

Abstract
Helicobacter pylori is a human gastroduodenal pathogen that leads to active chronic inflammation characterized by T-cell responses biased toward a Th1 phenotype. It has been accepted that H. pylori infection induces a Th17 response. At mucosal sites, dendritic cells (DCs) have the capacity to induce effector T cells. Here, we evaluate the role of DCs in the H. pylori-induced interleukin-17 (IL-17) response. Immunohistochemistry and immunofluorescence were performed on human gastric mucosal biopsy samples and showed that myeloid DCs in H. pylori-infected patients colocalized with IL-23- and that IL-17-producing lymphocytes were present in H. pylori-infected antral biopsy samples. In parallel, human monocyte-derived DCs stimulated in vitro with live H. pylori cells produced significant levels of IL-23 in the absence of IL-12 release. The subsequent incubation of H. pylori-infected DCs with autologous CD4+ T cells led to gamma interferon (IFN-γ) and IL-17 expression. The inhibition of IL-1 and, to a lesser extent, IL-23 inhibited IL-17 production by T cells. Finally, isogenic H. pylori mutant strains not expressing major virulence factors were less effective in inducing IL-1 and IL-23 release by DCs and IL-17 release by T cells than parental strains. Altogether, we can conclude that DCs are potent inducers of IL-23/IL-17 expression following H. pylori stimulation. IL-1/IL-23 as well as H. pylori virulence factors seem to play an important role in mediating this response.
Gram-negative Helicobacter pylori is a gastroduodenal pathogen identified as being the causative agent of a variety of disease including gastritis, peptic ulcer, gastric adenocarcinoma, and mucosa-associated lymphoma (23, 27, 41, 42). H. pylori infection of gastric mucosa leads to active chronic inflammation characterized by both a lymphocytic and neutrophil infiltrate with the induction of proinflammatory cytokines, mainly interleukin-1β (IL-1β), tumor necrosis factor alpha (TNF-α), IL-8, and IL-6 (13, 29).
The H. pylori-specific gastric mucosal T-cell response is predominantly a CD4+ T-cell response polarized toward a T-helper 1 (Th1) phenotype with increased levels of gamma interferon (IFN-γ) (4, 38, 55). Although profound, this immune response does not clear the bacteria, and indeed, the cytokines secreted are more associated with pathogenesis (38, 45). Furthermore, neutrophil responses are associated with tissue damage and ulceration (7, 60). The release of the neutrophil chemoattractant IL-8 by gastric epithelial cells was previously shown to depend on the expression of an H. pylori virulence factor: the cytotoxin-associated gene (cag) pathogenicity island (PAI) (14, 62). The cag PAI encodes the immunodominant protein CagA and the type IV secretion system, which serves to transfer the bacterial CagA protein and other soluble factors, such as peptidoglycans, to the cytoplasm of the host cell (9, 52). Strains expressing the cag PAI have been associated with a more severe inflammatory response than that induced by cag PAI-negative strains (12). The cellular recognition of cag PAI-positive strains is mediated via signaling through the host-intracellular pathogen recognition molecule NOD1 (nucleotide-binding oligomerization domain 1), leading to NF-κB activation and the induction of proinflammatory responses (58).
It was previously shown that H. pylori infection is also associated with a marked production of Th17 cytokines (2, 39, 44). By using real-time PCR and Western blotting, it was previously demonstrated that IL-17, a proinflammatory cytokine, is upregulated in H. pylori-infected stomach biopsy specimens in comparison to uninfected specimens (39). IL-17 is a cytokine that characterizes a distinct population of T cells, namely, Th17 (1, 28). IL-17 has been associated with chronic inflammatory conditions such as rheumatoid arthritis (10) and multiple sclerosis (37). In addition, IL-17 proinflammatory function leading to IL-8 stimulation raises the possibility that IL-17 may play a role during bacterial infections (39, 57). Major cytokines associated with the differentiation of human Th17 cells were identified to be IL-23, IL-1β, and IL-6 (11, 61). While IL-12 plays a key role in the differentiation of naïve T cells to Th1 cells, IL-23 promotes the expansion of Th17 cells. In contrast, IL-27, another IL-12 family member, has been shown to limit the development of Th17 cells (25). IL-12 and IL-23 are heterodimers with a shared subunit, p40. Both IL-23 and IL-12 are produced by activated antigen-presenting cells (APCs) such as DCs and macrophages (48, 53).
DCs, which play a central role in the induction of adaptive immune responses, are widely distributed in tissues, including gastrointestinal mucosa (32, 33), and were previously shown to be capable of migrating through epithelial tight junctions to gain access to the gastrointestinal lumen (33, 49). Furthermore, we and others have shown that H. pylori interactions with DCs trigger maturation and activation events that lead to the production of cytokines, which are important for the induction and regulation of immune responses (5, 18, 34, 43, 46).
Previous studies of DC activation by H. pylori have focused on the induction of the Th1-biased response. Much less is known about the mechanism of induction as well as the cells and cytokine stimuli responsible for the expression of IL-17 in Helicobacter infection. Here, we have reevaluated the role of DCs in the induction of immune responses to Helicobacter infection by addressing the interaction of H. pylori-infected DCs with CD4+ T lymphocytes in initiating a Th17 response.
MATERIALS AND METHODS
Helicobacter pylori and growth conditions.
H. pylori strains used were Cag PAI-positive H. pylori 26695 (NCTC12455), 84-183 (ATCC 53726), and 84-183 cagA and cagE isogenic mutants (5, 50). The strains were grown for 48 h on solid medium based on Columbia base agar (Oxoid Ltd., United Kingdom) supplemented with 10% (vol/vol) laked horse blood (Oxoid Ltd., United Kingdom) under a microaerobic atmosphere generated by Anaerocult C (Oxoid Ltd., United Kingdom). The number of bacteria was determined by the absorption at A550, with 0.8 optical density (OD) units corresponding to 108 CFU/ml. For the coculture experiments, the bacterial cells were used at a multiplicity of infection of 10 (26).
Generation of human monocyte-derived dendritic cells.
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats (National Blood Transfusion Centre, South Thames, United Kingdom) through Ficoll-Paque (PAA Laboratories Ltd., United Kingdom) gradient centrifugation. Monocyte-derived DCs were generated from CD14+ cells isolated by using CD14 beads (Miltenyi Biotec, Surrey, United Kingdom) as previously described (43). The purity of cells was greater than 96%. DCs were generated after 6 days of culture in the presence of recombinant human (rHu) IL-4 and granulocyte-macrophage colony-stimulating factor (rHu GM-CSF) (43). Immature DCs were routinely characterized by the expression of CD1a, CD11c, HLA-DR, CD80, CD83, CD86, CD40, and CD14 by using fluorescein isothiocyanate (FITC)-conjugated mouse anti-human monoclonal antibodies (MAbs) (Caltag Laboratories, United Kingdom).
DC stimulation and coculture with CD4+ T cells.
Autologous CD4+ T lymphocytes were purified from the CD14-negative population through negative selection as previously described (43). Briefly, CD4+ T cells were negatively selected by using a cocktail of antibodies against CD8, CD45RO, CD33 (Caltag), CD14, CD16, CD19, CD56 (Diaclone Research, Besaucon, France), and γδ-T-cell receptor (TCR) (Becton Dickinson). The labeled cells were removed by using BioMag goat anti-mouse IgG Fc (Qiagen, Germantown, MD). Naïve T-cell purity was analyzed by flow cytometry. CD4+ cells were more than 98% pure. DCs were collected on day 6 of culture in RPMI medium supplemented with 10% fetal calf serum (FCS) and 2 mmol/liter l-glutamine. Viable-cell counts were determined by trypan blue exclusion. DCs were cocultured with H. pylori at a 1:10 (DC-to-H. pylori cell) ratio, which was selected as the optimal ratio for DC maturation and proinflammatory cytokine secretion following a dose-dependent response (ranging from a 1:1 to a 1:100 DC-to-H. pylori cell ratio). Lipopolysaccharide (LPS) from Escherichia coli (serotype O55:B50) (Sigma, United Kingdom) was used at 100 ng/ml as a positive control for DC maturation. H. pylori-stimulated DCs were collected for RNA extraction or subsequent incubation with CD4+ T cells.
H. pylori-stimulated DCs and coculture with CD4+ T cells.
H. pylori-stimulated DCs were collected and washed twice in RPMI medium. Supernatant from H. pylori-stimulated DCs was collected for cytokine analyses, whereas the stimulated cells were then cultured with CD4+ T cells at a 1:10 (DC-to-CD4+) ratio for 5 days. The ratio was selected as optimal for cytokine secretion. Cells and supernatants were then collected for RNA extraction and cytokine analyses, respectively. In addition, cells were collected for intracellular cytokine staining (ICS).
Intracellular cytokine staining and FACS.
Day 5 H. pylori-stimulated DC cocultures with CD4+ T cells were restimulated with 4α-phorbol 12-myristate 13-acetate (PMA) (50 ng/ml) and ionomycin (1 μg/ml) (Sigma, United Kingdom) in the presence of monensin at 3 μM (Insight Biotech, United Kingdom). Cells were surface stained for CD3+ using mouse anti-human phycoerythrin (PE)-conjugated CD3 antibody (Caltag, United Kingdom). The cells were then fixed in 4% paraformaldehyde and permeabilized in 0.1% (wt/vol) saponin in 1% FCS-phosphate-buffered saline (PBS) followed by intracellular staining for IFN-γ and IL-17 by the addition of mouse anti-human allophycocyanin-conjugated IFN-γ (Insight Biotech, United Kingdom) and Alexa Fluor 488-conjugated IL-17 (Insight Biotech, United Kingdom), respectively. PE-IgG1, APC-IgG1, and Alexa Fluor 488-IgG1 were used as isotype controls. Cells were washed prior to immediate fluorescence-activated cell sorting (FACS) analyses. Three-color flow cytometric analyses were performed by using a FACSCalibur flow cytometer, and data were analyzed by using CELLQuest software (Becton Dickinson). A total of 10,000 gated events were analyzed for each cell population.
RNA extraction and RT-PCR.
RNA was extracted from CD4+ T cells cocultured with H. pylori-stimulated DCs by using an RNAaquous extraction kit (Ambion, Applied Biosystems, United Kingdom) according to the manufacturer's protocol. Reverse transcription (RT)-PCR was performed on the extracted RNA by using a RETROscript kit (Ambion, Applied Biosystems, United Kingdom) according to the manufacturer's instructions.
Quantification of IL-12/IL-23 and IL-17 expression by real-time PCR.
The real-time expression of IL-23/IL-12 and IL-17 was measured by TaqMan gene expression assays from Applied Biosystems using IL-12-specific subunit p35 (IL-12A; assay identification number Hs00168405_m1), IL-23-specific subunit p19 (IL-23A; assay identification number Hs00372324_m1), IL-12/23 common subunit p40 (IL-12B; assay identification number Hs00233688_m1), and IL-17 (assay identification number Hs00174383_m1). Human β-actin (assay identification number Hs00242273_m1) was used as the endogenous control. Quantitative amplification was carried out according to the manufacturer's instructions by using a Chromo 4 real-time PCR detector (MJ Research, Bio-Rad, Hertfordshire, United Kingdom). Gene expression levels were normalized to that of β-actin and expressed as −ΔΔCT (ΔΔCT = ΔCTstimulated cells − ΔCTunstimulated cells).
Effect of cytokines on IL-17 secretion.
Cytokines secreted from CD4+ T cells stimulated with H. pylori-treated DCs were blocked by the addition of 10 μg/ml of the following cytokine-neutralizing monoclonal antibodies during coculture of CD4+ T cells and H. pylori DCs: anti-TNF-α, anti-IL-4, anti-IFN-γ, anti-IL-6, anti-transforming growth factor β (TGF-β) (R&D Systems, United Kingdom), and anti-IL-23 (Insight Biotech, United Kingdom). IL-1 receptor antagonist (10 μg/ml) (kindly provided by K. Ray, GlaxoSmithKline, Stevenage, United Kingdom) was used to block the effect of IL-1. Blocking antibodies (goat IgG and mouse IgG isotypes) used in this study were referenced blocking antibodies from the manufacturers and were selected based on data from previously published reports (20, 21). Supernatants were collected after 5 days of incubation and tested for IL-17 and IFN-γ secretion by using an enzyme-linked immunosorbent assay (ELISA).
ELISA and MSD multiplex cytokine detection system.
The collected supernatants were tested for IL-23 and IL-12 secretion by using human IL-23 and IL-12 Ready-Set-Go ELISA (Insight Biotech, United Kingdom). A DuoSet ELISA kit was used to test for IL-17 and IFN-γ secretion (R&D Systems, United Kingdom). All ELISA experiments were performed in triplicate and according to the manufacturer's instructions. IL-1β was assessed by using the Meso Scale Discovery (MSD) detection system (Gaithersburg, MD) according to the manufacturer's instructions. The results were read by using the Sector Imager 6000 apparatus (Gaithersburg, MD).
Immunohistochemical analysis for detection of myeloid DCs.
Immunohistochemical analysis was performed on frozen antral gastric biopsy specimens from patients undergoing upper gastrointestinal endoscopy for dyspepsia after ethical committee approval and written consent. Patients were selected if they were over the age of 18 years and diagnosed with dyspepsia. Patients who had been on proton pump inhibitors or antibiotics within the previous 3 months were excluded. Biopsy sections from the patients were fixed in formalin, embedded in paraffin, and stained with Giemsa and hematoxylin and eosin (H&E) reagents prior to microscopic examination for the presence of the H. pylori and gastritis. All patients had chronic active gastritis that was demonstrated histologically. H. pylori positivity was assessed histologically and by using the Campylobacter-like organism (CLO) test (a standard urease-based test). Uninfected patients, confirmed by a negative CLO test and the absence of H. pylori organisms by histology, were used as controls. Sections were cut at 5 μm. Frozen sections were stained for the presence of myeloid DCs with mouse anti-human monoclonal antibody CD1c (BDCA-1) (Miltenyi Biotec, United Kingdom). Staining was demonstrated by using a routine avidin-biotin-horseradish peroxidase system.
Immunofluorescence and confocal microscopy.
Fresh material from antral biopsy samples was imbedded in optimal cutting temperature compound (OCT; VWR International, United Kingdom). Cryosectioning was performed at 7 μm. Primary antibodies were applied in pairs of rabbit anti-human IL-17 (Insight Biotech, United Kingdom) and mouse anti-human CD3 (Insight Biotech, United Kingdom), and mouse anti-human CD1c (BDCA-1) (Miltenyi Biotech, United Kingdom) and rabbit anti-human IL-23 (Insight Biotech, United Kingdom). This was followed by incubation with fluorescently conjugated secondary antibodies (Alexa Fluor 488 goat anti-mouse and Alexa Fluor 568 anti-rabbit [both from Molecular Probes, United Kingdom]). For nonspecific staining, a parallel set of sections was incubated with a mixture of equivalent rabbit and mouse IgGs (Vector Laboratories, United Kingdom) in place of the primary antibodies. The sections were analyzed by using a Leica TCS-NT laser scanning confocal microscope (Leica Microsystems, Germany). Where comparisons of staining levels were made, confocal settings were kept constant.
Statistical analysis.
Data were statistically analyzed by using the Student t test when normally distributed or by using the Mann-Whitney U test for nonparametric data. Statistical significance was assumed for P values of less than 0.05. All analyses were performed by using SPSS 14 software.
RESULTS
Detection of myeloid dendritic cells.
The recruitment of DCs during H. pylori infection was previously shown for murine models (31). To underline the importance of myeloid DCs in humans, we investigated the presence of myeloid DCs in gastric mucosal biopsy specimens of H. pylori-infected patients by immunohistochemistry. Frozen sections of endoscopic gastric mucosal biopsy specimens were stained by using myeloid DC marker CD1c (BDCA-1). Myeloid DCs were detected with clear dendritic cell-like extensions between the epithelial cells (Fig. 1). Dendritic cells were not detected in uninfected biopsy samples (data not shown for immunohistochemistry; uninfected sections are shown in Fig. 3E when tested for the presence of myeloid DCs using fluorescent staining).
FIG. 1.

Myeloid DCs are detected in H. pylori-infected human gastric mucosa. Shown is the immunohistochemistry of H. pylori-positive gastric stomach biopsy sections. Antral gastric biopsy sections were stained for the presence of myeloid DCs with mouse anti-human monoclonal antibody CD1c (BDCA-1) (Miltenyi Biotec, Surrey, United Kingdom) (original magnification, ×100).
FIG. 3.

Costaining of the myeloid cell marker CD1c (BDCA-1) (green) and IL-23 (red) in gastric antral biopsy samples. Confocal microscopy after immunofluorescence costaining showed that in samples from H. pylori-infected patients (A, B, and C), CD1c+ cells express IL-23 and thus appear yellow (arrows). (The yellow box in the top right corner of C is a zoomed section of the costaining indicated by the dashed arrow.) For uninfected patients (D, E, and F), no myeloid DC infiltrates were seen, and no IL-23 expression was detected. (The staining shown is representative of 3 biopsy samples from 3 H. pylori-infected and 3 uninfected patients.)
H. pylori-treated DCs induce IL-23 expression.
IL-23 has been reported to positively regulate IL-17 production by promoting Th17 cell population survival and expansion. Our focus on the role of DCs in the induction of IL-17 in H. pylori infection was based on our previous work showing that paraformaldehyde-fixed H. pylori-treated DCs express IL-23 (43). Here, we analyze the kinetics of IL-23/IL-12 mRNA expression in live H. pylori-infected DCs.
H. pylori cells were cocultured with DCs for 30 min, 120 min, 16 h, 48 h, 72 h, and 5 days for real-time analyses. LPS from E. coli, a well-known inducer of DC maturation, was used as a control for DC function (3, 22, 43). Real-time PCR was performed to analyze the expression of the common subunit p40, which combines with either the p35 or the p19 subunit to form a functional IL-12 or IL-23, respectively. As shown in Fig. 2A, there was no induction of the IL-12-specific p35 subunit at 30 min after incubation of DCs with H. pylori. At this time point, although there was no increase in levels of the IL-12 p35 subunit, the p19 subunit was already upregulated in H. pylori-treated DCs. The p19 expression levels at the different time points were consistently higher in H. pylori-stimulated DCs than in DCs treated with LPS (Fig. 2B). At its highest (overnight stimulation), the level of expression of the p19 subunit in H. pylori-treated DCs was significantly 3.5-fold higher than that of the LPS-treated DCs (P = 0.002) (Fig. 2A and B). On the other hand, the p35 subunit was 3.2-fold-more expressed in the LPS-treated DCs (P = 0.003) (Fig. 2B). The common subunit p40 was expressed similarly from both LPS- and H. pylori-treated DCs. Taken together, these results suggest that H. pylori cells elicit a robust expression of IL-23 from DCs and a low level of induction of IL-12.
FIG. 2.

H. pylori induces IL-23 expression in DCs. (A and B) Kinetics of IL-12/IL-23 expression from H. pylori (A)- and LPS (B)-stimulated DCs assessed by real-time PCR. Data are expressed as means ± SEM and are representative of five independent experiments; each experiment was done in triplicate. (C) Supernatants collected from H. pylori- and LPS-stimulated DCs at 1, 8, 24, 48, and 72 h and 5 days at a 1:10 DC-to-H. pylori cell ratio and 100 ng/ml LPS were analyzed by ELISA for IL-23 production. (D) Supernatants collected from H. pylori- and LPS-stimulated DCs at 8, 24, and 48 h at a 1:10 DC-to-H. pylori cell ratio and 100 ng/ml LPS were analyzed by ELISA for IL-12 production. Data are expressed as means ± SEM and are representative of five independent experiments.
To verify that IL-23 and IL-12 were indeed being produced, ELISAs were performed. IL-23 production was detectable from 8 h postincubation, peaked at 24 h, and started to decline thereafter (Fig. 2C). In line with the real-time analyses, LPS seemed to trigger no or a very low level of expression of IL-23 (Fig. 2C) and high levels of IL-12 in comparison to H. pylori-stimulated DCs (Fig. 2D). Accordingly and unless otherwise stated, for all subsequent experiments, DCs were preincubated overnight with H. pylori.
IL-23-positive myeloid DCs are present in gastric mucosal biopsy samples from H. pylori-infected patients.
We sought to investigate whether our in vitro findings that H. pylori stimulation of myeloid DCs induces IL-23 secretion could be detected in human gastric mucosal specimens. Here, we demonstrate that CD1c (myeloid DC marker) expression is colocalized with IL-23 production in H. pylori-infected biopsy samples (Fig. 3A, B, and C). Uninfected controls showed no presence of myeloid DCs or IL-23 secretion (Fig. 3D, E, and F).
H. pylori-treated DCs are potent inducers of IL-17 expression from CD4+ T cells.
IL-17 expression in H. pylori-infected patients was previously detected by using Western blot and RNA analyses (39). In our study, double immunofluorescence staining and confocal microscopy analyses were performed to assess the site of IL-17 expression at the cellular level by using rabbit anti-human IL-17-specific antibody. For H. pylori-positive patients, the IL-17 protein was detected in lymphoid cells (Fig. 4A, B, and C).
FIG. 4.

Confocal microscopy after double immunofluorescence labeling of CD3 (green) and IL-17 (red). The arrows show the colocalization of CD3+ and IL-17 (in yellow) seen in H. pylori-infected gastric mucosal biopsy samples (A, B, and C). In uninfected patients, although CD3+ cells were present, they appeared to be few in number and did not express IL-17 (D, E, and F) compared to the infected samples. (The staining is representative of biopsy experiments from 3 H. pylori-positive and 3 negative patients.)
Based on our data showing that H. pylori induces IL-23 expression in DCs together with the immunostaining detection of IL-17 expression in lymphoid cells, we next wished to determine whether H. pylori-stimulated DCs are capable of inducing IL-17 expression from autologous CD4+ T cells. CD4+ T cells were selected, as they represent the predominant component of the gastric infiltrates following H. pylori infection of humans and animal models (16, 17, 29, 45).
ICS showed that both IL-17 and IFN-γ were detectable in CD4+ T cells stimulated for 5 days with H. pylori-infected DCs (Fig. 5A). Proportions of IL-17-producing cells were significantly higher following stimulation with H. pylori-infected DCs than for unstimulated DCs (P = 0.001) (Fig. 5B).
FIG. 5.

H. pylori induces IL-17 expression from CD4+ T cells. (A) ICS of CD4+ T cells following coculture with H. pylori-stimulated DCs. At 5 days after coculture, CD4+ T lymphocytes in the presence of H. pylori-treated DCs were restimulated with PMA and ionomycin in the presence of monensin. Cells were stained for surface markers using anti-CD3-PE and for intracellular cytokines using IL-17-Alexa Fluor 488 (top left) and IFN-γ-APC (top right). Isotype controls used were IgG-Alexa Fluor 488 and IgG-APC (bottom left and right, respectively). (B) Comparison of cumulative data for ICS from CD4+ T cells stimulated with untreated or H. pylori-treated DCs (P = 0.001) (n = 7). (C) IL-17 expression from cocultures of H. pylori-treated DCs or unstimulated DCs with CD4+ T cells assessed by real-time PCR (cumulative data from 7 experiments) shows a significant increase in numbers of H. pylori-treated DCs compared to untreated DCs (P = 0.0002). The relative expression level of IL-17 is expressed as −δδCT. (Hp:DC, H. pylori-stimulated DCs).
IL-17 expression was confirmed by real-time PCR and ELISA. The detection of RNA following 24 h of coculture showed that H. pylori-treated DCs induced significant levels of IL-17 from CD4+ T cells compared to untreated DCs (P = 0.001) (Fig. 5C).
ELISA results correlated with the real-time data confirming the expression of IL-17 at the protein level. The expression was detectable at 48 h, increased after 3 days, and reached the highest level at 5 days (87.704 ± 17.52 pg/ml [mean of 7 independent experiments ± standard error of the mean {SEM}]). LPS-stimulated DCs induced virtually undetectable levels of IL-17, which was in line with previously reported data (11).
IL-1 and IL-23 are the most potent cytokines involved in H. pylori-mediated IL-17 production.
IL-23, IL-1β, and IL-6 are key factors in human Th17 cell development (11, 61). We investigated the role of these cytokines and other major H. pylori-DC-derived cytokines (6, 35) in the induction of IL-17 protein production. H. pylori-stimulated DCs were cocultured with CD4+ T cells in the presence of neutralizing antibodies, namely, anti-TNF-α, anti-IL-23, anti-IFN-γ, and anti-IL-6. As IL-1β and IL-1α bind to the same cell surface receptor (IL-1R), which mediates their biological activity (54), we decided to use an IL-1 receptor antagonist to block IL-1 receptor signaling. Anti-IL-4 was used as a control. The role of TGF-β in IL-17 differentiation is controversial, with some reports suggesting that neutralizing antibodies to TGF-β lead to an inhibition of the IL-17 response (40, 59) and others demonstrating that the presence of TGF-β inhibits Th17 differentiation (11, 61). We therefore tested the role of TGF-β in our system by using an anti-TGF-β neutralizing antibody. The most significant inhibitory effect was seen in the presence of IL-1 receptor antagonist (P < 0.0005) (Fig. 6). Anti-IL-23 caused a significant decrease in IL-17 production (P = 0.002) (Fig. 6). Neutralizing antibodies to TNF-α, IL-4, IFN-γ, IL-6, and TGF-β (Fig. 6) and higher concentrations of all antibodies did not have an effect on IL-17 production.
FIG. 6.
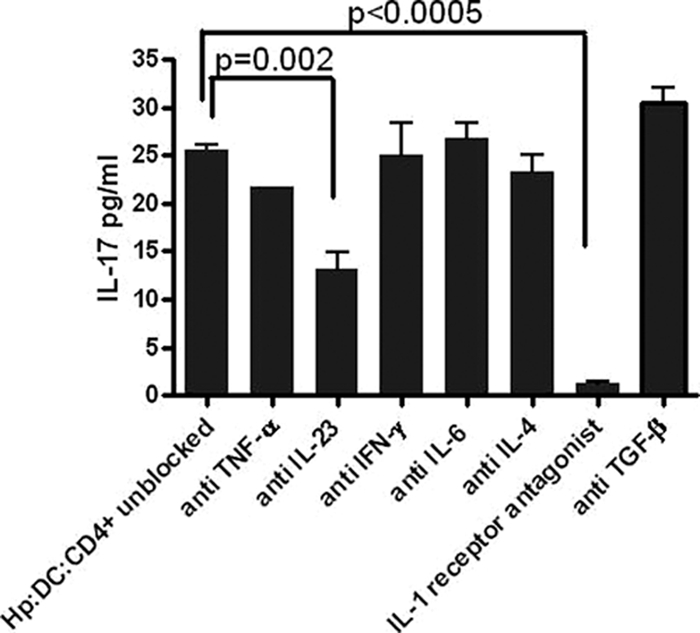
IL-1 and IL-23 influence IL-17 production from CD4+ T cells stimulated with H. pylori-treated DCs. Shown is the effect of cytokine neutralizing antibodies on the expression of IL-17. IL-17 expression was assessed by using ELISA on supernatants collected on day 5 from cocultured CD4+ T cells with H. pylori-stimulated DCs in the presence or absence of anti-TNF-α, anti-IL-23, anti-IFN-γ, anti-IL-6, anti-IL-4, IL-1 receptor antagonist, and anti-TGF-β (all used at 10 μg/ml) (data are representative of 3 independent experiments).
IL-17 induction requires an active cagA and type IV secretion system.
To evaluate any potential contribution of H. pylori virulence factors to IL-17 production, H. pylori 84-183 and its cagA and cagE isogenic mutants were tested. Following overnight culture, the phenotypic response of DCs to these bacterial cells was monitored. Maturation surface markers (CD86, CD40, and HLA-DR) were all upregulated compared to unstimulated DCs (Table 1). The cells showed no major differences in the expression profiles among the three bacterial preparations (Table 1). When tested for IL-17 and IFN-γ production by cocultured autologous CD4+T cells, the strain lacking CagA expression showed a marked decrease in the induction of IL-17 production by CD4+ T cells compared to the parental strain (P = 0.01) (Fig. 7A). Notably, an almost complete inhibition was detected following stimulation with the cagE mutant (P = 0.008) (Fig. 7A). However, there was a less pronounced effect on IFN-γ production by CD4+ T cells stimulated with DCs preincubated with these bacterial cells (Fig. 7B). Interestingly, when cytokines produced by the DCs in response to the parental cells were compared to those produced in response to the mutant bacterial cells, both IL-1β and IL-23 seemed to be produced with a pattern similar to that of IL-17 production; i.e., the smallest amounts of IL-1β and IL-23 production were seen in the CagE mutant-stimulated DCs (Fig. 7C and D), with differences in IL-23 production reaching statistical significance (Fig. 7D).
TABLE 1.
Upregulation of DC maturation markers following overnight stimulation with H. pylori strain 84-183 and its isogenic mutants determined by FACS analysesa
| Maturation marker | % of cells (MFI) |
||
|---|---|---|---|
| CD86 | CD40 | HLA-DR | |
| H. pylori | |||
| 84-183 parental strain | 85.1 (121) | 94.3 (139.9) | 89.3 (65.4) |
| 84-183 CagA mutant | 92 (143) | 91.4 (158) | 87.3 (60.6) |
| 84-183 CagE mutant | 89.7 (125.2) | 92 (145.9) | 87.6 (75.8) |
| Unstimulated DCs | 51.3 (86) | 82 (88.7) | 79 (50.3) |
Data are representative of three independent experiments. MFI, mean fluorescence intensity.
FIG. 7.

H. pylori cag PAI virulence factor affects IL-17 production. DCs were exposed to H. pylori 84-183 and its isogenic mutants (cagA and cagE mutants) and subsequently incubated with CD4+ T cells. (A and B) IL-17 (A) and IFN-γ (B) production assessed by ELISA after 5 days of coculture. (A) Preincubation of DCs with the mutant strains reduced the levels of IL-17 production significantly. (B) No significant differences in the production of IFN-γ were detected. (C and D) Of the cytokines produced following stimulation with the parental and mutant bacterial cells tested, IL-1β (C) and IL-23 (D) seemed to be produced with similar patterns where the mutant strains induced fewer of the cytokines than the parental strain (cumulative data from 4 experiments, each done in triplicate).
DISCUSSION
In this study, we provide evidence for the presence of myeloid DCs in the stomach of H. pylori-infected individuals. We also report for the first time the presence of IL-23-secreting CD1c+ DCs in H. pylori-infected gastric specimens. We also report a new role for DCs in the recognition of H. pylori by demonstrating their capacity to direct a Th17 response against this pathogen and show the presence of IL-17-secreting lymphocytes in the gastric mucosa during H. pylori infection.
We found previously that fixed H. pylori-stimulated DCs are capable of inducing IL-23 expression (43). We have further expanded this finding by assessing the kinetics of this expression at the protein and RNA levels by using live H. pylori-infected DCs. Our data indicate that myeloid DCs are a potent source of functional heterodimeric IL-23, as they produce both the common (p40) and the specific (p19) subunits at high levels. Our results are consistent with data from a recent report by Caruso and colleagues showing that IL-23 is overexpressed in H. pylori-infected patients and that its secretion is able to sustain IL-17 production (8). In contrast to a number of reports that had detected IL-12 production from H. pylori-infected DCs (26, 34) and although not consistent with the H. pylori Th1-associated response, we did not observe any detectable IL-12 production from H. pylori-stimulated DCs in our system. Interestingly and in line with our findings, Kao and colleagues have shown that H. pylori-secreted factors inhibit IL-12 secretion from dendritic cells (31). Having used live bacteria in our study, the previous report by Kao et al. seems to provide a plausible explanation for our observation. Nevertheless, we previously reported that low levels of IL-12 from H. pylori-stimulated DCs can be induced following CD40 ligation (43). Furthermore, other cells at the site of infection, such as macrophages and neutrophils, are capable of producing additional IL-12 (15, 56) and therefore provide the environment for Th1 development.
Our study shows that H. pylori-stimulated DCs subsequently activate autologous CD4+ T cells and induce IL-17 production. IL-17 proinflammatory activity is required to clear bacterial infections at mucosal surfaces, including the intestine and airway (30, 36, 63). In the case of H. pylori, Mizuno et al. showed previously that infection with H. pylori is associated with IL-17 production at the site of infection (44). Our data provide further evidence for the presence of IL-17-secreting T lymphocytes in H. pylori-colonized human gastric mucosa. The gastric expression of IL-17 was also detected in H. pylori-infected mice (2). It was previously suggested that through the induction of IL-8 expression, IL-17 plays a role in maintaining and expanding the acute inflammatory response during H. pylori infection (39, 51). With regard to disease outcome, IL-17 production seems to be associated with ulcerogenesis (44).
For humans, IL-1β and IL-23 were previously reported to be the most potent cytokines for controlling the expansion and maintenance of Th17 cells (61). In addition to IL-23, IL-1β is a well-recognized cytokine in H. pylori infection with profound effects on gastric physiology (47). Previously reported studies have linked host IL-1 gene polymorphism, which is associated with an enhanced production of IL-1β, to H. pylori-related gastric carcinoma development (19). In our system, we found that IL-1 and IL-23 seem to be the main players in the induction of the observed IL-17 production. This is consistent with data from a recent report showing that IL-23 contributes to sustaining IL-17 production in H. pylori-infected gastric mucosa (8). This further emphasizes the need to dissect the role of Th17 cells in H. pylori-associated pathology, particularly in relation to the development of gastric cancer itself.
In addition to the role of the secreted cytokines, we showed that H. pylori virulence factors are key elements in the induction of IL-17 production. Despite the induction of similar levels of DC maturation, cag PAI-positive strains, more frequently associated with severe polymorphonuclear (PMN) cell infiltrates in vivo (13), induce the highest levels of IL-17. This seems to be due in part to the action of the immunodominant protein CagA but mostly the absence of a CagE-dependent transfer of soluble factors from live bacterial cells but is also associated with IL-1β and IL-23 produced by stimulated DCs. It is well documented that an active type IV secretion system mediates the translocation of CagA and soluble peptidoglycan that signal through the NOD1 pathway in epithelial cells (58). Fritz and colleagues previously suggested that NOD1 signaling in conjunction with Toll-like receptor recognition is involved in the onset of Th17 (24). Our results seem to suggest a similar role for NOD1 signaling in H. pylori-induced IL-17 production. However, this needs to be further elucidated.
The production of IL-23/IL-17 in the presence of H. pylori is of direct interest for the process of gastric inflammation. The prevalence of H. pylori infection in the human population attests to the failure of the immune system to clear this pathogen. The intriguing question remains regarding why, rather than being protective, a strong immune response characterized by the coexpression of IFN-γ and IL-17 is further contributing to the pathology of the infection. An understanding of the interplay between T-cell subsets and APCs is likely to shed more light on this topic.
Acknowledgments
This work was supported by the Wexham Gastrointestinal Trust.
We thank A. Postgate and G. Mehta (Faculty of Medicine, Imperial College London) for their assistance in the collection of stomach biopsy samples. We also thank L. Lawrence (National Heart and Lung Institute, Imperial College London) for excellent technical assistance with the immunofluorescence staining. We thank R. Argent (Wolfson Digestive Diseases Centre, University of Nottingham, Nottingham, United Kingdom) and K. Robinson (Centre for Biomolecular Sciences, University of Nottingham, Nottingham, United Kingdom) for helpful discussions.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 16 November 2009.
REFERENCES
- 1.Aggarwal, S., and A. L. Gurney. 2002. IL-17: prototype member of an emerging cytokine family. J. Leukoc. Biol. 71:1-8. [PubMed] [Google Scholar]
- 2.Algood, H. M., J. Gallo-Romero, K. T. Wilson, R. M. Peek, Jr., and T. L. Cover. 2007. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol. Med. Microbiol. 51:577-586. [DOI] [PubMed] [Google Scholar]
- 3.Ausiello, C. M., G. Fedele, F. Urbani, R. Lande, B. Di Carlo, and A. Cassone. 2002. Native and genetically inactivated pertussis toxins induce human dendritic cell maturation and synergize with lipopolysaccharide in promoting T helper type 1 responses. J. Infect. Dis. 186:351-360. [DOI] [PubMed] [Google Scholar]
- 4.Bamford, K. B., X. Fan, S. E. Crowe, J. F. Leary, W. K. Gourley, G. K. Luthra, E. G. Brooks, D. Y. Graham, V. E. Reyes, and P. B. Ernst. 1998. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 114:482-492. [DOI] [PubMed] [Google Scholar]
- 5.Bebb, J. R., D. P. Letley, J. L. Rhead, and J. C. Atherton. 2003. Helicobacter pylori supernatants cause epithelial cytoskeletal disruption that is bacterial strain and epithelial cell line dependent but not toxin VacA dependent. Infect. Immun. 71:3623-3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergman, M. P., A. Engering, H. H. Smits, S. J. van Vliet, A. A. van Bodegraven, H. P. Wirth, M. L. Kapsenberg, C. M. Vandenbroucke-Grauls, Y. van Kooyk, and B. J. Appelmelk. 2004. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J. Exp. Med. 200:979-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaser, M. J. 1992. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 102:720-727. [DOI] [PubMed] [Google Scholar]
- 8.Caruso, R., D. Fina, O. A. Paoluzi, B. G. Del Vecchio, C. Stolfi, A. Rizzo, F. Caprioli, M. Sarra, F. Andrei, M. C. Fantini, T. T. Macdonald, F. Pallone, and G. Monteleone. 2008. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur. J. Immunol. 38:470-478. [DOI] [PubMed] [Google Scholar]
- 9.Censini, S., C. Lange, Z. Xiang, J. E. Crabtree, P. Ghiara, M. Borodovsky, R. Rappuoli, and A. Covacci. 1996. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. U. S. A. 93:14648-14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chabaud, M., J. M. Durand, N. Buchs, F. Fossiez, G. Page, L. Frappart, and P. Miossec. 1999. Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 42:963-970. [DOI] [PubMed] [Google Scholar]
- 11.Costa-Rodriguez, E. V., G. Napolitani, A. Lanzavecchia, and F. Sallusto. 2007. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 8:942-949. [DOI] [PubMed] [Google Scholar]
- 12.Covacci, A., S. Censini, M. Bugnoli, R. Petracca, D. Burroni, G. Macchia, A. Massone, E. Papini, Z. Xiang, and N. Figura. 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. U. S. A. 90:5791-5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crabtree, J. E. 1996. Immune and inflammatory responses to Helicobacter pylori infection. Scand. J. Gastroenterol. Suppl. 215:3-10. [PubMed] [Google Scholar]
- 14.Crabtree, J. E., S. M. Farmery, I. J. Lindley, N. Figura, P. Peichl, and D. S. Tompkins. 1994. CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J. Clin. Pathol. 47:945-950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Elios, M. M., A. Amedei, and G. Del Prete. 2003. Helicobacter pylori antigen-specific T-cell responses at gastric level in chronic gastritis, peptic ulcer, gastric cancer and low-grade mucosa-associated lymphoid tissue (MALT) lymphoma. Microbes Infect. 5:723-730. [DOI] [PubMed] [Google Scholar]
- 16.D'Elios, M. M., M. Manghetti, M. De Carli, F. Costa, C. T. Baldari, D. Burroni, J. L. Telford, S. Romagnani, and G. Del Prete. 1997. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 158:962-967. [PubMed] [Google Scholar]
- 17.Di Tommaso, A., Z. Xiang, M. Bugnoli, P. Pileri, N. Figura, P. F. Bayeli, R. Rappuoli, S. Abrignani, and M. T. De Magistris. 1995. Helicobacter pylori-specific CD4+ T-cell clones from peripheral blood and gastric biopsies. Infect. Immun. 63:1102-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drakes, M. L., S. J. Czinn, and T. G. Blanchard. 2006. Regulation of murine dendritic cell immune responses by Helicobacter felis antigen. Infect. Immun. 74:4624-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Omar, E. M., M. Carrington, W. H. Chow, K. E. McColl, J. H. Bream, H. A. Young, J. Herrera, J. Lissowska, C. C. Yuan, N. Rothman, G. Lanyon, M. Martin, J. F. Fraumeni, Jr., and C. S. Rabkin. 2000. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404:398-402. [DOI] [PubMed] [Google Scholar]
- 20.Evans, H. G., N. J. Gullick, S. Kelly, C. Pitzalis, G. M. Lord, B. W. Kirkham, and L. S. Taams. 2009. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc. Natl. Acad. Sci. U. S. A. 106:6232-6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans, H. G., T. Suddason, I. Jackson, L. S. Taams, and G. M. Lord. 2007. Optimal induction of T helper 17 cells in humans requires T cell receptor ligation in the context of Toll-like receptor-activated monocytes. Proc. Natl. Acad. Sci. U. S. A. 104:17034-17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fedele, G., L. Frasca, R. Palazzo, E. Ferrero, F. Malavasi, and C. M. Ausiello. 2004. CD38 is expressed on human mature monocyte-derived dendritic cells and is functionally involved in CD83 expression and IL-12 induction. Eur. J. Immunol. 34:1342-1350. [DOI] [PubMed] [Google Scholar]
- 23.Forman, D. 1991. Helicobacter pylori infection: a novel risk factor in the etiology of gastric cancer. J. Natl. Cancer Inst. 83:1702-1703. [DOI] [PubMed] [Google Scholar]
- 24.Fritz, J. H., L. Le Bourhis, G. Sellge, J. G. Magalhaes, H. Fsihi, T. A. Kufer, C. Collins, J. Viala, R. L. Ferrero, S. E. Girardin, and D. J. Philpott. 2007. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 26:445-459. [DOI] [PubMed] [Google Scholar]
- 25.Goriely, S., M. F. Neurath, and M. Goldman. 2008. How microorganisms tip the balance between interleukin-12 family members. Nat. Rev. Immunol. 8:81-86. [DOI] [PubMed] [Google Scholar]
- 26.Guiney, D. G., P. Hasegawa, and S. P. Cole. 2003. Helicobacter pylori preferentially induces interleukin 12 (IL-12) rather than IL-6 or IL-10 in human dendritic cells. Infect. Immun. 71:4163-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansson, L. E., O. Nyren, A. W. Hsing, R. Bergstrom, S. Josefsson, W. H. Chow, J. F. Fraumeni, Jr., and H. O. Adami. 1996. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N. Engl. J. Med. 335:242-249. [DOI] [PubMed] [Google Scholar]
- 28.Harrington, L. E., R. D. Hatton, P. R. Mangan, H. Turner, T. L. Murphy, K. M. Murphy, and C. T. Weaver. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123-1132. [DOI] [PubMed] [Google Scholar]
- 29.Hatz, R. A., G. Meimarakis, E. Bayerdorffer, M. Stolte, T. Kirchner, and G. Enders. 1996. Characterization of lymphocytic infiltrates in Helicobacter pylori-associated gastritis. Scand. J. Gastroenterol. 31:222-228. [DOI] [PubMed] [Google Scholar]
- 30.Hue, S., P. Ahern, S. Buonocore, M. C. Kullberg, D. J. Cua, B. S. McKenzie, F. Powrie, and K. J. Maloy. 2006. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203:2473-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kao, J. Y., S. Rathinavelu, K. A. Eaton, L. Bai, Y. Zavros, M. Takami, A. Pierzchala, and J. L. Merchant. 2006. Helicobacter pylori-secreted factors inhibit dendritic cell IL-12 secretion: a mechanism of ineffective host defense. Am. J. Physiol. Gastrointest. Liver Physiol. 291:G73-G81. [DOI] [PubMed] [Google Scholar]
- 32.Kelsall, B. L., and F. Leon. 2005. Involvement of intestinal dendritic cells in oral tolerance, immunity to pathogens, and inflammatory bowel disease. Immunol. Rev. 206:132-148. [DOI] [PubMed] [Google Scholar]
- 33.Kelsall, B. L., and W. Strober. 1997. Dendritic cells of the gastrointestinal tract. Springer Semin. Immunopathol. 18:409-420. [DOI] [PubMed] [Google Scholar]
- 34.Kranzer, K., A. Eckhardt, M. Aigner, G. Knoll, L. Deml, C. Speth, N. Lehn, M. Rehli, and W. Schneider-Brachert. 2004. Induction of maturation and cytokine release of human dendritic cells by Helicobacter pylori. Infect. Immun. 72:4416-4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kranzer, K., L. Sollner, M. Aigner, N. Lehn, L. Deml, M. Rehli, and W. Schneider-Brachert. 2005. Impact of Helicobacter pylori virulence factors and compounds on activation and maturation of human dendritic cells. Infect. Immun. 73:4180-4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kullberg, M. C., D. Jankovic, C. G. Feng, S. Hue, P. L. Gorelick, B. S. McKenzie, D. J. Cua, F. Powrie, A. W. Cheever, K. J. Maloy, and A. Sher. 2006. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 203:2485-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurasawa, K., K. Hirose, H. Sano, H. Endo, H. Shinkai, Y. Nawata, K. Takabayashi, and I. Iwamoto. 2000. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. 43:2455-2463. [DOI] [PubMed] [Google Scholar]
- 38.Lundgren, A., C. Trollmo, A. Edebo, A. M. Svennerholm, and B. S. Lundin. 2005. Helicobacter pylori-specific CD4+ T cells home to and accumulate in the human Helicobacter pylori-infected gastric mucosa. Infect. Immun. 73:5612-5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luzza, F., T. Parrello, G. Monteleone, L. Sebkova, M. Romano, R. Zarrilli, M. Imeneo, and F. Pallone. 2000. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J. Immunol. 165:5332-5337. [DOI] [PubMed] [Google Scholar]
- 40.Manel, N., D. Unutmaz, and D. R. Littman. 2008. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 9:641-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marshall, B. J. 1995. The 1995 Albert Lasker Medical Research Award. Helicobacter pylori. The etiologic agent for peptic ulcer. JAMA 274:1064-1066. [DOI] [PubMed] [Google Scholar]
- 42.McColl, K. E., and E. el-Omar. 1996. Helicobacter pylori and disturbance of gastric function associated with duodenal ulcer disease and gastric cancer. Scand. J. Gastroenterol. Suppl. 215:32-37. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell, P., C. Germain, P. L. Fiori, W. Khamri, G. R. Foster, S. Ghosh, R. I. Lechler, K. B. Bamford, and G. Lombardi. 2007. Chronic exposure to Helicobacter pylori impairs dendritic cell function and inhibits Th1 development. Infect. Immun. 75:810-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizuno, T., T. Ando, K. Nobata, T. Tsuzuki, O. Maeda, O. Watanabe, M. Minami, K. Ina, K. Kusugami, R. M. Peek, and H. Goto. 2005. Interleukin-17 levels in Helicobacter pylori-infected gastric mucosa and pathologic sequelae of colonization. World J. Gastroenterol. 11:6305-6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohammadi, M., J. Nedrud, R. Redline, N. Lycke, and S. J. Czinn. 1997. Murine CD4 T-cell response to Helicobacter infection: TH1 cells enhance gastritis and TH2 cells reduce bacterial load. Gastroenterology 113:1848-1857. [DOI] [PubMed] [Google Scholar]
- 46.Nishi, T., K. Okazaki, K. Kawasaki, T. Fukui, H. Tamaki, M. Matsuura, M. Asada, T. Watanabe, K. Uchida, N. Watanabe, H. Nakase, M. Ohana, H. Hiai, and T. Chiba. 2003. Involvement of myeloid dendritic cells in the development of gastric secondary lymphoid follicles in Helicobacter pylori-infected neonatally thymectomized BALB/c mice. Infect. Immun. 71:2153-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noach, L. A., N. B. Bosma, J. Jansen, F. J. Hoek, S. J. van Deventer, and G. N. Tytgat. 1994. Mucosal tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 production in patients with Helicobacter pylori infection. Scand. J. Gastroenterol. 29:425-429. [DOI] [PubMed] [Google Scholar]
- 48.Oppmann, B., R. Lesley, B. Blom, J. C. Timans, Y. Xu, B. Hunte, F. Vega, N. Yu, J. Wang, K. Singh, F. Zonin, E. Vaisberg, T. Churakova, M. Liu, D. Gorman, J. Wagner, S. Zurawski, Y. Liu, J. S. Abrams, K. W. Moore, D. Rennick, R. de Waal-Malefyt, C. Hannum, J. F. Bazan, and R. A. Kastelein. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13:715-725. [DOI] [PubMed] [Google Scholar]
- 49.Rescigno, M., M. Urbano, B. Valzasina, M. Francolini, G. Rotta, R. Bonasio, F. Granucci, J. P. Kraehenbuhl, and P. Ricciardi-Castagnoli. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2:361-367. [DOI] [PubMed] [Google Scholar]
- 50.Rittig, M. G., B. Shaw, D. P. Letley, R. J. Thomas, R. H. Argent, and J. C. Atherton. 2003. Helicobacter pylori-induced homotypic phagosome fusion in human monocytes is independent of the bacterial vacA and cag status. Cell. Microbiol. 5:887-899. [DOI] [PubMed] [Google Scholar]
- 51.Sebkova, L., A. Pellicano, G. Monteleone, B. Grazioli, G. Guarnieri, M. Imeneo, F. Pallone, and F. Luzza. 2004. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in Helicobacter pylori-infected human gastric epithelial cells. Infect. Immun. 72:5019-5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma, S. A., M. K. Tummuru, M. J. Blaser, and L. D. Kerr. 1998. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J. Immunol. 160:2401-2407. [PubMed] [Google Scholar]
- 53.Siegemund, S., N. Schutze, M. A. Freudenberg, M. B. Lutz, R. K. Straubinger, and G. Alber. 2007. Production of IL-12, IL-23 and IL-27p28 by bone marrow-derived conventional dendritic cells rather than macrophages after LPS/TLR4-dependent induction by Salmonella enteritidis. Immunobiology 212:739-750. [DOI] [PubMed] [Google Scholar]
- 54.Sims, J. E. 2002. IL-1 and IL-18 receptors, and their extended family. Curr. Opin. Immunol. 14:117-122. [DOI] [PubMed] [Google Scholar]
- 55.Sommer, F., G. Faller, P. Konturek, T. Kirchner, E. G. Hahn, J. Zeus, M. Rollinghoff, and M. Lohoff. 1998. Antrum- and corpus mucosa-infiltrating CD4(+) lymphocytes in Helicobacter pylori gastritis display a Th1 phenotype. Infect. Immun. 66:5543-5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trinchieri, G., and P. Scott. 1995. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions. Res. Immunol. 146:423-431. [DOI] [PubMed] [Google Scholar]
- 57.Vanaudenaerde, B. M., W. A. Wuyts, L. J. Dupont, D. E. Van Raemdonck, M. M. Demedts, and G. M. Verleden. 2003. Interleukin-17 stimulates release of interleukin-8 by human airway smooth muscle cells in vitro: a potential role for interleukin-17 and airway smooth muscle cells in bronchiolitis obliterans syndrome. J. Heart Lung Transplant. 22:1280-1283. [DOI] [PubMed] [Google Scholar]
- 58.Viala, J., C. Chaput, I. G. Boneca, A. Cardona, S. E. Girardin, A. P. Moran, R. Athman, S. Memet, M. R. Huerre, A. J. Coyle, P. S. DiStefano, P. J. Sansonetti, A. Labigne, J. Bertin, D. J. Philpott, and R. L. Ferrero. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5:1166-1174. [DOI] [PubMed] [Google Scholar]
- 59.Volpe, E., N. Servant, R. Zollinger, S. I. Bogiatzi, P. Hupe, E. Barillot, and V. Soumelis. 2008. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 9:650-657. [DOI] [PubMed] [Google Scholar]
- 60.Wallace, J. L. 1991. Possible mechanisms and mediators of gastritis associated with Helicobacter pylori infection. Scand. J. Gastroenterol. Suppl. 187:65-70. [PubMed] [Google Scholar]
- 61.Wilson, N. J., K. Boniface, J. R. Chan, B. S. McKenzie, W. M. Blumenschein, J. D. Mattson, B. Basham, K. Smith, T. Chen, F. Morel, J. C. Lecron, R. A. Kastelein, D. J. Cua, T. K. McClanahan, E. P. Bowman, and M. R. de Waal. 2007. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 8:950-957. [DOI] [PubMed] [Google Scholar]
- 62.Yamaoka, Y., M. Kita, T. Kodama, N. Sawai, and J. Imanishi. 1996. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology 110:1744-1752. [DOI] [PubMed] [Google Scholar]
- 63.Yen, D., J. Cheung, H. Scheerens, F. Poulet, T. McClanahan, B. McKenzie, M. A. Kleinschek, A. Owyang, J. Mattson, W. Blumenschein, E. Murphy, M. Sathe, D. J. Cua, R. A. Kastelein, and D. Rennick. 2006. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116:1310-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]