

Abstract
Tuberculosis remains a leading cause of human mortality. The emergence of strains of Mycobacterium tuberculosis, the causative agent, that are resistant to the major frontline antitubercular drugs increases the urgency for the development of new therapeutic agents. Sequencing of the M. tuberculosis genome revealed the existence of twenty cytochrome P450 enzymes, some of which are potential candidates for drug targeting. The recent burst of studies reporting microarray-based gene essentiality and transcriptome analyses under in vitro, ex vivo and in vivo conditions highlight the importance of selected P450 isoforms for M. tuberculosis viability and pathogenicity. Current knowledge of the structural and biochemical properties of the M. tuberculosis P450 enzymes and their putative redox partners is reviewed, with an emphasis on findings related to their physiological function(s) as well as their potential as drug targets.
Keywords: Mycobacterium tuberculosis, cytochrome P450, azole drugs, CYP51, CYP121, CYP130, CYP128, FprA, cyclodipeptide, sulfolipid
1. Tuberculosis prevalence and importance
Tuberculosis (TB)1 has been a scourge of mankind for nearly as long as recorded history exists. Egyptian mummies show evidence of skeletal decay caused by TB and have been shown to harbor Mycobacterium tuberculosis (Mtb) DNA[1]. Hippocrates, in “Of the epidemics” written in 400 BC, describes phthisis, a disease that is consistent with TB. Phthisis is the Greek work for consumption, the term by which tuberculosis was known for many centuries. In the Seventeenth Century, Paul Bunyan labeled tuberculosis the “Captain of all these Men of Death”, reflecting its preeminence as a cause of infectious death [2]. TB is still with us today as a major worldwide threat.
The discovery of Mtb, the causative agent of TB, was announced by Robert Koch in a famous lecture on March 24, 1882 [3]. In his lecture Koch, who received the Nobel Prize in 1905 for his discoveries, reminded his audience that one in seven human beings died of tuberculosis. The fifty years following Koch's groundbreaking work led to the development of the antituberculosis drugs that we still use to treat this disease, starting with isoniazid. The availability of effective drugs, together with improvements in economics, medicine and public health, have led to a gradual decrease in the incidence of TB in industrialized nations. However, worldwide, TB continues to be a leading cause of death, as the World Health Organization fact sheet for 2008 estimates that there were 9.2 million new TB cases in 2006, 1.7 million people died of the disease in that year, one-third of the world's population carries the latent (dormant) form of the disease, and one in ten people infected with latent Mtb bacilli will become sick with active TB in their lifetime [4]. Furthermore, TB has again become a concern in industrialized nations due to the emergence of resistant (DR), multiply resistant (MDR), and more recently extremely resistant (XDR) strains of Mtb.
The resumption of research on TB, driven by the reemergence of the disease as a concern in developing countries, has been greatly assisted by the sequencing of the Mtb genome [5]. This has facilitated a worldwide search for new drug targets in Mtb and renewed efforts to find new therapeutic approaches. As outlined below, the cytochrome P450 enzymes, of which there are twenty in this organism, are of potential interest in this context.
2. Cytochromes P450
2.1 Basic mechanism
At the center of P450 catalysis is a highly conserved protein scaffold, the “P450 fold”, working in unison with an active site heme (iron protoporphyrin IX) cofactor [6]. The typical P450 reaction is mono-oxygenation in which one of the oxygen atoms of molecular oxygen is inserted into an organic substrate while the second oxygen atom undergoes reduction to water. However, there are other typical P450-catalyzed reactions, including heteroatom oxidation and epoxidation [7]. For many years researchers have sought to understand how these ubiquitous hemeproteins can efficiently catalyze the oxidation of non-activated hydrocarbons with high stereo- and regio-specificity, whereas similar uncatalyzed reactions require harsh reaction conditions. Many aspects of the complex reaction cycle are agreed upon but others are not yet fully understood. A high valent iron(IV)-oxo intermediate radical π-cation species, Compound I, has been established as the critical species responsible for oxygen insertion in P450 enzymes [7-9].
The P450 reaction cycle is a complex orchestration of events that has been fine-tuned through evolution. In the resting state of the enzyme the heme iron is typically in octahedral coordination: the planar ligands are provided by the four nitrogen lone pairs of electrons from the heme skeleton and a strictly conserved cysteine thiolate acts as the fifth ligand and appears to largely modulate P450 reactivity [10]. The bound heme cofactor provides a good spectroscopic “handle” to observe changes within the active site. The cysteine thiolate gives rise to the characteristic Soret absorption at 450 nm when the ferrous form of the enzyme is complexed with carbon monoxide [11, 12]. In most cases a water molecule serves as the sixth axial ligand (distal ligand), but there are P450 enzymes for which the equilibrium is shifted towards not having a water coordinated to the iron atom [13]. Substrates typically displace the aqua ligand upon binding in the active site and the resulting spectroscopic signature is called a “Type-I” shift. Molecules such as azole inhibitors can also displace the aqua ligand but they often coordinate instead to the heme iron through lone pair of electrons from a heteroatom, which is called a “Type-II” spectral shift [14, 15]. A complete catalytic cycle involves transient changes of the iron oxidation state and coordination geometry that lead to activation and scission of the oxygen-oxygen bond accompanying oxygen insertion into its substrate. A typical reaction cycle is shown in Figure 1 and involves several consensus steps:
Substrate binding: the resting state of P450 enzymes is the ferric (III) low-spin state in which the iron is six coordinated, with the axial ligands provided by a cysteine thiolate and usually a water molecule. Upon substrate binding the bound water is expelled to produce a five-coordinated ferric high spin species.
Production of the five-coordinated high-spin species results in the iron atom moving out of plane of the porphyrin ring concomitant with elevation of the redox potential.
The elevated redox potential of the ferric 5-coordinated heme enables the first of two single electron reductions to occur, the first of which generates the ferrous 5-coordinated heme.
Molecular oxygen next binds to the ferrous enzyme and results in formation of the ferrous 6-coordinated dioxygen species.
A second electron reduction of the heme iron produces the ferric dioxo species.
Protonation of the ferric dioxo species produces the ferric peroxide complex called Compound 0. It is possible to bypass the preceding catalytic steps by supplying peroxide that can directly form Compound 0.
Delivery of a second solvent-derived proton leads to heterolytic cleavage of the O-O bond with the loss of water to generate the iron (IV) oxo-porphyrin π-cation radical termed Compound I (Cpd I).
Cpd I, abstracts a hydrogen atom from the substrate to form a ferryl-hydroxo species, called Compound II, and a substrate centered radical.
Subsequent “rebound” of the ferryl-hydroxo intermediate creates the ferric heme and product, regenerating the ferric low spin aqua complex.
Figure 1. General P450 catalytic cycle depicting oxygen activation and subsequent hydroxylation of substrate (RH) into product (ROH).
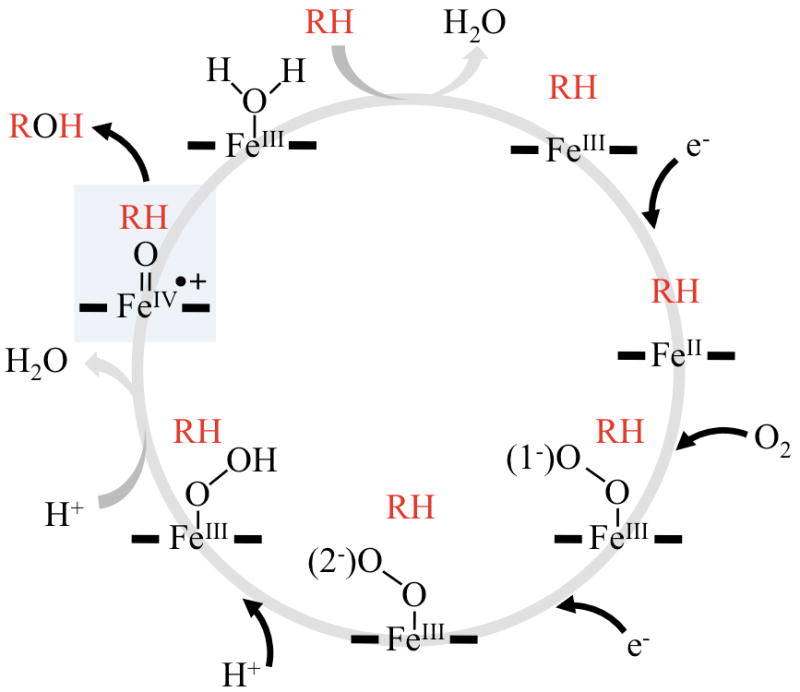
For simplicity the cysteine thiolate proximal ligand is omitted and thick horizontal lines denote the heme protoporphyrin framework. The shunt pathway is depicted by the dotted line and Compound I is highlighted in blue.
2.2 Electron donation to P450 enzymes
The P450 reaction cycle depicted in Figure 1 requires two precisely delivered electrons to the heme iron [16]. These reducing equivalents are most often provided by nicotinamide adenine dinucleotide NAD(P)H and their delivery to the active site is mediated by accessory redox proteins. Two primary types of redox systems have evolved to shuttle electrons to the heme iron [16]. The first kind, called Class I, employs ferredoxins (iron-sulfur cluster containing enzymes) or a flavodoxin (FMN-containing proteins) in conjunction with a separate reductase protein that uses flavin adenine dinucleotide (FAD) as its cofactor. The Class I systems are used by organisms with soluble P450 enzymes, such as bacteria, but they are also found in higher organisms, where they are associated with the mitochondrial membrane. The second type of redox system, called Class II, is exclusively membrane bound and possesses two different flavin-binding domains within a single polypeptide. The mammalian cytochrome P450 reductases (CPRs) are typical of a Class II P450 reductase system and these diflavin reductases, like the Type I systems, contains an FAD-bound reductase domain; however, in Type II systems they are fused to an FMN-containing domain, forming a single CPR redox protein. There are also examples of P450 enzymes, such as CYP102A1 (P450BM3), that contain a CPR-like domain fused to the P450 domain [17-19].
3. Mycobacterium tuberculosis P450 enzymes – genomic and proteomic insights
Identification of virulence factors is a critical step for understanding the biology of a pathogen. In this regard, the sequencing of the whole Mtb genome was a key step towards achievement of this goal [5]. The identification and study of operons can facilitate the assignment of specific genes to known biosynthetic pathways. Targeted gene disruption and transposon insertion mutagenesis (TRaSH) are widely used to unravel the function of individual proteins and to identify essential genes for in vitro, ex vivo and in vivo growth. Transcriptomic and proteomic studies are also used to study how Mtb cells respond to different conditions at the whole genome level. In the next sections, we summarize the most relevant data concerning Mtb P450 enzymes that was obtained from genome comparisons and transcriptional and protein expression profiles.
3.1 The genome of Mtb is rich in lipid-metabolizing and P450 enzymes
The cell envelope of Mtb is unique and is associated with its pathogenicity [20]. Figure 2 shows a schematic representation of the structure of the cell envelope. Two of its most prominent features are the presence of (a) arabinogalactan-mycolate covalently linked to the cell wall peptidoglycan via a phosphodiester bond located on the inner leaflet of the outer membrane [21-23] and (b) a free glycolipid called trehalose dimycolate (TDM) on the surface of the cells [24, 25]. These particular structures provide a thick layer of lipid on the outer part of the cell that protects it against antibiotics, toxic chemicals and the host's immune system [20]. Mycolic acids are the major constituents of this protective barrier and are essential for survival, virulence and antibiotic resistance [26]. Inhibitors of mycolic acid biosynthesis, such as isoniazid (INH), ethambutol (EMB) and pyrazinamide (PZA), are still in the frontline of antitubercular drugs [27]. The most remarkable substances intercalating the cell wall core and associated with the mycolic acids are the phosphatidylinositol-containing glycolipids, such as the lipomannans and the lipoarabinomannans [28]. In addition, among the unique features of the envelope of pathogenic mycobacteria are the complex lipids esterified with up to five multiple methyl-branched long-chain fatty acids. In Mtb, these lipids include the phenolphthiocerol and phthiocerol dimycocerosates (PDIM), and the trehalose ester families that include sulfatides (SL), diacyltrehaloses (DAT), triacyltrehaloses (TAT) and polyacyltrehaloses (PAT) [29].
Figure 2. Schematic representation of the cell envelope of Mtb.
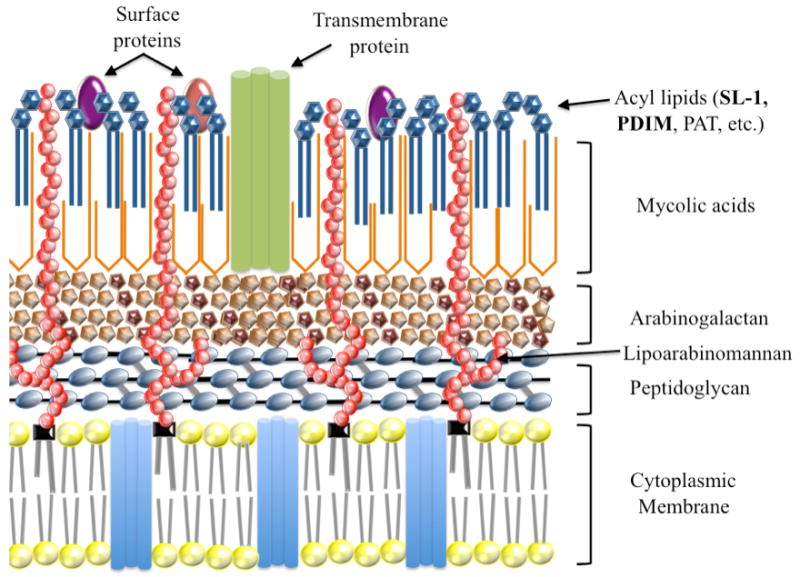
The sequence of the Mtb genome confirms the importance of lipid metabolism in this human pathogen [5]. From the 4,411,529 base pairs, a very large proportion of the coding capacity is devoted to the production of enzymes involved in lipogenesis [29-34] and lipolysis [35-37]. Interestingly, the sequence of the genome also revealed the existence of twenty putative cytochrome P450 enzymes, suggesting that at least some of these P450s could be involved in anabolism and/or catabolism of lipids. Of these, only CYP51B1 was functionally assigned based on its significant sequence similarity to eukaryotic CYP51 enzymes and its sterol 14α-demethylase catalytic activity. With the exception of CYP135A1 and CYP135B1, which show 40% identity, the percentage identity among the other Mtb P450 enzymes is much lower and averages around 30%. A simple phylogenetic analysis performed on the twenty Mtb and other bacterial P450s reveals that some of the Mtb P450 enzymes possess significant similarities with known hydroxylases of fatty acids and isoprenoid-derived compounds (Figure 3). For instance, CYP132, which also possesses low, but significant, similarity with the fatty-acid hydroxylases of the mammalian CYP4 family, clearly clusters with CYP102 (P450BM3) from Bacillus megaterium. Moreover, the phylogenetic analysis (Figure 3) indicates that CYP124A1, CYP125A1 and CYP126A1 are grouped with the α-terpineol hydroxylase CYP108 (P450terp) and linalool hydroxylase CYP111 (P450lin), whereas CYP143A1 clusters with the camphor hydroxylase CYP101 (P450cam).
Figure 3. Phylogenetic analysis of the amino acid sequences of the twenty Mtb P450 enzymes.

3.2 Comparison with other actinomycete genomes
At the time of the publication of the Mtb genome, the presence of twenty P450 enzymes was unprecedentedly high for a bacterial genome [5]. Currently, completed and on-going sequencing projects of related actinomycete genomes show that most of these bacteria are P450-rich and many Mtb P450 orthologs have been identified in them (Table S1, supporting material). The first striking observation is the large difference in the number of P450s in these genomes. Indeed, only one P450, sharing only 27% identity with Mtb CYP123A1, is found in Corynebacterium glutamicum, whereas the P450 complement of Mycobacterium gilvum and Mycobacterium sp. MCS is much more complex, as they possess 58 and 60 putative P450-encoding genes, respectively. While CYP123A1, CYP124A1, CYP125A1, CYP126A1, CYP136A1 and CYP51B1 are the most conserved P450s in actinomycetes, CYP121A1, CYP128A1, CYP141A1, and to lesser extent CYP135A1 appear to be unique to the Mtb complex, which includes three other TB-causing mycobacteria: Mycobacterium bovis, Mycobacterium africanum and Mycobacterium microti. These latter four P450 enzymes share low, albeit significant, identity (< 35%) with P450 enzymes from diverse streptomyces species, suggesting that they are not the result of horizontal transfers. On the other hand, it is intriguing that these three proteins have evolved to such a degree, presumably to accomplish unique and still unknown functions in Mtb.
3.3 Survey of transposon site hybridization mutagenesis, DNA microarray and proteomic analyses
TraSH studies were carried out in order to identify Mtb genes that are essential for growth both in vitro under laboratory conditions and in vivo during macrophage infection. It was found that a number of the genes essential for Mtb growth are conserved in the genome of M. leprae, which has undergone drastic gene reduction and decay [38]. A summary of TRaSH mutagenesis with the focus on Mtb P450 enzymes is presented in Table S2 (supporting material). Of interest is the report of the first screening of genes required for growth under normal laboratory conditions with the clinical H37Rv strain [38]. With the exception of cyp128A1, none of the other P450 enzymes was predicted to be essential for in vitro growth. Using a highly similar approach, Lamichhane and co-workers also screened another clinical strain, CDC1551 [39], and confirmed the non-essentiality of most P450 enzymes (Table S2, supporting material). However, cyp128A1 was not among the list of genes predicted with high probability to be essential in this second study. The discrepancies are probably due to limited gene coverage within their library. In a third study, the same library of mutants generated in H37Rv strain was screened for survival during infection in mice [40] and interestingly, only cyp125A1 was experimentally shown to be important for in vivo growth (Table S2, supporting material). The importance of CYP125A1 was independently confirmed using a similar approach [41]. Since a cyp128A1 mutant could not be initially obtained during in vitro growth, it can be only presumed that it plays an important role during infection (Table S2, supporting material). In a fourth study, it was shown that none of the P450-encoding genes, including cyp128A1, are required for adaptation and survival in macrophages. However, a detailed analysis of these microarray data highlights a significant attenuation effect for cyp123A1 and cyp128A1 mutants and very high levels of variability in the data obtained for cyp121A1, cyp123A1 and cyp137A1 (260 to 420-fold difference). These large variations indicate that the inactivation of genes encoding for proteins involved in key cellular functions may provokes stochastic gene expression, which has important consequences for cellular function, being beneficial or detrimental for normal stress responses and metabolism [42].
To date, cyp121A1 is the only P450-encoding gene that has been reported to be successfully knocked-out by homologous recombination [43]. However, the deletion of cyp121A1 was conditional to the preliminary introduction of a second copy of the gene in the chromosome. This requirement indicates that CYP121A1 is essential for growth under in vitro conditions. This result, which contradicts what was previously observed in the TRaSH studies [38, 39], will be further discussed later.
Many profiles of Mtb gene expression have been obtained under a variety of in vitro, ex vivo and in vivo conditions. An up-to-date summary of key facts on Mtb P450 enzymes is presented in Table 1. Of particular interest are the studies performed under conditions that mimic the dormant phase of Mtb infection [44-47]. In order to identify novel Mtb drug targets, a meta-analysis of multiple published data sets was performed using gene expression DNA microarray experiments that modeled infection leading to and including the dormant state, along with data from genome-wide insertional mutagenesis that examined gene essentiality [48]. Using a sophisticated scoring system a list of potential candidates for targeting by drugs that are classified according to different major metabolic pathways was established. It is noteworthy that none of the P450 enzymes is listed as such, but some of them obtained a good score, with CYP125A1, CYP144A1 and CYP123A1 at the top of the list. The CYP128A1-encoding gene was also shown to be up-regulated under conditions that simulate nutrient depletion during the latent state of infection [49]. Also of interest, the cyp124A1 gene was identified among a group of genes differentially expressed in mouse lungs, with an average 2-fold decrease in expression [50].
Table 1. Summary on experimental data and key facts about Mtb P450 enzymes.
| CYP/gene | Microarray/Proteomics and other key facts |
|---|---|
| CYP121A1 (Rv2276) | Induced in isoniazid- and thiolactomycin treated cells [164]. Partially or completely deleted in many clinical H37Rv isolates [93]. Inactivation by transposon-insertion mutagenesis confers resistance to beta-lactams [92]. Possible role in virulence [83, 165] |
| CY123A1 (Rv0766c) | Up-regulated by high temperatures and mRNA levels are higher in the phoP mutant [110]. Part of a putative operon with CYP51 and a ferredoxin. Protein detected in the membrane fraction [51]. Potential candidates for drug targeting [48]. |
| CYP124A1 (Rv2266) | Gene located near cyp128 and a sulfotransferase sft3. Expression repressed in infected mouse lungs [50]. |
| CYP125A1 (Rv3545c) | Induced in macrophage [46]. Essential for infection in mice [40]. Predicted to be in the kstR regulon [166]. Part of an operon with fadE28, fadE29, Rv3542c, Rv3541c and ltp2 [41]. Up-regulated during infection of dendritic cells [114]. Potential candidates for drug targeting [48]. |
| CYP126A1 (Rv0778) | Part of a putative operon with purB, a probable adenylosuccinate lyase PurB. Located near essential genes encoding enzymes involved in the de novo biosynthesis of purine. |
| CYP128A1 (Rv2268c) | Up-regulated after starvation. Part of a putative operon with sft3, a sulfotransferase involved in biosynthesis of a sulfolipid S881 [108]. |
| CYP130A1 (Rv1256c) | Absent from M. bovis and M. bovis BCG strains (deletion RD10) [84]. |
| CYP132A1 (Rv1394c) | Possible role in virulence. Transcription controlled by adjacent AraC transcription factor [167]. Induced after 30 min of post-diamide stress [168]. Up-regulated during infection of dendritic cells [114]. |
| CYP135A1 (Rv0327c) | Conserved only in Mtb complex strains and distantly related Nocardioides sp. Induced after 30 min of post-diamide stress [168]. |
| CYP135B1 (Rv0568) | Protein detected in the cytosol fraction [51]. |
| CYP136A1 (Rv3059) | Distantly related to CYP51. Possibly involved in lipid degradation. Located near a gene (fadE22) encoding for an acyl-CoA dehydrogenase. |
| CYP137A1 (Rv3685c) | Protein detected in the membrane fraction [51]. |
| CYP138A1 (Rv0136) | Up-regulated by high temperatures [109]. Up-regulated after 2h incubation in the presence of lung surfactant [111]. Up-regulated under iron-limitation conditions [169]. |
| CYP139A1 (Rv1666c) | Adjacent to pks17, pks9 and pks11 and a putative macrolide transporter [82]. |
| CYP140A1 (Rv1880c) | Closest relative to the only P450 enzyme in M. leprae. |
| CYP141A1 (Rv3121) | Absent from M. bovis and M. bovis BCG strains (RD5)[84]. Up-regulated after 2h incubation in the presence of lung surfactant [111]. |
| CYP142A1 (Rv3518c) | Predicted to be in the kstR regulon [166]. Pseudogene in M. bovis and M. bovis BCG due to a 2-bp deletion causing a frame-shift. Protein detected in the cell wall fraction [51] |
| CYP143A1 (Rv1785c) | Adjacent to a probable ferredoxin (Rv1786). |
| CYP144A1 (Rv1777) | Up-regulated during infection of dendritic cells [114]. Potential candidate for drug targeting [48]. |
| CYP51B1 (Rv0764c) | Shows sterol 14α-demethylase activity [63]. Possible role in host sterol/steroid metabolism. |
Proteomic approaches are also commonly used to study the biology of Mtb, as the cellular localization of enzymes may provide useful information on their physiological function(s). In this regard, a proteomics study of the different cellular fractions revealed the location of at least four P450 enzymes [51]. CYP135B1 peptides were detected in the cytosolic fraction, whereas CYP142A1 was found in the cell wall. Although bacterial P450 enzymes are not known to possess an N-terminal transmembrane domain and are predicted to be soluble, it is intriguing that CYP123A1 and CYP137A1 are actually associated with the membrane fraction.
Finally, in order to provide a more complete picture of the protein and gene expression of P450 enzymes in Mtb, it should be noted that there are also studies that have not shown any significant evidence of P450 expression (protein or gene) under various conditions [52-56]. Clearly more research is required to understand cytochrome P450 biochemistry in vivo and how it relates to Mtb growth, infection and persistence.
4. Structural and biochemical analysis of individual enzymes
Of the twenty Mtb P450 enzymes, only CYP51B1, CYP121A1 and CYP130A1 have been structurally and biochemically characterized. Recombinant CYP125A1 and CYP142A1 have also been expressed and purified from E. coli. In the following sections we will review CYP51B1, CYP121A1, CYP130A1, CYP125A1, CYP128A1, CYP124A1, and CYP142A1 separately. A last section will then summarize the current knowledge of the relatively uncharacterized Mtb P450 enzymes.
4.1 CYP51B1: the first member of the CYP51 family found in prokaryotes
The membranous eukaryotic CYP51B1 enzymes, which catalyze the 14α-demethylation of sterol substrates, are conserved in all phyla. Sterol 14α-demethylation, a general feature of sterol biosynthetic pathways in eukaryotes, has been known and studied for more than three decades [57-60]. At the dawn of the new millennium, the orthologous nature of the first prokaryotic CYP51-like gene in Mtb was confirmed phylogenetically [61] and biochemically [62, 63]. CYP51 is not unique to Mtb since it is conserved in the mycobacterium genus [64, 65] and other actinomycetes-related species (Table S1, supporting material). An ortholog was also found and characterized in an obligate methanotrophic gram-negative bacterium Methylococcus capsulatus [66]. This CYP51 protein represents a new class of P450 enzyme consisting of the P450 domain naturally fused to a ferredoxin domain at the C-terminus via an alanine-rich linker.
4.1.1 Spectroscopic features
Spectroscopic analyses performed on CYP51B1 expressed as a recombinant protein in E. coli are consistent with the presence of a cysteine thiolate and aqua-ligated heme iron atom [62, 67]. Reduction of the protein in the presence of carbon monoxide leads to formation of the ferrous-CO complex typical of a P450 protein with a Soret band at 448.5 nm, which denatures to form a P420 species with a Soret maximum at 421.5 nm. The P450 form is further destabilized at lower pH, consistent with protonation of the cysteine thiolate (Cys394) to a thiol that underlies the P450-P420 transition. On the other hand, the binding of the substrate analog, estriol, stabilizes the thiolate character of the heme proximal ligand [67, 68]. Analysis of reoxidized CYP51B1 demonstrates that the protonation of Cys394 is reversible [67].
4.1.2 Crystal structure of CYP51B1: the first member of the CYP51 family with a solved 3D-structure
CYP51B1 is an example of a P450 enzyme in which conformational flexibility and substrate access are not restricted to the FG region, as opposed to what is commonly reported for a number of bacterial P450 enzymes [69-72] and mammalian CYP2B4 [73-75]. Indeed, the crystal structures of ligand-free, 4-phenylimidazole-, fluconazole- and estriol-bound CYP51B1 reveal a substrate access opening through a highly bent I-helix and an extended conformation of the B-C loop stretched over the protein surface [76, 77]. In the ligand-free state, the C helix is disordered and no electron density is observed for residues 91–100. In contrast, in the 4-phenylimidazole bound structure, the C helix is well defined, whereas co-crystallization with fluconazole results again in loss of the C-helix structure. Although the FG substrate channel is also apparent in the CYP51B1 structures, its entrance is closed at the surface [77]. However, motion in the FG region detected by fluorescence resonance energy transfer (FRET) spectroscopy upon the binding of lanosterol suggests that large-scale conformational changes may also be functionally important [78]. Movements of the BC- and FG-regions are thought to be generally important for the dynamics of the entry/exit of the substrate/product during catalysis [79].
4.1.3 Substrate Specificity of CYP51B1
The enzymatic activity of CYP51B1 was demonstrated in vitro [58, 63, 80, 81]. CYP51B1, just like the related isoforms from plants and Trypanosoma brucei, exhibits specificity for the C4-monomethylated sterol obtusifoliol, whereas the fungi/animal and Trypanosoma cruzi isoforms prefer C4-dimethylsterols. The structure of estriol-bound CYP51B1 allowed determination of both the orientation of the substrate within the active site and the protein contacts with the invariant 3β-hydroxyl group of the sterol, and identified Phe78 as a key discriminator between 4α-methylated and 4α,4β-dimethylated substrates. As pointed out previously [82], the relevance of this activity to Mtb biology is not clear, since a complete sterol biosynthetic pathway does not exist in this organism.
4.2 CYP130: absence in Mycobacterium bovis
CYP130A1 is absent from the virulent M. bovis strain and its avirulent counterpart M. bovis BCG (Table S1, supporting material), suggesting that it is not essential for growth, but may be relevant for virulence and infectivity of Mtb towards the human host [83]. Indeed, the chromosomal segment encompassing Rv1255c-Rv1257c constitutes the region of difference 10 (RD10, Behr's nomenclature), one of the 16 RDs found in M. bovis BCG [84, 85]. The surrounding genes, Rv1255c and Rv1257c, encode for a TetR-like transcription factor and a putative FAD-containing lactate dehydrogenase enzyme, respectively. The sequence and the genomic organization of the genes encoding these two proteins are also well maintained in all actinomycetes species possessing CYP130A1 (Table S1, supporting material), with the exception of the distantly-related Rhodococcus sp. RHA1, suggesting that these three genes may constitute a functional unit. Moreover, in recent studies, the cellular immune response was evaluated in response to antigens encoded by genes predicted in eleven RDs, including RD10 [86]. Specifically, the exposure of peripheral blood mononuclear cells to synthetic peptides covering the sequences of RD10 resulted in the secretion of a higher amount of the Th1 cytokine, IFNγ, compared to the amount of the Th2 cytokine, IL-10. High IFNγ/IL-10 ratios were proposed to strongly correlate with protection and TB cure, whereas low ratios correlate with disease severity [86, 87]. In light of these observations, the results of these studies suggest that RD10 plays a role in mediating protection against Mtb rather than contributing to virulence, and thus could be useful in searching for new vaccine candidates.
4.2.1 Crystal structures of the ligand-free and econazole-bound protein
Recently, our laboratory reported purification and determination of the crystal structure of CYP130A1 in the ligand-free and econazole-bound forms [88]. In this P450 enzyme, econazole was shown to coordinate directly to the heme iron via an imidazole nitrogen atom, albeit with a slight distortion from linearity of the Fe-N (azole) bond with respect to the Fe-S (cysteine) linkage. Ligand-free CYP130 crystallizes in an “open” conformation as a monomer, whereas the econazole-bound form crystallizes in a “closed” conformation as a dimer (Figure 4A). Conformational changes enabling the “open-closed” transition involve repositioning of the BC-loop and the F and G helices that envelop the inhibitor in the binding site and reshape the protein surface (Figure 4B). Crystal structure analysis also showed that a portion of the BC-loop relocates as much as 18 Å between the open and closed conformations (Figure 4B). The binding of econazole to CYP130A1 involves a conformational change and is driven by hydrophobic interactions with amino acid residues in the active site as well as coordination to the heme iron.
Figure 4. Crystal structure of ligand-free and econazole-bound CYP130A1.

(A) The dimerization interface of the econazole-bound structure (2000 Å3) is formed largely via interactions between the G helices in anti-parallel orientations, overlapping N termini of the I helices, and multiple contacts in the BC-loop region. The monomers are colored in green and blue, heme is in blue and red, and econazole in yellow. (B) Superimposition of the active site of ligand-free (green cartoon) and econazole-bound (cyan cartoon) structures. The residues P87, M89 and Y392 are in contact with the ligand. The heme for the ligand-free form is in red while that of the econazole-bound form is in blue. The econazole ligand is colored in yellow. The pdb files for the ligand-free and econazole-bound structures are 2UUQ and 2UVN, respectively.
4.2.2 Biophysical characterization of the interactions with antifungal azole drugs
The interactions between CYP130A1 and the antifungal azole drugs, econazole, clotrimazole, miconazole and ketoconazole were studied by UV-vis electronic absorption spectroscopy and isothermal titration calorimetry (ITC) [88]. Spectrophotometric titrations revealed that CYP130A1 binds miconazole with virtually the same binding affinity (Table 3) as econazole (1.70 - 1.93 μM), while clotrimazole and ketoconazole bind with somewhat lower affinities (13.3 - 48.0 μM). This makes it a plausible target for this class of therapeutic drugs. Binding of econazole and clotrimazole exhibits positive cooperativity that may reflect a propensity of CYP130A1 to associate into a dimeric structure. Using ITC, the binding of the azole inhibitors was found to be the result of a sequential two-step, entropy-driven endothermic process. We attribute the second binding step to conformational changes associated with CYP130A1 dimerization.
Table 3. Comparison of M. tuberculosis MIC values for selected azole antifungal drugs with the relevant KD values for Mtb CYP121A1, CYP130A1 and CYP51B1.
| Azole |
KD, μM (Hill coefficient (n), if applicable) |
MIC (μM) |
References | ||
|---|---|---|---|---|---|
| CYP51B1 | CYP121A1 | CYP130A1 | |||
| Clotrimazole | < 1.0 | < 0.2 | 13.3 ± 0.6 n = 1.95 ± 0.05 |
0.38 33a |
[43, 88, 117, 132, 135] |
| Econazole | 0.77 ± 0.04 | < 0.2 | 1.93 ± 0.03 n = 1.37 ± 0.02 |
0.25 16a |
[43, 88, 132, 135] |
| Ketoconazole | 19 ± 1.9 | 3.3 ± 0.3 | 48.0 ± 1.5 | 30 | [43, 88, 89] |
| Miconazole | 0.59 ± 0.03 | < 0.2 | 1.70 ± 0.21 | 16.6a | [43, 88, 135] |
4.3 CYP121A1: the first Mtb P450 enzyme with an identified substrate
CYP121A1 is exclusively found in strains of the Mtb complex (Table S1, supporting material) and constitutes, among the twenty P450 proteins, one of the most logical candidates for evaluation as a potential drug target. CYP121A1 has been expressed in E. coli and the protein has been purified to homogeneity [89]. Very tight binding affinity (< 0.2 μM) was measured with a variety of azole antifungal drugs, confirming CYP121A1 as a plausible Mtb target for azole drugs (Table 3).
Until recently, the catalytic activity of CYP121A1 remained unknown. However, it has now been shown that a cyclodipeptide cyclo(L-Tyr-L-Tyr) (cYY) molecule binds to CYP121A1 and is converted into a single major product in a CYP121A1 activity assay utilizing spinach ferredoxin and ferredoxin reductase as the electron donor partners [90]. NMR spectroscopic analysis of the reaction product confirmed that CYP121A1 catalyzes the formation of an intramolecular C-C bond between two tyrosyl carbon atoms of cYY (Figure 5C). The cyp121A1 gene forms a functional operon with Rv2275, which was shown to encode the cyclodipeptide synthetase enzyme responsible for the formation of cYY [91]. Also of interest, a CYP121A1-deficient mutant generated by transposon-insertion mutagenesis was shown to be resistant to β-lactams [92].
Figure 5. Structures of the active sites of the (A) fluconazole- and (B) cYY-bound CYP121A1 complexes and (C) the chemical structures of the proposed substrate and reaction product in the CYP121A1-catalyzed reaction.

For the fluconazole-bound structure, a conformer (conformer B) in which the ligand is not coordinated was chosen. The respective ligands are colored in yellow. The water molecules are labeled w1 to w5. In the fluconazole-bound structure, the azole moiety of the ligand occupies the position of w2. The pdb files for the fluconazole- and the cYY-bound structure are 2IJ7 and 3G5H, respectively.
4.3.1 Spectroscopic features
Spectroscopic analyses performed on recombinant CYP121A1 are consistent with the prototypical presence of a cysteine thiolate- and aqua-ligated heme iron atom in the ferric state [89]. As expected, the protein binds CO in the ferrous state to give the typical P450 form. As observed for CYP51B1, the P450 form of CYP121A1 is converted to a P420 species that is accentuated, but reversible, at lower pH, suggesting that protonation of the cysteine thiolate (Cys345) to a thiol occurs [68]. Laser photoexcitation of NAD(P)H was found to reduce the heme iron of CYP121A1 on the microsecond time scale. The rates of formation of the ferrous-CO complexes were determined across a range of CO concentrations and the reaction transients were found to be biphasic. Moreover, a hyperbolic dependence on CO concentration was observed, consistent with the presence of a binding site in ferric CYP121A1 for a non-coordinated CO molecule [68].
4.3.2 CYP121A1: an essential P450 enzyme for Mtb viability?
Attempts to inactivate the CYP121A1-encoding gene by homologous recombination were unsuccessful unless a second copy of the gene was first introduced in the chromosome, suggesting that CYP121A1 is essential for growth under normal laboratory conditions [43]. This last result contradicts the screening of Mtb H37Rv and CDC1551 mutants for viability using the TRaSH method [38, 39]. It was also reported that the Rv2275-cyp121A1 genes are partially or completely deleted in many clinical isolates of the H37Rv strain [93]. This discrepancy with the TRaSH studies could be explained by a negative polarity effect on the transcription of both the Rv2275 and cyp121A1 genes, resulting in the inactivation of both genes simultaneously. On the other hand, it is possible that the generation of an unmarked in-frame deletion [43] could potentially result in the detrimental accumulation of cYY, the Rv2275 reaction product. However, the role of this molecule in Mtb biology and virulence remains to be determined.
4.3.3 CYP121A1: crystal structures of the ligand-free, azole-bound and substrate-bound protein
Crystal structures of CYP121A1 were obtained for the ligand-free, fluconazole-bound and cYY-bound forms at a resolution of 1.06, 1.6 and 1.46 Å, respectively [90, 94, 95]. For the ligand-free form, this unprecedented level of resolution for a P450 structure revealed novel information relating to P450 architecture. Indeed, the heme was shown to bind in two orientations, corresponding to a 180° “flip” of the heme cofactor. Despite a large active site cavity, the region in the vicinity of the heme on the distal side is constricted, primarily as a consequence of the location of residues Arg386 and Ser237 that are H-bonded to each other and to the water molecule coordinated to the heme iron atom as a sixth ligand. For the fluconazole-bound structure, the ligand is found in two binding modes (A and B). In mode A, the azole moiety of fluconazole is coordinated to the heme iron, whereas in mode B, the azole group is slightly shifted away and bridged to the heme iron via a water molecule that is also the sixth iron ligand. As shown in Figure 5, the similarities between the structure of fluconazole-bound in mode B and the cYY-bound form are striking, with the ligands displaying the same topology. Water molecules are also observed in similar positions within the fluconazole-bound structures, with the exception of water molecule (w2) that is instead occupied by the azole group of fluconazole.
4.3.4 CYP125A1: a role in host cholesterol catabolism?
The importance of CYP125A1 was revealed by studies that showed that cyp125A1 and its associated operon (Rv3545c-Rv3540c) were induced and required during the infection of macrophages by Mtb, and that these genes are required for the survival of Mtb in infected mice [40, 41, 45, 46]. In addition, other recent work has discovered a cluster of genes in the Mtb-related soil bacterium Rhodococcus sp. strain RHA1 involved in the degradation of cholesterol [96]. Since the majority of those genes are well conserved in the Mtb genome, it is perhaps not surprising to learn from two studies [97, 98] that Mtb is capable of in vitro growth using cholesterol as a sole source of carbon. Collectively, these data suggest a key role for CYP125A1 in the survival of Mtb in the macrophage and indicate that CYP125A1 may oxidize cholesterol or other steroid-like substrates.
CYP125A1 has been expressed heterologously and the purified ferrous enzyme displays the typical Soret absorption features of P450 enzymes upon binding of CO [99, 100]. As opposed to other recombinant Mtb P450 enzymes purified so far, the ferric form is mostly in the high-spin state with a prominent Soret band centered at 390 nm. In addition, CYP125A1, like nearly all P450 enzymes, binds azole drugs (econazole, bifonazole, miconazole) with relatively high affinities (< 3 μM, unpublished data), but interestingly, as type-I ligands and not through coordination to the heme iron atom [100].
4.3.5 CYP128A1: an essential P450 enzyme with a role in sulfolipid biosynthesis?
Another exciting development with respect to Mtb P450 enzymes is related to sulfolipid biosynthesis. Mtb produces a variety of lipids, some of which are involved in modulating the immune response within infected individuals and others that encase the organism within a robust physical barrier [101-104]. Work by Bertozzi and coworkers identified several sulfotransferases within the Mtb genome [103, 105]. A sulfated metabolite, called S881, was shown to be located on the outer cell wall where it acts as a negative regulator of virulence in the mouse model of TB [106, 107]. It was also shown that the biosynthesis of S881 is strictly dependent on a PAPS (3′-phosphoadenosine 5′-phosphosulfate)-dependent sulfotransferase Sft3 (Rv2267c) [106] that is located in a predicted three gene operon containing cyp128A1 [5, 108]. The chemical structure of S881 (Figure 6) was elucidated by Holsclaw and co-workers and shown to be a menaquinone-like molecule (MK-9 DH-2) containing a linear chain consisting of nine isoprenoid units appended to a napthoquinone ring. Instead of a double bond, the second isoprenoid unit was shown to be fully saturated. A biosynthetic scheme was proposed [108], in which CYP128A1 first hydroxylates the terminal isoprene unit of MK-9 (DH-2) thus activating it for nucleophilic PAPS-dependent sulfation (Figure 6). A transposon mutant library demonstrated that CYP128A1 was essential for viability under laboratory conditions [38], but essentiality has not been established under relevant in vivo conditions. Alignment with other Mtb P450 enzymes shows that CYP128A1 and CYP136A1 each contain a stretch of approximately 100 “extra” amino acids at their respective N-termini, but it is unclear whether these polypeptide motifs are necessary for activity in vitro or in vivo. Efforts to express the soluble, folded CYP128A1 protein have, thus far, been unsuccessful (J.B. Johnston and P. R. Ortiz de Montellano, unpublished data).
Figure 6. Biosynthesis of S881.

As proposed by Holsclaw et al. the first synthetic step would be the hydroxylation of the terminal ω-position of MK-9 (DH2) by CYP128, followed by PAPS-dependent sulfation by Sft3 to form the S881 sulfolipid.
4.3.6 CYP124: a highly conserved bacterial P450 enzyme
Two other Mtb P450 enzymes CYP124A1 (Rv2266) and CYP121 (Rv2277) are located near the sft3/cyp128A1 probable operon. CYP121A1 is unlikely to be related to S881 biosynthesis given both its demonstrated ability to catalyze carbon-carbon bond formation in cyclodityrosine and its proximity to the cyclodipeptide synthase CDPs (Rv2275) [90, 91], but there is a possibility that CYP124A1 is involved. CYP124A1 flanks the sulfotransferase Sft3, albeit not in the same putative operon. Various strains of Mtb, other actinomycetes, as well as proteobacteria contain genes closely related to CYP124A1. Among the twenty Mtb CYP enzymes, CYP125A1 displays the closest homology to CYP124A1 (38%), followed by CYP126A1 with (34%). That CYP124A1 is conserved not only in mycobacteria but also across several other species suggests that it provides an important activity. The gene directly upstream of CYP124A1 (Rv2265) displays homology to the major facilitator class of proteins involved in transport across the cell membrane.
CYP124A1 has been expressed heterologously and the purified ferrous enzyme displays the typical Soret absorption features of P450 enzymes when coordinated to carbon monoxide. In addition, CYP124A1 like nearly all P450 enzymes binds azole drugs through coordination to the heme iron. Efforts underway in this laboratory to characterize CYP124A1 include identifying substrates and inhibitors, understanding the in vivo role of CYP124A1 through gene deletions, and making sense of the biochemical data from a structural standpoint (J. B. Johnston, L. M. Podust, and P. R. Ortiz de Montellano, manuscript in preparation).
4.3.7 CYP142A1 is also part of large cluster of genes involved in lipid degradation
We have expressed CYP142A1 heterologously and the purified protein binds CO in the ferrous form to give the typical Soret absorption features of P450 enzymes [99]. CYP142A1 binds the antifungal azole drugs econazole, clotrimazole, miconazole and bifonazole with high affinity (submicromolar, Ouellet and Ortiz de Montellano, unpublished data). Very recently, we reported that this protein forms a tight complex with NO in both the ferric and ferrous states [99].
4.4. More poorly characterized Mtb P450 enzymes
CYP123A1 (Rv0766c) is highly conserved among actinomycetes and proteobacteria and was found to be up-regulated in Mtb at elevated temperatures [109]. On the other hand, CYP123A1 was shown to be down-regulated in a PhoPR two-component system mutant strain of Mtb [110]. CYP141 and CYP138A1 were shown by Schwab and coworkers to be up-regulated following a brief (2 h) exposure to lung surfactant, suggesting a possible role for these during the initial stages of infection [111]. In addition, CYP141A1 and FprA are located within the Mtb genome near both a gene cluster involved in molybdopterin cofactor biosynthesis as well as the DevS/DevR regulon. Interestingly, molybdopterin is required for nitrate reductase and other enzymes involved in anaerobic metabolism and several genes involved in molybdopterin cofactor biosynthesis have also been shown to be up-regulated in the lung during mouse infection [50]. CYP138A1 is up-regulated during heat shock response at elevated temperatures [109].
Also among poorly characterized Mtb P450 enzymes is CYP126A1, which appears to be highly conserved across actinomycetes, including many pathogenic and non-pathogenic strains. This suggests that CYP126A1 and other closely conserved P450 enzymes such as CY123A1, CYP124A1, CYP125A1, CYP130A1, CYP142A1, CYP143A1, and CYP143A1 may perform important general functions (Table S1, supporting material). On the other hand, the uniqueness of CYP121A1, CYP128A1, CYP141A1, CYP132A1, and CYP135A1 within Mtb suggests that they perform more specialized functions (Table S1, supporting material). The latest report on CYP121A1 and its potential role in the biosynthesis of cyclodipeptides supports such an idea [90, 91].
Little can be inferred from the available data for the remaining Mtb P450 enzymes. CYP135B1 shows close sequence homology to CYP135A1 but little else is obvious. CYP136A1 is distant relative of sterol demethylase (CYP51) enzymes, but it is unclear if such a functional assignment is relevant. CYP137A1 is located in the genome close to one of the essential WhiB-like transcriptional regulatory proteins, WhiB4, which may act in response to redox changes or metabolic shifts [112]. CYP139 is also eukaryotic-like and notably conserved among pathogenic mycobacterial strains but absent from non-pathogenic strains. CYP139A1 is flanked by three polyketide synthase gene clusters (pks17, pks9, and pks11) and situated also adjacent to a macrolide transport protein, suggesting a possible connection of CYP139A1 in oxidative tailoring of a nascent polyketide prior to export from the cell [82]. Of the three pks gene clusters, only pks17 has been shown to be essential through screening of a transposon-mutant library [38]. The gene pks17 and the pks8 cluster are implicated in the biosynthesis of methyl-branched unsaturated fatty acids [113]. The pks11 cluster in Mtb shows homology to iterative Type III polyketide synthases [101] such as chalcone synthase. CYP143A1 and CYP144A1 are each strongly conserved across many actinomycete species including pathogenic and non-pathogen strains. CYP144A1 along with CYP125A1 and CYP132A1, was shown by transcriptional profiling to be expressed preferentially in Mtb-infected human dendritic cells after 18 h [114], providing yet another link between pathogenesis and Mtb P450 enzymes. CYP143A1 is located in the same genomic region as CY144A1, but there is little that is obvious about their potential functions. A putative ferredoxin encoded by the Rv1786 gene is located downstream of CYP143A1.
5. Mtb cytochrome P450 redox partners
The genome of Mtb encodes several ferredoxin and ferredoxin-NAD(P)H reductase-like proteins, a combination of which, in principle, should support the catalytic activity of each of the twenty putative P450 enzymes (Table 2). These redox proteins often display promiscuity in their ability to mediate electron transfer to P450 enzymes, and the ratio of P450 genes to potential redox partners within the genome also suggests this. To date only two, CYP51B1 [63] and CYP121A1, of the twenty Mtb P450 enzymes have a demonstrated catalytic activity. Exogenous spinach ferredoxin/ferredoxin reductase proteins were shown to support cyclodipeptide carbon-carbon bond formation with CYP121A1 [90]. However, the endogenous ferredoxin (Rv0763c) and ferredoxin-NADPH reductase (FprA, Rv3106) were used to reconstitute the catalytic activity of CYP51B1 [63]. Given the proximity of CYP51B1 (Rv0764c) to the ferredoxin (Rv0763c), it is perhaps not surprising that it supports CYP51B1 activity. Zanno and coworkers further demonstrated that Fdx, when reconstituted with FdrA, also supported CYP51B1 catalytic activity [115]. They found that FdrA preferentially binds and utilizes NADH over NADPH as a source of electrons. It should be noted that CYP143A1 (Rv1785c) and a putative ferredoxin (Rv1786) are also in adjacent locations in the genome, which suggests that they may be native P450/ferredoxin partners.
Table 2. Mtb cytochrome P450 redox partners.
| Protein (Rv #) | Comments |
|---|---|
| FdX (Rv0763c) | 3Fe-4S ferredoxin; supports catalysis with CYP51; mediates electron transfer from FprA; part of putative operon containing cyp123 and cyp51 [63, 115]. |
| Rv1786 (Rv1786) | putative ferredoxin; located near CYP143 (Rv1785c) [83]. |
| FdxA (Rv2007c) | putative ferredoxin, predicted 3Fe-4S or 4Fe-4S; cluster, induced by hypoxia and low pH in vitro; structure available of related (87% identical) M. smegmatis FdxA [116, 117, 120]. |
| FdxB (Rv3554) | putative fusion of ferredoxin and ferredoxin reductase domains. |
| FdxC (Rv1177) | predicted to have both 3Fe-4S and 4Fe-4S clusters; required for optimal growth in vitro, structure available of related M. smegmatis FdxA (87% identical) [38, 116]. |
| FdxD (Rv3503c) | putative ferredoxin. |
| FprA (Rv3106) | ferredoxin reductase, reduces Rv0763c to support CYP51 activity, homology to mammalian AdR, structure available [121-124, 128, 170] |
| FprB (Rv0886) | putative fusion of a ferredoxin and FprA-like domains |
| FdrA (Rv0688) | flavoprotein reductase, reduces FdX to support CYP51 activity [171]. |
Both Mtb FdxA and FdxC show high sequence homology to M. smegmatis FdxA [116]. Mtb FdxA (Rv2007c) was shown to be induced under hypoxic conditions [117] and at low pH [118]. Such conditions may mimic those encountered upon phagocytosis of Mtb. The structure of M. smegmatis FdxA demonstrated that it belongs to the class of 7Fe-ferredoxins, i.e., it contains both a 3Fe-4S and 4Fe-4S cluster. Due to the phylogenetic relationship among 7Fe and 8Fe ferredoxins it is expected that Mtb FdxA and FdxC will be structurally related [116, 119]. Further, the high sequence homology between Mtb FdxC and M. smegmatis FdxA argues that it may be the biologically relevant redox partner for Mtb FprA [120]. FdxC was shown to be required for optimal growth in vivo [38], suggesting an important role in shuttling electrons to a yet unidentified P450.
The best characterized Mtb P450-related redox protein is FprA, an NADPH-ferredoxin reductase, which is structurally and functionally related to the mammalian adrenodoxin reductase (AdR) family by approximately 40% sequence identity [116, 120-128]. Mtb FprA mediates electron transfer from NADPH to various endogenous and exogenous ferredoxin proteins and preferentially utilizes NADPH over NADH [120]. High-resolution crystal structures for FprA have been obtained for both the oxidized (1.05 Å) and reduced (1.25 Å) states. These are the first structures of Mtb P450 redox components [128]. An unusual discovery from this work was the presence of a covalently bound NADP+ cofactor [128] that was subsequently shown to be indicative of a general reaction catalyzed by other mammalian-like adrenodoxin-reductases. However, the in vivo relevance of this finding remains unclear [123].
The structural studies have furthered our understanding of the conserved critical active site residues of FprA. In particular, the importance of a histidine (His57) for efficient hydride transfer was demonstrated by site-directed mutagenesis and structural studies of the mutant enzyme [124]. Munro and coworkers also highlighted the critical role of conserved arginine residues in cofactor (FAD) reactivity [121] and Pennati and coworkers measured the interaction of FprA and NADPH by elevating the salt concentration, such that association/dissociation rate constants could be determined [122]. Furthermore, they were able to establish the rates of hydride transfer from NADPH to the enzyme-bound FAD cofactor [122]. To further understand the correlation between in vitro and in vivo experiments, Bhatt and coworkers studied the effects of cations and pH on FprA and concluded that the environment in which Mtb grows and proliferates will likely impact the structural properties of FprA [127].
Less is known about the remaining P450-related redox proteins in Mtb. Three other putative ferredoxin genes have been annotated. Interestingly FdxB appears to be a fusion of ferredoxin and ferredoxin reductase domains but FdxD encodes a ferredoxin. FprB is another fusion protein found in the Mtb genome, although the domains in this case are ferredoxin and an FprA-like domain. Additional work is needed to unravel the complex biochemistry of each of the potential redox couples, including delineating cofactor preferences and stoichiometries, as well as validating their redox properties with relevant substrates. In fact, linking the remaining 18 P450 enzymes with any specific catalytic activity would be very useful. In addition to understanding the biochemistry and mechanistic details of the redox partners of each of the Mtb P450 enzymes, it will be necessary to establish under what conditions in vivo, if any, they are essential for organism viability.
6. Inhibition of Mtb P450 enzymes
Although antibiotics against Mtb have decreased the incidence of tuberculosis in the human population significantly, the emergence of drug-resistant strains renders many current treatments ineffective. Therefore, it is urgent to identify biochemical pathways in Mtb that can serve as targets for new anti-mycobacterial drugs. In this regard, the numerous P450 enzymes maintained in the Mtb genome suggest that at least some of them play important roles in viability and virulence and consequently may be potential candidates for drug targeting.
Increasing biochemical, genetic, proteomic, and computational data are becoming available for the complex pathogen Mtb. A recently published perspective [129] highlights one strategy to harness the large volume of Mtb-related data using the TDR Targets database that prioritizes potential drug targets in pathogenic organisms. When applied to Mtb, six of the top eight (out of more than 1,500) ranked drug target candidates) involved P450 enzymes: CYP123A1, CYP124A1, CYP125A1, CYP130A1, CYP140A1, and CYP142A1. Also of interest were the rankings of CYP126A1, CYP128A1, and CYP51B1, which were included among the top 50 candidate enzymes out of more than 1,500. In total, nine of twenty Mtb P450 enzymes were placed near the top of a prioritized list of drug targets, further impetus for continued research on these intriguing enzymes.
6.1 Inhibition by antifungal azole drugs
The discovery of CYP51B1, a fungal sterol 14α-demethylase homolog, in the Mtb genome raised the possibility that antifungal azole compounds might also target CYP51B1 and other P450 enzymes to kill Mtb cells. Antifungal azole drugs were later shown to exhibit an antimycobacterial activity against Mtb and the closely related species, M. bovis and M. smegmatis, with econazole and clotrimazole being the most potent [43, 130-135]. An inhibitory activity of econazole and clotrimazole on glycopeptidolipid (GPLs) biosynthesis in M. smegmatis has been demonstrated [136]. The authors of this study proposed that an unidentified P450 enzyme may be involved in hydroxylation of the N-acyl chain in GPL biosynthesis. It should be pointed out that GPLs are not known to be found in Mtb, hence azoles drugs must target other P450 enzymes in this organism. Azole drugs coordinate tightly to the heme iron of Mtb CYP51B1, CYP130A1 and CYP121A1 [88, 133, 135], with the latter exhibiting higher affinities (submicromolar). This suggests that CYP121A1 is a particularly sensitive enzyme target for azole drugs [135], although high affinity is not the only factor that must be considered in evaluating the potential role of a P450 enzyme as a target. Furthermore, analysis of the whole Mtb P450 complement may yet reveal the existence of even better candidates than those that have already been investigated. As discussed earlier, the 3-D structures of CYP51B1-fluconazole, CYP121A1-fluconazole and CYP130A1-econazole have been determined [76, 88, 95].
Although azole drugs have proven their antimycobacterial potential under in vitro, ex vivo and in vivo conditions against murine tuberculosis, the bioavailability of this class of drugs through the oral route is problematic. In this regard, the chemotherapeutic potential of nanoencapsulated azoles against murine tuberculosis was explored and the results demonstrated that this approach may reduce the dosing by 15-fold [137]. Despite this improvement in bioavailability, the use of azole compounds (imidazoles and triazoles) as therapeutics remains problematic, as they also interfere with the normal activities of the human P450 enzymes [138-141]. Indeed, azole antifungal drugs exhibit a wide range and variety of drug-drug interactions, as they are substrates for and inhibitors of human liver P450 enzymes [142, 143]. The inhibition or induction of P450 enzymes may alter the pharmacokinetic profile of co-administered drugs, an interaction that should be avoided, whenever possible, because it can lead to toxicity or loss of efficacy [144-149].
A recent report has challenged the hypothesis that P450 enzymes are the target for azole drugs in Mtb. Indeed, the sequencing of all P450-encoding genes in an azole-resistant mutant of M. bovis BCG did not reveal the presence of any mutations, indicating that P450 enzymes are not involved in the mechanism of resistance [56]. Moreover, DNA microarrays were used to investigate the genomic response of wild-type Mtb and a bifonazole-resistant (Bif1) mutant. Surprisingly, no P450-encoding genes were differentially expressed between the two strains. Instead, only three genes, Rv0678, Rv0677c and Rv0676c, exhibited higher levels of expression in the Bif1 mutant compared to the wild-type strain [56]. Interestingly, Rv0677c and Rv0676c encode the mycobacterial membrane proteins MmpS5 and MmpL5, which have been predicted to be part of the resistance–nodulation–division (RND) family of transporters, whose role in drug efflux in Gram-negative bacteria has been well-documented [150]. Also of interest is a report on the inhibition of succinate dehydrogenase in Mtb lysates by econazole and clotrimazole [53], suggesting that this activity may also constitute a plausible target through, for instance, a direct interaction with the putative heme-containing domain of this multidomain enzyme. In this regard, the inhibition of mitochondrial functions by antifungal azoles drugs has also been reported [151].
6.2 Development of new P450 inhibitor scaffolds
New scaffolds for inhibitors of CYP51B1 were identified by high-throughput screenings (HTS) of libraries of organic compounds [152, 153]. The HTS assay was based on the changes in the P450 spectral signature associated with the either substrate binding (type I) or inhibitor binding (type II) in the active site. Of the 20,000 compounds that were screened, three top hits, alpha-ethyl-N-4-pyridinyl-benzeneacetamide (EPBA), 2-(benzo[d]-2,1,3-thiadiazole-4-sulfonyl)-2-amino-2-phenyl-N-(pyridinyl-4)-acetamide (BSPPA), and 4,4′-dihydroxybenzophenone (DHBP), were identified and analyzed spectrophotometrically. EPBA and BSPPA are both type-II ligand hits that were found to coordinate to the heme iron of CYP51B1 with higher affinity (5-fold) than fluconazole. Moreover, EPBA was shown to be specific for CYP51B1, as the two other Mtb P450 isoforms, CYP125A1 and CYP130A1, used as references bind this compound with 50- and 400-fold less affinity, respectively. On the other hand, DHBP binds as a substrate-like ligand (type-I) with an apparent affinity (29 μM) that is 3-fold higher than that measured for estriol (100 μM). Crystal structures of the CYP51B1 in complexes with EPBA-, BSPPA- and DHBP have been obtained at a resolution of 1.53, 1.50 and 1.95 Å, respectively.
Interestingly, significant inhibitory activities of EPBA and DHPB on Mtb growth in liquid medium were also observed. The killing activity of DHPB was also tested in the mouse macrophage model, resulting in a significant mycobactericidal effect. However, the high concentrations of drugs used in these assays are well above (100-fold) those for the most common antitubercular drugs, e.g. isoniazid [154] and rifampycin [155]. Moreover, the targeting of CYP51B1 is questionable, as no sterol biosynthetic pathway has been identified in this organism and the cyp51B1 gene has been shown to be dispensable for Mtb growth under in vitro conditions [38, 39].
Substantial in vitro antimycobacterial activity of new mono- disubstituted 1,3,4-oxadiazol-2(3H)-one derivatives has recently been reported [156, 157]. Although the actual target and the binding mode of these molecules have yet to be established, molecular modeling experiments suggested that these compounds posses all the necessary features to bind to Mtb CYP51B1. Among these, as observed for EBPA and BSPPA [153], the rigid planar structure of the pyridyl-substituted oxadiazolone ring would enable some of these molecules to coordinate to the heme group of CYP51B1 [157].
In very recent work, we have crystallized and determined the atomic structure of Mtb CYP130 in complex with two arylamine compounds identified from a HTS assay [158]. Arylamines and heterocyclic arylamines are widespread in the environment as industrial chemicals, food additives and drugs. The x-ray structure analysis revealed that the compounds bind in the active site by Fe-coordination and H-bonding of the arylamine group to the carbonyl oxygen of Gly243. Moreover, the topology of the CYP130A1 active site favors angular coordination of the arylamine group over the orthogonal coordination of azoles. A role was suggested for the conserved Ala(Gly)243-Gly244 motif in the I-helix in modulating both the binding affinity of the axial water ligand and the ligand selectivity.
6.3 Nitric oxide
During the initial growth infection stage of Mtb, •NO produced by host macrophages inhibits heme-containing terminal cytochrome oxidases, inactivates iron/sulfur proteins, and promotes entry into latency [159-162]. The potential of •NO as an inhibitor of Mtb cytochrome P450 enzymes, as represented by CYP130A1, CYP51B1, and two poorly characterized enzymes, CYP125A1 and CYP142A1, has also been evaluated [99]. Using UV-visible absorption, resonance Raman, and stopped-flow spectroscopy, we investigated the reactions of •NO with these heme proteins in their ferric resting form. •NO coordinates tightly to CYP125A1 and CYP142A1 (submicromolar) and with a lower affinity (micromolar) to CYP130A1 and CYP51A1. Anaerobic reduction of the ferric-NO species with sodium dithionite resulted in the formation of two spectrally distinct classes of five-coordinate ferrous-NO complexes. Exposure of these species to O2–revealed that the ferrous-NO forms of CYP125A1 and CYP142A1 are labile and convert back to the ferric state within a few minutes, whereas ferrous CYP130A1 and CYP51A1 bind •NO very tightly. Physiological concentrations (approximately 1 μM), of •NO would impair the activity of CYP130A1 and CYP51B1, whereas CYP125A1 and CYP142A1 would be more resistant. Selective P450 enzyme inhibition may contribute to the inhibitory effects of •NO on Mtb growth.
7. Summary and perspectives
Recent progress in the biochemical and structural characterization of Mtb P450 enzymes and their accessory redox partners provides key insights on their function(s), but much remains to be done. The large fraction of the Mtb genome devoted to lipid metabolism indicates that both catabolic and anabolic pathways are central to its infectivity and to its survival in the human host. In this regard, the available data suggests novel roles for at least some of the isoforms in the modification of endogenous (CYP128A1) and host (CYP51B1 and CYP125A1) lipids. An intriguing role for CYP121A1 in the biosynthesis of cyclodipeptides was very recently demonstrated and represents a good example of how analysis of an operon or surrounding genes may help to discover new functions. Another interesting perspective is the possible involvement of two P450 enzyme isoforms that are absent in M. bovis BCG (CYP130A1 and CYP141A1) in modulation of the host immune response, which could lead to the development of new vaccines.
TRaSH mutagenesis and transcriptional studies have highlighted important functions for at least two P450 enzymes (CYP125A1 and CYP128A1) that could potentially be targeted. However, the results of these studies are only suggestive and must be interpreted with caution. The physiological function(s) of every Mtb P450 enzyme and its impact under ex vivo and in vivo conditions must be investigated by generating individual “knockout” strains. The identification and characterization of P450 enzymes that play a role during the dormant phase of Mtb infection is also a high priority. In this context, the specific inhibition of pathogen P450 enzyme isoforms remains a major challenge. As an alternative to targeted inhibition of P450 activities, the demonstrated capability of some human P450 enzyme isoforms in the anaerobic bioreductive activation of prodrugs [163] can also be envisaged as a route for utilization of the Mtb P450 enzymes in treatment of the infection.
More work must be done on characterization of the putative Mtb P450 redox partner proteins in order to reconstitute P450 activity assays, as these are required to test possible substrates (natural or artificial) and to validate hits obtained from the screening of inhibitor libraries. Crystallization of these redox partners, alone or in complex with P450 enzymes, will provide useful insights on protein-protein interactions.
Supplementary Material
Acknowledgments
The authors acknowledge the support from National Institutes of Health grant AI074824 for their research on the Mycobacterium tuberculosis P450 system.
Footnotes
Abbreviations used: BSPPA, 2-(benzo[d]-2,1,3-thiadiazole-4-sulfonyl)-2-amino-2-phenyl-N-(pyridinyl-4)-acetamide; Cpd I, Compound I; CYP, cytochrome P450; cYY, cyclodityrosine (L-Tyr, L-tyr); DHBP, 4,4′-dihydroxybenzophenone; EPBA, alpha-ethyl-N-4-pyridinyl-benzeneacetamide; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; ITC, isothermal titration calorimetry; Mtb, Mycobacterium tuberculosis; NAD(P)H, reduced nicotinamide adenine dinucleotide cofactor; PAPS, 3′-phosphoadenosine 5′-phosphosulfate; TB, tuberculosis; TRaSH, transposon site hybridization.
References
- 1.Zink AR, Sola C, Reischl U, Grabner W, Rastogi N, Wolf H, Nerlich AG. J Clin Microbiol. 2003;41:359–367. doi: 10.1128/JCM.41.1.359-367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunyan J. The Life and Death of Mr Badman. New York: 1900. [Google Scholar]
- 3.Koch R. Berl Klin Wochenschr. 1882;15:221–230. [Google Scholar]
- 4.W.H.O. (WHO), World Health Organization Facts Sheets on Tuberculosis. 2009 [Google Scholar]
- 5.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, B CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 6.Poulos TL, Johnson EF. In: Cytochrome P450 Structure, mechanism, and biochemistry. Ortiz de Montellano PR, editor. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 87–114. [Google Scholar]
- 7.Ortiz de Montellano PR, editor. Cytochrome P450 Structure, mechanism, and biochemistry. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 8.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 9.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 10.Auclair K, Moenne-Loccoz P, Ortiz de Montellano PR. J Am Chem Soc. 2001;123:4877–4885. doi: 10.1021/ja0040262. [DOI] [PubMed] [Google Scholar]
- 11.Omura T, Sata R. J Biol Chem. 1965;239:2370–2378. [PubMed] [Google Scholar]
- 12.Yu CA, Gunsalus IC. J Biol Chem. 1974;249:102–106. [PubMed] [Google Scholar]
- 13.Guengerich FP, Johnson WW. Biochemistry. 1997;36:14741–14750. doi: 10.1021/bi9719399. [DOI] [PubMed] [Google Scholar]
- 14.Jefcoate CR. Methods Enzymol. 1978;52:258–279. doi: 10.1016/s0076-6879(78)52029-6. [DOI] [PubMed] [Google Scholar]
- 15.Schenkman JB, Remmer H, Estabrook RW. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- 16.Paine MJI, Scrutton NS, Munro AW, Gutierrez A, Roberts GCK, Wolf CR. In: Cytochrome P450 Structure, mechanism, and biochemistry. Ortiz de Montellano PR, editor. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 115–148. [Google Scholar]
- 17.Kelly SL, Kelly DE, Jackson CJ, Warrilow AGS, Lamb DC. In: Cytochrome P450 Structure, mechanism, and biochemistry. Ortiz de Montellano PR, editor. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 585–617. [Google Scholar]
- 18.McLean KJ, Sabri M, Marshall KR, Lawson RJ, Lewis DG, Clift D, Balding PR, Dunford AJ, Warman AJ, McVey JP, Quinn AM, Sutcliffe MJ, Scrutton NS, Munro AW. Biochem Soc Trans. 2005;33:796–801. doi: 10.1042/BST0330796. [DOI] [PubMed] [Google Scholar]
- 19.Munro AW, Girvan HM, McLean KJ. Biochim Biophys Acta. 2007;1770:345–359. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 20.Takayama K, Wang C, Besra GS. Clin Microbiol Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lederer E, Adam A, Ciorbaru R, Petit JF, Wietzerbin J. Mol Cell Biochem. 1975;7:87–104. doi: 10.1007/BF01792076. [DOI] [PubMed] [Google Scholar]
- 22.McNeil M, Daffe M, Brennan PJ. J Biol Chem. 1991;266:13217–13223. [PubMed] [Google Scholar]
- 23.Misaki A, Seto N, Azuma I. J Biochem. 1974;76:15–27. doi: 10.1093/oxfordjournals.jbchem.a130540. [DOI] [PubMed] [Google Scholar]
- 24.Noll H, Bloch H. J Biol Chem. 1955;214:251–265. [PubMed] [Google Scholar]
- 25.Noll H, Bloch H, Asselineau J, Lederer E. Biochim Biophys Acta. 1956;20:299–309. doi: 10.1016/0006-3002(56)90289-x. [DOI] [PubMed] [Google Scholar]
- 26.Barry CE, 3rd, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Prog Lipid Res. 1998;37:143–179. doi: 10.1016/s0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- 27.Zhang M, Yue J, Yang YP, Zhang HM, Lei JQ, Jin RL, Zhang XL, Wang HH. J Clin Microbiol. 2005;43:5477–5482. doi: 10.1128/JCM.43.11.5477-5482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hunter SW, Brennan PJ. J Biol Chem. 1990;265:9272–9279. [PubMed] [Google Scholar]
- 29.Jackson M, Stadthagen G, Gicquel B. Tuberculosis. 2007;87:78–86. doi: 10.1016/j.tube.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Tong L. Cell Mol Life Sci. 2005;62:1784–1803. doi: 10.1007/s00018-005-5121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schweizer E, Hofmann J. Microbiol Mol Biol Rev. 2004;68:501–517. doi: 10.1128/MMBR.68.3.501-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhatt A, Fujiwara N, Bhatt K, Gurcha SS, Kremer L, Chen B, Chan J, Porcelli SA, Kobayashi K, Besra GS, Jacobs WR., Jr Proc Natl Acad Sci USA. 2007;104:5157–5162. doi: 10.1073/pnas.0608654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhatt A, Molle V, Besra GS, Jacobs WR, Jr, Kremer L. Mol Microbiol. 2007;64:1442–1454. doi: 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- 34.Trivedi OA, Arora P, Vats A, Ansari MZ, Tickoo R, Sridharan V, Mohanty D, Gokhale RS. Mol Cell. 2005;17:631–643. doi: 10.1016/j.molcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Gelbard A, Goldman DS. Arch Biochem Biophys. 1961;94:228–235. doi: 10.1016/0003-9861(61)90034-0. [DOI] [PubMed] [Google Scholar]
- 36.Goldman DS, Gelbard A. Arch Biochem Biophys. 1959;83:360–370. doi: 10.1016/0003-9861(59)90044-x. [DOI] [PubMed] [Google Scholar]
- 37.Mahadevan U, Padmanaban G. Biochem Biophys Res Commun. 1998;244:893–897. doi: 10.1006/bbrc.1998.8354. [DOI] [PubMed] [Google Scholar]
- 38.Sassetti CM, Boyd DH, Rubin EJ. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 39.Lamichhane G, Zignol M, Blades NJ, Geiman DE, Dougherty A, Grosset J, Broman KW, Bishai WR. Proc Natl Acad Sci USA. 2003;100:7213–7218. doi: 10.1073/pnas.1231432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sassetti CM, Rubin EJ. Proc Natl Acad Sci USA. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang JC, Harik NS, Liao RP, Sherman DR. J Infect Dis. 2007;196:788–795. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- 42.Raj A, van Oudenaarden A. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLean KJ, Carroll P, Lewis DG, Dunford AJ, Seward HE, Neeli R, Cheesman MR, Marsollier L, Douglas P, Smith WE, Rosenkrands I, Cole ST, Leys D, Parish T, Munro AW. J Biol Chem. 2008;283:33406–33416. doi: 10.1074/jbc.M802115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hampshire T, Soneji S, Bacon J, James BW, Hinds J, Laing K, Stabler RA, Marsh PD, Butcher PD. Tuberculosis. 2004;84:228–238. doi: 10.1016/j.tube.2003.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rengarajan J, Bloom BR, Rubin EJ. Proc Natl Acad Sci USA. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voskuil MI, Visconti KC, Schoolnik GK. Tuberculosis. 2004;84:218–227. doi: 10.1016/j.tube.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Murphy DJ, Brown JR. BMC Infect Dis. 2007;7:84. doi: 10.1186/1471-2334-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Mol Microbiol. 2002;43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 50.Dubnau E, Chan J, Mohan VP, Smith I. Infect Immun. 2005;73:3754–3757. doi: 10.1128/IAI.73.6.3754-3757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mawuenyega KG, Forst CV, Dobos KM, Belisle JT, Chen J, Bradbury EM, Bradbury AR, Chen X. Mol Biol Cell. 2005;16:396–404. doi: 10.1091/mbc.E04-04-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bacon J, James BW, Wernisch L, Williams A, Morley KA, Hatch GJ, Mangan JA, Hinds J, Stoker NG, Butcher PD, Marsh PD. Tuberculosis. 2004;84:205–217. doi: 10.1016/j.tube.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 53.Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE., 3rd J Biol Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 54.Dahl JL, Kraus CN, Boshoff HI, Doan B, Foley K, Avarbock D, Kaplan G, Mizrahi V, Rubin H, Barry CE., 3rd Proc Natl Acad Sci USA. 2003;100:10026–10031. doi: 10.1073/pnas.1631248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denkin S, Byrne S, Jie C, Zhang Y. Arch Microbiol. 2005;184:152–157. doi: 10.1007/s00203-005-0037-9. [DOI] [PubMed] [Google Scholar]
- 56.Milano A, Pasca MR, Provvedi R, Lucarelli AP, Manina G, Ribeiro AL, Manganelli R, Riccardi G. Tuberculosis. 2009;89:84–90. doi: 10.1016/j.tube.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 57.Akhtar M, Alexander K, Boar RB, McGhie JF, Barton DH. Biochem J. 1978;169:449–463. doi: 10.1042/bj1690449b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lepesheva GI, Waterman MR. Biochim Biophys Acta. 2007;1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitropoulos KA, Gibbons GF, Reeves BE. Steroids. 1976;27:821–829. doi: 10.1016/0039-128x(76)90141-0. [DOI] [PubMed] [Google Scholar]
- 60.Trzaskos JM, Bowen WD, Shafiee A, Fischer RT, Gaylor JL. J Biol Chem. 1984;259:13402–13412. [PubMed] [Google Scholar]
- 61.Yoshida Y, Noshiro M, Aoyama Y, Kawamoto T, Horiuchi T, Gotoh O. J Biochem. 1997;122:1122–1128. doi: 10.1093/oxfordjournals.jbchem.a021870. [DOI] [PubMed] [Google Scholar]
- 62.Aoyama Y, Horiuchi T, Gotoh O, Noshiro M, Yoshida Y. J Biochem. 1998;124:694–696. doi: 10.1093/oxfordjournals.jbchem.a022167. [DOI] [PubMed] [Google Scholar]
- 63.Bellamine A, Mangla AT, Nes WD, Waterman MR. Proc Natl Acad Sci USA. 1999;96:8937–8942. doi: 10.1073/pnas.96.16.8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackson CJ, Lamb DC, Marczylo TH, Parker JE, Manning NL, Kelly DE, Kelly SL. Biochem Biophys Res Commun. 2003;301:558–563. doi: 10.1016/s0006-291x(02)03078-4. [DOI] [PubMed] [Google Scholar]
- 65.Pietila MP, Vohra PK, Sanyal B, Wengenack NL, Raghavakaimal S, Thomas CF., Jr Am J Respir Cell Mol Biol. 2006;35:236–242. doi: 10.1165/rcmb.2005-0398OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jackson CJ, Lamb DC, Marczylo TH, Warrilow AG, Manning NJ, Lowe DJ, Kelly DE, Kelly SL. J Biol Chem. 2002;277:46959–46965. doi: 10.1074/jbc.M203523200. [DOI] [PubMed] [Google Scholar]
- 67.McLean KJ, Warman AJ, Seward HE, Marshall KR, Girvan HM, Cheesman MR, Waterman MR, Munro AW. Biochemistry. 2006;45:8427–8443. doi: 10.1021/bi0601609. [DOI] [PubMed] [Google Scholar]
- 68.Dunford AJ, McLean KJ, Sabri M, Seward HE, Heyes DJ, Scrutton NS, Munro AW. J Biol Chem. 2007;282:24816–24824. doi: 10.1074/jbc.M702958200. [DOI] [PubMed] [Google Scholar]
- 69.Park SY, Yamane K, Adachi S, Shiro Y, Weiss KE, Maves SA, Sligar SG. J Inorg Biochem. 2002;91:491–501. doi: 10.1016/s0162-0134(02)00446-4. [DOI] [PubMed] [Google Scholar]
- 70.Podust LM, Kim Y, Arase M, Neely BA, Beck BJ, Bach H, Sherman DH, Lamb DC, Kelly SL, Waterman MR. J Biol Chem. 2003;278:12214–12221. doi: 10.1074/jbc.M212210200. [DOI] [PubMed] [Google Scholar]
- 71.Yano JK, Koo LS, Schuller DJ, Li H, Ortiz de Montellano PR, Poulos TL. J Biol Chem. 2000;275:31086–31092. doi: 10.1074/jbc.M004281200. [DOI] [PubMed] [Google Scholar]
- 72.Zerbe K, Pylypenko O, Vitali F, Zhang W, Rouset S, Heck M, Vrijbloed JW, Bischoff D, Bister B, Sussmuth RD, Pelzer S, Wohlleben W, Robinson JA, Schlichting I. J Biol Chem. 2002;277:47476–47485. doi: 10.1074/jbc.M206342200. [DOI] [PubMed] [Google Scholar]
- 73.Gay SC, Sun L, Maekawa K, Halpert JR, Stout CD. Biochemistry. 2009 doi: 10.1021/bi9003765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD. Proc Natl Acad Sci USA. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR. J Biol Chem. 2004;279:27294–27301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 76.Podust LM, Poulos TL, Waterman MR. Proc Natl Acad Sci USA. 2001;98:3068–3073. doi: 10.1073/pnas.061562898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Podust LM, Yermalitskaya LV, Lepesheva GI, Podust VN, Dalmasso EA, Waterman MR. Structure. 2004;12:1937–1945. doi: 10.1016/j.str.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 78.Lepesheva GI, Seliskar M, Knutson CG, Stourman NV, Rozman D, Waterman MR. Arch Biochem Biophys. 2007;464:221–227. doi: 10.1016/j.abb.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lampe JN, Floor SN, Gross JD, Nishida CR, Jiang Y, Trnka MJ, Ortiz de Montellano PR. J Am Chem Soc. 2008;130:16168–16169. doi: 10.1021/ja8071463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lepesheva GI, Nes WD, Zhou W, Hill GC, Waterman MR. Biochemistry. 2004;43:10789–10799. doi: 10.1021/bi048967t. [DOI] [PubMed] [Google Scholar]
- 81.Lepesheva GI, Zaitseva NG, Nes WD, Zhou W, Arase M, Liu J, Hill GC, Waterman MR. J Biol Chem. 2006;281:3577–3585. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]
- 82.McLean KJ, Munro AW. Drug Metab Dispos. 2008;40:427–446. doi: 10.1080/03602530802186389. [DOI] [PubMed] [Google Scholar]
- 83.McLean KJ, Dunford AJ, Neeli R, Driscoll MD, Munro AW. Arch Biochem Biophys. 2007;464:228–240. doi: 10.1016/j.abb.2007.03.026. [DOI] [PubMed] [Google Scholar]
- 84.Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Small PM. Science. 1999;284:1520–1523. doi: 10.1126/science.284.5419.1520. [DOI] [PubMed] [Google Scholar]
- 85.Gordon SV, Brosch R, Billault A, Garnier T, Eiglmeier K, Cole ST. Mol Microbiol. 1999;32:643–655. doi: 10.1046/j.1365-2958.1999.01383.x. [DOI] [PubMed] [Google Scholar]
- 86.Al-Attiyah R, Mustafa AS. Infect Immun. 2008;76:4190–4198. doi: 10.1128/IAI.00199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jamil B, Shahid F, Hasan Z, Nasir N, Razzaki T, Dawood G, Hussain R. Tuberculosis. 2007;87:279–287. doi: 10.1016/j.tube.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 88.Ouellet H, Podust LM, de Montellano PR. J Biol Chem. 2008;283:5069–5080. doi: 10.1074/jbc.M708734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McLean KJ, Cheesman MR, Rivers SL, Richmond A, Leys D, Chapman SK, Reid GA, Price NC, Kelly SM, Clarkson J, Smith WE, Munro AW. J Inorg Biochem. 2002;91:527–541. doi: 10.1016/s0162-0134(02)00479-8. [DOI] [PubMed] [Google Scholar]
- 90.Belin P, Le Du MH, Fielding A, Lequin O, Jacquet M, Charbonnier JB, Lecoq A, Thai R, Courcon M, Masson C, Dugave C, Genet R, Pernodet JL, Gondry M. Proc Natl Acad Sci USA. 2009;106:7426–7431. doi: 10.1073/pnas.0812191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gondry M, Sauguet L, Belin P, Thai R, Amouroux R, Tellier C, Tuphile K, Jacquet M, Braud S, Courcon M, Masson C, Dubois S, Lautru S, Lecoq A, Hashimoto S, Genet R, Pernodet JL. Nat Chem Biol. 2009;5:414–420. doi: 10.1038/nchembio.175. [DOI] [PubMed] [Google Scholar]
- 92.Danilchanka O, Mailaender C, Niederweis M. Antimicrob Agents Chemother. 2008;52:2503–2511. doi: 10.1128/AAC.00298-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, Hannan M, Goguet de la Salmoniere YO, Aman K, Kato-Maeda M, Small PM. Proc Natl Acad Sci USA. 2004;101:4865–4870. doi: 10.1073/pnas.0305634101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leys D, Mowat CG, McLean KJ, Richmond A, Chapman SK, Walkinshaw MD, Munro AW. J Biol Chem. 2003;278:5141–5147. doi: 10.1074/jbc.M209928200. [DOI] [PubMed] [Google Scholar]
- 95.Seward HE, Roujeinikova A, McLean KJ, Munro AW, Leys D. J Biol Chem. 2006;281:39437–39443. doi: 10.1074/jbc.M607665200. [DOI] [PubMed] [Google Scholar]
- 96.Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD. Proc Natl Acad Sci USA. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pandey AK, Sassetti CM. Proc Natl Acad Sci USA. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yam KC, D'Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, Ly LH, Converse PJ, Jacobs WR, Jr, Strynadka N, Eltis LD. PLoS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ouellet H, Lang J, Couture M, Ortiz de Montellano PR. Biochemistry. 2009;48:863–872. doi: 10.1021/bi801595t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ouellet H, Ortiz de Montellano PR. manuscript in preparation [Google Scholar]
- 101.Gokhale RS, Saxena P, Chopra T, Mohanty D. Nat Prod Rep. 2007;24:267–277. doi: 10.1039/b616817p. [DOI] [PubMed] [Google Scholar]
- 102.Brennan PJ, Crick DC. Curr Top Med Chem. 2007;7:475–478. doi: 10.2174/156802607780059763. [DOI] [PubMed] [Google Scholar]
- 103.Schelle MW, Bertozzi CR. ChemBioChem. 2006;7:1516–1524. doi: 10.1002/cbic.200600224. [DOI] [PubMed] [Google Scholar]
- 104.Onwueme KC, Vos CJ, Zurita J, Ferreras JA, Quadri LE. Prog Lipid Res. 2005;44:259–302. doi: 10.1016/j.plipres.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 105.Mougous JD, Green RE, Williams SJ, Brenner SE, Bertozzi CR. Chem Biol. 2002;9:767–776. doi: 10.1016/s1074-5521(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 106.Mougous JD, Senaratne RH, Petzold CJ, Jain M, Lee DH, Schelle MW, Leavell MD, Cox JS, Leary JA, Riley LW, Bertozzi CR. Proc Natl Acad Sci USA. 2006;103:4258–4263. doi: 10.1073/pnas.0510861103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.ten Bokum AMC, Movahedzadeh F, Frita R, Bancroft GJ, Stoker NG. Trends Microbiol. 2008;16:436–441. doi: 10.1016/j.tim.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 108.Holsclaw CM, Sogi KM, Gilmore SA, Schelle MW, Leavell MD, Bertozzi CR, Leary JA. ACS Chem Biol. 2008;3:619–624. doi: 10.1021/cb800145r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, Young DB, Butcher PD. Microbiol. 2002;148:3129–3138. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- 110.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. Mol Microbiol. 2006;60:312–330. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 111.Schwab U, Rohde KH, Wang Z, Chess PR, Notter RH, Russell DG. Microb Pathog. 2009;46:185–193. doi: 10.1016/j.micpath.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soliveri JA, Gomez J, Bishai WR, Chater KF. Microbiology. 2000;146(Pt 2):333–343. doi: 10.1099/00221287-146-2-333. [DOI] [PubMed] [Google Scholar]
- 113.Dubey VS, Sirakova TD, Cynamon MH, Kolattukudy PE. J Bacteriol. 2003;185:4620–4625. doi: 10.1128/JB.185.15.4620-4625.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tailleux L, Waddell SJ, Pelizzola M, Mortellaro A, Withers M, Tanne A, Castagnoli PR, Gicquel B, Stoker NG, Butcher PD, Foti M, Neyrolles O. PLoS ONE. 2008;3:e1403. doi: 10.1371/journal.pone.0001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zanno A, Kwiatkowski N, Vaz AD, Guardiola-Diaz HM. Biochim Biophys Acta. 2005;1707:157–169. doi: 10.1016/j.bbabio.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 116.Ricagno S, de Rosa M, Aliverti A, Zanetti G, Bolognesi M. Biochem Biophys Res Commun. 2007;360:97–102. doi: 10.1016/j.bbrc.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 117.Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. Proc Natl Acad Sci USA. 2001;98:7534–7539. doi: 10.1073/pnas.121172498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fisher MA, Plikaytis BB, Shinnick TM. J Bacteriol. 2002;184:4025–4032. doi: 10.1128/JB.184.14.4025-4032.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fukuyama K, Matsubara H, Tsukihara T, Katsube Y. J Mol Biol. 1989;210:383–398. doi: 10.1016/0022-2836(89)90338-0. [DOI] [PubMed] [Google Scholar]
- 120.Fischer F, Raimondi D, Aliverti A, Zanetti G. Eur J Biochem. 2002;269:3005–3013. doi: 10.1046/j.1432-1033.2002.02989.x. [DOI] [PubMed] [Google Scholar]
- 121.Sabri M, Dunford AJ, McLean KJ, Neeli R, Scrutton NS, Leys D, Munro AW. Biochem J. 2009;417:103–112. doi: 10.1042/BJ20080466. [DOI] [PubMed] [Google Scholar]
- 122.Pennati A, Zanetti G, Aliverti A, Gadda G. Biochemistry. 2008;47:3418–3425. doi: 10.1021/bi702250h. [DOI] [PubMed] [Google Scholar]
- 123.Rosa Md, Pennati A, Pandini V, Monzani E, Zanetti G, Aliverti A. FEBS J. 2007;274:3998–4007. doi: 10.1111/j.1742-4658.2007.05934.x. [DOI] [PubMed] [Google Scholar]
- 124.Pennati A, Razeto A, de Rosa M, Pandini V, Vanoni MA, Mattevi A, Coda A, Aliverti A, Zanetti G. Biochemistry. 2006;45:8712–8720. doi: 10.1021/bi060369m. [DOI] [PubMed] [Google Scholar]
- 125.McLean KJ, Clift D, Lewis DG, Sabri M, Balding PR, Sutcliffe MJ, Leys D, Munro AW. Trends Microbiol. 2006;14:220–228. doi: 10.1016/j.tim.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 126.Shukla N, Bhatt AN, Aliverti A, Zanetti G, Bhakuni V. FEBS J. 2005;272:2216–2224. doi: 10.1111/j.1742-4658.2005.2005.04645.x. [DOI] [PubMed] [Google Scholar]
- 127.Bhatt AN, Shukla N, Aliverti A, Zanetti G, Bhakuni V. Prot Sci. 2005;14:980–992. doi: 10.1110/ps.041162705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bossi RT, Aliverti A, Raimond D, Fischer F, Zanetti G, Ferrari D, Tahallah N, Maier CS, Heck AJR, Rizzi M, Mattevi A. Biochemistry. 2002;41:8807–8818. doi: 10.1021/bi025858a. [DOI] [PubMed] [Google Scholar]
- 129.Aguero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, Carmona S, Carruthers IM, Chan AW, Chen F, Crowther GJ, Doyle MA, Hertz-Fowler C, Hopkins AL, McAllister G, Nwaka S, Overington JP, Pain A, Paolini GV, Pieper U, Ralph SA, Riechers A, Roos DS, Sali A, Shanmugam D, Suzuki T, Van Voorhis WC, Verlinde CL. Nat Rev Drug Discov. 2008;7:900–907. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ahmad Z, Sharma S, Khuller GK. FEMS Microbiol Lett. 2005;251:19–22. doi: 10.1016/j.femsle.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 131.Ahmad Z, Sharma S, Khuller GK. FEMS Microbiol Lett. 2006;261:181–186. doi: 10.1111/j.1574-6968.2006.00350.x. [DOI] [PubMed] [Google Scholar]
- 132.Ahmad Z, Sharma S, Khuller GK. FEMS Microbiol Lett. 2006;258:200–203. doi: 10.1111/j.1574-6968.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- 133.Guardiola-Diaz HM, Foster LA, Mushrush D, Vaz AD. Biochem Pharmacol. 2001;61:1463–1470. doi: 10.1016/s0006-2952(01)00571-8. [DOI] [PubMed] [Google Scholar]
- 134.Jackson CJ, Lamb DC, Kelly DE, Kelly SL. FEMS Microbiol Lett. 2000;192:159–162. doi: 10.1111/j.1574-6968.2000.tb09375.x. [DOI] [PubMed] [Google Scholar]
- 135.McLean KJ, Marshall KR, Richmond A, Hunter IS, Fowler K, Kieser T, Gurcha SS, Besra GS, Munro AW. Microbiology. 2002;148:2937–2949. doi: 10.1099/00221287-148-10-2937. [DOI] [PubMed] [Google Scholar]
- 136.Burguiere A, Hitchen PG, Dover LG, Dell A, Besra GS. Microbiology. 2005;151:2087–2095. doi: 10.1099/mic.0.27938-0. [DOI] [PubMed] [Google Scholar]
- 137.Ahmad Z, Sharma S, Khuller GK. Nanomedicine. 2007;3:239–243. doi: 10.1016/j.nano.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 138.Maurice M, Pichard L, Daujat M, Fabre I, Joyeux H, Domergue J, Maurel P. FASEB J. 1992;6:752–758. doi: 10.1096/fasebj.6.2.1371482. [DOI] [PubMed] [Google Scholar]
- 139.Tassaneeyakul W, Birkett DJ, Miners JO. Xenobiotica. 1998;28:293–301. doi: 10.1080/004982598239579. [DOI] [PubMed] [Google Scholar]
- 140.Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM. Drug Metab Dispos. 2002;30:314–318. doi: 10.1124/dmd.30.3.314. [DOI] [PubMed] [Google Scholar]
- 141.Balding PR, Porro CS, McLean KJ, Sutcliffe MJ, Marechal JD, Munro AW, Visser SP. J Phys Chem A. 2008 doi: 10.1021/jp802087w. [DOI] [PubMed] [Google Scholar]
- 142.Bruggemann RJ, Alffenaar JW, Blijlevens NM, Billaud EM, Kosterink JG, Verweij PE, Burger DM. Clin Infect Dis. 2009;48:1441–1458. doi: 10.1086/598327. [DOI] [PubMed] [Google Scholar]
- 143.Hollenberg PF. Drug Metab Rev. 2002;34:17–35. doi: 10.1081/dmr-120001387. [DOI] [PubMed] [Google Scholar]
- 144.Bohmer GM, Drollmann A, Gleiter CH, Nave R. Clin Pharmacokinet. 2008;47:343–349. doi: 10.2165/00003088-200847050-00005. [DOI] [PubMed] [Google Scholar]
- 145.Damanhouri Z, Gumbleton M, Nicholls PJ, Shaw MA. J Antimicrob Chemother. 1988;21:187–194. doi: 10.1093/jac/21.2.187. [DOI] [PubMed] [Google Scholar]
- 146.Ervine CM, Matthew DE, Brennan B, Houston JB. Drug Metab Dispos. 1996;24:211–215. [PubMed] [Google Scholar]
- 147.Mazzu AL, Lasseter KC, Shamblen EC, Agarwal V, Lettieri J, Sundaresen P. Clin Pharmacol Ther. 2000;68:391–400. doi: 10.1067/mcp.2000.110537. [DOI] [PubMed] [Google Scholar]
- 148.Swaisland HC, Ranson M, Smith RP, Leadbetter J, Laight A, McKillop D, Wild MJ. Clin Pharmacokinet. 2005;44:1067–1081. doi: 10.2165/00003088-200544100-00005. [DOI] [PubMed] [Google Scholar]
- 149.Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Clin Pharmacokinet. 2000;38:111–180. doi: 10.2165/00003088-200038020-00002. [DOI] [PubMed] [Google Scholar]
- 150.Poole K. J Mol Microbiol Biotechnol. 2001;3:255–264. [PubMed] [Google Scholar]
- 151.Rodriquez RJ, Acosta D., Jr J Biochem Toxicol. 1996;11:127–131. doi: 10.1002/(SICI)1522-7146(1996)11:3<127::AID-JBT4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 152.Eddine AN, von Kries JP, Podust MV, Warrier T, Kaufmann SH, Podust LM. J Biol Chem. 2008;283:15152–15159. doi: 10.1074/jbc.M801145200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Podust LM, Kries JPv, Eddine AN, Kim Y, Yermalitskaya LV, Kuehne R, Ouellet H, Warrier T, Altekoster M, Lee JS, Rademann J, Oschkinat H, Kaufmann SHE, Waterman MR. Antimicrob Agents Chemother. 2007;51:3915–3923. doi: 10.1128/AAC.00311-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mohamad S, Ibrahim P, Sadikun A. Tuberculosis. 2004;84:56–62. doi: 10.1016/j.tube.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 155.Taniguchi H, Aramaki H, Nikaido Y, Mizuguchi Y, Nakamura M, Koga T, Yoshida S. FEMS Microbiol Lett. 1996;144:103–108. doi: 10.1111/j.1574-6968.1996.tb08515.x. [DOI] [PubMed] [Google Scholar]
- 156.Mamolo MG, Zampieri D, Vio L, Fermeglia M, Ferrone M, Pricl S, Scialino G, Banfi E. Bioorg Med Chem. 2005;13:3797–3809. doi: 10.1016/j.bmc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 157.Zampieri D, Mamolo MG, Laurini E, Fermeglia M, Posocco P, Pricl S, Banfi E, Scialino G, Vio L. Bioorg Med Chem. 2009;17:4693–4707. doi: 10.1016/j.bmc.2009.04.055. [DOI] [PubMed] [Google Scholar]
- 158.Podust LM, Ouellet H, Ortiz de Montellano PR. submitted manuscript. [Google Scholar]
- 159.Brunori M. Trends Biochem Sci. 2001;26:21–23. doi: 10.1016/s0968-0004(00)01698-4. [DOI] [PubMed] [Google Scholar]
- 160.Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Infect Immun. 1995;63:736–740. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nathan C, Shiloh MU. Proc Natl Acad Sci USA. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shiloh MU, Nathan CF. Curr Opin Microbiol. 2000;3:35–42. doi: 10.1016/s1369-5274(99)00048-x. [DOI] [PubMed] [Google Scholar]
- 163.Nishida CR, Ortiz de Montellano PR. J Med Chem. 2008;51:5118–5120. doi: 10.1021/jm800496s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Betts JC, McLaren A, Lennon MG, Kelly FM, Lukey PT, Blakemore SJ, Duncan K. Antimicrob Agents Chemother. 2003;47:2903–2913. doi: 10.1128/AAC.47.9.2903-2913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gao Q, Kripke K, Arinc Z, Voskuil M, Small P. Tuberculosis. 2004;84:188–196. doi: 10.1016/j.tube.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 166.Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B, Frita R, Ten Bokum A, Besra GS, Lott JS, Stoker NG. Mol Microbiol. 2007;65:684–699. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Recchi C, Sclavi B, Rauzier J, Gicquel B, Reyrat JM. J Biol Chem. 2003;278:33763–33773. doi: 10.1074/jbc.M305963200. [DOI] [PubMed] [Google Scholar]
- 168.Mehra S, Kaushal D. J Bacteriol. 2009 doi: 10.1128/JB.00064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bacon J, Dover LG, Hatch KA, Zhang Y, Gomes JM, Kendall S, Wernisch L, Stoker NG, Butcher PD, Besra GS, Marsh PD. Microbiology. 2007;153:1435–1444. doi: 10.1099/mic.0.2006/004317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.McLean KJ, Scrutton NS, Munro AW. Biochem J. 2003;372:317–327. doi: 10.1042/BJ20021692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zanno A, Kwiatkowski N, Vaz ADN, Guardiola-Diaz HM. Biochimica et Biophysica Acta. 2005;1707:157–169. doi: 10.1016/j.bbabio.2004.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.