 immunolabeling (C). Rare nuclei are cD1 immunoreactive (D, arrows), whereas many nuclei are cD2 immunolabeled in both the VZ and the SVZ (E). The distribution of cD2-immunopositive nuclei overlaps the distribution of Tbr2-immunolabeled cells (F). Diagram of cD2 versus cD1 usage in embryonic cortex (G–I). Precursor cells label with Pax6 (green cells), Tbr2 (purple cells), or Tbr1 (blue cells). cD2 (red nucleus) is expressed in Pax6+ radial glia undergoing symmetric stem divisions (G) to expand the RG population OR in Pax6+ cells transitioning (yellow nucleus, T) to intermediate progenitors (purple, Tbr2+; red, cD2+) that divide symmetrically in the SVZ (H). In contrast, cD1+ radial glial cells undergo asymmetric divisions in the VZ, giving rise to a postmitotic cell (blue, Tbr1+) and a renewed radial glial cell (I). Note that cells lacking both cD1 and cD2 (white nuclei) exit the cycle to become postmitotic, Tbr1+ (blue) cells. IFL, Inner fiber layer; iSVZ, inner subventricular zone; CP, cortical plate.
Discussion
Previous histological data show that cD1 and cD2 predominate in distinct neuroepithelial populations and that cD2 localizes to SVZ in which transit-amplifying divisions occur (Glickstein et al., 2007a,b). The present study provides a number of additional insights into D-cyclin regulation of cortical histogenesis. First, it was formerly possible that cD2 is expressed only in IPCs. The present study shows that cD2 is expressed as cells transition from RGCs to IPCs. Second, cD1 is not expressed in IPCs. Third, it was formerly possible that no IPC could arise without cD2. The present study shows that cD2 is not an absolute requirement for Tbr2+/BrdU+ IPCs to appear.
Fourth, cD2 is important for the expansion of IPC numbers. The location of cD2 labeling, dual expression with Tbr2, and cD2-deficit effects on the size of the precursor pool subsets including Pax6+/Tbr2+ and Tbr2+/BrdU+ cells indicate that cD2 is the D-type cyclin used in these expansive IPC divisions. This assertion is further supported by the degree of microcephaly, greater thinning of superficial cortical layers, and significant reduction in the SVZ population in the cD2−/− compared with cD1 nulls (Figs. 3, 6). The additional loss of Pax6+ progenitors in cD2 nulls, the continued expression of cD2 in Pax6−/− cortex (Holm et al., 2007), and in Tbr2−/− cortex (Fig. 3) further localizes cD2 function to cells transitioning between RGCs and IPCs and is consistent with the effect of cD2 loss on both progenitor pools. It is also consistent with the more pronounced impact of cD2 loss on IPCs, in which abventricular mitoses are significantly diminished in cD2 null embryonic cortex at E14.5, whereas ventricular mitoses, the majority of which occur in cell cycles that use cD1, are no different from wild-type siblings. The effect of cD2 loss on IPCs is no less dramatic than that of Insm1 (insulinoma-associated-1) protein that was reported recently to have a role in expansion of basal progenitors in developing neocortex (Farkas et al., 2008), yet Insm1 inactivation only reduces the Tbr2+ IPC population (Tbr2+/PH3 dual-labeled cells across both basal and apical locations) by ∼50% (Farkas et al., 2008). This compares well with our data that IPCs still arise in the absence of cD2 but with a nearly 40% decrease in Tbr2+/BrdU+ dual-labeled cells. Together, data presented here indicate that cD2 is not necessary for IPCs to emerge but that cD2 is used in progenitors as they transition into and expand their IPC numbers.
Fifth, the present manuscript shows that cD2 regulates not only the length of G1-phase but also influences the length of S-phase (Ts), which has been postulated to be important in the regulation of primate cortex lamination. Sixth, the Tbr2-CKO expresses cD2 even in the absence of Tbr2, suggesting that cD2 emerges temporally before Tbr2. However, because a smaller population of Tbr2+ cells still arises in cD2−/− cortex, there must be cD2-independent routes to Tbr2 expression, and cD2 is important in cortex primarily to promote the expansion of the IPC pool. Finally, cD2 expression predominates in the human SVZ at 19–20 weeks gestation, supporting a significant role for this protein in the expansion of brain size over the course of species evolution.
Cell cycle parameters estimated here were in line with those reported for C57BL/6J mice (Hodge et al., 2004; Chakrabarti et al., 2007) but longer than reported in CD1 mice (Takahashi et al., 1995a). Both estimates of TG2+M and Tc were calculated using independent groups within each genotype and were increased to a similar extent (∼20%) in both wild-type strains compared with CD1 mice at the same age (Takahashi et al., 1995a). Strain-dependent factors may account for the longer estimates of cell cycle times in C57BL/6J mice observed here and by others. Regardless of differences in wild-type values compared with the literature, cD2+/+ and nulls showed fundamental differences in cell cycle parameters determined in a well controlled study.
The cumulative BrdU labeling experiments were instructive. Despite the relatively small proportion of cD2-expressing cells in the apical progenitor pool, loss of cD2 expression had a measurable impact on cell cycle dynamics in the VZ. Cell cycle length (Tc) tended to be prolonged in the cD2 nulls but not in cD1−/− mice. G1-phase (TG1) was prolonged a striking 4 h in embryos without cD2, confirming lack of cD1 induction in the absence of cD2. Conversely, the unchanged TG1 or Ts in cD1 nulls supported the effective compensation of cD1 loss by cD2 induction. TG1 lengthening was consistent with an unopposed increase in expression of the cyclin-dependent kinase (cdk) inhibitor p27, just as acute induction of p27 prolonged TG1 in transgenic mice (Mitsuhashi et al., 2001). However, subsequent direct measurement of cell cycle phases in p27 overexpressors and knock-outs has led to the consensus that modulation of p27 levels increases the rate of cell cycle exit during neurogenesis without significantly changing Tc, TG1, or Ts (Tarui et al., 2005). Thus, the observed effect of cD2 loss on G1 may result from combinatorial changes in p27, phosphorylated Rb, and/or other cell cycle components yet to be identified. One caveat is that previous data investigating p27 overexpression was gathered between E12 and E14, whereas the present experiments in cD2 nulls examined the interval between E14.5 and E16, when p27 effects on cell cycle parameters may be more robust. The increased fraction of cells leaving the cycle in cD2 nulls was consistent with the measured lengthening of TG1, because pharmacological inhibition of G1-active cdks prolongs G1, pushing cortical progenitors out of cycle (Calegari and Huttner, 2003; Calegari et al., 2005).
A striking insight from the BrdU cumulative labeling was the contraction of S-phase (Ts) in cD2 nulls. This was unexpected, because G1-active cD2 is not known to have effects on S-phase. Loss of cD2 resulted in a “catch up” so that Tc in the cD2 nulls was close to wild type. One possible mechanism for shortening of Ts could be that a cyclin E-cdk2 buildup needed to overcome the higher set point created by elevated p27 (unopposed by D-cyclin) served to propel the cell through S-phase, once the G1-S checkpoint was traversed. Although immunohistochemical staining for cyclin E was attempted in the cD2 nulls, localization with the available anti-cyclin E antibodies did not permit a conclusive determination. Similarly, existing phospho-cdk2 antibodies cross react with other cdks (e.g., cdk5). Thus, tests of this hypothesis await more specific/sensitive reagents. Nevertheless, the determination that absence of cD2 significantly lengthens TG1 and shortens Ts in neural progenitors is unique in the examination of cell cycle mouse mutants and suggests that events in both G1- and S-phases may be important to the influence exerted by cD2. Indeed, changes in Ts across the period of cortical neurogenesis have been observed in primates, in which biphasic modulation of Ts has been related to cortical expansion and “laminar elaboration” (Kornack and Rakic, 1998; Dehay and Kennedy, 2007). That cD2 is the predominant D-cyclin expressed in the human SVZ at 19–20 weeks gestation indicates the evolutionary importance of cD2 in larger mammals for whom expansive intermediate progenitor divisions have been proposed to enable the generation of larger, convoluted, cerebral cortices (Noctor et al., 2008; Pontious et al., 2008). It may be that molecular programs associated with cD2 allow for changes in Tc and Ts sufficient to be detected in cD2-deficient mouse cortex and that are essential in primate corticogenesis.
These data fit a model in which cyclin D2 is required for expansive cortical cell divisions (Fig. 7G–I). In Pax6-expressing apical progenitors, a relatively small number of cells using cD2 undergo symmetric stem divisions to enlarge the VZ progenitor pool (Fig. 7G). Asymmetric divisions driven by cD1 maintain the population by generating a daughter that will leave the VZ to become a neuron, glial, or intermediate progenitor cell and another daughter that replenishes the radial glial pool (Fig. 7H,I). Cells becoming Tbr2-expressing intermediate progenitors use cD2 for that critical symmetric transit amplifying division (Fig. 7H). Without either cD1 or cD2, progenitors exit the cell cycle and differentiate (Fig. 7H,I), evident in the increased cell cycle exit in cD2−/− cortex. The preponderance of cD2 expression in human embryonic SVZ strongly supports this hypothesized requirement of cD2 to promote expansive transit amplifying divisions. The fact that double knock-out of cD2 and cD1 is not compensated by cD3 expression in brain (Ciemerych et al., 2002) and that cortical neuroepithelial cells still proliferate in the absence of all D-cyclins (Kozar et al., 2004) suggest that cD3 does not play a significant role in either cD2 or cD1 null embryonic cortex. The disparity in cD2 expression between the VZ and SVZ is consistent with the volume differences between the ventricles and convoluted cortex, and cell number differences from the deep to superficial cortical layers needed to construct a “gyral” brain (Kriegstein et al., 2006; Noctor et al., 2008). Thus, cD2 may be a key evolutionary determinant of cortical size and organization among species.
Footnotes
This work was supported by National Institutes of Health Grant PO1NS048120 (M.E.R.). We are indebted to Dr. Chai-An Mao and Dr. William Klein (University of Texas, M. D. Anderson Cancer Center, Houston, TX) for generously providing fixed embryo brains from Tbr2fx/+::Foxg1-Cre × Tbr2fx/fx intercrosses used in this study.
References
- Arnold SJ, Huang GJ, Cheung AF, Era T, Nishikawa S, Bikoff EK, Molnár Z, Robertson EJ, Groszer M. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 2008;22:2479–2484. doi: 10.1101/gad.475408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baala L, Briault S, Etchevers HC, Laumonnier F, Natiq A, Amiel J, Boddaert N, Picard C, Sbiti A, Asermouh A, Attié-Bitach T, Encha-Razavi F, Munnich A, Sefiani A, Lyonnet S. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet. 2007;39:454–456. doi: 10.1038/ng1993. [DOI] [PubMed] [Google Scholar]
- Calegari F, Huttner WB. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci. 2003;116:4947–4955. doi: 10.1242/jcs.00825. [DOI] [PubMed] [Google Scholar]
- Calegari F, Haubensak W, Haffner C, Huttner WB. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci. 2005;25:6533–6538. doi: 10.1523/JNEUROSCI.0778-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS, Jr, Goto T, Tarui T, Takahashi T, Bhide PG, Nowakowski RS. Cell output, cell cycle duration and neuronal specification: a model of integrated mechanisms of the neocortical proliferative process. Cereb Cortex. 2003;13:592–598. doi: 10.1093/cercor/13.6.592. [DOI] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci. 2007;27:11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Duan RS, Zhu Y, Folkesson R, Albanese C, Winblad B, Zhu J. Increased cyclin E expression may obviate the role of cyclin D1 during brain development in cyclin D1 knockout mice. J Neurochem. 2005;92:1281–1284. doi: 10.1111/j.1471-4159.2004.02934.x. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;16:3277–3289. doi: 10.1101/gad.1023602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci. 2007;8:438–450. doi: 10.1038/nrn2097. [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas LM, Haffner C, Giger T, Khaitovich P, Nowick K, Birchmeier C, Pääbo S, Huttner WB. Insulinoma-associated 1 has a panneurogenic role and promotes the generation and expansion of basal progenitors in the developing mouse neocortex. Neuron. 2008;60:40–55. doi: 10.1016/j.neuron.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hébert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, Slack RS. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 2002;21:3337–3346. doi: 10.1093/emboj/cdf338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Yu Q, Sicinska E, Das M, Bronson RT, Sicinski P. Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proc Natl Acad Sci U S A. 2001;98:194–199. doi: 10.1073/pnas.011522998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickstein SB, Desteno DA, Hof PR, Schmauss C. Mice lacking dopamine D2 and D3 receptors exhibit differential activation of prefrontal cortical neurons during tasks requiring attention. Cereb Cortex. 2005;15:1016–1024. doi: 10.1093/cercor/bhh202. [DOI] [PubMed] [Google Scholar]
- Glickstein SB, Alexander S, Ross ME. Differences in cyclin d2 and d1 protein expression distinguish forebrain progenitor subsets. Cereb Cortex. 2007a;17:632–642. doi: 10.1093/cercor/bhk008. [DOI] [PubMed] [Google Scholar]
- Glickstein SB, Moore H, Slowinska B, Racchumi J, Suh M, Chuhma N, Ross ME. Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development. 2007b;134:4083–4093. doi: 10.1242/dev.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz M, Barde YA. Radial glial cells defined and major intermediates between embryonic stem cells and CNS neurons. Neuron. 2005;46:369–372. doi: 10.1016/j.neuron.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55:223–233. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hodge RD, D'Ercole AJ, O'Kusky JR. Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J Neurosci. 2004;24:10201–10210. doi: 10.1523/JNEUROSCI.3246-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Kowalczyk TD, Wolf SA, Encinas JM, Rippey C, Enikolopov G, Kempermann G, Hevner RF. Intermediate progenitors in adult hippocampal neurogenesis: Tbr2 expression and coordinate regulation of neuronal output. J Neurosci. 2008;28:3707–3717. doi: 10.1523/JNEUROSCI.4280-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm PC, Mader MT, Haubst N, Wizenmann A, Sigvardsson M, Götz M. Loss- and gain-of-function analyses reveal targets of Pax6 in the developing mouse telencephalon. Mol Cell Neurosci. 2007;34:99–119. doi: 10.1016/j.mcn.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME. Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development. 1999;126:1927–1935. doi: 10.1242/dev.126.9.1927. [DOI] [PubMed] [Google Scholar]
- Kornack DR, Rakic P. Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci U S A. 1998;95:1242–1246. doi: 10.1073/pnas.95.3.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, Akashi K, Sicinski P. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, Martínez-Cerdeño V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci. 2006;7:883–890. doi: 10.1038/nrn2008. [DOI] [PubMed] [Google Scholar]
- Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10:201–212. doi: 10.1016/0896-6273(93)90311-e. [DOI] [PubMed] [Google Scholar]
- Lu M, Zhang RL, Zhang ZG, Yang JJ, Chopp M. Linkage of cell cycle kinetics between embryonic and adult stroke models: an analytical approach. J Neurosci Methods. 2007;161:323–330. doi: 10.1016/j.jneumeth.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Mitsuhashi T, Aoki Y, Eksioglu YZ, Takahashi T, Bhide PG, Reeves SA, Caviness VS., Jr Overexpression of p27Kip1 lengthens the G1 phase in a mouse model that targets inducible gene expression to central nervous system progenitor cells. Proc Natl Acad Sci U S A. 2001;98:6435–6440. doi: 10.1073/pnas.111051398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Kriegstein AR. Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J Comp Neurol. 2008;508:28–44. doi: 10.1002/cne.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HdF. Ed 2. New York: Oxford UP; 1991. The fine structure of the nervous system: neurons and their supporting cells. [Google Scholar]
- Pontious A, Kowalczyk T, Englund C, Hevner RF. Role of intermediate progenitor cells in cerebral cortex development. Dev Neurosci. 2008;30:24–32. doi: 10.1159/000109848. [DOI] [PubMed] [Google Scholar]
- Quinn JC, Molinek M, Martynoga BS, Zaki PA, Faedo A, Bulfone A, Hevner RF, West JD, Price DJ. Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol. 2007;302:50–65. doi: 10.1016/j.ydbio.2006.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Hof PR. Recommendations for straightforward and rigorous methods of counting neurons based on a computer simulation approach. J Chem Neuroanat. 2000;20:93–114. doi: 10.1016/s0891-0618(00)00066-1. [DOI] [PubMed] [Google Scholar]
- Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron. 2008;60:56–69. doi: 10.1016/j.neuron.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, Ivanova NB, Stifani S, Morrisey EE, Temple S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci. 2006;9:743–751. doi: 10.1038/nn1694. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–630. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature. 1996;384:470–474. doi: 10.1038/384470a0. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr Cell cycle parameters and patterns of nuclear movement in the neocortical proliferative zone of the fetal mouse. J Neurosci. 1993;13:820–833. doi: 10.1523/JNEUROSCI.13-02-00820.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995a;15:6046–6057. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr Early ontogeny of the secondary proliferative population of the embryonic murine cerebral wall. J Neurosci. 1995b;15:6058–6068. doi: 10.1523/JNEUROSCI.15-09-06058.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS. Overexpression of p27 Kip 1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex. 2005;15:1343–1355. doi: 10.1093/cercor/bhi017. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Zecevic N, Chen Y, Filipovic R. Contributions of cortical subventricular zone to the development of the human cerebral cortex. J Comp Neurol. 2005;491:109–122. doi: 10.1002/cne.20714. [DOI] [PMC free article] [PubMed] [Google Scholar]