

Abstract
Many flavoproteins catalyze the oxidation of primary and secondary amines, with the transfer of a hydride equivalent from a carbon -nitrogen bond to the flavin cofactor. Most of these amine oxidases can be classified into two structural families, the D -amino acid oxidase/sarcosine oxidase family and the monoamine oxidase family. This review discusses the present understanding of the mechanisms of amine and amino acid oxidation by flavoproteins, focusing on these two structural families.
Keywords: flavoproteins, enzyme mechanisms, monoamine oxidase, D-amino acid oxidase
Introduction
The isoalloxazine ring of FAD and FMN is the most versatile of the redox active cofactors in the cell [1, 2]. It is capable of one and two electron transfers, so that flavoproteins frequently serve as intermediaries between obligate one and two electron donors and acceptors in electron transport chains and individual enzymes. Its redox potential is suitable for metabolism of a wide range of biological molecules by interconverting single and double bonds, so that flavoprotein oxidases and dehydrogenases play critical roles in oxidative metabolism. The reduced form can react with oxygen to form a peroxyflavin capable of hydroxylating activated aromatic rings, so that flavoprotein hydroxylases are common in the catabolism of aromatic metabolites [3]. Finally, the oxidized form can react with nucleophiles to form N5 and C4a adducts, further expanding the potential reactions of flavoenzymes [4]. This review will focus on the present understanding of the mechanisms by which flavoproteins oxidize carbon-nitrogen bonds. The substrates are mostly amino acids and primary or secondary amines (Scheme 1). Oxidation of the CN single bond to a double bond is commonly followed by hydrolysis of the Schiff base, so that the overall reaction is loss of ammonium from primary amines and cleavage of secondary amines. Most commonly, the reduced flavin is oxidized by O2 with the formation of H2O2, so that the enzyme is an oxidase. In some cases of amino acid oxidation the H2O2 reacts with the oxidized substrate in the enzyme active site, forming an amide plus CO2 [5, 6].
Scheme 1.

Historically, mechanistic studies of flavoprotein -catalyzed amine oxidation have focused on the substrates. As discussed below, this has resulted in different mechanistic proposals for the oxidation of amino acids, primary and secondary amines. However, with the enormous increase in the number of protein structures in recent years, it has become apparent that most flavoprotein amine oxidases can be grouped into one of two structural families. The monoamine oxidase (MAO) family (Figure 1) contains MAO A and B [7, 8], the L-amino acid oxidases (LAAO) [9, 10], the spermine and polyamine oxidases [11, 12], and the histone lysine demethylase LSD1 [13, 14]. The D-amino acid oxidase (DAAO) family (Figure 2) contains DAAO [15] and enzymes that oxidize glycine or N-methylated amino acids [16–20]. Thus, both families of amine oxidases contain members that oxidize amino acids, primary amines, and secondary amines. While a single protein fold can be used to catalyze a range of reactions, as demonstrated by the versatility of the TIM barrel enzymes [21, 22], it is a reasonable expectation that enzymes that have similar structures and catalyze similar reactions use similar catalytic mechanisms. Consequently, in the following discussion, mechanistic results are considered for each family as a whole rather than by substrate.
Figure 1.

Structures of representative member of monoamine oxidase family of flavin amine oxidases. The figures were constructed using the pdb files 1yy5, 1f8s, 2h94, and 1gos.
Figure 2.

Structures of representative member of D-amino acid oxidase family of flavin amine oxidases. The figures were constructed using the pdb files 1eli, 1kif, 1pj6, and 1ng4.
The D-Amino Acid Oxidase Family
D-Amino Acid Oxidase
Pig kidney DAAO was one of the first flavoproteins to be discovered [23], and it has served as a paradigm for understanding mechanisms of flavoprotein oxidases ever since. The more recently discovered glycine oxidases have similar structure to DAAOs from pig kidney and yeast and also oxidize D-alanine but not L-alanine [16, 24]; they are therefore likely to have the same mechanism. Kinetic studies of DAAO were key to establishing the common kinetic mechanism of flavoprotein oxidases shown in Scheme 2[1]. In the reductive half-reaction, a hydride equivalent is transferred from the substrate to the flavin to yield the oxidized substrate bound to the reduced enzyme. With most enzyme-substrate combinations, this is sufficiently favorable energetically that enzyme reduction is effectively irreversible. The product can then dissociate from the enzyme, which subsequently reacts with oxygen to regenerate the oxidized enzyme (lower path in Scheme 2) or oxygen can react with the reduced enzyme before the product dissociates (upper path) followed by product dissociation from the oxidized enzyme. In either case stopped -flow experiments allow the reductive half-reaction to be studied directly by monitoring the flavin spectrum after mixing enzyme and substrate in the absence of oxygen. In no case has an intermediate been detected between the oxidized enzyme with substrate bound and the reduced enzyme with product bound.
Scheme 2.

The oxidation of an amine (Scheme 1) involves the net loss of two electrons, as the CN single bond is converted to a double bond, and of two protons, one from the carbon and one from the positively charged nitrogen. The ability of the flavin to accept one or two electrons and to form adducts with nucleophiles in combination with the various possible sequences for loss of protons and electrons from the substrate allow for an impressive variety of possible mechanisms; virtually all have been considered for this enzyme [25]. To limit the possibilities, Neims et al. [26] examined the kinetics of pig kidney DAAO with a series of ring-substituted phenylglycines as substrates. They found a biphasic correlation between the value of the substrate and the Vmax value, which increased with a ρ value of 5.4 to a limiting value. This was interpreted as evidence that the substrate α-hydrogen was removed as a proton. Subsequently, Hamilton [27] proposed that oxidation of an amine or alcohol could occur by nucleophilic attack of the heteroatom on the flavin C4a position followed by loss of the α-hydrogen as a proton (Scheme 3). However, with most substrates for DAAO, the rate-limiting step is product release, not CH bond cleavage [28, 29], so that the Vmax value does not reflect chemistry. Thus, the relevance of this value to the mechanism of amino acid oxidation is unclear. Pollegioni et al. [30] later characterized the reaction of DAAO from T. variabilis with substituted phenylglycines and found a ρ value of 0.73 for the correlation of the rate constant for flavin reduction with the σ+ value of the substituent. This value is more likely to reflect the transition state for CH bond cleavage.
Scheme 3.

In 1978 Walsh et al. [31] described the ability of DAAO to catalyze elimination of HCl from β-Cl-alanine in addition to the normal oxidation of this substrate to β-Cl-pyruvate. This was interpreted as additional evidence for proton removal to form a substrate carbanion as the initial step in catalysis. The relative amounts of oxidation and HCl varied with the oxygen concentration, with the normal catalytic reaction predominating at high concentrations and HCl elimination at low concentrations. The mechanism of Scheme 4 was proposed to account for these results; the data did not address the mechanism by which electrons would be transferred from the carbanion to the flavin, although various substrate-flavin adducts were considered. To account for the effect of the oxygen concentration on the partitioning between oxidation and elimination of HCl, it was necessary to propose that the reaction of the carbanion intermediate with the flavin was readily reversible. Subsequent studies of the elimination reaction with α-amino-β-Cl-butyrate established that the hydrogen originally on the substrate α-carbon is found at the β-carbon in a fraction of the α-ketoglutarate formed in the elimination reaction [32]. For the mechanism of Scheme 4, this would require that tautomerization of the initial eneamine to the imino acid and its hydrolysis to the keto acid occur in the active site, in contrast to the behavior of other substrates, where the imino acid product dissociates from the enzyme before the keto acid is formed [33].
Scheme 4.

An alternative mechanism which accounts for both the effect of the oxygen concentration on the partitioning between elimination and oxidation and the retention of the α-hydrogen at the β-carbon of the product is shown in Scheme 5. Here the amino acid substrate is oxidized to an imino acid by the normal catalytic mechanism. The reduced flavin can then react with O2to form the oxidized enzyme, followed by dissociation of the β-Cl-imino acid. Alternatively, the reduced flavin can act as a hydride donor for an SN2 reaction in which the chloride is the leaving group to produce the oxidized enzyme and the β-Cl-imino acid. This mechanism accounts for both the oxygen dependence of the product and the shift of the α-hydrogen to the β-carbon of the product. If the reduced flavin acts as a donor to the imino acid in the elimination reaction, it is likely to act as a hydride acceptor from the amino acid in the normal reaction. Stronger evidence that amino acid oxidation by DAAO involves direct transfer of a hydride from the substrate α-carbon to the flavin was provided by studies of the enzyme containing 5-deazaFAD in place of the native flavin [34]. This modified form of the cofactor is an obligatory two electron donor/acceptor that is competent for hydride transfer but not radical reactions [35]. The reaction of [α-3H]alanine with 5-deazaFAD DAAO yielded tritium -labeled reduced flavin (Scheme 6). When the labeled enzyme was then reacted with pyruvate plus ammonia, which would react in solution to form the imino acid, [α-3H]alanine was reformed. These observations are consistent with hydride transfer from the amino acid to the flavin as the mechanism.
Scheme 5.

Scheme 6.

Despite the result with the 5-deazaFAD-containing enzyme, the observation of a DAAO-catalyzed HCl elimination reaction led to the carbanion mechanism for DAAO being generally accepted; it was subsequently extended to flavoproteins catalyzing similar reactions, such as flavocytochrome b2[36]and lactate monooxygenase[37]. However, this mechanism became less tenable when the three-dimensional structure of DAAO was solved in 1996 [15, 38]. This showed that the active site lacks an appropriate base to abstract the substrate -proton. Subsequent structures of the yeast enzyme with D-alanine at 1.2 Å and with D-3,3,3-trifluoroalanine at 1.47 Å showed the substrate Cα to be 3.2–3.4 Å from the flavin N5 with the α-hydrogen pointing toward the flavin N5 [39]. These structures are more in line with the expectations of a mechanism in which a hydride is transferred directly from the amino acid α-carbon to the flavin. The ρ value of 0.73 from the subsequent re-analysis of the kinetics of DAAO with substituted phenylglycines [30] provides additional support for a hydride transfer mechanism.
A variety of kinetic isotope effects have been used to probe the mechanism of DAAO. This approach has the advantage that the mechanism is studied directly with normal substrates rather than utilizing substrates of unusual reactivity that can undergo nonphysiological reactions. With D-serine as substrate for DAAO, cleavage of the substrate CH bond is fully rate-limiting for the reductive half-reaction. This was established by showing that the deuterium isotope effect of 4.5 on the kcat/Km value and on the rate constant for flavin reduction is pH -independent and equal to that calculated from the tritium isotope effect of 8.7 when one accounts for the difference in mass of the two isotopes [40]. As a result, any isotope effect on these kinetic parameters reports on the structure of the transition state for CH bond cleavage. In 2000, Kurtz et al. [41] reported measurement of 15N kinetic isotopes on the oxidation of D-serine by DAAO. The 15N isotope effect on the kcat/Km value for D-serine is pH -dependent, decreasing from 1.018 at pH 7.5 to 0.994 at pH 10.4 [42]. This pH effect reflects the fact that a proton is lost from the substrate nitrogen in going from the free amino acid to the enzyme-bound imino acid [43]. The proton is not lost as the CH bond is broken, since there is no solvent isotope effect on the V/K value with D-serine [40]. While one could propose that the proton is lost in a pre-equilibrium after the amino acid binds, there is no active site base to accept the proton. Instead, the anionic form of the amino acid must be the active form of the substrate. The pKa of an amino group increases slightly upon replacement of the nitrogen with 15N [44], so that the fraction of the substrate with a neutral amino group is higher for the 15N-containing amino acid. The equilibrium isotope effect on deprotonation of the amino group is 1.023 [45–47]. At pH 7.5, where the amino acid is predominantly in the zwitterionic form, the measured 15N isotope effect for DAAO is a combination of this equilibrium isotope effect and the kinetic isotope effect on the CH bond cleavage step. At pH 10.4, the amino acid is predominantly in the anionic form so that the equilibrium between the zwitterion and the anion is not involved and the intrinsic isotope effect on the bond cleavage step is seen. This inverse isotope effect of 0.994 is due to the rehybridization of the amino group nitrogen from sp3 to sp2 as the substrate CH bond is broken. In the carbanion mechanism, rehybridization of the nitrogen does not occur in the same step as CH bond cleavage, but it would in a hydride transfer mechanism. Thus, the 15N kinetic isotope effects are not compatible with a carbanion intermediate, but do support hydride transfer. Subsequent computational studies have also supported direct transfer of a hydride from the anionic amino acid to the flavin as the lowest energy pathway [48].
This convergence of structural, kinetic, and computational approaches has established the mechanism shown in Scheme 7 for DAAO. The anionic form of the amino acid substrate, which constitutes ~2% of the total at neutral pH, is the productive form. Oxidation of this species occurs by direct transfer of a hydride from the amino acid Cα to the flavin without the direct involvement of amino acid side chains in catalysis.
Scheme 7.

Sarcosine Oxidase
A number of flavoproteins catalyze the oxidation of N -methylated amino acids. The reaction differs from that of DAAO in that the bond between the amino nitrogen and the methyl group is oxidized (Scheme 1, reaction II) rather than the bond between the amino nitrogen and the α-carbon. The structures of sarcosine oxidase and other family members show that the substrate binds in the opposite orientation to that in DAAO (Figure 3), providing a ready explanation for the altered substrate specificity. Mechanistically, the best characterized family member is sarcosine oxidase, in which the substrate is N -methylglycine [17], but N, N-dimethylglycine oxidase, N-methyltryptophan oxidase, an archaeal proline dehydrogenase, and NikD catalyze similar reactions and have similar structures [18, 19, 49, 50]. The FAD in these enzymes is covalently attached to the protein through the flavin C8 methyl group.
Figure 3.
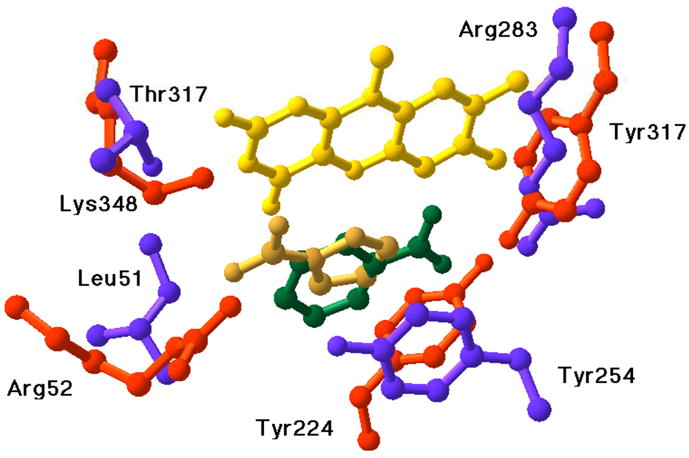
Overlay of the active sites of pig kidney D-amino acid oxidase (red residues) with benzoate (green) and monomeric sarcosine oxidase (purple residues) with pyrrole-2-carboxylate (gold). The flavins in the two proteins were superimposed to create the alignment.
Several different mechanisms have been proposed for the transfer of a hydride equivalent from the methyl group of an N -methyl amino acid by sarcosine oxidase and related enzymes [51, 52]. Scheme 8 summarizes the various proposals, although slight variations on each have also been suggested. Transfer of a single electron from the nitrogen to the flavin (mechanism I) would generate an aminium radical that would readily lose a proton to form a carbon-based radical; transfer of a second electron to the flavin completes the reaction. This mechanism requires that the enzyme greatly alter the redox potentials of the flavin and amine to overcome the unfavorable thermodynamics of the original electron transfer. Mechanism II is reminiscent of the original proposal of Hamilton (Scheme 3) in which nucleophilic attack of the amine on the flavin forms an adduct; the flavin then acts as an electron sink, allowing removal of a proton from the methyl group as the adduct collapses to form reduced flavin and oxidized substrate. Mechanism III shows a hydride transfer mechanism similar to that discussed above for DAAO. In addition, a carbanion mechanism in which a proton is removed from the methyl group to initiate the reaction has been proposed [53], but the high pKa of the methyl group and the evidence against a carbanion mechanism for DAAO has limited consideration of such a mechanism for sarcosine oxidase.
Scheme 8.

Studies by Jorns and coworkers [52, 54, 55] of the effect of pH on the reductive half-reaction of the monomeric sarcosine oxidase are consistent with the enzyme being able to bind the substrate whether the nitrogen is neutral or charged, but further reaction only occurring from the anionic substrate. Thus, the anionic substrate is the active form, as is the case with DAAO. While both the radical and the nucleophilic mechanisms of Scheme 8 involve intermediates, no intermediates have been detected in several studies of the reductive half reactions of sarcosine oxidase and related enzymes. Instead, formation of the oxidized substrate and reduced flavin occurs in a single step, even with deuterated substrates [16, 53, 54, 56]. In the case of the tetrameric sarcosine oxidase this involves quantum mechanical tunneling of the hydrogen [53], but this observation alone does not provide insight into the mechanism of cleavage of the substrate CH bond.
The monomeric sarcosine oxidase is slowly inactivated by N -cyclopropyl glycine [57]. In the process a modified flavin is formed that is unstable, but it can be reduced by sodium borohydride to a form sufficiently stable for structure determination [58]. The electron density and mass of this modified flavin is consistent with formation of a five member ring involving the flavin N5 and C4a. Since opening of cyclopropyl rings during a redox reaction is typically taken as evidence for a radical intermediate [59–61], the mechanism of Scheme 9 was proposed. The relevance of this reaction to the mechanism of oxidation of sarcosine during turnover was questioned at the time because the inactivation reaction was much slower than normal turnover [58]. In addition, both cholesterol oxidase and methanol oxidase are inactivated by cyclopropyl compounds [62, 63], but the evidence favors a hydride transfer mechanism for the glucose oxidase/cholesterol oxidase/methanol oxidase family of enzymes [64–66]. In the case of cholesterol oxidase, an adduct similar to that formed by sarcosine oxidase is also formed, although in that case it was proposed to involve the flavin C6 and N5 atoms [63].
Scheme 9.

Sarcosine is a slow substrate for N -methyltryptophan oxidase (MTOX). Since this suggested that chemical steps rather than product release would be rate-limiting with this substrate, kinetic isotope effects were measured to probe the mechanism of CH bond cleavage. With [2H3-methyl]-sarcosine as the substrate, the deuterium isotope effect on the steady state kinetic parameters and on the rate constant for flavin reduction (k2 in Scheme 2) is 7.2 [67], establishing that CH bond cleavage occurs in the rate-determining step and that isotope effects reflect the structure of the transition state for this step. This deuterium isotope effect is identical to that seen with the tetrameric sarcosine oxidase [53], suggesting that the structures of the transition states for CH bond cleavage are similar for the two enzymes. There is no solvent isotope effect on the kinetic parameters for MTOX; this is consistent with only the anionic form of the substrate binding productively, as with DAAO and sarcosine oxidase. The 15N kinetic isotope effect on the kcat/Ksarcosine value was determined by mass spectral analysis of both the residual sarcosine and the product glycine [47]. The effect is pH dependent, with a value of 1.017 at pH 7.5 that decreases with increasing pH. As is the case with DAAO, this is due to the isotope effect on the pKa of the substrate nitrogen; correction for this equilibrium isotope effect of 1.023 gives the pH -independent 15N kinetic isotope effect on the CH bond cleavage step, 0.995 if the values are determined from the residual sarcosine and 0.994 if from the product glycine.
The patterns of isotope effects for DAAO and sarcosine oxidase/MTOX are similar: a significant deuterium isotope effect, no solvent isotope effect, and an inverse 15N isotope effect at high pH. These isotope effects are qualitatively informative about the structure of the transition states for cleavage of the substrate CH bonds in the reactions catalyzed by these enzymes, establishing that there is no solvent -exchangeable proton in flight and that the nitrogen is changing bond order as the CH bond is broken. Ab initio methods have been used to calculate the deuterium and 15N isotope effects for the mechanisms proposed in Scheme 8 to ascertain if any could be eliminated by the experimental results [47]. To be able to carry out the calculations, it was necessary to use a simplified model, with dimethylamine as the substrate and alloxazine in place of the flavin. Calculation of deuterium isotope effects is difficult, due to the significant contribution of quantum mechanical tunneling to the isotope effect [68–70]. In contrast, heavy atom effects are much less effected by tunneling and can therefore be more readily calculated. For radical mechanisms such as mechanism I of Scheme 8, the calculated 15N effect is 0.994–0.996, with the variation depending on details of the model. All versions of the nucleophilic mechanism (II) are predicted to yield 15N isotope effects of ~1.02, irrespective of whether the reaction is stepwise or concerted. Finally, a hydride transfer mechanism (III) is predicted to give an 15N isotope effect of 0.992–0.994, again depending on details of the model. The calculated values for mechanism II are clearly different from the experimental values for MTOX, while the values for radical and hydride reactions cannot be used to rule out either of these mechanisms. However, the calculations also provide insight into the energetics of these reactions, with the caveat that the thermodynamics could be different in the enzyme active site. Radical mechanisms exhibit significantly greater barriers than do hydride transfer mechanisms, consistent with the >1 V difference in the one-electron redox potentials of an amine and a flavin [71]. The much more favorable thermodynamics of hydride transfer and the absence of evidence for a flavin semiquinone in the reductive half-reaction establishes hydride transfer as the most likely mechanism for members of the DAAO family.
The Monoamine Oxidase Family
Monoamine oxidase (MAO) A and B catalyze the oxidation of endogenous amine neurotransmitters such as serotonin and dopamine and a wide range of dietary amines and drugs. Consequently, these enzymes have been the subject of intense study for decades. MAO A and B have the same overall structure [7, 8], but MAO A has a more open active site, resulting in differences in substrate specificities for the two forms [72]. However, their catalytic mechanisms are assumed to be the same. The two mechanisms that have been most discussed are shown in Scheme 10. They are clearly similar to the first two mechanisms proposed for sarcosine oxidase (Scheme 8), with mechanism I involving a radical intermediate and mechanism II nucleophilic attack of the amine substrate on the flavin C4a position. Historically, a hydride transfer mechanism for MAO has been disregarded, although it is clearly a possibility.
Scheme 10.

Most of the support for a radical mechanism for MAO comes from studies with inhibitors containing cyclopropyl or cyclobutyl rings. McEwen et al. [73] initially reported that trans-2-phenylcyclopropylamine is a time-dependent inhibitor of MAO. Paech et al. [74] subsequently showed that the compound is also a substrate and that inactivation results in covalent attachment of the inhibitor to the protein rather than the covalently bound FAD. They proposed that inhibition was due to nucleophilic attack of a protein thiol on the cyclopropyl imine product or its hydrolysis product (Scheme 11, paths a and b). Silverman showed that a number of primary and secondary cyclopropyl amines will inactivate MAO [75]; different inhibitors labeled the protein or the flavin or both. These results were taken as support for a mechanism of inactivation involving one-electron transfers (Scheme 12) [60], although it was proposed at that time that a protein rather than a flavin radical reacted with the inhibitor. Silverman extended this mechanism to the normal catalytic mechanism, proposing that oxidation of amines by MAO involved radical intermediates as shown in Mechanism I of Scheme 10 [75]. However, trans-2-phenyl(aminomethyl)cyclopropane (1 in Scheme 13) does not inactivate MAO or produce ring-opened products [76], even though production of the carbon radical adjacent to the cyclopropyl ring should result in rapid ring opening. To test the possibility that radical collapse is faster than opening of the cyclopropyl ring, aminomethylcubane (2) was examined as a mechanism -based inhibitor, since the rate constant for opening of the ring in the radical is much greater than that for a cyclopropyl ring [77]. No direct evidence for a ring-opened product could be obtained in this case either [76]. These results suggested that a radical intermediate is not necessarily involved in inactivation of MAO by cyclopropyl compounds.
Scheme 11.

Scheme 12.

Scheme 13.

Detection of the flavin semiquinone during the MAO reaction would provide direct support for a radical mechanism. A transient flavin semiquinone is not seen in the visible absorbance spectrum of MAO A or B during reduction with deuterated benzylamines [78, 79]; if it forms at all it therefore does not accumulate significantly. Yelekci et al. [80] reported that a radical could be trapped if a nitrone spin trap were included during turnover with benzylamine. However, accumulation of the radical was very slow, occurring over hours, and the amount was not quantified. DeRose et al. [81] subsequently reported that a flavin radical could be detected in resting MAO B, but the EPR spectrum of the radical was unaffected by substrates. Stable flavin radicals have also been detected in other simple flavoprotein oxidases, such as methanol oxidase [82] and choline oxidase [83], where they have been shown not to be involved in catalysis, so that a stable flavin semiquinone does not demonstrate that catalysis involves a radical species. A variation on the radical mechanism of Scheme 10 has been proposed that involves an amino acid radical which transiently accepts an electron from the amine cation radical [60]. In this mechanism, inactivation by trans-2-phenylcylopropylamine would be due to radical recombination of this radical with the inhibitor radical formed upon opening of the cyclopropyl ring. Consistent with such a mechanism, Rigby et al. [84] reported that reduction of MAO A by one electron produces a species with an epr spectrum consistent with a tyrosyl radical. However, site-directed mutagenesis of MAO B [85] and the related enzyme polyamine oxidase [86] has shown that none of the conserved tyrosines is essential [86]. While Cys365 is modified upon inactivation of MAOB by trans-2-phenylcylopropylamine [87], this residue is located on the surface of the protein far from the active site [7]. It is unlikely that a carbon-centered radical would have a sufficiently long lifetime to diffuse this far before reacting, but modification of a residue far from the active site is fully consistent with the original mechanism proposed by Paech et al. [74] shown in Scheme 11.
The structure of MAO B inactivated with trans-2-phenylcylopropylamine has been determined. The electron density around the flavin is consistent with the structure in Scheme 14 [88], in contrast to the prediction of Scheme 12. This structure is similar to that of a proposed intermediate in the inactivation of sarcosine oxidase by cyclopropyl glycine (Scheme 9); the presence of the phenyl ring must block the ring closure seen with that enzyme. Given the similarity of the adduct formed upon inactivation of MAO by this cyclopropyl amine to those formed with flavoproteins for which data support hydride transfer, one must conclude that inactivation of a given flavoprotein by a cyclopropyl ring-containing substrate does not provide unambiguous support for a radical mechanism for oxidation of normal substrates by MAO. Thus, there is no direct structural or spectroscopic evidence for a radical intermediate in the MAO reaction.
Scheme 14.

If the MAO reaction does not involve radical intermediates, one must propose either that cyclopropyl amines are not oxidized by the normal catalytic pathway or that inactivation by these compounds does not involve a radical intermediate. Sherry and Abeles [62] proposed the non-radical mechanism in Scheme 15 for inactivation of methanol oxidase by cyclopropyl alcohol, and an analogous mechanism was proposed to explain the inactivation of cholesterol oxidase by a cyclopropyl compound [63]. In the normal reaction of both enzymes, an active site base removes the hydroxyl proton to form the alkoxide prior to hydride transfer to the flavin [89]; a similar reaction is clearly possible for an uncharged amine even in the absence of a base. An alternative possibility is that inactivation is due to a reaction between the reduced flavin and the oxidized substrate. The cyclopropyl imine formed upon oxidation of a cyclopropyl amine is similar to an allene in reactivity, and electrophiles can react with reduced flavins at both N5 and C4a [2]. Attack of C4a of the reduced flavin on the cyclopropyl imine formed upon oxidation of trans 2-phenylcyclopropylamine would generate the flavin adduct seen in the structure of MAO B (Scheme 16). A similar reaction in the case of sarcosine oxidase and cyclopropylglycine would be consistent with the structure of the modified flavin in that enzyme, since it would form the same nonradical intermediate shown in Scheme 9. As described below, an analogous attack of N5 of the reduced flavin in LSD1 on the imine formed upon oxidation of a propargyl amine substrate has been proposed to explain inactivation in that case [90].
Scheme 15.

Scheme 16.

Edmondson and colleagues have been the strongest proponent of a nucleophilic mechanism (Scheme 10, mechanism II) for amine oxidation by MAO and related enzymes [72]. As originally proposed by Hamilton (Scheme 3), a flavin adduct forms prior to abstraction of the substrate hydrogen as a proton. However, large isotope effects are seen on the kcat/Km value with deuterated benzylamine [78]; this requires that no irreversible steps precede CH bond cleavage, so that adduct formation must be reversible. Moreover, no intermediate is seen upon reduction of MAO with deuterated benzylamine, so that formation of the adduct must be sufficiently unfavorable that it does not accumulate. Finally, there is no obvious base in the active site of MAO to abstract a proton from the substrate [91]. These observations led to the version of the mechanism shown in Scheme 10, in which the flavin N5 is the base, with proton abstraction occurring as the substrate nitrogen adds to the flavin [72] rather than in separate steps as in Schemes 3 and 8. The pKa for the neutral N5 in reduced flavins has been estimated to be greater than 20 [92].
Direct evidence for formation of a substrate-flavin adduct is lacking for MAO, although an N5 adduct is seen in the structure of MAO B inactivated with N(2-aminoethyl)-p-chlorobenzamide [88]. A similar adduct is seen in the structure of maize polyamine oxidase after inactivation with N1-ethyl-N11-[(cycloheptyl)methyl]-4,8-diazaundecane [7]. In that case formation of the adduct was proposed to occur in a reaction between reduced flavin and the aldehyde produced from the oxidized substrate rather than during the normal catalytic reaction. Support for the nucleophilic mechanism has instead been indirect, relying primarily on the effects of the amine structure on rate constants. The rate constant for reduction of MAO A by a series of ring-substituted benzylamines correlates well with the value of the substituent, yielding a ρ value of 2.0; the deuterium kinetic isotope effects are large and probably the intrinsic values with these substrates [78], so that CH bond cleavage occurs in the rate-limiting step. The ρ value for a hydride transfer mechanism is typically between -1 and 1 [93], similar to the expectations for a radical mechanism [94]. As a result, the ρ value of 2.0 has been taken as evidence for an electron -rich transition state due to removal of the substrate hydrogen as a proton [72]. However, when the same substituted benzylamines are used as substrates for MAO B, large deuterium kinetic isotope effects are again seen on the rate constant for flavin reduction, but this parameter correlates best with the volume of the substituent [95]. A similar correlation has been found between the volume of the substituent and the rate constant for reduction of MAO A by substituted phenylethylamines [96]. The lack of a correlation between the rate constant for reduction of MAO B by substituted benzylamines with the electron donating ability of the substituent has been rationalized by the restricted active site of the enzyme preventing proper alignment of the orbitals of the aromatic ring with the CH bond being cleaved [72].
One complication in interpreting the results with MAO A is that the kinetics of reduction by substituted benzylamines were analyzed at pH 7.5. Both the rate constants for reduction of MAO A by amines and the kcat/Km values are maximal ~ pH 8.5 – 9 [97], consistent with a requirement for the neutral amine for catalysis. The pKa values for benzylamines decrease with the electron-donating abilities of substituents on the aromatic ring [98]. Thus, an increasing electron donation by the substituent will result in an increasing fraction of the benzylamine substrate being unprotonated, whether in solution or in the enzyme active site. This effect could increase the observed value significantly. Indeed, with mouse polyamine oxidase with ring-substituted N,N′-dibenzyl-1,4-diaminobutanes as substrates, the value increases by 0.5 upon going from pH 6.6 to pH 8.6 [99].
Nitrogen isotope effects have been measured for MAO B with benzylamine as substrate [100]. The effect is normal at neutral pH, where the protonated form of the substrate predominates, with a value of 1.0069. Correction for the 15N equilibrium isotope effect on the amine deprotonation yielded a value of 0.985, slightly more inverse than predicted for hydride transfer (0.992–0.994) and radical mechanisms (0.994–0.006) and significantly different from the prediction for a nucleophilic mechanism (1.02). Whether the lower value for the isotope effect for MAO versus DAAO reflects actual differences in the transition states for CH bond cleavage between the two structural families is not clear.
Thus, while radical and nucleophilic mechanisms for MAO have received the most attention to date, the data supporting them are not conclusive. The lack of detectable intermediates during the reductive half-reaction of MAO [79], the insensitivity of the rate constant for reduction of MAO B by benzylamines [95], and the 15N isotope effect are fully consistent with a hydride transfer mechanism for these enzymes. In addition, ab initio computational analyses of flavin-catalyzed amine oxidation found the transition states for radical and nucleophilic mechanisms to be much higher in energy than that for hydride transfer [47]. As discussed below studies of the mechanisms of other family members are also fully consistent with hydride transfer as the mechanism. In light of the absence of direct evidence for radical or nucleophilic mechanisms for MAO, a hydride transfer mechanism is a reasonable possibility for this enzyme also.
The L-amino acid oxidases are also in the MAO structural family; the amino acid substrates binds in a mirror image fashion to the substrate in DAAO, consistent with similar mechanisms but opposite stereochemistry [10]. Tryptophan monooxygenase is an L-amino acid oxidase that catalyzes the oxidative deamination of amino acids [101, 102]. With alanine as substrate, cleavage of the substrate CH bond is rate-limiting, based on the expression of the intrinsic deuterium isotope effect of 6.3 in the kinetic parameters [67]. No intermediates between oxidized and reduced enzyme are seen during reduction with deuterated amino acids [103], in line with the observations for all other flavoprotein amine oxidases. The pattern of isotope effects for this enzyme resembles that for DAAO: a lack of a solvent isotope effect and a pH -sensitive 15N kinetic isotope effect which is inverse (0.992) at high pH. In light of similar results for the two enzymes, a hydride transfer mechanism seems most reasonable for the L-amino acid oxidases. A similar conclusion has been recently drawn based on structural considerations [9]
The spermine and polyamine oxidases catalyze oxidation of polyamines such as spermine and spermidine, with the latter preferring the N-acetylated forms [104, 105]. Structures are available for the maize enzyme [11] and for the yeast Fms1 [12], clearly establishing the homology to MAO. Again, no intermediates have been detected between fully oxidized and fully reduced enzymes during reduction by substrates [86, 106]. In the case of mouse polyamine oxidase, the pH dependence of the reductive half-reaction establishes that the polyamine nitrogen in the bond being oxidized must be neutral for catalysis [107], in line with mechanistic expectations. The rate constants for reduction of the enzyme by a series of ring-substituted N,N′-dibenzyl-1,4-diaminobutanes show a correlation with both the σ+ value (ρ= −0.59) and the volume of the substituent at the pH optimum of 8.6 [99]. At pH 6.6, the ρ value is close to zero (0.09) while the contribution of the volume is affected little by pH. These results are consistent with a hydride transfer mechanism in which a neutral substrate nitrogen is required and demonstrate the need to consider the effects of pH on enzyme activity when analyzing substrate reactivity.
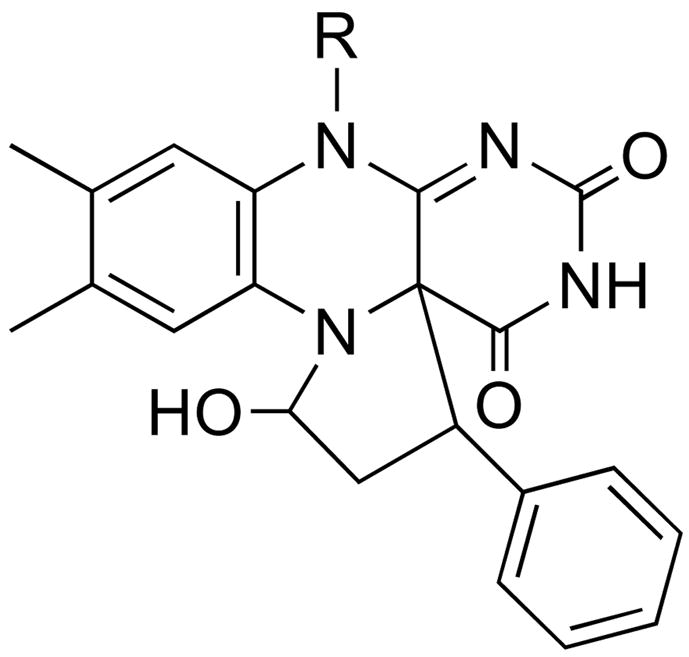
LSD1 is a recently discovered member of the flavoprotein amine oxidase family that catalyzes the oxidative deamination of mono-and dimethylated lysine residues in the N-terminus of histone H3 [108, 109]. Its structure clearly shows the homology to MAO (Figure 1) with the addition of a SWIRM domain [13, 14]. Mechanistic studies of this enzyme have utilized a peptide based on the N -terminal ~21 residues of histone H3 [110] due to the difficulty of preparing the homogeneously methylated protein. Attachment of a propargyl moiety to Lys4 of the peptide substrate yields a mechanism-based inhibitor, whereas replacement with a cyclopropyl moiety to yield the cyclopropyl amine does not [90]. (A number of other flavoprotein oxidases, including DAAO [111] and tryptophan monooxygenase [112], are also inactivated by compounds containing propargyl groups.) Mass spectral analyses of the inactivated LSD1 and the structure of the enzyme inactivated with an N -methyl, N -propargyl Lys4 peptide establish that inactivation involves formation of an N5 flavin adduct [90, 113]. The mechanism of Scheme 17 (R = H, CH 3), in which inactivation occurs after amine oxidation, was proposed to account for the results. The adduct formed in this case clearly resembles those formed upon inactivation of sarcosine oxidase and MAO by cyclopropyl compounds, suggesting similar mechanisms account for the inactivation of those enzymes also. Trans-2-phenylcyclopropylamine, an inhibitor of MAO, also inactivates LSD1, modifying the flavin [113, 114]. Direct structure determination of the inactivated enzyme established the structure in Scheme 18 [113]. The structure resembles that of the adduct formed upon inactivation of sarcosine oxidase by N -cyclopropyl glycine (Scheme 9), suggesting that a common mechanism is responsible.
Scheme 17.

Scheme 18.
The MAO family of amine oxidases clearly shows more variety in substrate structure than the DAAO family. Still, mechanistic studies of both families lead to similar conclusions. Carbanions are unlikely to be intermediates in the oxidation of amines by flavoproteins. While evidence for radical intermediates comes from studies with cyclopropyl substrates, no direct evidence for a radical intermediate in catalysis has been reported. Moreover, alternative mechanisms can explain inactivation by these compounds, and the energetics for formation of the proposed radicals are unfavorable. In the case of nucleophilic mechanisms, the evidence is indirect in that no adduct has been detected for any family and a ρ value greater than one has only been reported for MAO A. Moreover, the nitrogen isotope effects for MAO and tryptophan monooxygenase are significantly different from the value predicted for nucleophilic mechanisms. In contrast, the results for all members of the MAO family, including the lack of intermediates and the values of isotope effects, can be accommodated by a hydride transfer mechanism. Thus, the preponderance of evidence supports hydride transfer as the general mechanism for amine oxidation by members of both the MAO and the DAAO family.
Other Amine Oxidases
There are several other flavoproteins that can catalyze the oxidation of CN bonds but do not fall into the structural families discussed so far. Trimethylamine dehydrogenase is an iron-sulfur flavoprotein that catalyzes the oxidation of trimethylamine to dimethylamine and formaldehyde. The redox cofactors, including the 6-cysteinyl FMN, lie in a TIM-barrel N-terminal domain [115] that is placed by SCOP [116] in the same family as the flavin hydroxy acid oxidases and Old Yellow Enzyme. As with the enzymes described above, the active form of the substrate is the neutral amine. The mechanism of this enzyme has not been firmly established, but a nucleophilic mechanism such as that proposed for MAO has been proposed [117]. Glutamate synthase, which similarly contains the flavin cofactor and an iron -sulfur center in a TIM-barrel domain, catalyzes the reverse of the oxidation reaction discussed here, using reduced flavin to convert an iminoglutarate intermediate to glutamate; a hydride transfer mechanism has been proposed for this enzyme [118].
Flavoenzymes in the vanillyl alcohol oxidase family catalyze the oxidation of a variety of alcohol substrates. These enzymes contain covalently linked FAD, either through a 8α-N1-histidinyl linkage or through both 8α-N1-histidinyl and 6-S-cysteinyl linkages [119]. The parent member of the family, vanillyl alcohol oxidase, is most active with 4-hydroxy-benzyl alcohols as substrates, but will also catalyze the oxidation of 4-hydroxybenzylamine [120]. For alcohols as substrates, the proposed mechanism involves transfer of a hydride equivalent from the benzylic hydrogen of the phenolate to form a quinone methide [121]. A similar mechanism is likely for amines (Scheme 19) [120].
Scheme 19.

Nitroalkane oxidase [122] cleaves the CN bond in nitroalkanes to form nitrite and the respective aldehyde or ketone [123]. The enzyme has a similar fold to acyl-CoA dehydrogenases, including the position of the active site base, although the substrate binding sites are located on different sides of the two enzymes [124]. The mechanisms of both involve removal of the substrate α-proton by an active site base [125, 126]. However, in NAO this is followed by nucleophilic attack of the substrate carbanion on the flavin N5 (Scheme 20). The kinetic competency of the flavin adduct 3 has been established by trapping it with cyanide during turnover [127], and the structure of the trapped adduct has been determined [128]. Nitroalkane oxidase is unusual in that cleavage of the substrate CH bond by proton abstraction and the involvement of a flavin adduct as an intermediate have both been clearly established with this enzyme.
Scheme 20.

Flavoprotein Alcohol Oxidases
Separate families of flavoproteins catalyze the oxidation of alcohols and of α-hydroxy acids. Choline oxidase, cholesterol oxidase, glucose oxidase, and methanol oxidase are all well-studied enzymes with similar structures [129–131] that oxidize their substrates to the respective alcohols. The consensus for this family of enzymes is that oxidation of the alcohol occurs by hydride transfer from the alkoxide form of the substrate [65, 132]. The flavoproteins that oxidize α-hydroxy acids include glycolate oxidase, lactate monooxygenase, flavocytochrome b2, and mandelate dehydrogenase and make up a separate structural family [133–136]. Although their mechanism of oxidation has been controversial [137, 138], structural, mechanistic, and computational studies support a mechanism similar to that of the alcohol oxidases, hydride transfer from the alkoxide form of the substrate [139–141].
Conclusions
The mechanism of oxidation of amines by flavoproteins has been controversial for decades, and multiple mechanisms still have support. Still, the combination of structural, mechanistic, and computational studies is most consistent with a hydride transfer mechanism for these enzymes. This would make hydride transfer the general mechanism for oxidation of both amines and alcohols by flavoproteins.
Acknowledgments
The work from my lab described here would not have been possible without the contributions of my collaborators and former students and postdoctoral fellows. Generous support from the NIH (GM47291) and The Welch Foundation (AQ-1245) is also gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Palmer G, Massey V. In: Biological oxidation. Singer TP, editor. John Wiley and Sons; New York: 1968. pp. 263–300. [Google Scholar]
- 2.Müller F. In: Chemistry and Biochemistry of Flavoenzymes. Muller F, editor. I. CRC Press; Boca Raton: 1991. pp. 1–71. [Google Scholar]
- 3.Massey V. J Biol Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 4.Massey V, Müller F, Feldberg R, Schuman M, Sullivan PA, Howell LG, Mayhew SG, Matthews RG, Foust GP. J Biol Chem. 1969;244:3999–4006. [PubMed] [Google Scholar]
- 5.Kosuge T. Methods Enzymol. 1970;17A:446–449. [Google Scholar]
- 6.Koyama H. J Biochem. 1982;92:1235–1240. doi: 10.1093/oxfordjournals.jbchem.a134041. [DOI] [PubMed] [Google Scholar]
- 7.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Nat Struct Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 8.De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Proc Nat Acad Sci USA. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faust A, Niefind K, Hummel W, Schomburg D. J Mol Biol. 2007;367:234–248. doi: 10.1016/j.jmb.2006.11.071. [DOI] [PubMed] [Google Scholar]
- 10.Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A. EMBO J. 2000;19:4204–4215. doi: 10.1093/emboj/19.16.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A. Structure. 1999;7:265–276. doi: 10.1016/s0969-2126(99)80037-9. [DOI] [PubMed] [Google Scholar]
- 12.Huang Q, Liu Q, Hao Q. J Mol Biol. 2005;348:951–959. doi: 10.1016/j.jmb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Stavropoulos P, Blobel G, Hoelz A. Nat Struct Mol Biol. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Proc Natl Acad Sci USA. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Proc Natl Acad Sci USA. 1996;93:7496–7501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Settembre EC, Dorrestein PC, Park J, Augustine AM, Begley TP, Ealick SE. Biochemistry. 2003;42:2971–2981. doi: 10.1021/bi026916v. [DOI] [PubMed] [Google Scholar]
- 17.Trickey P, Wagner MA, Jorns MS, Mathews FS. Structure. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 18.Leys D, Basran J, Scrutton NS. EMBO J. 2003;22:4038–4048. doi: 10.1093/emboj/cdg395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.F.B. Proteins: Structure, A., Bonamore A, Franceschini S, Fiorillo A, Boffi A, Colotti G. Proteins: Struct Funct Bioinform. 2008;71:2065–2075. doi: 10.1002/prot.21898.
- 20.Khanna P, Schuman Jorns M. Biochemistry. 2001;40:1441–1450. doi: 10.1021/bi0024411. [DOI] [PubMed] [Google Scholar]
- 21.Hegyi H, Gerstein M. J Mol Biol. 1999;288:147–164. doi: 10.1006/jmbi.1999.2661. [DOI] [PubMed] [Google Scholar]
- 22.Sterner R, Hocker B. Chem Rev. 2005;105:4038–4055. doi: 10.1021/cr030191z. [DOI] [PubMed] [Google Scholar]
- 23.Warburg O, Christian W. Biochem Zeit. 1938;298:150–168. [Google Scholar]
- 24.Job V, Marcone GL, Pilone MS, Pollegioni L. J Biol Chem. 2002;277:6985–6993. doi: 10.1074/jbc.M111095200. [DOI] [PubMed] [Google Scholar]
- 25.Porter DJT, Bright HJ. In: Flavins and Flavoproteins. Singer TP, editor. Elsevier Scientific Publishing Company; Amsterdam: 1976. pp. 225–237. [Google Scholar]
- 26.Neims AH, De Luca DC, Hellerman L. Biochemistry. 1966;5:203–213. doi: 10.1021/bi00865a027. [DOI] [PubMed] [Google Scholar]
- 27.Brown LE, Hamilton GA. J Am Chem Soc. 1970;92:7225–7227. doi: 10.1021/ja00727a049. [DOI] [PubMed] [Google Scholar]
- 28.Porter DJT, Voet JG, Bright HJ. J Biol Chem. 1977;252:4464–4473. [PubMed] [Google Scholar]
- 29.Fitzpatrick PF, Massey V. J Biol Chem. 1982;257:12916–12923. [PubMed] [Google Scholar]
- 30.Pollegioni L, Blodig W, Ghisla S. J Biol Chem. 1997;272:4924–4934. doi: 10.1074/jbc.272.8.4924. [DOI] [PubMed] [Google Scholar]
- 31.Walsh CT, Schonbrunn A, Abeles RH. J Biol Chem. 1971;246:6855–6866. [PubMed] [Google Scholar]
- 32.Massey V, Ghisla S, Ballou DP, Walsh CT, Cheung YT, Abeles RH. Flavins and flavoproteins. 1976:199–212. [Google Scholar]
- 33.Porter DJT, Bright HJ. Biochem Biophys Res Commun. 1972;46:571–577. doi: 10.1016/s0006-291x(72)80177-3. [DOI] [PubMed] [Google Scholar]
- 34.Hersh LB, Jorns MS. J Biol Chem. 1975;250:8728–8734. [PubMed] [Google Scholar]
- 35.Edmondson D, Ghisla S. In: Methods Mol Biol. Chapman SK, Reid GA, editors. Humana Press Inc; Totowa: 1999. pp. 157–179. [DOI] [PubMed] [Google Scholar]
- 36.Lederer F, Mathews FS. In: Flavins and Flavoproteins, 1987. Edmondson DE, McCormick DB, editors. Walter de Gruyter & Co; Berlin: 1987. pp. 133–142. [Google Scholar]
- 37.Walsh C, Lockridge O, Massey V, Abeles R. J Biol Chem. 1973;248:7049–7054. [PubMed] [Google Scholar]
- 38.Mizutani H, Miyahara I, Hirotsu K, Nishina Y, Shiga K, Setoyama C, Miura R. J Biochem. 1996;120:14–17. doi: 10.1093/oxfordjournals.jbchem.a021376. [DOI] [PubMed] [Google Scholar]
- 39.Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone MS, Ghisla S. Proc Natl Acad Sci USA. 2000;97:12463–12468. doi: 10.1073/pnas.97.23.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denu JM, Fitzpatrick PF. Biochemistry. 1994;33:4001–4007. doi: 10.1021/bi00179a029. [DOI] [PubMed] [Google Scholar]
- 41.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. J Am Chem Soc. 2000;122:12896–12897. [Google Scholar]
- 42.Kurtz K. Studies on the mechanism of D-amino acid oxidase. Texas A&M University; College Station, TX: 2000. [Google Scholar]
- 43.Fitzpatrick PF, Massey V. J Biol Chem. 1982;257:9958–9962. [PubMed] [Google Scholar]
- 44.Rishavy MA, Cleland WW. Can J Chem. 1999;77:967–977. [Google Scholar]
- 45.Rabenstein DL, Mariappan SVS. J Org Chem. 1993;58:4487–4489. [Google Scholar]
- 46.Pehk T, Kiirend E, Lippmaa E, Ragnarsson U, Grehn L. J Chem Soc Perkin Trans. 1997;2:445–450. [Google Scholar]
- 47.Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tilocca A, Gamba A, Vanoni MA, Fois E. Biochemistry. 2002;41:14111–14121. doi: 10.1021/bi020309q. [DOI] [PubMed] [Google Scholar]
- 49.Carrell CJ, Bruckner RC, Venci D, Zhao G, Jorns MS, Mathews FS. Structure. 2007;15:928–941. doi: 10.1016/j.str.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuge H, Kawakami R, Sakuraba H, Ago H, Miyano M, Aki K, Katunuma N, Ohshima T. J Biol Chem. 2005;280:31045–31049. doi: 10.1074/jbc.C500234200. [DOI] [PubMed] [Google Scholar]
- 51.Scrutton NS. Nat Prod Rep. 2004;21:722–730. doi: 10.1039/b306788m. [DOI] [PubMed] [Google Scholar]
- 52.Zhao G, Jorns MS. Biochemistry. 2006;45:5985–5992. doi: 10.1021/bi0600852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harris RJ, Meskys R, Sutcliffe MJ, Scrutton NS. Biochemistry. 2000;39:1189–1198. doi: 10.1021/bi991941v. [DOI] [PubMed] [Google Scholar]
- 54.Zhao G, Jorns MS. Biochemistry. 2002;41:9747–9750. doi: 10.1021/bi020285n. [DOI] [PubMed] [Google Scholar]
- 55.Zhao G, Jorns MS. Biochemistry. 2005;44:16866–16874. doi: 10.1021/bi051898d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ralph EC, Fitzpatrick PF. Biochemistry. 2005;44:3074–3081. doi: 10.1021/bi047716h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao G, Qu J, Davis FA, Jorns MS. Biochemistry. 2000;39:14341–14347. doi: 10.1021/bi001421w. [DOI] [PubMed] [Google Scholar]
- 58.Chen ZW, Zhao G, Martinovic S, Jorns MS, Mathews FS. Biochemistry. 2005;44:15444–15450. doi: 10.1021/bi0515422. [DOI] [PubMed] [Google Scholar]
- 59.Fitzpatrick PF, Villafranca JJ. J Am Chem Soc. 1985;107:5022–5023. [Google Scholar]
- 60.Silverman RB. J Biol Chem. 1983;258:14766–14769. [PubMed] [Google Scholar]
- 61.Hanzlik RP, Kishore V, Tullman R. J Med Chem. 1979;22:759–760. doi: 10.1021/jm00193a002. [DOI] [PubMed] [Google Scholar]
- 62.Sherry B, Abeles RH. Biochemistry. 1985;24:2594–2605. doi: 10.1021/bi00332a002. [DOI] [PubMed] [Google Scholar]
- 63.McCann AE, Sampson NS. J Am Chem Soc. 2000;122:35–39. [Google Scholar]
- 64.Menon V, Hsieh CT, Fitzpatrick PF. Bioorganic Chem. 1995;23:42–53. [Google Scholar]
- 65.Brinkley DW, Roth JP. J Am Chem Soc. 2005;127:15720–15721. doi: 10.1021/ja056025l. [DOI] [PubMed] [Google Scholar]
- 66.Fan F, Gadda G. J Am Chem Soc. 2005:2067–2074. doi: 10.1021/ja044541q. [DOI] [PubMed] [Google Scholar]
- 67.Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qian W, Hammes-Schiffer S. J Chem Phys. 2006;125:184101–184111. [Google Scholar]
- 69.Fernandez-Ramos A, Truhlar DG. J Chem Theory Comp. 2005;1:1063–1078. doi: 10.1021/ct050153i. [DOI] [PubMed] [Google Scholar]
- 70.Mavri J, Liu H, Olsson MHM, Warshel A. J Phys Chem B. 2008;112:5950–5954. doi: 10.1021/jp0758420. [DOI] [PubMed] [Google Scholar]
- 71.Fu Y, Liu L, Yu HZ, Wang YM, Guo QX. J Amer Chem Soc. 2005;127:7227–7234. doi: 10.1021/ja0421856. [DOI] [PubMed] [Google Scholar]
- 72.Edmondson DE, Binda C, Mattevi A. Arch Biochem Biophys. 2007;464:269–276. doi: 10.1016/j.abb.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McEwen CM, Jr, Sasaki G, Jones DC. Biochemistry. 1969;8:3952–3962. doi: 10.1021/bi00838a011. [DOI] [PubMed] [Google Scholar]
- 74.Paech C, Salach JI, Singer TP. J Biol Chem. 1980;255:2700–2704. [PubMed] [Google Scholar]
- 75.Silverman RB. Acc Chem Res. 1995;28:335–342. [Google Scholar]
- 76.Silverman RB, Zhou JP, Eaton PE. J Am Chem Soc. 1993;115:8841–8842. [Google Scholar]
- 77.Choi SY, Eaton PE, Newcomb M, Yip YC. J Am Chem Soc. 1992;114:6326–6329. [Google Scholar]
- 78.Miller JR, Edmondson DE. Biochemistry. 1999;38:13670–13683. doi: 10.1021/bi990920y. [DOI] [PubMed] [Google Scholar]
- 79.Miller JR, Edmondson DE, Grissom CB. J Am Chem Soc. 1995;117:7830–7831. [Google Scholar]
- 80.Yelekci K, Lu X, Silverman RB. J Am Chem Soc. 1989;111:1138–1140. [Google Scholar]
- 81.DeRose VJ, Woo JCG, Hawe WP, Hoffman BM, Silverman RB, Yelekci K. Biochemistry. 1996;35:11085–11091. doi: 10.1021/bi960749f. [DOI] [PubMed] [Google Scholar]
- 82.Mincey T, Tayrien G, Mildvan AS, Abeles RH. Proc Natl Acad Sci USA. 1980;77:7099–7101. doi: 10.1073/pnas.77.12.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghanem M, Fan F, Francis K, Gadda G. Biochemistry. 2003;42:15179–15188. doi: 10.1021/bi035435o. [DOI] [PubMed] [Google Scholar]
- 84.Rigby SE, Hynson RM, Ramsay RR, Munro AW, Scrutton NS. J Biol Chem. 2005;280:4627–4631. doi: 10.1074/jbc.M410596200. [DOI] [PubMed] [Google Scholar]
- 85.Geha RM, Chen K, Wouters J, Ooms F, Shih JC. J Biol Chem. 2002;277:17209–17216. doi: 10.1074/jbc.M110920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Royo M, Fitzpatrick PF. Biochemistry. 2005;44:7079–7084. doi: 10.1021/bi050347k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhong B, Silverman RB. J Am Chem Soc. 1997;119:6690–6691. [Google Scholar]
- 88.Binda C, Li M, Hubalek F, Restelli N, Edmondson D, Mattevi A. Proc Natl Acad Sci USA. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sampson NS, Vrielink A. Acc Chem Res. 2003;36:713–722. doi: 10.1021/ar9800587. [DOI] [PubMed] [Google Scholar]
- 90.Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Biochemistry. 2007;46:6892–6902. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Binda C, Mattevi A, Edmondson DE. J Biol Chem. 2002;277:23973–23976. doi: 10.1074/jbc.R200005200. [DOI] [PubMed] [Google Scholar]
- 92.Macheroux P, Ghisla S, Sanner C, Ruterjans H, Muller F. BMC Biochem. 2005;6:26. doi: 10.1186/1471-2091-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kudo T, Higashide T, Ikedate S, Yamatake H. J Org Chem. 2005;70:5157–5163. doi: 10.1021/jo050435a. [DOI] [PubMed] [Google Scholar]
- 94.Pryor WA, Lin TH, Stanley JP, Henderson RW. J Am Chem Soc. 1973;95:6993–6998. [Google Scholar]
- 95.Walker MC, Edmondson DE. Biochemistry. 1994;33:7088–7098. doi: 10.1021/bi00189a011. [DOI] [PubMed] [Google Scholar]
- 96.Nandigama RK, Edmondson DE. Biochemistry. 2000;39:15258–15265. doi: 10.1021/bi001957h. [DOI] [PubMed] [Google Scholar]
- 97.Dunn RV, Marshall KR, Munro AW, Scrutton NS. FEBS Journal. 2008;275:3850–3858. doi: 10.1111/j.1742-4658.2008.06532.x. [DOI] [PubMed] [Google Scholar]
- 98.Blackwell LFFA, Miller IJ, Topsom RD, Vaughan J. J Chem Soc. 1964:3588–3591. [Google Scholar]
- 99.M. Henderson Pozzi, V. Gawandi, P.F. Fitzpatrick, manuscript in preparation.
- 100.S. MacMillar, D.E. Edmondson, O. Mattson, manuscript in preparation.
- 101.Emanuele JJ, Jr, Heasley CJ, Fitzpatrick PF. Arch Biochem Biophys. 1995;316:241–248. doi: 10.1006/abbi.1995.1034. [DOI] [PubMed] [Google Scholar]
- 102.Sobrado P, Fitzpatrick P. Biochemistry. 2003;42:13826–13832. doi: 10.1021/bi035299n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Emanuele JJ, Jr, Fitzpatrick PF. Biochemistry. 1995;34:3716–3723. doi: 10.1021/bi00011a029. [DOI] [PubMed] [Google Scholar]
- 104.Vujcic S, Diegelman P, Bacchi CJ, Kramer DL, Porter CW. Biochem J. 2002;367:665–675. doi: 10.1042/BJ20020720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu T, Yankovskaya V, McIntire WS. J Biol Chem. 2003;278:20514–20525. doi: 10.1074/jbc.M302149200. [DOI] [PubMed] [Google Scholar]
- 106.Bellelli A, Angelini R, Laurenzi M, Federico R. Arch Biochem Biophys. 1997;343:146–148. doi: 10.1006/abbi.1997.0129. [DOI] [PubMed] [Google Scholar]
- 107.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. Biochemistry. 2009;48:1508–1516. doi: 10.1021/bi802227m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. FEBS Lett. 2005;579:2203–2207. doi: 10.1016/j.febslet.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 109.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 110.Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. J Biol Chem. 2005;280:41360–41365. doi: 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- 111.Marcotte P, Walsh C. Biochemistry. 1976;15:3070–3076. doi: 10.1021/bi00659a021. [DOI] [PubMed] [Google Scholar]
- 112.Gadda G, Dangott LJ, Johnson WH, Jr, Whitman CP, Fitzpatrick PF. Biochemistry. 1999;38:5822–5828. doi: 10.1021/bi982777z. [DOI] [PubMed] [Google Scholar]
- 113.Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA, Yu Biochemistry. 2007;46:8058–8065. doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- 114.Schmidt DMZ, McCafferty DG. Biochemistry. 2007;46:4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 115.Barber M, Neame P, Lim L, White S, Matthews F. J Biol Chem. 1992;267:6611–6619. [PubMed] [Google Scholar]
- 116.Murzin AG, Brenner SE, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 117.Rohlfs RJ, Hille R. J Biol Chem. 1994;269:30869–30879. [PubMed] [Google Scholar]
- 118.Vanoni MA, Curti B. Arch Biochem Biophys. 2005;433:193–211. doi: 10.1016/j.abb.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 119.Leferink NGH, Heuts DPHM, Fraaije MW, van Berkel WJH. Arch Biochem Biophys. 2008;474:292–301. doi: 10.1016/j.abb.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 120.Fraaije MW, Veeger C, Van Berkel WJH. Eur J Biochem. 1995;234:271–277. doi: 10.1111/j.1432-1033.1995.271_c.x. [DOI] [PubMed] [Google Scholar]
- 121.Fraaije MW, Van Berkel WJH. J Biol Chem. 1997;272:18111–18116. doi: 10.1074/jbc.272.29.18111. [DOI] [PubMed] [Google Scholar]
- 122.Fitzpatrick PF, Orville AM, Nagpal A, Valley MP. Arch Biochem Biophys. 2005;433:157–165. doi: 10.1016/j.abb.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 123.Gadda G, Fitzpatrick PF. Arch Biochem Biophys. 1999;363:309–313. doi: 10.1006/abbi.1998.1081. [DOI] [PubMed] [Google Scholar]
- 124.Nagpal A, Valley MP, Fitzpatrick PF, Orville AM. Biochemistry. 2006;45:1138–1150. doi: 10.1021/bi051966w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Valley MP, Fitzpatrick PF. Biochemistry. 2003;42:5850–8566. doi: 10.1021/bi034061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ghisla S, Thorpe C. Eur J Biochem. 2004;271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 127.Valley MP, Tichy SE, Fitzpatrick PF. J Am Chem Soc. 2005;127:2062–2066. doi: 10.1021/ja043542f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Héroux A, Bozinovski DM, Valley MP, Fitzpatrick PF, Orville AM. Biochemistry. 2009;48:3407–3416. doi: 10.1021/bi8023042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vrielink A, Lloyd LF, Blow DM. J Mol Biol. 1991;219:533–554. doi: 10.1016/0022-2836(91)90192-9. [DOI] [PubMed] [Google Scholar]
- 130.Hecht HJ, Kalisz HM, Hendle J, Schmid RD, Schomburg D. J Mol Biol. 1993;229:153–172. doi: 10.1006/jmbi.1993.1015. [DOI] [PubMed] [Google Scholar]
- 131.Kiess M, Hecht HJ, Kalisz HM. Eur J Biochem. 1998;252:90–99. doi: 10.1046/j.1432-1327.1998.2520090.x. [DOI] [PubMed] [Google Scholar]
- 132.Gadda G. Biochemistry. 2008;47:13745–13753. doi: 10.1021/bi801994c. [DOI] [PubMed] [Google Scholar]
- 133.Lindqvist Y, Branden CI. Proc Natl Acad Sci USA. 1985;82:6855–6859. doi: 10.1073/pnas.82.20.6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mathews FS, Xia Z-X. In: Flavins and Flavoproteins 1987. Edmondson DE, McCormick DB, editors. Walter de Gruyter; Berlin: 1987. pp. 123–132. [Google Scholar]
- 135.Sukumar N, Xu Y, Gatti DL, Mitra B, Mathews FS. Biochemistry. 2001;40:9870–9878. doi: 10.1021/bi010938k. [DOI] [PubMed] [Google Scholar]
- 136.Cunane LM, Barton JD, Chen ZW, Le KH, Amar D, Lederer F, Mathews FS. Biochemistry. 2005;44:1521–1531. doi: 10.1021/bi048616e. [DOI] [PubMed] [Google Scholar]
- 137.Lederer F. In: Flavins and Flavoproteins 1996. Stevenson KJ, Massey V, Williams CH, editors. University of Calgary Press; 1996. pp. 545–553. [Google Scholar]
- 138.Fitzpatrick PF. Bioorg Chem. 2004;32:125–139. doi: 10.1016/j.bioorg.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 139.Mowat CG, Wehenkel A, Green AJ, Walkinshaw MD, Reid GA, Chapman SK. Biochemistry. 2004;43:9519–9526. doi: 10.1021/bi049263m. [DOI] [PubMed] [Google Scholar]
- 140.Sobrado P, Fitzpatrick PF. Biochemistry. 2003;42:15208–15214. doi: 10.1021/bi035546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tabacchi G, Zucchini D, Caprini G, Gamba A, Lederer F, Vanoni MA, Fois E. FEBS Journal. 2009;276:2368–2380. doi: 10.1111/j.1742-4658.2009.06969.x. [DOI] [PubMed] [Google Scholar]